

Abstract
Dentate gyrus granule cells acutely dissociated from hippocampal slices obtained from chronic temporal lobe epilepsy (TLE) patients displayed a high-voltage activated (HVA) Ca2+ conductance with a pronounced Ca2+-dependent inactivation.
Inactivation time constants and peak HVA Ca2+ current (ICa) amplitudes did not differ between perforated patch and whole-cell recordings without added exogenous Ca2+ buffers, indicating that the Ca2+-dependent characteristics of ICa inactivation were well preserved in whole-cell recordings.
Inactivation time constants correlated with whole-cell ICa, and were increased when Ca2+ was replaced with Ba2+ in the external solution or 5 mm BAPTA was added to the pipette solution.
In recordings without added exogenous Ca2+ buffers, the time course of ICa inactivation was comparable between human TLE and kindled rat granule cells. Conversely, the time course of ICa in human TLE granule cells loaded with 5 mm intracellular BAPTA resembled that observed in buffer-free recordings from control rat neurones.
The loss of a putative intraneuronal Ca2+ buffer, the Ca2+-binding protein calbindin (CB), from human granule cells during TLE may result in the pronounced Ca2+-dependent ICa inactivation. This process could serve a neuroprotective role by significantly decreasing Ca2+ entry during prolonged trains of action potentials known to occur during seizures.
The spatio-temporal profile of intraneuronal free [Ca2+] is highly regulated because it controls vital cellular functions, such as excitability, plasticity and gene expression (Kennedy, 1989; Ghosh & Greenberg, 1995), under physiological conditions and chronic disease (Gibbons, Brorson, Bleakman, Chard & Miller, 1993). Ligand-gated ion channels, voltage-gated Ca2+ channels (VGCCs) and Ca2+ extruding pumps regulate the net Ca2+ flux across the cell membrane, while intracellular Ca2+ stores and buffers control the dynamics of intracellular free [Ca2+] (Miller, 1991).
A negative feedback mechanism between Ca2+ entry and intracellular [Ca2+] is provided by the Ca2+-dependent inactivation of Ca2+ currents (ICa) (Brehm & Eckert, 1978). Basal levels of inactivation have been shown to increase with elevated intracellular [Ca2+] (Branchaw, Banks & Jackson, 1997) and inactivation rates of ICa are sensitive to exogenous Ca2+ buffers added intracellularly (Chad, 1989). The precise mechanism whereby Ca2+ entry mediates the inactivation of ICa is unknown. For L-type channels (α1C) inactivation requires an intact C-terminus of the protein. Binding of Ca2+ to an EF-hand like region in this terminus (de Leon et al. 1995), or to another short sequence nearby (Zhou, Olcese, Qin, Noceti, Birnbaumer & Stefani, 1997) is thought to reduce the probability of channel opening. Therefore, a disease-related change in the endogenous Ca2+ buffering or in the affinity of the Ca2+ binding site responsible for inactivation would be expected to control the gain of the negative feedback.
An increase in this feedback on Ca2+ entry through high-voltage activated (HVA) ICa is associated with the loss of the Ca2+ binding protein calbindin D−28k (CB) in rat dentate gyrus granule cells (DGGCs) during kindling, an animal model of epilepsy (Köhr, Lambert & Mody, 1991). It was hypothesized that CB normally acts as a local Ca2+ buffer which competes for entering Ca2+ ions with the binding site responsible for inactivation. A recent histochemical study (Maglóczky, Halász, Vajda, Czirják & Freund, 1997) has demonstrated the selective loss of CB from granule cells in the human hippocampus in temporal lobe epilepsy (TLE) patients. Therefore, we wanted to find out whether a similar functional relationship might exist between intraneuronal Ca2+ buffering and ICa inactivation in human TLE granule cells following the loss of CB. Our experiments in human granule cells acutely dissociated from TLE patients reveal a strongly inactivating Ca2+ conductance with its inactivation sensitive to intraneuronal Ca2+ buffering.
METHODS
Patient data
Surgical specimens were obtained from ten TLE patients (5 males, 5 females) who underwent therapeutic surgical resection of their hippocampal formation. Their average age at the time of surgery was 35 ± 4 years (mean ±s.e.m.). Informed consent was obtained from all patients for post-operative electrophysiological analysis of the resected tissue. The study was conducted under the guidance of the Declaration of Helsinki (1989) and the informed consent approved by the UCLA Human Subject Protection Committee (protocol HSPC no. 93-11-642-11).
Cell preparation
Transverse hippocampal slices (450 μm thick) were prepared from human hippocampi by standard procedures (Isokawa, Avanzini, Finch, Babb & Levesque, 1991) and were stored in an oxygenated (95 % CO2−5 % O2) and warmed (32°C) chamber filled with bicarbonate-based artificial cerebrospinal fluid (ACSF) of the following composition (mm): 126 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 10 glucose, 26 NaHCO3 and 1 pyruvic acid. The dissociation procedure was similar to the one described previously (Köhr & Mody, 1991). Briefly, slices were subjected to a 30 min protease (pronase E, type XIV, Sigma) treatment (2 mg (ml ACSF)−1) prior to isolation of the dentate gyrus under a dissection microscope in ice-cold Hepes-based ACSF. Mechanical trituration with fire-polished Pasteur pipettes yielded a cell suspension which was transferred into a culture dish and washed with recording medium after a 10 min settling period.
Control and kindled rat granule cells were dissociated in the same way. The kindling protocol and the preparation of rat slices were done as previously described (Köhr & Mody, 1991). Rats were anaesthetized using 75 mg kg−1 sodium pentobarbitone (i.p.) prior to undertaking the surgical procedures for electrode implantation. The depth of anaesthesia was ensured by the lack of the pinch-paw reflex which lasted in excess of 30 min following the injection. As the total length of the surgical procedures was less than 15 min, no supplemental anaesthesia was required during surgery. After surgery, the wound was treated with the topical antibacterial/anaesthetic unguent Neosporin (Warner-Lambert, Morris Plains, NJ, USA). Daily during the post-operative week, and weekly during the stimulation sessions, bacitracin zinc ointment (E. Fougera & Co., Melville, NY, USA) was applied to the area around the wound to avoid possible infections. The animals were anaesthetized using 75 mg kg−1 sodium pentobarbitone (i.p.) before the decapitation preceding the preparation of brain slices.
Recording solutions
The external recording medium contained (mm): 106 NaCl, 2.5 KCl, 2 MgCl2, 3 CsCl, 25 tetraethylammonium chloride (TEA), 5 4-aminopyridine, 1 pyruvic acid, 10 glucose, 20 Hepes, 0.001 TTX and either 5 mm CaCl2 or 5 mm BaCl2. Final osmolarity was 290–300 mosmol l−1. The internal solution consisted of (mm): 110 CsCl, 3 TEA, 10 Hepes and an ATP-regenerating system with 4 Mg-ATP, 0.1 Tris-GTP, 25 creatine phosphate; and 50 U ml−1 phosphocreatine kinase (Sigma). Final osmolarity was 265-275 mosmol l−1. In the experiments using 5 mm BAPTA inside the pipette, CsCl was reduced to adjust for the change in osmolarity. In perforated patch experiments amphotericin B (Sigma) was dissolved in DMSO (3 mg (50 μl)−1) and added to the pipette solution (1 mg amphotericin (ml recording solution)−1) (Rae, Cooper, Gates & Watsky, 1991) which consisted of (mm): 90 caesium aspartate, 20 CsCl, 20 Hepes, 20 glucose and 20 TEA. All solutions were adjusted to pH 7.25. All recordings were performed at room temperature (19-22°C). All chemicals, except those indicated otherwise, were purchased from Fluka (Buchs, Switzerland).
Data acquisition and analysis
Recordings from a total of twenty visually identified granule cells were accepted for analysis. Since the cells were acutely isolated from the dissected dentate gyrus and since only cells that looked similar to truncated human DGGCs in situ were used for recording, we assume that the data originate from one population only, i.e. the DGGCs.
Voltage-clamped currents were recorded with an Axopatch 200A amplifier (Axon Instruments) and analysed with the WinWCP 1.6c program (courtesy of J. Dempster, University of Strathclyde, Glasgow, UK). Pipettes were pulled from 1.5 mm o.d., 1.1 mm i.d. borosilicate glass capillaries. Currents were low-pass filtered at 5 kHz with an 8-pole Bessel filter and acquired at 15 kHz. Whole-cell recordings (n= 16) yielded access resistances (Rss) of 21 ± 2 MΩ (mean ±s.e.m.), which were compensated by 60-90 %. Whole-cell recording began > 5 min after rupture of the patch. Perforated patch recordings (n= 4) yielded stable Rs values of 48 ± 5 MΩ, approximately 10 min after seal formation. The Rs values were compensated by 80-90 %. Thus, the remaining uncompensated steady-state error in voltage control was less than 3 mV, as calculated by ΔV=ImaxRs(100 - % compensation). The liquid junction potential was fairly small (< 3 mV) (Neher, 1992) and was not compensated for. Cell capacitances were estimated by the transient cancellation procedure to be 9 ± 1.2 pF. All measurements were made on linear leak-subtracted currents (P/4 procedure; Bezanilla & Armstrong, 1977). All traces displayed in the figures have been subtracted in this manner.
Steady-state activation curves were fitted (minimal χ2 Levenberg-Marquart algorithm) by the following single Boltzmann function: g/gmax=[1 + exp((V½ - V)/K)]−1 where g is the conductance (Ipeak/(Vstep - ECa)), gmax is the maximum of g, Ipeak is the peak current amplitude during a potential step Vstep, V½ is the voltage at half-maximal G, ECa is the Ca2+ equilibrium potential and K is a constant.
The decay phases of the currents were individually fitted (starting from the peak) using the same non-linear least-squares algorithm by a mono-exponential decay function with variable offset: I(t) =aexp(-t/τ) +C, which yielded best fits for a, c and τ.
The level of statistical significance was set to P < 0.05 and was determined using Student's t tests.
RESULTS
Voltage-clamped inward currents (Fig. 1A) were elicited by stepping the potential from a holding potential of −60 mV in the whole-cell mode using recording solutions designed to isolate the Ca2+ conductance. Cadmium (50 μm, n= 3 cells, data not shown) blocked the currents, identifying them as Ca2+ currents. The peak ICavs.V plot was bell shaped and peaked at +10 mV (−308 ± 22 pA), which is characteristic of well-clamped calcium currents. Since a single Boltzmann function yielded a good fit (χ2= 0.00083) to the steady-state activation curve (Fig. 1B), we conclude that the predominant conductance activated by voltage steps from −60 mV is of the high-threshold type (HVA).
Figure 1. Human TLE granule cells possess a HVA calcium conductance which inactivates in a Ca2+-dependent fashion.

A, whole-cell voltage-clamped Ca2+ currents were elicited from a −60 mV holding potential by steps of 160 ms duration delivered every 8 s. The currents (○) were fitted with mono-exponential decay functions (continuous lines). See Methods for details. For the maximal ICa: τ= 44 ms, a= −265 pA, c= −160 pA. B, the normalized peak conductance g is plotted as a function of the membrane potential step and fitted with a single Boltzmann function (dashed line). The voltage at half-maximal activation (V1/2) is −6.5 ± 0.5 mV. C, the peak Ca2+ current amplitude and the decay time constant τ for the whole-cell recordings (n= 8) are plotted as a function of step voltages. Note the correlation between current amplitudes and decay time constants.
To find out whether the currents inactivated in a Ca2+-dependent manner, ICa traces were fitted with a mono-exponential decay function (Fig. 1A), and the resulting decay time constants (τ) were plotted as a function of the step potential (Fig. 1C). The τ had a U-shaped dependency on membrane voltage, indicating that calcium entry was correlated with current inactivation, which is a characteristic of the Ca2+-dependent inactivation process of Ca2+ currents (Brehm & Eckert, 1978).
In order to avoid imposing an exogenous Ca2+ buffering capacity on the neurones, in control whole-cell recordings, we omitted any exogenous Ca2+ buffers from the pipette solutions (Köhr & Mody, 1991). To test whether the Ca2+-dependent ICa inactivation recorded in the buffer-free whole-cell mode corresponds to unperturbed endogenous Ca2+ buffering conditions, we also recorded calcium currents in the perforated patch mode (Fig. 2A), which provides electrical but not diffusional access to the cell. Based on the calculated steady-state voltage error, the shape of the rising phase of ICa, the steady-state activation curve (Fig. 2B), and the I-V plot (Fig. 2C) the quality of the voltage clamp appears to have been adequate for the experiments in spite of the high Rs in such recordings. As shown in Fig. 2C, the inactivation time constant τ and the peak ICa amplitude were not significantly different between the perforated and whole-cell recordings (perforated: for Vstep=+10 mV, Ipeak= −273 ± 57 pA, τ= 40 ± 1.4 ms, n= 4; whole-cell: for Vstep=+10 mV, Ipeak= −308 ± 22 pA, τ= 42.4 ± 4.5 ms, n= 8).
Figure 2. Inactivation of HVA ICa in perforated patch recordings that preserve the endogenous Ca2+ buffering.

A, the perforated patch method was used to elicit Ca2+ currents which were analysed in the same way as the whole-cell currents. The fit to the maximal ICa yielded the following values: τ= 49 ms, a = −234 pA, c = −114 pA. B, the normalized peak conductance g is plotted as a function of the step potential and fitted with a single Boltzmann function (dashed line). The V½ is −4.3 ± 0.4 mV, not significantly different from the V½ obtained for the whole-cell recordings. C, the peak Ca2+ current amplitude and the decay time constant (τ) for perforated patch recordings (n= 4) are plotted as functions of step voltages. Note the similarity to the I-V and τvs.V relationships obtained for the whole-cell recordings (Fig. 1C).
Having shown that the inactivation process is intact in buffer-free whole-cell recording, we further probed the Ca2+ dependence of ICa inactivation in this configuration by perfusing the cells with equimolar Ba2+ substituted for Ca2+ (n= 6) or added 5 mm BAPTA, a rapid Ca2+ chelator, to the pipette solution (n= 5). The normalized current traces in Fig. 3A show that both conditions effectively reduced the ICa inactivation. BAPTA increased the decay time constant associated with the maximal ICa to 66 ± 4 ms and Ba2+ increased it to 85 ± 17 ms. Both values were significantly larger than the control τ. Furthermore, under these recording conditions, τ no longer displayed the pronounced U-shaped dependency on membrane voltage (Fig. 3B). The addition of BAPTA to the internal solution also affected the ICa amplitudes, which were significantly larger (Ipeak= 429 ± 45 pA) than in control recordings. The peak I-V curve for Ba2+ currents (Fig. 3B) reflects the characteristic left shift in IBa activation and increased channel permeability for Ba2+ over Ca2+ (McDonald, Pelzer, Trautwein & Pelzer, 1994).
Figure 3. Inactivation of Ca2+ currents is Ca2+ dependent and sensitive to intracellular Ca2+ buffering.
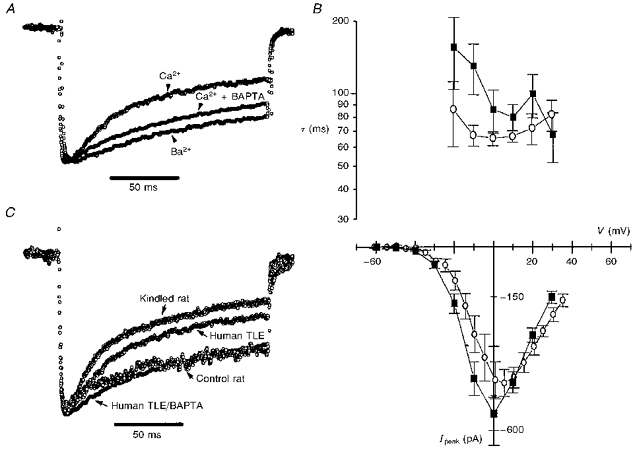
A, the whole-cell currents were recorded under different ionic and intracellular buffering conditions, and are shown here normalized. One trace (control current) was recorded with 5 mm Ca2+ in the extracellular solution and no Ca2+ buffer in the internal solution, the second was obtained with 5 mm Ca2+ outside and 5 mm BAPTA inside the pipette, and the third was recorded after equimolar replacement of Ca2+ with Ba2+ in the extracellular solution and no Ca2+ buffer inside. B,I-V plot for the Ba2+ (▪) and Ca2+ with BAPTA inside (○) currents and the voltage dependence of the associated decay time constants τ. Note that the τ values are elevated for the Ba2+ and BAPTA cases compared with the Ca2+ case and have lost the characteristic U-shape. C, the time course of inactivation of HVA Ca2+ currents is similar in granule cells obtained from human TLE and experimental animal model of TLE (kindling). Representative traces of normalized whole-cell Ca2+ currents recorded from epileptic human, control and kindled rat granule cells are superimposed. The recording conditions are the same as those described in Fig. 1A.
Without access to human control granule cells which have been shown to contain CB (Maglóczky et al. 1997), we compared the ICa recorded in human TLE neurones to Ca2+ currents recorded in DGGCs acutely dissociated using the same protocols from hippocampal slices prepared from kindled and control rats. Representative HVA Ca2+ current traces recorded under identical whole-cell voltage-clamp conditions from epileptic human, control and kindled (epileptic) rat DGGCs are shown superimposed in Fig. 3C. The time course of ICa inactivation is comparable between kindled rat and epileptic human and between control rat and 5 mm BAPTA-loaded epileptic human dentate gyrus granule cells.
DISCUSSION
We have described a significant Ca2+-dependent component contributing to the inactivation of HVA calcium currents in human granule cells obtained from TLE patients. Moreover, we have compared the time course of current inactivation in human epileptic, kindled and control rat DGGCs.
Two previous studies have examined calcium currents in acutely dissociated human neurones obtained from TLE patients (Sayer, Brown, Schwindt & Crill, 1993). Unlike Beck, Steffens, Heinemann & Elger (1997b), we did not detect a T-type ICa component. This lack may be due to minor differences in the dissociation procedure which may differentially preserve a T-type conductance possibly located on truncated dendrites. Alternatively, the holding potentials employed in our study may have inactivated a large proportion of the T channels. This latter possibility is, however, not supported by our recordings (n= 2) in which we failed to observe a T-type conductance in spite of a −90 mV holding potential. Given the scarcity of human dissociated neurones, we did not use specific pharmacological tools to separate the underlying Ca2+ conductances. While in guinea-pig DGGCs at least four different ICa components (T, L, N and P) were demonstrated (Eliot & Johnston, 1994), under our experimental conditions the predominant source of Ca2+ influx into dissociated human granule cells is a HVA ICa composed of up to three Ca2+ conductances. Our recordings also differ from those of Beck et al. (1997b) in the substantially smaller peak IBa densities (60 ± 6 pA pF−1versus the 144.3 ± 24.3 pA pF−1 in their report). This difference may stem from their use of 10 mm intracellular EGTA, thereby possibly lowering resting [Ca2+] and consequently basal Ca2+-dependent inactivation (see below). However, the absolute peak ICa amplitudes in the presence of intracellular BAPTA in our recordings are quite similar to the currents measured by Sayer et al. (1993) in neocortical neurones.
Previous studies of Ca2+ currents in human neurones have not addressed the Ca2+ dependence of ICa inactivation. We specifically examined this issue in light of the anatomical study of Maglóczky et al. (1997) describing the loss of CB, a putative intraneuronal Ca2+ buffer (Chard et al. 1995; Li, Decavel & Hatton, 1995; Airaksinen, Eilers, Garaschuk, Thoenen, Konnerth & Meyer, 1997). The inactivation of Ca2+ currents observed in our recordings fulfils all criteria established for the Ca2+ dependency of ICa inactivation (Chad, 1989): (1) the presence of a U-shaped relationship between inactivation time constants and membrane voltages, (2) the correlation between the amount of Ca2+ entry and τ, (3) the suppression of inactivation by enhancing internal Ca2+ buffering, and (4) the divalent ion specificity of inactivation (Ca2+ > Ba2+).
In addition to slowing the decay of ICa, the intracellular presence of Ca2+-free BAPTA is expected to reduce resting free [Ca2+]. This would tend to diminish the level of basal ICa inactivation by resting [Ca2+] and should lead to an increase in the peak ICa. Indeed, intracellular BAPTA significantly increased peak ICa amplitudes. Since this effect of BAPTA was not observed in kindled rat neurones (Köhr & Mody, 1991), it might reflect a difference in the endogenous steady-state Ca2+ buffering between human TLE and kindled rat DGGCs.
Our findings are of interest in the context of TLE, in which CB is selectively lost from the DGGCs (Maglóczky et al. 1997). As shown by our study, a functional consequence of this loss may be an increased negative feedback on Ca2+ entry through HVA channels. A similar outcome was postulated for the kindled rat model, where CB is also selectively lost from the DGGCs (Baimbridge, 1992) and ICa inactivation rates are enhanced (Köhr & Mody, 1991). The comparable time courses of ICa inactivation (Fig. 3C) and the restored inactivation by exogenous internal Ca2+ buffering in human TLE and kindled rat neurones (Köhr & Mody, 1991)underscore the similarities between cellular alterations in human TLE and kindled DGGCs.
Whereas CB is not expected to affect the resting level of free [Ca2+] in normal human DGGCs, it might critically restrict the dynamic free [Ca2+] profile at the point of Ca2+ entry since it is present in a high concentration (as much as a few hundred micromolar), has four or six functional EF-hand Ca2+ binding domains (Chard, Bleakman, Christakos, Fullmer & Miller, 1993), and is associated with membranes (Winsky & Kuznicki, 1995). Thus, CB may effectively compete for entering Ca2+ ions with the EF-hand Ca2+ binding site on the channel (de Leon et al. 1995) or with other Ca2+ binding sites with similar affinities (Zhou et al. 1997) thought to be responsible for inactivation.
According to our studies, an epilepsy-associated loss of CB may be responsible for the enhanced negative feedback on ICa. This process may represent a neuroprotective mechanism aimed at reducing potentially cytotoxic Ca2+ loads following prolonged trains of action potentials known to occur during seizures. Furthermore, a decrease in endogenous Ca2+ buffering could promote the activation of a Ca2+-dependent K+ conductance in epileptic DGGCs (Beck, Clusmann, Kral, Schramm, Heinemann & Elger, 1997a) thereby invoking another mechanism to protect the neurones from hyperexcitability.
Acknowledgments
This study was supported by NIH/NINDS grant NS27528 to I. M. The human tissue was kindly provided by Dr Itzak Fried (Division Of Neurosurgery, UCLA) and Dr Masako Isokawa (Brain Research Institute, UCLA), members of the NINDS Program Project ‘A Clinical Research Program for the Partial Epilepsies’ (Dr Jerome Engel Jr, Program Director). We appreciate B. Oyama's technical assistance. We would like to thank David DiGregorio for helpful comments on the manuscript.
References
- Airaksinen MS, Eilers J, Garaschuk O, Thoenen H, Konnerth A, Meyer M. Ataxia and altered dendritic calcium signaling in mice carrying a targeted null mutation of the calbindin-D28k gene. Proceedings of the National Academy of Sciences of the USA. 1997;94:1488–1493. doi: 10.1073/pnas.94.4.1488. 10.1073/pnas.94.4.1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baimbridge KG. Calcium-binding proteins in the dentate gyrus. Epilepsy Research Supplement. 1992;7:211–220. [PubMed] [Google Scholar]
- Beck H, Clusmann H, Kral T, Schramm J, Heinemann U, Elger CE. Potassium currents in acutely isolated human hippocampal dentate granule cells. Journal of Physiology. 1997a;498:73–85. doi: 10.1113/jphysiol.1997.sp021842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beck H, Steffens R, Heinemann U, Elger CE. Properties of voltage-activated Ca2+ currents in acutely isolated human hippocampal granule cells. Journal of Neurophysiology. 1997b;77:1526–1537. doi: 10.1152/jn.1997.77.3.1526. [DOI] [PubMed] [Google Scholar]
- Bezanilla F, Armstrong CM. Inactivation of the sodium channel. I. Sodium current experiments. Journal of General Physiology. 1977;70:549–566. doi: 10.1085/jgp.70.5.549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branchaw JL, Banks MI, Jackson MB. Ca2+- and voltage-dependent inactivation of Ca2+ channels in nerve terminals of the neurohypophysis. Journal of Neuroscience. 1997;17:5772–5781. doi: 10.1523/JNEUROSCI.17-15-05772.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brehm P, Eckert R. Calcium entry leads to inactivation of calcium channel in Paramecium. Science. 1978;202:1203–1206. doi: 10.1126/science.103199. [DOI] [PubMed] [Google Scholar]
- Chad J. Inactivation of calcium channels. Comparative Biochemistry and Physiology. 1989;A 93:95–105. doi: 10.1016/0300-9629(89)90196-5. [DOI] [PubMed] [Google Scholar]
- Chard PS, Bleakman D, Christakos S, Fullmer CS, Miller RJ. Calcium buffering properties of calbindin D28k and parvalbumin in rat sensory neurones. Journal of Physiology. 1993;472:341–357. doi: 10.1113/jphysiol.1993.sp019950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chard PS, Jordan J, Marcuccilli CJ, Miller RJ, Leiden JM, Roos RP, Ghadge GD. Regulation of excitatory transmission at hippocampal synapses by calbindin D28k. Proceedings of the National Academy of Sciences of the USA. 1995;92:5144–5148. doi: 10.1073/pnas.92.11.5144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon M, Wang Y, Jones L, Perez-Reyes E, Wei X, Soong TW, Snutch TP, Yue DT. Essential Ca2+-binding motif for Ca2+-sensitive inactivation of L-type Ca2+ channels. Science. 1995;270:1502–1506. doi: 10.1126/science.270.5241.1502. [DOI] [PubMed] [Google Scholar]
- Eliot LS, Johnston D. Multiple components of calcium current in acutely dissociated dentate gyrus granule neurons. Journal of Neurophysiology. 1994;72:762–777. doi: 10.1152/jn.1994.72.2.762. [DOI] [PubMed] [Google Scholar]
- Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science. 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Gibbons SJ, Brorson JR, Bleakman D, Chard PS, Miller RJ. Calcium influx and neurodegeneration. Annals of the New York Academy of Sciences. 1993;679:22–33. doi: 10.1111/j.1749-6632.1993.tb18286.x. [DOI] [PubMed] [Google Scholar]
- Isokawa M, Avanzini G, Finch DM, Babb TL, Levesque MF. Physiologic properties of human dentate granule cells in slices prepared from epileptic patients. Epilepsy Research. 1991;9:242–250. doi: 10.1016/0920-1211(91)90058-n. 10.1016/0920-1211(91)90058-N. [DOI] [PubMed] [Google Scholar]
- Kennedy MB. Regulation of neuronal function by calcium. Trends in Neurosciences. 1989;12:417–420. doi: 10.1016/0166-2236(89)90089-1. 10.1016/0166-2236(89)90089-1. [DOI] [PubMed] [Google Scholar]
- Köhr G, Lambert CE, Mody I. Calbindin-D28K (CaBP) levels and calcium currents in acutely dissociated epileptic neurons. Experimental Brain Research. 1991;85:543–551. doi: 10.1007/BF00231738. [DOI] [PubMed] [Google Scholar]
- Köhr G, Mody I. Endogenous intracellular calcium buffering and the activation/inactivation of HVA calcium currents in rat dentate gyrus granule cells. Journal of General Physiology. 1991;98:941–967. doi: 10.1085/jgp.98.5.941. 10.1085/jgp.98.5.941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZH, Decavel C, Hatton GI. Calbindin-D28k: role in determining intrinsically generated firing patterns in rat supraoptic neurones. Journal of Physiology. 1995;488:601–608. doi: 10.1113/jphysiol.1995.sp020993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald TF, Pelzer S, Trautwein W, Pelzer DJ. Regulation and modulation of calcium channels in cardiac, skeletal, and smooth muscle cells. Physiological Reviews. 1994;74:365–507. doi: 10.1152/physrev.1994.74.2.365. [DOI] [PubMed] [Google Scholar]
- Maglóczky Z, Halász P, Vajda J, Czirják S, Freund TF. Loss of calbindin-D28k immunoreactivity from dentate granule cells in human temporal lobe epilepsy. Neuroscience. 1997;76:377–385. doi: 10.1016/s0306-4522(96)00440-x. 10.1016/S0306-4522(96)00440-X. [DOI] [PubMed] [Google Scholar]
- Miller RJ. The control of neuronal Ca2+ homeostasis. Progress in Neurobiology. 1991;37:255–285. doi: 10.1016/0301-0082(91)90028-y. 10.1016/0301-0082(91)90028-Y. [DOI] [PubMed] [Google Scholar]
- Neher E. Correction for liquid junction potentials in patch clamp experiments. Methods in Enzymology. 1992;207:123–131. doi: 10.1016/0076-6879(92)07008-c. [DOI] [PubMed] [Google Scholar]
- Rae J, Cooper K, Gates P, Watsky M. Low access resistance perforated patch recordings using amphotericin B. Journal of Neuroscience Methods. 1991;37:15–26. doi: 10.1016/0165-0270(91)90017-t. 10.1016/0165-0270(91)90017-T. [DOI] [PubMed] [Google Scholar]
- Sayer RJ, Brown AM, Schwindt PC, Crill WE. Calcium currents in acutely isolated human neocortical neurons. Journal of Neurophysiology. 1993;69:1596–1606. doi: 10.1152/jn.1993.69.5.1596. [DOI] [PubMed] [Google Scholar]
- Winsky L, Kuznicki J. Distribution of calretinin, calbindin D28k, and parvalbumin in subcellular fractions of rat cerebellum: Effects of calcium. Journal of Neurochemistry. 1995;65:381–388. doi: 10.1046/j.1471-4159.1995.65010381.x. [DOI] [PubMed] [Google Scholar]
- Zhou J, Olcese R, Qin N, Noceti F, Birnbaumer L, Stefani E. Feedback inhibition of Ca2+ channels by Ca2+ depends on a short sequence of the C terminus that does not include the Ca2+-binding function of a motif with similarity to Ca2+-binding domains. Proceedings of the National Academy of Sciences of the USA. 1997;94:2301–2305. doi: 10.1073/pnas.94.6.2301. 10.1073/pnas.94.6.2301. [DOI] [PMC free article] [PubMed] [Google Scholar]