

Abstract
The actions of the neuropeptide nociceptin on the calcium channel currents (IBa) of acutely dissociated rat periaqueductal grey (PAG) neurons were examined using whole-cell patch clamp techniques. These effects were compared with those of opioid receptor agonists and the GABAB receptor agonist baclofen.
Neurons from young adult rats (23 to 56 days old) expressed predominantly ω-conotoxin GVIA (N-type)- and ω-agatoxin IVA (P/Q-type)-sensitive IBa, together with smaller amounts of nimodipine-sensitive current and current resistant to all three blockers. There was proportionately more N-type IBa in neurons from female rats and proportionately more resistant current in neurons from male rats.
Nociceptin (EC50, 5 nm) and baclofen (EC50, 0.8 μm) inhibited IBa in all PAG neurons, while the opioid agonist methionine enkephalin (met-enkephalin; 300 nm-10 μm) inhibited IBa in 40 % of neurons. The effects of met-enkephalin were reversed by the μ-opioid antagonist CTAP, and mimicked by the μ-opioid agonist DAMGO (300 nm-3 μm). The δ-opioid agonists DPDPE and deltorphin II, and the κ-opioid agonist U69593, did not affect IBa in any neuron. The actions of nociceptin were not mimicked or blocked by the opioid antagonist naloxone or the nociceptin analogue [desPhe1]-nociceptin.
The effects of nociceptin and baclofen on IBa were blocked by pretreatment of the neurons with pertussis toxin (500 ng ml−1, 8 h).
Nociceptin predominantly inhibited the N-type (EC50, 2 nm; maximum inhibition, 50 %) and P/Q-type (EC50, 7 nm; maximum inhibition, 33 %) IBa while having little effect on the L-type and R-type IBa.
These results are consistent with the previously described actions of nociceptin, baclofen and μ-opioids in PAG slices, whereby they couple to increases in an inwardly rectifying K+ conductance. These agonists thus have the potential to modulate the function of PAG neurons via a number of different cellular effectors.
The midbrain periaqueductal grey (PAG) plays a pivotal role in the integration of an animal's response to threat, stress and pain (reviewed by Bandler & Shipley, 1994). A number of lines of evidence suggest that the PAG is an important central site of action of opioid drugs. Microinjection of opioid agonists such as morphine into subregions of the PAG produces analgesia in animals (summarized by Yaksh & Rudy, 1978), and electrical stimulation of the PAG produces an analgesic response sensitive to opioid receptor antagonists (Akil, Mayer & Liebeskind, 1976). There is also considerable evidence that the PAG is critically involved in the responses of an animal to withdrawal of opioid drugs (reviewed in Christie, Williams, Osborne & Bellchambers, 1997).
Immunohistochemical studies have shown that the rat PAG contains μ-, δ- and κ-opioid receptors (Kalyuzhny, Arvidsson, Wu & Wessendorf, 1996; Mansour, Burke, Pavlic, Akil & Watson, 1996), as well as a recently identified member of the opioid receptor family, the opioid receptor-like receptor (ORL1) (Anton, Fein, To, Silberstein & Evans, 1996; reviewed in Henderson & McKnight, 1997). Both μ- and δ-opioid receptor activation inhibits adenylyl cyclase activity in PAG membrane preparations (Noble & Cox, 1996); however, only μ-opioid receptor activation has been shown to affect the membrane properties of PAG neurons or synaptic transmission within the PAG. μ-Opioid agonists increase an inwardly rectifying potassium conductance in a subpopulation of PAG neurons in vitro (Chieng & Christie 1994) and μ-opioids also inhibit both GABAergic and glutamatergic synaptic inputs to all PAG neurons (Vaughan & Christie 1997). While the present study was in progress it was also reported that μ-opioids inhibit voltage-dependent calcium channel activity in some PAG neurons acutely dissociated from neonate rats (Kim, Rhee & Akaike, 1997).
In contrast to μ-receptor activation, the putative endogenous agonist for ORL1, nociceptin/orphanin FQ (Meunier et al. 1995; Reinscheid et al. 1995) was recently shown to increase an inwardly rectifying potassium conductance in all PAG neurons, but to modulate GABAergic and glutamatergic synaptic inputs to only some PAG neurons (Vaughan, Ingram & Christie, 1997). Furthermore, nociceptin microinjection into the PAG attenuated the analgesic effects of an earlier microinjection of morphine (Morgan, Grisel, Robbins & Grandy, 1997). Because of the important role that the PAG plays in both opioid analgesia and withdrawal, we have sought to extend our understanding of the actions of opioids and nociceptin by examining the effects of these agonists on the voltage-dependent currents of acutely dissociated PAG neurons from young adult rats. In this study we have examined the regulation of voltage-dependent calcium channel currents in PAG neurons, and compared the actions of nociceptin and μ-opioids with that of the GABAB receptor agonist baclofen, which also directly hyperpolarizes all PAG neurons in vitro (Chieng & Christie, 1995). Portions of this work have been presented in abstract form to the Society for Neuroscience (Connor & Christie, 1997).
METHODS
Tissue dissociation
Sprague-Dawley rats of either sex (postnatal days 23-56) were anaesthetized with halothane and then killed by cervical dislocation. Midbrain slices (between 290 and 320 μm thick) containing the periaqueductal grey were cut with a vibratome in ice-cold physiological saline of composition (mm): NaCl, 126; KCl, 2.5; MgCl2, 1.2; CaCl2, 2.4; NaH2PO4, 1.2; NaHCO3, 24; and glucose, 11; gassed with 95 % O2-5 % CO2 and stored for 30 min at 35°C. Slices were cut in either the horizontal or the coronal plane. The dissociation procedures were based on those outlined in Ingram, Wilding, McCleskey & Williams (1997). The slices were transferred to a dissociation buffer of composition (mm): Na2SO4, 82; K2SO4, 30; Hepes, 10; MgCl2, 5; glucose, 10; containing 20 units ml−1 papain, pH 7.3 and incubated for 2-3 min at 35°C. The slices were then placed in fresh dissociation buffer containing 1 mg ml−1 bovine serum albumin (BSA) and 1 mg ml−1 trypsin inhibitor. The periaqueductal grey region was subdissected from each slice with a fine tungsten wire and the cells dissociated from the slices by gentle trituration through a series of silanized Pasteur pipettes with fire-polished tips of decreasing size. The cells were plated onto plastic culture dishes and kept at room temperature in dissociation buffer. Cells remained viable for at least 10 h after dissociation.
Recordings of currents through Ca2+ channels were made using standard whole-cell patch clamp techniques (Hamill, Marty, Neher, Sakmann & Sigworth, 1981) at room temperature (22-24°C). Cells were perfused in solution containing (mm): TEACl, 140; BaCl2, 4; CsCl, 2.5; Hepes, 10; glucose, 10; BSA, 0.05 %; pH 7.3. Recordings were made with fire-polished borosilicate pipettes of between 2 and 4 MΩ resistance when filled with intracellular solution of the following composition (mm): CsCl, 110; MgATP, 5; Na2GTP, 0.2; EGTA, 10; CaCl2, 2; and Hepes, 10; pH 7.3. The peak calcium channel current in each cell was determined by stepping the membrane potential from a holding potential of -90 mV to potentials between -60 and +60 mV, usually for 30 ms, in 10 mV increments. Following this procedure the peak current was evoked every 30 s, and monitored for at least a further 2 min before drugs or toxins were applied. The inhibition by drugs or toxins was quantified by measuring the current amplitude isochronically with the peak of the control calcium channel current. In a few cells, where quantification of current components was not attempted, the cells were stepped to 0 mV and nociceptin applied after the current had stabilized for at least 2 min. Cells in which the calcium channel current declined in the absence of drug treatment were discarded and their current-voltage relationships were not used in subsequent analysis of the population characteristics of the PAG cells. Whole-cell capacitance and series resistance were compensated manually by nulling the capacitive transient evoked by a 20 mV pulse from -90 mV. The series resistance was between 3 and 10 MΩ, with an average value of 5 MΩ; series resistance compensation of at least 80 % was used in all experiments. An approximate value of whole-cell capacitance was read from the amplifier capacitance compensation circuit (Axopatch 1D, Axon Instruments). Leak current was subtracted on-line using a P/8 protocol; typically the leak conductance was less than 1 nS. Evoked calcium channel currents were sampled at 5-10 kHz and recorded on hard disk for later analysis. Data were collected and analysed off-line with the pCLAMP suite of programs (Axon Instruments). Cells were exposed to drugs and toxins via a series of flow pipes positioned above the cells. All data are expressed as means ±s.e.m., unless otherwise indicated.
Drugs and chemicals
Nociceptin (Phe-Gly-Gly-Phe-Thr-Gly-Ala-Arg-Lys-Ser-Ala-Arg-Lys-Leu-Ala-Asn-Gln) was synthesized and purified by Chiron Mimotopes (Clayton, Victoria, Australia). des[Phe1]-Nociceptin was obtained from Phoenix Pharmaceuticals (Mountain View, CA, USA). Buffer salts were from BDH Australia or Sigma Australia. Papain was from Worthington Biochemical Corporation (Freehold, NJ, USA). DAMGO (Tyr-D-Ala-Gly-N-methyl-Phe-Gly-ol enkephalin), DPDPE ([D-Pen2,5]-enkephalin), BSA and trypsin inhibitor (Type II-O) were from Sigma Australia. ω-Conotoxin GVIA and ω-conotoxin MVIIC were from Auspep (Melbourne, Australia). Methionine enkephalin (met-enkephalin) was from either Sigma or Auspep. Baclofen, CTAP (D-Phe-Cys-Tyr-D-Trp-Arg-Thr-Pen-Thr-NH2), deltorphin II, naloxone hydrochloride, nimodipine and U69593 were from Research Biochemicals International (Natick, MA, USA). ω-Agatoxin IVA was from the Peptide Institute (Osaka, Japan).
RESULTS
When PAG neurons were stepped from a holding potential of -90 mV to potentials between -60 and +60 mV the inward currents in most cells began to activate at about -40 mV and were invariably greatest at membrane potentials between -10 and +10 mV (Fig. 1, n= 268). There appeared to be very little low voltage-activated current in PAG neurons. About 20 % of cells displayed detectable inward current at -60 mV; this current represented less than 5 % of the peak current in these neurons and was not examined further in this study. The inward currents in PAG neurons were sensitive to Cd2+, a non-selective blocker of high voltage-activated calcium channels. At a concentration of 10 μm, Cd2+ blocked 92 ± 2.3 % (n= 12) of the peak inward current, and at 30 μm, Cd2+ blocked 98.3 ± 0.5 % (Fig. 1, n= 9) of the current. This suggests that the inward current observed under our recording conditions was current through calcium channels. The peak calcium channel currents (IBa), evoked by steps from -90 to -10 or 0 mV, generally displayed only a mild decline during the test pulse, with the current at the end of a 100 ms step being 82 ± 1 % (n= 103) of the peak current.
Figure 1. Periaqueductal grey (PAG) neurons express several calcium channel types.
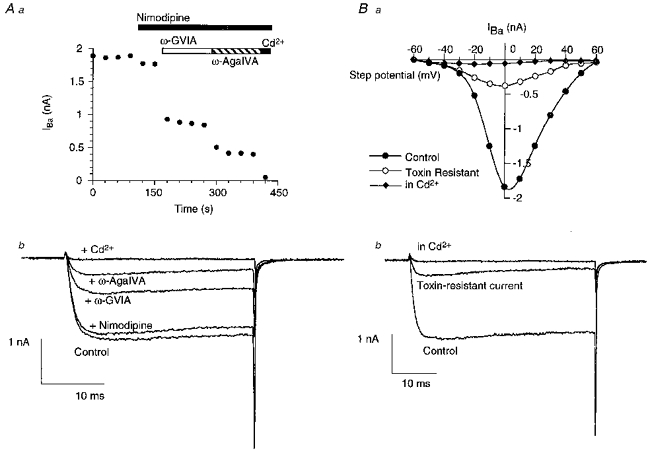
A, calcium channel currents (IBa) were elicited by repetitively stepping the membrane potential from -90 to 0 mV. a, time plot of the peak amplitude of the calcium channel current following the consecutive application of the L-type calcium channel antagonist nimodipine (3 μm), the N-type calcium channel antagonist ω-conotoxin GVIA (1 μm), the P/Q-type calcium channel antagonist ω-agatoxin IVA (500 nm) and the non-selective calcium channel blocker Cd2+ (30 μm). b, selected traces from the same experiment, showing the calcium channel current remaining after application of each antagonist. B, in another typical PAG neuron, calcium currents were elicited by stepping the membrane potential from -90 mV to a series of potentials between -60 and +60 mV. A, a plot of the peak inward current at each test potential, determined in the absence of calcium channel blockers (•), in the presence of nimodipine (3 μm), ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (500 nm) (○), and in the presence of Cd2+ (30 μm) (♦). b, selected traces from the same experiment, showing the inward currents elicited by a step from -90 to 0 mV in each condition.
The peak current density was similar in male and female rats: -257 ± 11 pA pF−1 (n= 113) in males and -263 ± 7 pA pF−1 (n= 155) in females. There did not appear to be any difference in current density between the youngest and oldest rats used in this study. The peak current density in cells from rats aged 23-25 days was -253 ± 17 pA pF−1 (n= 22) and in cells from rats aged 51-56 days, it was -240 ± 13 pA pF−1 (n= 18).
A number of calcium channel types have been identified on the basis of their differential sensitivity to drugs and toxins from animal venoms. Application of a high concentration of the L-type calcium channel blocker nimodipine (3 μm) inhibited the whole-cell current by 11 ± 1 % (range, 5-22 %, n= 15; Figs 1 and 2). This inhibition was readily reversible on washing the cells. The inhibition of the whole-cell current by nimodipine was not affected by prior exposure of the cells to a combination of the N-type calcium channel inhibitor ω-conotoxin GVIA (1 μm) and the P/Q-type calcium channel inhibitor ω-agatoxin IVA (50-500 nm), and was 11 ± 1 % (n= 7). There was no difference in the proportion of nimodipine-sensitive current in neurons from male (12 ± 5 %, n= 3) and female (11 ± 1 %, n= 13) rats.
Figure 2. Summary of the relative contributions of various calcium channel types to the peak IBa in PAG neurons.
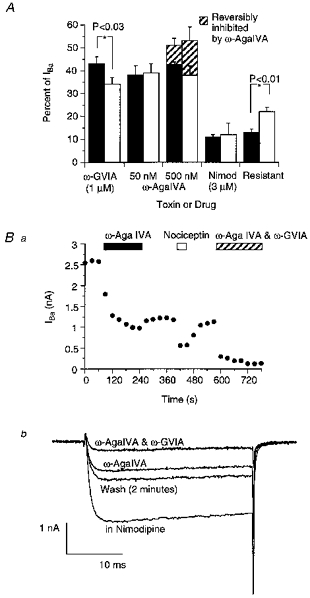
A shows that, overall, similar amounts of the IBa in PAG neurons are irreversibly inhibited by ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (50-500 nm). However, there is significantly more ω-conotoxin GVIA-sensitive current in neurons from female (▪) than male (□) rats. Conversely, there is significantly more current resistant to selective calcium channel antagonists in male than female rats (see text for numbers). The highest concentration of ω-agatoxin IVA tested (500 nm) reversibly inhibited a portion of the current in neurons from both male and female rats. This is illustrated in B a, a time plot of the peak amplitude of IBa during the application of ω-agatoxin IVA (500 nm) and its washout. The cell was subsequently exposed to nociceptin (100 nm) and then ω-agatoxin IVA (500 nm) and ω-conotoxin GVIA (1 μm) together. The neuron was stepped from -90 to 0 mV and nimodipine (3 μm) was present throughout the experiment. b, selected traces from the same experiment illustrate the partial reversal of the effects of ω-agatoxin IVA (500 nm), and the subsequent virtual abolition of the current by application of ω-agatoxin IVA (500 nm) and ω-conotoxin GVIA (1 μm) together.
Application of the N-type calcium channel inhibitor, ω-conotoxin GVIA (1 μm), produced an average of 39 ± 2 % (range, 21-65 %, n= 29) inhibition of the peak IBa in PAG neurons (Fig 1 and Fig 2). The inhibition was generally complete within 30 s and appeared to be irreversible. A total of forty-five cells were exposed to ω-conotoxin GVIA (1 μm) and of these neurons only one had IBa completely resistant to the peptide. There was significantly more ω-conotoxin GVIA-sensitive current in neurons from female (43 ± 3 %, n= 15) than male (34 ± 3 %, n= 14; P < 0.03, Student's unpaired t test) rats. Following application of the P/Q-type calcium channel blocker ω-agatoxin IVA (50-500 nm), ω-conotoxin GVIA (1 μm) still inhibited 36 ± 2 % (pooled data; 6 neurons from females, 5 neurons from males) of the whole-cell current.
Acute application of the P/Q-type calcium channel blocker ω-agatoxin IVA (50 nm) produced 38 ± 3 % (range, 11-57 %, n= 21) inhibition of the whole-cell IBa in PAG neurons (Fig 1 and Fig 2). The inhibition developed more slowly than that produced by ω-conotoxin GVIA, and often required 3-4 min to reach a steady state. Washing the cells for up to 10 min produced no significant reversal of the effects of ω-agatoxin IVA (50 nm; reversal was 3.5 ± 3 % of the inhibition, n= 4). However, application of a 10-fold higher concentration of ω-agatoxin IVA reversibly inhibited an additional component of the whole-cell IBa. ω-Agatoxin IVA (500 nm) inhibited 51 ± 3 % (range, 28-72 %, n= 15) of the peak IBa; however, washing the cells produced a rapid, partial reversal of the inhibition (reversal of 23 ± 3 %, n= 10), which was complete within 2 min (Fig. 2). The amount of current irreversibly inhibited by 500 nmω-agatoxin IVA was 40 ± 4 % of the whole-cell current, which is not different from the amount irreversibly inhibited by 50 nmω-agatoxin IVA. The reversible inhibition of a component of IBa by 500 nmω-agatoxin IVA persisted in the continued presence of 3 μm nimodipine (reversal of 21 ± 5 %, n= 5), and following perfusion of the cells with 1 μmω-conotoxin GVIA (reversal of 19 ± 4 %, n= 5), indicating that the ω-agatoxin IVA was not likely to be affecting either the L-type or N-type calcium channels in a reversible manner. There was no difference in the amount of current sensitive to ω-agatoxin IVA (50 nm) in neurons from female (38 ± 4 %, n= 14) versus male (39 ± 4 %, n= 7) rats. Following application of the N-type calcium channel blocker ω-conotoxin GVIA (1 μm), ω-agatoxin IVA (50 nm) still inhibited 32 ± 4 % (n= 8) of the whole-cell current.
Most of the IBa in PAG neurons was inhibited by nimodipine, ω-conotoxin GVIA and ω-agatoxin IVA but there was clearly a component that was resistant to a combination of all these blockers (Fig. 1). In twenty-one cells exposed to nimodipine (3 μm), ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (50-500 nm), the resistant current comprised between 4 and 22 % of the peak whole-cell current (average, 16 ± 2 %; Fig. 2). In some cells (e.g. Figs 1 and 2) the resistant current was quantified in the continued presence of ω-agatoxin IVA (500 nm), nimodipine (3 μm), and ω-conotoxin GVIA (1 μm); in others the ω-agatoxin IVA was washed on before the nimodipine and/or ω-conotoxin GVIA and thus the resistant current represented that unblocked following exposure to ω-agatoxin IVA. In the continued presence of ω-agatoxin IVA (500 nm), the resistant current comprised 10 ± 1 % (n= 10) of the total current while the resistant current after prior exposure to ω-agatoxin IVA comprised 20 ± 2 % (n= 11) of the total current. Taken together with the experiments outlined above, these data imply that the IBa component reversibly blocked by high concentrations of ω-agatoxin IVA is of the ‘resistant’ type. The resistant current peaked between -10 and 0 mV (n= 5) and declined by 54 ± 10 % (n= 6) over the course of a 60 ms pulse to -10 or 0 mV. There was more resistant current in neurons from male (22 ± 2 %, n= 7) than female (13 ± 2 %, n= 14; P < 0.01) rats. This is consistent with the results outlined above demonstrating that there were no sex differences in the amounts of L- and P/Q-type current in PAG neurons but that there was significantly less N-type current in neurons from males than females.
The component of IBa resistant to ω-conotoxin GVIA, ω-agatoxin IVA and nimodipine also appeared to be resistant to ω-conotoxin MVIIC, another blocker of P/Q-type calcium channels. Application of a combination of ω-conotoxin MVIIC (5 μm), ω-conotoxin GVIA (1 μm) and nimodipine (3 μm) inhibited the peak IBa of PAG neurons by 79 ± 2 % (range, 72-84 %, n= 7). The current remaining following this treatment also declined considerably over the course of a 60 ms pulse to -10 or 0 mV, by 46 ± 4 % (n= 7).
Modulation of calcium channel currents by nociceptin
Nociceptin inhibited IBa in almost all PAG neurons (113 of 114) tested (Fig. 3). This inhibition of IBa by nociceptin reversed on washout. A concentration-response relationship for nociceptin inhibition of PAG IBa was determined by application of one or more concentrations of nociceptin to cells stepped repetitively from -90 mV to the membrane potential that evoked the largest IBa in each neuron (either -10 or 0 mV; Fig. 4). Desensitization of the inhibition of IBa by nociceptin was not observed during brief applications of nociceptin at concentrations less than 1 μm. A logistic function fitted to the concentration-response relationship for nociceptin inhibition of IBa gave a -log EC50 (pEC50) of 8.3 ± 0.1 and a Hill slope for the curve of 0.9 ± 0.1. The maximum inhibition of IBa was 52 %, in the presence of 300 nm nociceptin.
Figure 3. Modulation of PAG IBa by neuropeptides and baclofen.
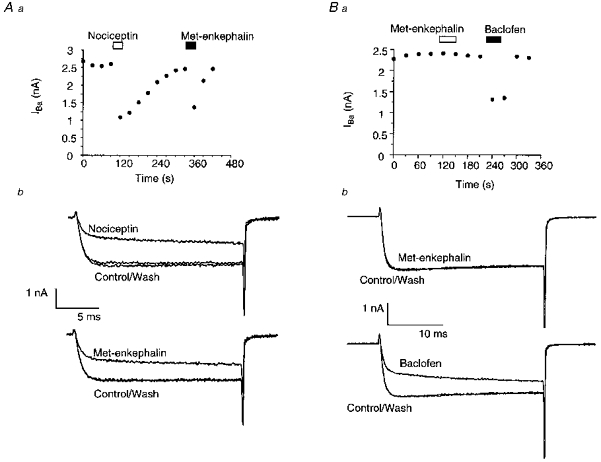
IBa was elicited by repetitively stepping the membrane potential from -90 to 0 mV. Aa, a time plot of the peak amplitude of IBa illustrating the effects of application of the neuropeptide nociceptin (100 nm) and the opioid receptor agonist met-enkephalin (10 μm). b, selected traces from the same experiment, showing the inhibition of IBa by each agonist. Nociceptin inhibited the IBa in virtually all neurons. B shows that in another neuron, met-enkephalin is ineffective at modulating IBa. a, a time plot of the peak amplitude of IBa illustrating the effects of application of met-enkephalin (300 nm) and the GABAB receptor agonist baclofen (10 μm). b, selected traces from the same experiment. Baclofen inhibited IBa in virtually all PAG neurons.
Figure 4. Characteristics of nociceptin and baclofen modulation of IBa.

A, concentration-response relationships for nociceptin (•) and baclofen (○) inhibition of the peak IBa in PAG neurons. Each point represents at least 5 cells tested. The EC50 for nociceptin was 5 nm, for baclofen 0.8 μm. B, the effects of both nociceptin (30 nm; a) and baclofen (3 μm; b) were blocked by pretreatment of PAG neurons with pertussis toxin (500 ng ml−1 for 8 h). The example traces were elicited by repetitively stepping the membrane potential of different PAG neurons from -90 to 0 mV. C, nociceptin inhibited IBa over a range of membrane potentials. IBa were elicited by stepping the membrane potential from -90 mV to potentials between -60 and +50 mV. a, a plot of the peak inward current at each test potential, in the absence (•) and presence (○) of nociceptin (30 μm). b, selected traces from the same experiment, showing the IBa elicited by a step from -90 to 0 mV in each condition.
The inhibition of IBa by nociceptin was usually, but not always, associated with a pronounced slowing of the activation of the currents (Fig. 3). At 30 nm, nociceptin increased the time to peak of IBa in nineteen of twenty-two cells; the reduction in the peak current was 38 % in cells where the time to peak was increased and 35 % in those where it was not. It was not possible to quantify the nociceptin-induced change in time to peak in all cells because in some cases the rise time of the current was slowed such that the current had not reached its peak by the end of the voltage step (up to 60 ms). However, in eleven cells, nociceptin (30 nm) increased the time to peak from 4.9 ± 0.3 to 20.2 ± 3.4 ms. When nociceptin was applied at approximately its EC50 concentration, 3 nm, the time to peak of the calcium channel current was increased at least 2-fold in four out of six cells.
There are no selective or potent antagonists available for the receptors for nociceptin; however, the non-selective opioid receptor antagonist naloxone failed to affect the inhibition of the calcium channel currents by a submaximally effective concentration of nociceptin (Fig. 5). Naloxone (10 μm) applied for 2 min did not affect IBa in PAG neurons. When nociceptin (30 nm) was subsequently applied in the continued presence of naloxone, IBa was inhibited by 38 ± 6 % (n= 5), which is not different from the inhibition of IBa by nociceptin (30 nm) alone in parallel experiments (40 ± 4 %, n= 4). The nociceptin analogue, des[Phe1]-nociceptin, which has a very low affinity for the nociceptin receptor, failed to mimic or occlude the actions of nociceptin (Fig. 5). des[Phe1]-Nociceptin (10 μm) applied for 2 min did not significantly inhibit IBa in PAG neurons (inhibition was 2 ± 3 %, n= 5). When nociceptin (30 nm) was subsequently applied in the continued presence of des[Phe1]-nociceptin, IBa was inhibited by 38 ± 3 % (n= 4), which was not different from the inhibition of IBa by nociceptin (30 nm) alone in parallel experiments (40 ± 4 %, n= 4).
Figure 5. Effects of antagonists and analogues on the responses to nociceptin and met-enkephalin.

The example traces were elicited by repetitively stepping the membrane potential of different PAG neurons from -90 mV to 0 mV. A, the effects of nociceptin (30 nm) are not mimicked or blocked by high concentrations of the nociceptin analogue [desPhe1]-nociceptin (10 μm; a) or the non-selective opioid receptor antagonist naloxone (10 μm; b). B, the effects of met-enkephalin (300 nm) are reversed by the selective μ-opioid receptor antagonist CTAP (1 μm). a, a time plot of the peak amplitude of IBa illustrating the effects of application of met-enkephalin and subsequent co-application of CTAP. b, selected traces from the same experiment.
Modulation of calcium channel currents by baclofen and opioids
Application of the GABAB receptor agonist baclofen also reversibly inhibited IBa in virtually all PAG neurons (54 of 55 cells tested; Fig. 3). The pEC50 for baclofen was 6.1 ± 0.1, with a maximum inhibition of 43 % at 30 μm (Fig. 4). The inhibition of IBa by baclofen was also associated with pronounced increase in the time to peak of the calcium channel current; in the presence of 3 μm baclofen the time to peak increased from 6.2 ± 0.7 to 19.2 ± 2.5 ms.
The opioid receptor agonist met-enkephalin (300 nm-10 μm) reversibly inhibited IBa in eighteen of forty-three PAG neurons tested (Fig. 3). Inhibition of peak IBa by 300 nm met-enkephalin was 35 ± 3 % (in 7 of 22 cells), and 10 μm met-enkephalin inhibited IBa by 47 ± 6 % (in 11 of 21 cells). The inhibition of IBa by 300 nm met-enkephalin could be rapidly and completely reversed by the selective μ-opioid receptor antagonist CTAP (1 μm; reversal, 100 ± 11 %, n= 6; Fig. 5). The selective μ-receptor agonist DAMGO (300 nm-3 μm) inhibited IBa in six of twenty-one PAG neurons; DAMGO at 300 nm inhibited the peak IBa by 33 ± 7 % (n= 3). Neither the selective κ-opioid receptor agonist U69593 (1 μm, n= 13), nor the selective δ-opioid receptor agonists DPDPE (300 nm-1 μm, n= 6) and deltorphin II (300 nm-3 μm, n= 12), inhibited IBa in any PAG neurons tested. The inhibition of IBa by met-enkephalin (300 nm) was also associated with a pronounced increase in the time to peak of IBa from 5.9 ± 0.5 to 17.3 ± 2 ms (n= 7).
Pretreatment of PAG neurons with pertussis toxin (PTX, 500 ng ml−1, 8 h, 35°C) largely abolished the effects of nociceptin and baclofen on IBa (Fig. 4). The IBa of PTX-treated cells in the presence of nociceptin (30 nm) was 99 ± 2 % (n= 8) of the pre-drug current. In cells from the same animals incubated for 8 h at 35°C in the absence of PTX, nociceptin (30 nm) inhibited IBa by 42 ± 5 % (n= 5). After treatment with PTX, baclofen (3 μm) inhibited IBa by 4 ± 1 % (n= 4), compared with an inhibition of 42 ± 4 % in untreated cells.
Nociceptin inhibited the major components of IBa current to different extents (Fig. 6). It is not possible to selectively block the R-type current, but a predominantly N-type current was obtained by treating the cells with ω-agatoxin IVA (100-500 nm) and continuous perfusion with nimodipine (3 μm). Nociceptin inhibited the predominantly N-type current with a pEC50 of 8.7 ± 0.1 and a maximum inhibition of 50 ± 7 % in the presence of 100 nm nociceptin. Conversely, a predominantly P/Q-type current could be obtained by treating the cells with ω-conotoxin GVIA (1 μm) and continuous perfusion of nimodipine (3 μm). Nociceptin inhibited the predominantly P/Q-type current with a pEC50 of 8.2 ± 0.1 and a maximum inhibition of 33 ± 6 % in the presence of 100 nm nociceptin.
Figure 6. Nociceptin differentially inhibits the components of IBa.

The example traces were elicited by repetitively stepping the membrane potential of different PAG neurons from -90 to 0 mV, in the absence and presence of nociceptin. Aa, nociceptin strongly inhibited the predominantly N-type IBa isolated by exposing neurons to ω-agatoxin IVA (100 nm) and nimodipine (3 μm). b, nociceptin also inhibited the predominantly P/Q-type IBa isolated by exposing neurons to ω-conotoxin GVIA (1 μm) and nimodipine (3 μm). c, nociceptin only modestly inhibited the L-type and resistant IBa isolated by exposing neurons to ω-conotoxin GVIA (1 μm) and ω-agatoxin IVA (100 nm). B, concentration-response relationships for nociceptin inhibition of the predominantly N-type IBa (•; EC50, 2 nm) and predominantly P/Q-type IBa (○; EC50, 7 nm).
Nociceptin did not substantially modulate the IBa that remained after treatment of the cells with blockers of N- and P/Q-type calcium channels. Following treatment with ω-agatoxin IVA (200 nm) and ω-conotoxin GVIA (1 μm), nociceptin (100-300 nm) inhibited IBa by 15 ± 3 %, n= 10. This inhibition reversed by approximately 60 % on washout of nociceptin. The IBa that remained following treatment of the cells with ω-agatoxin IVA, ω-conotoxin GVIA and nimodipine declined in the presence of nociceptin (300 nm-1 μm) by 11 ± 2 %; but this inhibition did not reverse on washout of nociceptin. The modest nature of the decreases caused by high concentrations of nociceptin precluded further quantitative study of these effects.
DISCUSSION
Nociceptin and the GABAB agonist baclofen both inhibited the calcium channel currents in almost all dissociated PAG neurons, while μ-opioid receptor activation only inhibited calcium channel currents in 40 % of neurons. These results are consistent with previous studies showing that nociceptin (Vaughan et al. 1997) and baclofen (Chieng & Christie 1995), increase an inwardly rectifying potassium conductance in virtually all neurons in PAG slices, while μ-opioids only increase the potassium conductance in a subpopulation of PAG neurons in slices (Chieng & Christie 1994). μ-Opioids have previously been shown to inhibit the calcium channels in a similar proportion of neurons dissociated from the PAG of neonate rats (Kim et al. 1997). Immunohistochemical and biochemical studies indicate that the PAG contains both δ- and κ-opioid receptors (Kalyuzhny et al. 1996; Mansour et al. 1996; Noble & Cox, 1996), but neither δ- nor κ-receptor agonists affected the calcium channel currents in the PAG. This finding is intriguing, but is consistent with the lack of effect of δ- and κ-opioids in PAG slices (Chieng & Christie, 1994; Vaughan & Christie, 1997). It is likely that nociceptin acted via ORL1 to inhibit calcium channels, although this cannot be demonstrated unequivocally in the absence of selective antagonists for ORL1. Nociceptin did not act via μ, δ- or κ-opioid receptors because high concentrations of the general opioid antagonist naloxone did not affect the actions of nociceptin, and the inactive nociceptin analogue des[Phe1]-nociceptin failed to mimic or occlude the effects of nociceptin (Matthes, Seward, Kieffer & North, 1996; Henderson & McKnight, 1997).
PAG neurons expressed a range of pharmacologically distinguishable calcium channel types. Under the experimental conditions of this study, the predominant current components were irreversibly inhibited by either ω-conotoxin GVIA or ω-agatoxin IVA. In contrast to the findings of Pearson, Sutton, Scott & Dolphin (1995) in cerebellar granule cells, we found no evidence for significant overlap between the current components inhibited by ω-agatoxin IVA and either ω-conotoxin GVIA or nimodipine. It has been shown that the inhibition of N-type channels by ω-conotoxin GVIA (Stocker, Nadasdi, Aldrich & Tsien, 1997) and P/Q-type channels by ω-agatoxin IVA (Mintz, Adams & Bean, 1992) can be reversed by either strong membrane hyperpolarizations or depolarizations, respectively, but the voltage protocols employed in this study were unlikely to induce any significant reversal of the ‘irreversible’ effects of these toxins. Irreversible inhibition by ω-conotoxin GVIA is generally accepted to define N-type calcium channels, which are formed by the Class 1B α-subunit (Dubel et al. 1992). Although a rapidly reversible block of some L-type calcium channels (Class D) by ω-conotoxin GVIA has been reported (see Williams et al. 1992, and references therein), we found no evidence for this in PAG neurons. In any case, there was only a small amount of nimodipine-sensitive, presumably L-type current in PAG neurons.
ω-Agatoxin IVA sensitivity is generally accepted to define P/Q-type calcium channels (Mintz et al. 1992; Randall & Tsien, 1995; but see Pearson et al. 1995). P-type channels are very sensitive to ω-agatoxin IVA (Mintz et al. 1992) and 50 nm should be enough to block all the channels. The presence of Q-type channels was tested with a 10-fold higher concentration of toxin (Randall & Tsien, 1995). ω-Agatoxin IVA at 500 nm did inhibit more of the whole-cell current, but this inhibition was rapidly reversible on washout, and the irreversible inhibition by 500 nmω-agatoxin IVA was not different from that caused by 50 nmω-agatoxin IVA. The block of Q-type channels by ω-agatoxin IVA in cerebellar granule cells is partly reversible (Randall & Tsien, 1995), but this recovery is slow (a time constant between 7 and 12 min) and seen with all concentrations of ω-agatoxin IVA tested. In PAG neurons there was no significant reversal of the effects of 50 nmω-agatoxin IVA, and the additional effects of 500 nmω-agatoxin IVA completely reversed within 2 min of washout, indicating that the additional effects of 500 nmω-agatoxin IVA could not be attributed to actions at Q-type channels. In any case, as both P-type and Q-type channels are probably formed by the same α subunit, α1A (Mori et al. 1991), it seems prudent to refer to the ω-agatoxin IVA-sensitive current in PAG neurons as P/Q-type.
The portion of the whole-cell current reversibly inhibited by ω-agatoxin IVA (500 nm) seemed to represent part of the otherwise toxin-resistant, or R-type current (Zhang et al. 1993). A similar amount of resistant current was apparent in the presence of combinations of either ω-conotoxin GVIA, ω-agatoxin IVA and nimodipine or ω-conotoxin GVIA, ω-conotoxin MVIIC and nimodipine, implying that the resistant current was not likely to be Q-type, which is relatively insensitive to ω-agatoxin IVA (Randall & Tsien, 1995). It is not at present possible to define the R-type current(s) pharmacologically, although it is thought that at least some R-type currents may be encoded by the α1E pore-forming subunit (Soong, Stea, Hodson, Dubel, Vincent & Snutch, 1993). The types of calcium channel current present in PAG neurons from young adult (23-56 days) rats generally appear similar to those found in neurons from neonate animals (10-16 days) by Kim et al. 1997, where N-, P/Q-, L- and R-type currents were found, although the very different recording conditions utilized in that study make direct comparisons difficult.
There was significantly less N-type current in male rats than females, with a correspondingly greater proportion of R-type current in the males. The physiological implications of this finding are difficult to assess without knowledge of which calcium channel types contribute to neurotransmitter release by PAG neurons, or which channel types may be involved in the generation of calcium-dependent after-hyperpolarizations in PAG cells. At present, nothing is known about either of these phenomena. It is clear, however, that different calcium channel types can be modulated differently by neurotransmitters (see below).
Nociceptin predominantly inhibited the N- and P/Q-type calcium channel currents in PAG neurons, which is different from its actions in hippocampal CA3 pyramidal cells, where it significantly inhibited the N-, P/Q-, L- and R-type calcium channel currents (Knoflach, Reinscheid, Civelli & Kemp, 1996). Although both the predominantly N- and predominantly P/Q-type currents were potently inhibited by nociceptin, it appeared to be more potent and have a greater maximal effect on the predominantly N-type current versus the predominantly P/Q-type current. A greater maximal effect of neurotransmitter receptor agonists on N-type versus P/Q-type currents has been observed in several rat neuronal cell types, including small spinal cord neurons treated with baclofen (Mintz & Bean, 1993) and adrenal chromaffin cells treated with ATP (Currie & Fox, 1997). In the present study, nociceptin inhibition of the N-type currents was also found to be more potent than the inhibition of P/Q-type currents. The differences in maximal effect are likely to arise from differences in the intrinsic sensitivity to G-proteins of the N- and P/Q-type channels themselves. μ-Opioid receptors co-expressed in Xenopus laevis oocytes with the pore-forming subunits for N-type (α1B) and P/Q-type (α1A) channels inhibited the α1B currents much more strongly than the α1A currents (Bourinet, Soong, Stea & Snutch, 1996), in good agreement with the results found with native N-type and P/Q-type channels in PAG neurons. Bourinet et al. (1996) also found that μ-opioid receptor activation did not significantly modulate the currents produced by the putative R-type (α1E) pore-forming subunit. Selective neurotransmitter modulation of native N-type and P/Q-type channels rather than L-type or R-type channels is a common finding in neuronal cells (Dolphin, 1995). It should be noted that our study utilized Ba2+ as a charge carrier in order to minimize current run-down mediated by Ca2+-dependent processes, and it is likely that any Ca2+-dependent modulatory pathways utilized by the receptors examined here were suppressed under these conditions. The possibility of additional, physiologically relevant mechanisms of calcium channel modulation in PAG neurones cannot be excluded.
The nociceptin-mediated inhibition of the calcium channel current in the PAG, hippocampus and human neuroblastoma cell line SH-SY5Y (Connor, Yeo & Henderson, 1996) were all mediated via pertussis toxin-sensitive G-proteins. In the hippocampus and PAG the inhibition was characterized by a marked slowing of the time to peak of the calcium channel current. These characteristics are consistent with the receptor for nociceptin utilizing the ubiquitous G-protein βγ subunit-mediated pathway for inhibiting high voltage-activated calcium channels, and thus the inhibition of calcium channels by nociceptin/ORL1 is likely to be a common finding. In contrast to the PTX-sensitive effects outlined above, nociceptin inhibition of the T-type calcium channel of rat dorsal root ganglion cells (Abdulla & Smith, 1997) appears not to be mediated by G-proteins. In these cells nociceptin inhibition of the high voltage-activated currents was sensitive to manipulations designed to interfere with G-protein coupling, but the nociceptin modulation of the T-type current was not, so it seems that nociceptin can act to inhibit calcium channels through different pathways in the same cell. The T-type current did not make a major contribution to the whole-cell currents recorded either in the present study or that of Knoflach et al. (1996), so the effect of nociceptin on this current has not yet been studied in central neurons.
It is not clear how the different effects of nociceptin, baclofen and μ-opioids on the various substrates within the PAG correlate with the physiological functions of the region, particularly the analgesia that arises in the PAG. Microinjection of either baclofen (Levy & Proudfit, 1979) or μ-opioids into the PAG can elicit analgesia, but co-injection of a high dose of nociceptin with μ-opioids in the PAG blocks the analgesic actions of opioids (Morgan et al. 1997). The effects of nociceptin microinjection alone on PAG function have not been determined (Morgan et al. 1997). However, given the similarities between the actions of baclofen and nociceptin on neurons within the PAG, it might be predicted that microinjection of nociceptin alone would be analgesic, and that co-application of baclofen with a μ-opioid agonist would block the μ-opioid-mediated analgesia.
This study demonstrates that the opioid-like peptide nociceptin inhibits calcium channel currents in essentially all PAG neurons, and does so through a receptor with a pharmacological profile consistent with that of ORL1. It provides further evidence that nociceptin utilizes the same cellular effectors as μ-opioids but affects a different subpopulation of neurons, which probably gives rise to the different behavioural effects of nociceptin when compared with μ-opioids. The overall effects of nociceptin on single cells within the PAG are most similar to those of the GABAB receptor agonist baclofen, which suggests that nociceptin is likely to be an important regulator of a wide range of functions in the PAG.
Acknowledgments
We would like to thank Susan Ingram for her help in developing the dissociation procedures, and Keith Rippon for his excellent technical assistance. This study was supported by the National Health and Medical Research Council of Australia. M. C. is the recipient of a Rolf Edgar Lake Fellowship from the Faculty of Medicine, University of Sydney.
References
- Abdulla FA, Smith PA. Nociceptin inhibits T-type Ca2+ channel current in rat sensory neurons by a G-protein-independent mechanism. Journal of Neuroscience. 1997;17:8721–8728. doi: 10.1523/JNEUROSCI.17-22-08721.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akil H, Mayer DJ, Liebeskind JC. Antagonism of stimulation-produced analgesia by naloxone, a narcotic antagonist. Science. 1976;191:961–962. doi: 10.1126/science.1251210. [DOI] [PubMed] [Google Scholar]
- Anton B, Fein J, To T, Silberstein L, Evans CJ. Immunohistochemical localization of ORL-1 in the central nervous system of the rat. Journal of Comparative Neurology. 1996;368:229–251. doi: 10.1002/(SICI)1096-9861(19960429)368:2<229::AID-CNE5>3.0.CO;2-5. 10.1002/(SICI)1096-9861(19960429)368:2<229::AID-CNE5>3.3.CO;2-G. [DOI] [PubMed] [Google Scholar]
- Bandler R, Shipley M. Columnar organization in the midbrain periaqueductal gray: modules for emotional expression. Trends in Neurosciences. 1994;17:379–388. doi: 10.1016/0166-2236(94)90047-7. [DOI] [PubMed] [Google Scholar]
- Bourinet E, Soong TW, Stea A, Snutch TP. Determinants of the G-protein-dependent opioid modulation of neuronal calcium channels. Proceedings of the National Academy of Sciences of the USA. 1996;93:1486–1491. doi: 10.1073/pnas.93.4.1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ. Hyperpolarization by opioids acting on μ-receptors of a subpopulation of rat periaqueductal grey neurones in vitro. British Journal of Pharmacology. 1994;113:121–128. doi: 10.1111/j.1476-5381.1994.tb16183.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chieng B, Christie MJ. Hyperpolarization by GABAB receptor agonists in midbrain periaqueductal grey neurones in vitro. British Journal of Pharmacology. 1995;116:1583–1588. doi: 10.1111/j.1476-5381.1995.tb16376.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christie MJ, Williams JT, Osborne PB, Bellchambers C. Where is the locus in opioid withdrawal. Trends in Pharmacological Sciences. 1997;18:134–140. doi: 10.1016/s0165-6147(97)01045-6. [DOI] [PubMed] [Google Scholar]
- Connor MA, Christie MJ. Modulation of the calcium channel currents of acutely dissociated rat periaqueductal grey neurons. Society for Neuroscience Abstracts. 1997;23:127. [Google Scholar]
- Connor M, Yeo A, Henderson G. Nociceptin inhibits Ca2+ channel currents and elevates intracellular Ca2+ in the SH-SY5Y human neuroblastoma cell line. British Journal of Pharmacology. 1996;118:205–207. doi: 10.1111/j.1476-5381.1996.tb15387.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Currie KPM, Fox AP. Comparison of N- and P/Q-type voltage-gated calcium channel current inhibition. Journal of Neuroscience. 1997;17:4570–4579. doi: 10.1523/JNEUROSCI.17-12-04570.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Voltage-dependent calcium channels and their modulation by neurotransmitters and G-proteins. Experimental Physiology. 1995;80:1–36. doi: 10.1113/expphysiol.1995.sp003825. [DOI] [PubMed] [Google Scholar]
- Dubel SJ, Starr TVB, Hell J, Ahlijanian MK, Enyeart JJ, Caterall WA, Snutch TP. Molecular cloning of the α1 subunit of an ω-conotoxin-sensitive calcium channel. Proceedings of the National Academy of Sciences of the USA. 1992;89:5058–5062. doi: 10.1073/pnas.89.11.5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Henderson G, McKnight AT. The orphan opioid receptor and its endogenous ligand-nociceptin/orphanin FQ. Trends in Pharmacological Sciences. 1997;18:293–301. [PubMed] [Google Scholar]
- Ingram S, Wilding TJ, McCleskey EW, Williams JT. Efficacy and kinetics of opioid action on acutely dissociated neurons. Molecular Pharmacology. 1997;52:136–143. doi: 10.1124/mol.52.1.136. [DOI] [PubMed] [Google Scholar]
- Kalyuzhny AE, Arvidsson U, Wu W, Wessendorf MW. μ-Opioid and δ-opioid receptors are expressed in brainstem antinociceptive circuits: studies using immunocytochemistry and retrograde tract tracing. Journal of Neuroscience. 1996;16:6490–6503. doi: 10.1523/JNEUROSCI.16-20-06490.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim CJ, Rhee J-S, Akaike N. Modulation of high-voltage activated Ca2+ channels in the rat periaqueductal grey neurons by μ-type opioid agonist. Journal of Neurophysiology. 1997;77:1418–1424. doi: 10.1152/jn.1997.77.3.1418. [DOI] [PubMed] [Google Scholar]
- Knoflach F, Reinscheid RK, Civelli O, Kemp JA. Modulation of voltage-gated calcium channels by orphanin FQ in freshly dissociated hippocampal neurons. Journal of Neuroscience. 1996;16:6657–6664. doi: 10.1523/JNEUROSCI.16-21-06657.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levy RA, Proudfit HK. Analgesia produced by microinjection of baclofen and morphine at brainstem sites. European Journal of Pharmacology. 1979;57:43–55. doi: 10.1016/0014-2999(79)90102-x. [DOI] [PubMed] [Google Scholar]
- Mansour A, Burke S, Pavlic RJ, Akil H, Watson SJ. Immunohistochemical localization of the cloned κ1 receptor in the rat CNS and pituitary. Neuroscience. 1996;71:671–690. doi: 10.1016/0306-4522(95)00464-5. [DOI] [PubMed] [Google Scholar]
- Matthes H, Seward EP, Kieffer B, North RA. Functional selectivity of orphanin FQ for its receptor coexpressed with potassium channel subunits in Xenopus laevis oocytes. Molecular Pharmacology. 1996;50:447–450. [PubMed] [Google Scholar]
- Meunier J-C, Mollereau C, Toll L, Suaudeau C, Moisand C, Alvinerie P, Butour J-L, Guillemot J-C, Ferrara P, Monsarrat B, Mazarguil H, Vassart G, Parmentier M, Costentin J. Isolation and structure of the endogenous agonist of opioid receptor-like ORL1 receptor. Nature. 1995;377:532–535. doi: 10.1038/377532a0. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- Mintz IM, Bean BP. GABAB receptor inhibition of P-type Ca2+ channels in central neurons. Neuron. 1993;10:889–898. doi: 10.1016/0896-6273(93)90204-5. [DOI] [PubMed] [Google Scholar]
- Morgan MA, Grisel JE, Robbins CS, Grandy DK. Antinociception mediated by the periaqueductal grey is attenuated by orphanin FQ. NeuroReport. 1997;8:3431–3434. doi: 10.1097/00001756-199711100-00003. [DOI] [PubMed] [Google Scholar]
- Mori Y, Friedrich T, Kim M-S, Mikami A, Nakai J, Ruth P, Bosse E, Hofmann F, Flockerzi V, Furuichi T, Mikoshiba K, Imoto K, Tanabe T, Numa S. Primary structure and functional expression from complementary DNA of a brain calcium channel. Nature. 1991;350:398–402. doi: 10.1038/350398a0. [DOI] [PubMed] [Google Scholar]
- Noble F, Cox BM. Differential desensitization of μ- and δ-opioid receptors in selected neural pathways following chronic morphine treatment. British Journal of Pharmacology. 1996;117:161–169. doi: 10.1111/j.1476-5381.1996.tb15169.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson HA, Sutton KG, Scott RH, Dolphin AC. Characterization of Ca2+ channel currents in cultured rat cerebellar granule neurones. Journal of Physiology. 1995;482:493–509. doi: 10.1113/jphysiol.1995.sp020535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Randall A, Tsien RW. Pharmacological dissection of multiple types of Ca2+ channel currents in rat cerebellar granule neurons. Journal of Neuroscience. 1995;15:2995–3012. doi: 10.1523/JNEUROSCI.15-04-02995.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinscheid RK, Nothacker H-P, Bourson A, Ardati A, Henningsen RA, Bunzow JR, Grandy DK, Langen H, Monsma FJ, Civelli O. Orphanin FQ: a neuropeptide that activates an opioidlike G protein-coupled receptor. Science. 1995;270:792–794. doi: 10.1126/science.270.5237.792. [DOI] [PubMed] [Google Scholar]
- Soong TW, Stea A, Hodson CD, Dubel SJ, Vincent SR, Snutch TP. Structure and functional expression of a member of the low voltage-activated calcium channel family. Science. 1993;260:1133–1136. doi: 10.1126/science.8388125. [DOI] [PubMed] [Google Scholar]
- Stocker JW, Nadasdi L, Aldrich RW, Tsien RW. Preferential interaction of ω-conotoxins with inactivated N-type Ca2+ channels. Journal of Neuroscience. 1997;17:3002–3013. doi: 10.1523/JNEUROSCI.17-09-03002.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Christie MJ. Presynaptic inhibitory action of opioids on synaptic transmission in the rat periaqueductal grey in vitro. Journal of Physiology. 1997;498:463–472. doi: 10.1113/jphysiol.1997.sp021872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan CW, Ingram SL, Christie MJ. Actions of the ORL1 receptor ligand nociceptin on membrane properties of rat periaqueductal grey neurons in vitro. Journal of Neuroscience. 1997;17:996–1003. doi: 10.1523/JNEUROSCI.17-03-00996.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Rudy TA. Narcotic analgetics: CNS sites and mechanisms of action as revealed by intracerebral injection techniques. Pain. 1978;4:299–359. doi: 10.1016/0304-3959(77)90145-2. [DOI] [PubMed] [Google Scholar]
- Williams ME, Feldman DH, McCue AF, Brenner R, Velicelebi G, Ellis SB, Harpold MM. Structure and functional expression of α1, α2, and β subunits of a novel human neuronal calcium channel subtype. Neuron. 1992;8:71–84. doi: 10.1016/0896-6273(92)90109-q. [DOI] [PubMed] [Google Scholar]
- Zhang J-F, Randall AD, Ellinor PT, Horne WA, Sather WA, Tanabe T, Schwarz TL, Tsien RW. Distinctive pharmacology and kinetics of cloned neuronal Ca2+ channels and their possible counterparts in mammalian CNS neurons. Neuropharmacology. 1993;32:1075–1088. doi: 10.1016/0028-3908(93)90003-l. [DOI] [PubMed] [Google Scholar]