

Abstract
The whole-cell patch clamp was employed to study Na+-K+ pump current (Ip) in acutely isolated myocytes. α-Adrenergic receptors were activated with noradrenaline (NA) after blocking β-adrenergic receptors with propranolol. Ip was measured as the current blocked by strophanthidin (Str).
Activation of α-receptors by NA increased Ip in a concentration-dependent manner. The K0.5 depended on intracellular calcium ([Ca2+]i), however maximal stimulation did not. At 15 nm[Ca2+]i the K0.5 was 219 nm NA whereas at 1.4 μm [Ca2+]i it was 3 nm.
The voltage dependence of Ip was not shifted by NA at either high or low [Ca2+]i. At each voltage, maximal stimulation of Ip was 14–15 %.
Staurosporine (St), an inhibitor of protein kinase C (PKC), eliminated the α-receptor-mediated stimulation of Ip at either high or low[Ca2+]i.
The stimulation of Ip was independent of changes in intracellular sodium or external potassium concentrations, and did not reflect a change in affinity for Str.
Phenylephrine, methoxamine and metaraminol, three selective α1-adrenergic agonists, stimulate Ip in a similar manner to NA. Stimulation of Ip by NA was eliminated by prazosin, an α1-antagonist, but was unaffected by yohimbine, an α2-antagonist.
We conclude noradrenaline activates ventricular α1-receptors, which are specifically coupled via PKC to increase Na+-K+ pump current. The sensitivity of the coupling is [Ca2+]i dependent, however the maximal increase in pump current is [Ca2+]i and voltage independent.
The Na+-K+ pump is an electrogenic ion transporter in the plasma membrane of all mammalian cells. Its primary role is to maintain low intracellular sodium and high intracellular potassium; however, in the heart it has other important functions. It hyperpolarizes the cardiac cell by producing a net outward current, Ip, that has a significant effect on the duration of the plateau phase of the cardiac action potential (Gadsby & Cranefield, 1980). Modulation of Ip therefore affects myocardial functions like rhythm, conduction, and force of contraction.
The sympathetic nervous system is a major regulator of the Na+-K+ pump in cardiac muscle, as previously reported by Falk & Cohen (1984) and Desilets & Baumgarten (1988), and in work from our laboratory. For example, β-receptor activation by isoprenaline (Iso) at low [Ca2+]i inhibited Ip whereas at high [Ca2+]i inhibition was eliminated but Iso shifted the I-V relationship of Ip in the negative direction on the voltage axis (Gao, Mathias, Cohen, Shi & Baldo, 1996). Both effects were mediated by a phosphorylation step via the cAMP-dependent PKA pathway (Gao, Mathias, Cohen & Baldo, 1992; Gao, Cohen, Mathias & Baldo, 1994). Acetylcholine (ACh) has no effect on Ip in the absence of Iso, but reverses the β-effects (Gao, Mathias, Cohen & Baldo, 1997a).
During the last decade, there has been growing evidence for modulation of Ip by α-adrenergic receptors. Studies in canine Purkinje myocytes and fibres (Shah, Cohen, Rosen, 1988; Zaza, Kline & Rosen, 1990), rat papillary muscle (Ertl, Jahnel, Nawrath, Carmeliet & Vereecke, 1991), rat atrial muscle (Terzic, Anagnostopoulos & Vogel, 1991), beating guinea-pig ventricular muscle (Wilde & Kleber, 1991), and rat ventricular myocytes (Williamson, Kennedy, Seifen, Lindemann & Stimers, 1993) suggest α-activation stimulates Ip. But all these studies used indirect measurements, such as the ouabain-sensitive hyperpolarization, change in holding current or change in [Na+]i induced by α-activation. Moreover, Tohse, Hattori, Nakaya & Kanno (1987a) reported that stimulation of the Na+-K+ pump was not involved in the α-mediated hyperpolarization in quiescent rat papillary muscle. Thus the data are indirect and in some cases contradictory. The purpose of this study is to measure directly changes in pump current resulting from α-activation, to determine whether these changes are specifically or indirectly coupled to α-receptors and to determine which α-receptor subtype is involved and which intracellular cascade mediates the effect.
METHODS
Studies were performed on single ventricular myocytes that were enzymatically isolated from guinea-pig hearts (Gao et al. 1992). Male guinea-pigs, weighing 300-500 g, were killed with sodium pentobarbitone solution (1 ml of 360 mg ml−1) by peritoneal injection. The isolated cells were stored for use the following day at 4°C in Kraft-Brühe (KB) solution containing (mm): K2HPO4, 30; KCl, 83; MgSO4, 5; sodium pyruvic acid, 5; β-hydroxybutyric acid, 5; creatine, 5; taurine, 20; glucose, 10; Hepes, 5; EGTA, 0.5; KOH, 2; Na2-ATP, 5; pH 7.2.
Cells were placed in a 0.65 ml lucite bath, which could be superfused at room temperature (25-27°C) with a solution flow of 3 ml min−1. Solutions were changed with a manual valve leading to the bath; the dead space was 0.15 ml. An Axopatch-1B amplifier (Axon Instruments) and the whole-cell patch-clamp technique were employed to observe cell membrane current. Patch pipette resistances were 1-3 MΩ prior to sealing. Myocytes were clamped to 0 mV and Ip was defined as the inward shift in holding current observed during bath application of 0.5 mm strophanthidin (Str), a selective and reversible inhibitor of the Na+-K+ pump. All patch-clamp data were displayed on a digital storage oscilloscope and recorded on computer disk for later analysis. The sampling rate was 500 ms point−1 and the data were low-pass filtered at 10 Hz during steady-state recording. For each experiment both control and test pump currents were obtained in the same cell and the ratio of the test Ip to control Ip was calculated. All values are given as means ± standard deviation.
The patch electrode solution contained (mm): sodium aspartic acid, 50; potassium aspartic acid, 20; CsOH, 30; TEACl, 20; MgSO4, 5; Hepes, 5; EGTA, 11; glucose, 10; Na2-ATP, 5; GTP, 0.2; CaCl2, 1 (giving a free [Ca2+]i of 15 nm), and pH was adjusted to 7.2 using βCsOH. The superfusion solutions include control and test solution. The former was Tyrode solution containing (mm): NaCl, 137.7; KCl, 5.4; NaOH, 2.3; MgCl2, 1; glucose, 10; Hepes, 5; BaCl2, 2; CdCl2, 1; propranolol, 0.1; pH 7.4. The latter was prepared by adding α-agonist, α-agonist plus α-antagonist and/or the pump current inhibitor to the control solution. With the exception of the concentration-dependence studies, the α-agonist in test solutions was 10 μm noradrenaline (NA; Sigma), 100 μm phenylephrine (Sigma), 100 μm metaraminol (Sigma), or 100 μm methoxamine (Sigma). The α1-antagonist was 10 μm prazosin (Pz). The α2-antagonist was 1 μm yohimbine (Yo). The inhibitor of PKC was 1.5 μm staurosporine (St). The inhibitor of Ip was 0.5 mm strophanthidin (Str). Staurosporine and strophanthidin were prepared as stock solutions by dissolving them in dimethyl sulphoxide (DMSO) and diluting to the final concentrations with external solution before the start of the experiment. The final concentration of DMSO was less than 0.41 %, which does not affect Ip (Gadsby & Nakao, 1989).
Pipette [Na+] of 60 mm was used to nearly saturate the intracellular Na+-binding sites of the Na+-K+ pump (Nakao & Gadsby, 1989; Gao et al. 1995). Moreover, this increases our signal-to-noise ratio and minimizes the effect of changes in intracellular [Na+] on Ip. The Cs+ and TEA+ in the pipette solution and Ba2+ in the external solution were used to eliminate K+ conductance. Cd2+ in the external solution was to block Ca2+ channel current (carried by Ba2+) and the Na+-Ca2+ exchanger. Propranolol was included in the superfusion solution to block the effects of β-receptors. Some experiments were performed with 100 mm instead of 60 mm pipette [Na+], or 1.4 μm (high calcium) instead of 15 nm Ca2+ (low calcium) in the pipette solution, or 15.4 mm external KCl (high [K+]o) instead of 5.4 mm (normal [K+]o), or 1 mm Str instead of 0.5 mm Str. For the 100 mm pipette [Na+], the pipette [K+] and [Cs+] were each reduced by 20 mm and replaced with [Na+]. The high calcium was achieved using 10 mm instead of 1 mm pipette CaCl2 with 11 mm EGTA (Gao et al. 1992). The high [K+]o was achieved by substituting KCl for external NaCl. The high Str was simply added to the external Tyrode solution.
RESULTS
NA increases Ip in a concentration-dependent manner
After whole-cell recording was initiated, a period of at least 5 min was required for the pipette and intracellular contents to come to steady state (Gao et al. 1992). When steady state was achieved, 0.5 mm Str was applied and the holding current shifted inward as Ip was blocked (Fig. 1A). This change in holding current was reversed upon washout of Str and was considered to reflect control Ip(con). After washout and once the current had again stabilized, a test concentration of NA was applied. This resulted in a small outward shift in holding current, probably due to activation of Ip. After a new steady state was achieved, 0.5 mm Str was again applied. This application resulted in a larger inward current shift as Ip(NA) was blocked. We examined the concentration dependence of the NA-induced increase in Ip at 0 mV and 15 nm[Ca2+]i. Figure 1A shows four original recordings of holding current in the presence of different concentrations of NA (0.1, 1.0, 5.0 and 10 μm). The records shown in Fig. 1A are the first two applications of Str taken from more complex records involving several interventions (see Fig. 8 or Table 1 for some examples). For all the experiments summarized in Fig. 1B, we were able to determine that the NA effect was reversible and there was no systematic run-down of Ip. In five cells, we extended the period of application of 10 μm NA to approximately 4 min before applying Str. The effect on Ip, however, did not appear to be time dependent, since the mean stimulation (14.5 ± 1 %) was not significantly different from that determined with shorter applications (see Fig. 1B). Figure 1B illustrates the mean data from a range of NA concentrations. The continuous line shows the best fit of a standard one-to-one binding curve that was used to estimate the concentration of NA at half-maximal stimulation (K0.5). Clearly, there are many steps between NA application and Ip stimulation so this is an effective K0.5 for the stimulation and does not necessarily reflect NA binding. The maximal stimulation of Ip (at 10 μm NA in 8 cells) was 14.4 ± 2 %. The K0.5 for the effect of NA was 219 nm.
Figure 1. The concentration dependence of NA-mediated stimulation of Ip.
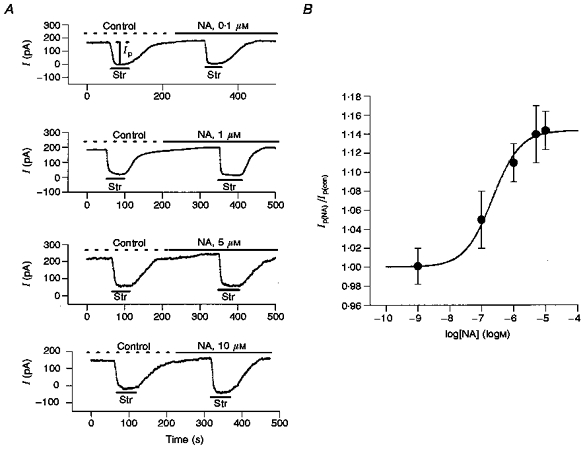
A, holding currents measured in different concentrations of NA. The myocyte membrane voltage was held at 0 mV. The control solution was normal Tyrode solution plus Ba2+, Cd2+ and propranolol (see Methods); the pipette solution contained 15 nm Ca2+. In each trace, the upper horizontal line indicates the time of application of the control solution (Control) and the noradrenaline-containing solution (NA). The lower horizontal bars show the time of application of 0.5 mm strophanthidin (Str). In the upper trace, the vertical bar illustrates the measured Ip amplitude. In the 4 experiments shown, the effect of NA was to increase Ip in each cell by approximately the mean values graphed in panel B. B, the concentration-response curve for the stimulation of Ip by NA. The ratios of Ip in NA to those in control solution (Ip(NA)/Ip(con)) were 1.00 ± 0.02 in 0.001 μm NA in 7 experiments, 1.05 ± 0.03 in 0.1 μm NA in 13 experiments, 1.11 ± 0.02 in 1 μm NA in 7 experiments, 1.14 ± 0.03 in 5 μm NA in 6 experiments, 1.14 ± 0.02 in 10 μm NA in 8 experiments. Error bars indicate s.d.
Figure 8. The effects of NA on Ip are eliminated by prazosin (Pz), a specific α1-antagonist, but unaffected by yohimbine (Yo), a specific α2-antagonist.
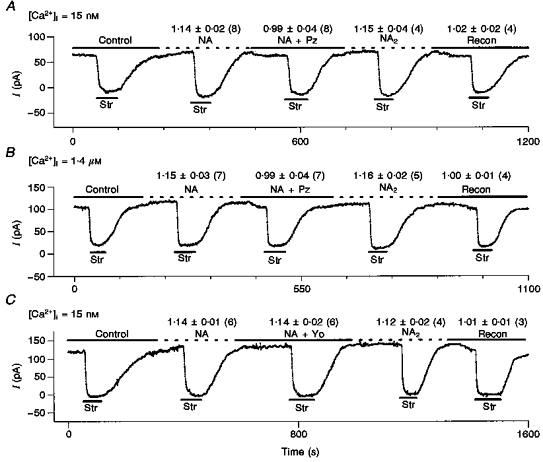
Above each test condition, we report: mean Ip(test)/Ip(con)±s.d. (number of cells). A, a typical record in low (15 nm) [Ca2+]i. We measured control Ip(con), applied 10 μm NA and measured Ip(NA), added 10 μm prazosin to the NA-containing solution and measured Ip(NA+Pz), washed out the prazosin and remeasured Ip(NA2), and lastly washed out the NA and remeasured control Ip(recon), In all experiments reported, we measured Ip(con), Ip(NA) and IP(NA+Pz), but in half of these we lost the patch before obtaining our second measurements Ip(NA2) and Ip(recon). B, the same protocol and information are illustrated except [Ca2+]i was 1.4 μm and NA was 1 μm. C, the protocol is the same as in A, except the α2-antagonist yohimbine (Yo) is substituted for Pz. However, 1 μm Yo does not antagonize the NA-mediated stimulation of Ip. These data provide direct evidence that the effects of NA on Ip in ventricle are mediated by the α1-receptor subtype. Moreover, in the 13 cells in which we were able to record Ip(NA2)/Ip(con), the ratio is within 1 % of the initial Ip(NA)/Ip(con), and in the 11 cells in which we completed the protocol, Ip(recon) is within 2 % of the initial Ip(con). Thus, our measurement of the NA effect is reproducible and run-down of Ip is negligible.
Table 1.
The α1-antagonist prazosin (Pz) eliminates the stimulation of Ip by α1-agonists
| α-Agonist (A) | Ip(A)/Ip(con) | Ip(A+Pz)/Ip(con) | Ip(A2)/Ip(con) | Ip(recon)/Ip(con) |
|---|---|---|---|---|
| Phenylephrine, 100 μm | 1.14 ± 0.04 (6) | 1.01 ± 0.05 (6) | 1.13 ± 0.05 (5) | 1.02 ± 0.03 (5) |
| Methoxamine, 100 μm | 1.13 ± 0.03 (17) | 0.99 ± 0.04 (17) | 1.14 ± 0.05 (8) | 1.00 ± 0.03 (8) |
| Metaraminol, 100 μm | 1.15 ± 0.05 (8) | 1.00 ± 0.04 (8) | 1.14 ± 0.07 (7) | 1.02 ± 0.02 (7) |
The increase in Ip is specifically coupled to α-adrenergic activation
In most experiments, we measured Ip using 0.5 mm Str, which yields 93-96 % pump inhibition (Gadsby & Nakao, 1989). Thus, one non-specific mechanism of stimulation could be a NA-induced change in affinity of the Na+-K+ pump for Str, though one would expect such a stimulation to be limited to less than 7 %. Nevertheless, we tested this possibility by increasing the concentration of Str to 1 mm and again determining the effect of 10 μm NA on Ip. Figure 2 illustrates the pump current measured with 0.5 mm and 1 mm Str before and after application of NA. In 1 mm Str the ratio Ip(NA)/Ip(con) was 1.14 ± 0.04 (9 cells), which is not significantly different from the Ip(NA)/Ip(con) determined with 0.5 mm Str (1.14 ± 0.02, in 8 cells), suggesting that the stimulatory effect of NA on Ip is not caused by a NA-induced change in the affinity of Na+-K+ pump for Str.
Figure 2. NA effects on Ip are not due to a change in the affinity of Na+-K+ pumps for Str.
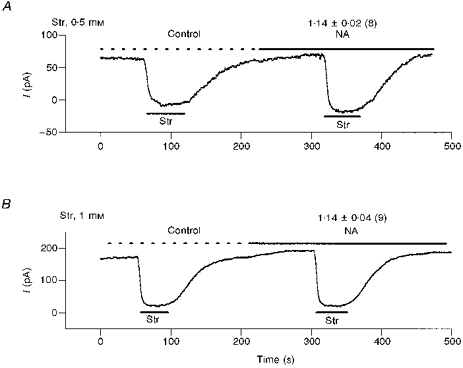
A, in 8 cells we used 0.5 mm Str to inhibit Ip and record control pump current (Ip(con)) and pump current in 10 μm NA (Ip(NA)). The ratio Ip(NA)/Ip(con) was 1.14 ± 0.02. B, in 9 cells we used 1.0 mm Str to inhibit Ip and record control pump current (Ip(con)) and pump current in 10 μm NA (Ip(NA)). The ratio Ip(NA)/Ip(con) was 1.14 ± 0.04. Thus, the Str-measured stimulation of Ip by NA does not depend on the concentration of Str used to block Ip. This suggests the stimulation is not through an effect of NA on the affinity of the Na+-K+ pump for Str.
Since Ip is a saturable function of intracellular sodium ([Na+]i), Ip might be influenced by changes in [Na+]i. However, in these experiments we used patch pipettes containing 60 mm[Na+], which should nearly saturate the internal Na+-binding sites of the Na+-K+ pump in guinea-pig ventricular myocytes (Nakao & Gadsby, 1989; Gao et al. 1995). This should prevent changes in Ip due to NA-related effects on membrane Na+ conductance and consequent changes in [Na+]i. Moreover, insofar as [Na+]i is saturating, the NA effect we observed (Ip stimulation) is not due to an increase in the intracellular Na+ affinity of the pump. To verify that our observations are [Na+]i independent, we repeated the 10 μm NA study in five cells with pipette [Na+] at 100 mm. In the presence of NA, Ip was increased by 14.4 ± 1 %, which is the same result obtained with 60 mm pipette [Na+].
Ip is also a saturable function of extracellular potassium ([K+]o) (Nakao et al. 1989; Gao et al. 1995), so Ip might be influenced by local accumulation/depletion of K+ in the transverse tubules. Gao et al. (1995) estimated changes in tubular K+ to be small, of the order of 0.1 mm, however no direct measurement was made. Hence we tested the possible stimulation of Ip due to a NA-related increase in tubular K+. Figure 3 illustrates three typical experiments carried out in 15.4 mm external KCl, which should nearly saturate the external K+ site of the Na+-K+ pump. If a NA-related increase in membrane K+ conductance were causing sufficient tubular K+ accumulation to stimulate Ip in normal 5.4 mm[K+]o, the higher [K+]o should eliminate the stimulation. However, the mean Ip(NA)/Ip(con) at 15.4 mm[K+]o was 1.06 ± 0.02 in 0.1 μm NA (8 cells); 1.11 ± 0.02 in 1 μm NA (6 cells); 1.15 ± 0.02 in 10 μm NA (6 cells); and the approximate K0.5 for the effect of NA was 166 nm. These data are not significantly different from those obtained in 5.4 mm[K+]o. We therefore conclude the stimulation of Ip by NA is not due to an increase in tubular K+ concentration.
Figure 3. NA effects on Ip are not due to changes in extracellular K+ concentration that might occur in the lumen of the transverse tubular system (T-system).
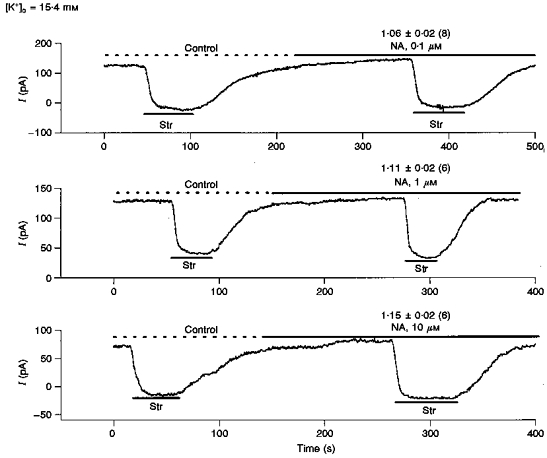
In Fig. 1, the dependence of Ip on NA was recorded with 5.4 mm[K+]o. If this observed stimulation of Ip was due to NA-related accumulation of K+ in the T-system, then increasing [K+]o to 15.4 mm should nearly saturate the K+-binding sites and eliminate the stimulation. We examine here, in 15.4 mm[K+]o, the stimulation of Ip by 0.1 μm NA (upper trace), 1.0 μm NA (middle trace) and 10 μm NA (lower trace). The mean Ip(NA)/Ip(con)±s.d. (number of cells) are shown above the NA-stimulated Ip for each concentration of NA. The maximal stimulation is about 15 % with a K0.5 of approximately 166 μm, which is nearly the same as recorded in 5.4 mm[K+]o. We conclude the effects of NA on Ip are not due to local T-system accumulation of K+.
The α-induced increase in Ip is [Ca2+]i dependent, voltage independent and mediated by PKC
We have previously reported that [Ca2+]i alters the effect of β-activation on Ip (Gao et al. 1992). The experiments summarized in Fig. 4 were designed to examine whether the stimulatory effect of α-activation on Ip is [Ca2+]i dependent. We buffered [Ca2+]i in the electrode solution to 1.4 μm free [Ca2+] (Gao et al. 1992), and recorded the effect of different concentrations of NA on Ip in the same manner as shown in Fig. 1. Figure 4 illustrates the mean data from a range of NA concentrations. The continuous lines show the best fit to the standard one-to-one binding curve at 1.4 μm and 15 nm[Ca2+]i. With 1.4 μm[Ca2+]i, the maximal stimulation of Ip (obtained at 1 μm NA) was 14.7 %. The K0.5 for the effect was at 3 nm NA. In comparison with data recorded at low [Ca2+]i, the maximal stimulation of Ip by NA was essentially unaltered, but the K0.5 was significantly reduced and the concentration-response relationship was significantly shifted to the left. These results suggest [Ca2+]i increases the sensitivity of some steps in the cascade leading to stimulation of Ip.
Figure 4. [Ca2+]i affects the sensitivity of the NA-induced stimulation of Ip but not maximal stimulation.
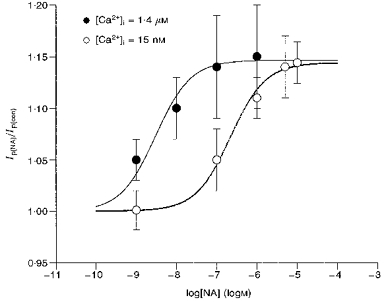
Using the same protocol shown in Fig. 1 but with [Ca2+]i buffered to 1.4 μm, we recorded the stimulation of Ip by 0.001, 0.01, 0.1 and 1.0 μm NA. The mean ratio of Ip(NA)/Ip(con)±s.d. (number of cells) was, respectively, 1.05 ± 0.02 (10), 1.10 ± 0.03 (8), 1.14 ± 0.05 (7) and 1.15 ± 0.03 (7). These data are shown as •. For purposes of comparison we also plot the data from Fig. 1 as ○. Based on the curve fit of a single binding site model (continuous lines) the maximal stimulation of Ip in 1.4 μm[Ca2+]i was 14.7 % whereas in 15 nm[Ca2+]i it was 14.4 %, and the K0.5 in 1.4 μm[Ca2+]i was 3 nm whereas in 15 nm[Ca2+]i the K0.5 was 219 nm. These data show that the maximal stimulation of 14–15 % is independent of [Ca2+]i but the sensitivity to NA increases nearly two orders of magnitude when [Ca2+]i is high. Moreover, the [Ca2+]i independence of maximal stimulation indicates propranolol has effectively blocked β-activation, since at 15 nm[Ca2+]i, β-activation inhibits Ip by about 25 %, whereas at 1.4 μm[Ca2+]i, β-activation has almost no effect at holding voltage of 0 mV. There is no indication of these β-mediated effects in the data shown here.
We have previously reported that the effects of β-adrenergic activation on Ip are voltage dependent (Gao et al. 1996). At low [Ca2+]i, β-activation inhibits Ip by approximately 25 % at all voltages, whereas at high [Ca2+]i the inhibition is eliminated but the voltage dependence of Ip is shifted by about 30 mV in the negative direction. We therefore examined the voltage dependence of the effects of α-activation on Ip when [Ca2+]i is either low (15 nm) or high (1.4 μm). The results are summarized in Fig. 5. The ramp protocol for determining the voltage dependence of Ip is illustrated in Fig. 5A and described in the legend. The left-hand panel of Fig. 5B shows the voltage dependence of Ip when [Ca2+]i is low. The upper squares (▪) are maximally NA-stimulated Ip and the lower circles (•) are the control pump currents recorded from the same cells. The right-hand panel shows that the ratio of NA-stimulated Ip to control is voltage independent when [Ca2+]i is low. Figure 5C shows the results of the same protocol when [Ca2+]i is high. Again, the ratio is voltage independent. These data suggest α-activation by NA does not shift the voltage dependence of Ip.
Figure 5. The voltage dependence of Ip is not shifted by NA when [Ca2+]i is either high or low.
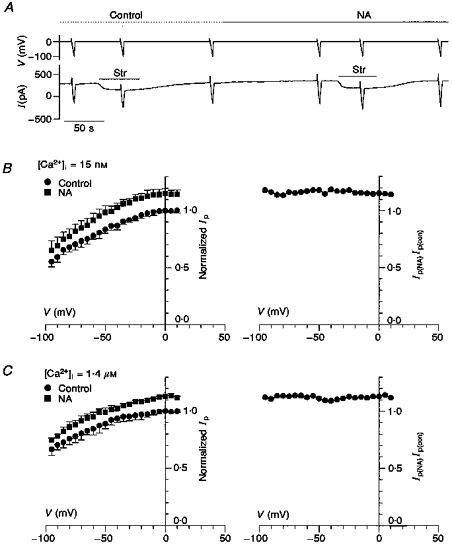
A, the ramp protocol for determining the voltage dependence of Ip. In the upper voltage trace, the cell is held at 0 mV, stepped to +20 mV then ramped to -100 mV in a period of 5 s. The lower trace is the current response to the voltage clamp protocol. The ramp protocol was done in the absence of Str where the response included the background membrane current plus Ip, in the presence of Str where the response was only the background membrane current, and lastly again in the absence of Str to verify the cell was not running down. This protocol was repeated in the presence of NA (10 μm NA in low [Ca2+]i and 1 μm NA in high [Ca2+]i). B, the voltage dependence of Ip is not shifted by NA when [Ca2+]i is low (15 nm). Ip was determined by subtracting the ramp current response in the presence of Str from that in the absence of Str. In each cell, Ip was normalized by its value at 0 mV in the absence of NA (in 5 cells the mean pump current was 170 ± 44 pA). This procedure removes cell-to-cell variation in pump density and membrane area. The normalized pump currents were averaged, and in the left-hand panel normalized Ip in the presence of NA is shown by ▪, whereas the normalized Ip in the absence of NA (Control) is shown by •. The right-hand panel graphs the ratio of the two curves shown in the left-hand panel. The lack of voltage dependence for the ratio indicates a uniform NA stimulation of about 15 % with no voltage shift. C, the voltage dependence of Ip is not shifted by NA when [Ca2+]i is high (1.4 μm). The data were recorded and analysed as described in B. In 4 cells the mean pump current at 0 mV was 160 ± 40 pA. Again, the lack of voltage dependence for the ratio of Ip(NA)/Ip(con) indicates a uniform NA stimulation with no voltage shift.
In a previous study (Gao, Mathias, Cohen, Sun & Baldo, 1997b), we found activation of PKC stimulates Ip in guinea-pig ventricular myocytes. Moreover, for a given stimulation of Ip, the concentration of PKC activator (such as phorbol 12-myristate13-acetate (PMA) and 1,2-dioctanoyl-sn-glycerol (diC8)) depends on [Ca+ 2]i. The PKC inhibitor, staurosporine, eliminated the effect of PMA or diC8 on Ip. In the present study, we have also found that the concentration of NA required for a given stimulation of Ip is [Ca2+]i dependent. These observations suggest that the α-effect on Ip might be mediated by PKC. To test this hypothesis, we used the protocol shown in Fig. 6. Ip was first measured in control conditions as the Str blockable current, then, in the same cell, 10 μm NA was applied and Ip remeasured; lastly, still in the same cell, 1.5 μm St was added to the NA-containing solution and Ip was measured a third time. As seen in the numbers above the currents, in 15 nm[Ca2+]i the ratio of Ip(NA)/Ip(con) was 1.15 ± 0.02 whereas the ratio Ip(NA+St)/Ip(con) was 1.00 ± 0.04. Similarly, in 1.4 μm[Ca2+]i in 7 cells, the ratio Ip(NA)/Ip(con) was 1.14 ± 0.02 but application of staurosporine eliminated the stimulation and Ip(NA+St)/Ip(con) was 1.02 ± 0.02. Thus PKC inhibition eliminated α-mediated stimulation of Ip. This result supports the hypothesis that the α-mediated effect on Ip is through PKC activation, which depends synergistically on [Ca2+]i.
Figure 6. The PKC inhibitor staurosporine eliminates the stimulation of Ip by α-activation when [Ca2+]i is either high or low.
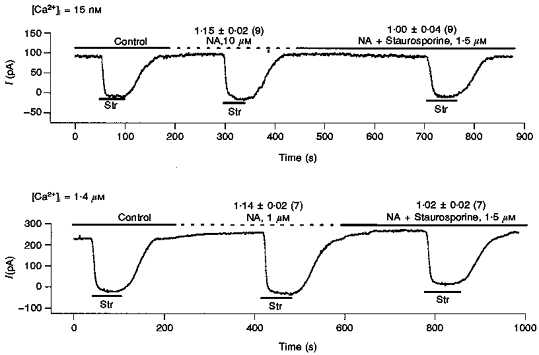
Using 0.5 mm Str, we first measured control Ip(con), then applied 10 μm NA and measured Ip(NA) and finally, with 10 μm NA present, we applied St and measured Ip(NA+St). In the upper trace with [Ca2+]i= 15 nm, in 9 cells we found Ip(NA)/Ip(con) was 1.15 ± 0.02 and Ip(NA+St)/Ip(con) was 1.00 ± 0.04. In the lower trace with 1.4 μm[Ca2+]i, in 7 cells we found Ip(NA)/Ip(con) was 1.14 ± 0.02 and Ip(NA+St)/Ip(con) was 1.02 ± 0.02. Thus, the stimulation of Ip by α-activation appears to be entirely through PKC activation, which is known to be [Ca2+]i dependent and is therefore the most likely source of the [Ca2 +]i dependence of the NA effect. Moreover, since the ratio Ip(NA+St)/Ip(con) is essentially unity, these data confirm propranolol is blocking any NA-induced β-mediated effects (see the legend to Fig. 4), as these are through PKA and not PKC (Gao et al. 1994).
The increase in Ip is coupled to the α1-receptor subtype
The literature suggests α-agonists increase Ip in other cardiac cells via α1-adrenergic receptors (Shah et al. 1988; Zaza et al. 1990; Ertl et al. 1991; Williamson et al. 1993). However, NA, the natural α-agonist, is not selective among the receptor subtypes. Thus, the next set of experiments utilized α1-specific agonists to compare their effects on Ip with that of NA. The α1-selective adrenergic agonists chosen were phenylephrine (PE), methoxamine (Meth), and metaraminol (Meta). The concentrations selected (100 μm for each) were the highest effective concentrations reported in previous studies. Figure 7 shows three recordings of holding current in the presence of 100 μm PE, Meth, and Meta, respectively. After the measurement of control Ip(con), the application of these selective α1-adrenergic agonists resulted in an outward shift in holding current similar to that observed with NA. Moreover, after a new steady state was achieved, 0.5 mm Str showed agonist pump current, Ip(A), was larger than Ip(con), and this increase in Ip reversed upon removal of the agonist. The ratio of agonist (A) to control pump current, Ip(A)/Ip(con), is listed above the agonist in Fig. 7 and the number of cells is in parentheses. The ratio of Ip(recon)/Ip(con) is also listed above the recontrol currents in Fig. 7. We were able to complete the third application of Str in twelve of the thirty-two experiments, hence the number of cells (shown in parentheses) for the recontrol is less than for the agonist, but where the patch lasted long enough to record a recontrol current there was no indication of run-down. With application of any of the three α1-adrenergic agonists, both the time course of the change in holding current and the increase in Ip are similar to those induced by NA. This implies the α-adrenergic stimulatory effect of NA on Ip is mediated via α1-adrenergic receptors.
Figure 7. Specific α1-agonists affect Ip in the same manner as NA.
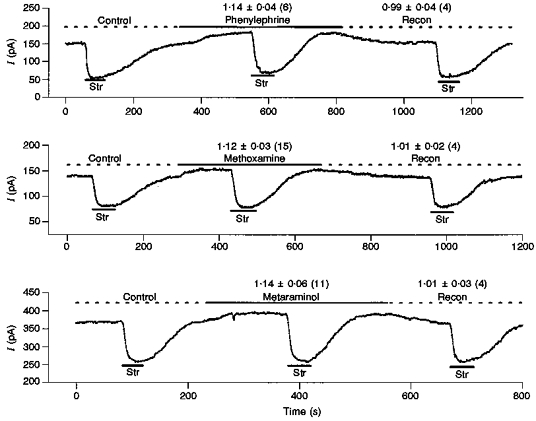
The 3 α1-specific agonists tested were phenylephrine (100 μm), methoxamine (100 μm) and metaraminal (100 μm). In all experiments [Ca2+]i was 15 nm. In each experiment we measured pump current in control Ip(con) then α1-agonist Ip(A) and if the patch survived long enough, we remeasured control Ip(recon). Above the traces we list: mean Ip(A)/Ip(con)±s.d. (number of cells). The effects of these α-agonists are indistinguishable from those of NA, suggesting NA acts in ventricle through the α1-receptor subtype.
If the α-adrenergic-mediated stimulation of Ip is via α1-receptors, prazosin (Pz), a selective α1-receptor antagonist, should completely abolish the effect. As shown in Fig. 8A, 10 μm Pz antagonizes the effects of NA on Ip in low [Ca2+]i and Fig. 8B shows the same result in high [Ca+ 2]i. We attempted to complete this same protocol with each of the α1-agonists (Table 1), however, we often lost the patch before the entire protocol could be completed. In all experiments reported in Table 1, we measured control Ip(con), agonist Ip(A) and agonist plus prazosin Ip(A+Pz). In a subset of cells we were able to remeasure agonist Ip(A2) and recontrol Ip(recon). All these results are summarized in Table 1 where mean ±s.d. values are given and the number of cells used is in parentheses. Although Pz is selective for α1-receptors, a sufficiently high concentration might bind to and competitively inhibit α2-receptors. As a control for this possibility, we used the α2-antagonist yohimbine (Yo) to demonstrate α2-receptors are not involved. Figure 8C shows the result in low [Ca2+]i. We did not repeat the study in high [Ca2+]i. The protocol is the same as with Pz except 1 μm Yo is used as the antagonist. The result is quite different, however, as the mean value of Ip(NA+Yo)/Ip(con) is the same as Ip(NA)/Ip(con), so the α2-antagonist Yo has no significant effect on the NA-mediated stimulation of Ip whereas the α1-antagonist Pz blocks the effect. These data support the hypothesis that the effects of α-activation on Ip are mediated by the α1-receptor subtype. Moreover, Fig. 8 and Table 1 document that, when the patch is stable, we can reproducibly measure α-mediated stimulation of Ip with no detectable run-down.
DISCUSSION
We have utilized the whole-cell patch-clamp technique to directly measure Ip in isolated guinea-pig ventricular myocytes. We have found the NA-mediated increase in Ip is specifically coupled to activation of α1-adrenergic receptors. The first evidence for α-adrenergic stimulation of cardiac Na+-K+ pumps was in canine Purkinje myocytes, where Shah et al. (1988) showed the phenylephrine-induced change in holding current is eliminated by dihydro-ouabain. Following this report, Zaza et al. (1990) reported α-adrenergic agonists reduce intracellular Na+ activity, a finding also consistent with stimulation of Na+-K+ pumps in Purkinje myocytes. Moreover, in isolated rat papillary muscle, hyperpolarization of the resting membrane potential induced by α-agonists was abolished in the presence of ouabain (Ertl et al. 1991), suggesting α-activation stimulates the Na+-K+ pump in this tissue as well. More recently, Williamson et al. (1993) observed an α-receptor-mediated increase in holding current in isolated rat ventricular myocytes, and this increase was eliminated by ouabain. The above results indirectly support α-adrenergic stimulation of the Na+-K+ pump. Our results are consistent with the above and more directly implicate the ventricular Na+-K+ pump as one specific target of α1-adrenergic activation. Moreover, we have shown direct evidence that all the α-effects on the Na+-K+ pumps are through PKC-mediated phosphorylation.
At present, the steps in signal transfer between ventricular α1-receptor activation and pump stimulation are largely unknown. In Purkinje myocytes the first step appears to involve a pertussis toxin-sensitive Gq protein (Shah et al. 1988). Several studies in cardiac myocytes have suggested PKC is responsible for many events caused by activation of α1-adrenergic receptors; for example, stimulation of the chloride conductance (Walsh, 1991; Ackerman & Clapham, 1993), K+ current (Tohse et al. 1987b; Tohse, Nakaya & Kanno, 1992), or Na+-H+ antiport (Gambassi, Spurgeon, Lakatta, Blank & Capogrossi, 1992). In ventricular myocytes, Gao et al. (1997b) show that PMA, a synthetic PKC activator, increases Ip, and its effect depends on [Ca2+]i, but staurosporine, a potent PKC inhibitor, eliminates the effect. These data suggest PMA and free [Ca2+]i act synergistically to activate PKC and increase Ip. In the present study, α1-adrenergic activation, like PMA, increases Ip, depends on free [Ca2+]i and its stimulatory effect on Ip is eliminated by staurosporine. This strongly suggests the effect of α1-adrenergic activation on Ip is also mediated by activation of PKC. In general, α1-receptors are thought to be coupled through a G protein to activate phospholipase C (PLC), which cleaves phosphatidylinositol 4,5-diphosphate into inositol 1,4,5-trisphosphate (InsP3) and diacylglycerol (DAG). InsP3 causes the release of intracellular Ca2+, then [Ca2+]i and DAG synergistically activate PKC (Nosek, Williams, Zeigler & Godt, 1986; Berridge, 1987, 1993; Scholz et al. 1988; Okumura, Kawai, Hashimoto, Ito, Ogawa & Satake, 1988). At the molecular level, PKC is thought to be activated when it associates with the plasma membrane and contacts DAG. Intracellular Ca2+ binds to PKC and helps anchor it to the membrane, hence a lower concentration of membrane DAG can activate PKC when [Ca2+]i is elevated (Berridge, 1987; Mosior & McLaughlin, 1991).
The above summary provides the following hypothesis for our results: in cardiac ventricular myocytes, activation of α1-adrenergic receptors is coupled, probably via a G protein as in other cells, to activation of PLC, which cleaves phosphatidylinositol to InsP3 and DAG. In the whole-cell patch-clamp mode, [Ca2+]i is strongly buffered by 11 mm EGTA, thus InsP3 has little effect on [Ca2+]i but DAG activates membrane-associated PKC. When we buffer [Ca2+]i to 1.4 μm, the association of PKC with the plasma membrane is stabilized and a relatively low membrane concentration of DAG can activate all the available PKC. As a consequence, activation of a small fraction of the α1-receptors by NA is sufficient to generate enough DAG to fully activate PKC. When we buffer [Ca2+]i to 15 nm, the association of PKC with the plasma membrane is much briefer, hence a relatively high membrane concentration of DAG is required to fully activate PKC. In this situation, a much higher concentration of NA is needed to activate a larger fraction of the α-receptors, G proteins and PLC to generate sufficient DAG to fully activate PKC. In this model, the [Ca2+]i-dependent saturable step is activation of PKC. Activation of PKC results in phosphorylation of target proteins, perhaps the Na-K+ pumps or perhaps a regulatory protein of the Na+-K+ pumps. In either event, PKC mediated phosphorylation leads to an increase in the maximal rate of Na+-K+-ATPase activity.
Although this model is consistent with the results of the present study and conforms with the conventional view of PKC activation, there are some questions that need to be addressed. First, Gao et al. (1997b) found PMA stimulates Ip by about 30 %, which is twice the stimulation elicited by α-activation, yet the PMA stimulation was eliminated by staurosporine, suggesting it was entirely mediated by PKC. It is possible that PMA has a larger effect on the maximal kinase activity of PKC, or that PMA has access to a pool of PKC inaccessible to DAG. Otherwise, this observation appears inconsistent with PKC activation being the saturable step in the experiments reported here. Second, Chibalin, Vasilets, Hennekes, Pralong & Geering (1992) have shown PKC phosphorylates the α-subunit of the Na+-K+ pump, and Vasilets & Schwartz (1992), as well as Bertorello, Aperia, Walaas, Nairn & Greengard (1991) found in vitro PKC-mediated phosphorylation of the Na+-K+pump inhibits ATPase activity whereas in ventricular myocytes, we find PKC activation stimulates ATPase activity. In ventricular myocytes, the steps following activation of PKC are apparently more complex than simple, direct phosphorylation of the pump.
Regardless of the details of the pathway, the coupling of ventricular α1-activation to an increase in Ip is physiologically important. With sympathetic activation, NA levels in the heart increase to values in the range of the [NA]-Ip response curves shown in Fig. 4 (Fujii & Vatner, 1986). As a result of sympathetic activity, heart rate increases, plateau Ca2+ current increases and diastolic interval decreases. The increase in rate is due to sympathetic effects on pacemaker cells. So in ventricular cells, sympathetic effects are to increase the plateau Ca2+ current, which tends to prolong action-potential duration, so outward currents are also increased to maintain a more constant duration of the action potential and allow a sufficient diastolic interval to fill the ventricles. To understand how the Na+-K+ pump contributes to these results, one has to consider the effects of both β- and α-adrenergic activation, their relation to [Ca2+]i and [Na+]i, and the relation of [Ca2+]i to the α- and β-mediated effects on Ip. This is obviously a complex system capable of a variety of responses to differing conditions. However, one can hypothesize an idealized situation in which there is a relatively low initial [Ca2+]i and sympathetic activation saturates both α- and β-receptors. When sympathetic tone increases, initially α- and β-effects on Ip are antagonistic and there is little change in Ip; however, the increases in Ca2+ current and heart rate cause a rise in [Ca2+]i and [Na+]i. The increase in [Na+]i causes a steady-state increase in Ip, moreover the increase in [Ca2+]i causes both β- and α-activation to synergistically further increase Ip. Since Ip is a net outward current, its increase tends to shorten the action potential and prevent an excessively short diastolic interval. Lastly, the increased Ip is a negative feedback brake on the increase in [Na+]i as well as [Ca2+]i, which depends on Na+-Ca2+ exchange. The role of the Na+-K+ pump in controlling intracellular ion concentrations is widely appreciated; however, its role as a major current shaping the cardiac action potential is not so widely discussed. In these guinea-pig ventricular myocytes, the action potential plateau repolarizes at a rate of about 85 mV s−1 and a typical cell has a capacitance of 180 pF, which gives a net outward current during the plateau of 15 pA. The total Na+-K+ pump current under normal physiological conditions and at 0 mV is on average about 100 pA, so a 15 % increase in Ip will double the rate of repolarization if all else remains equal. Thus, α- and β-mediated changes in Ip should have significant effects on the action potential shape and duration.
Acknowledgments
This work was supported by grants HL 28958, HL 54031 and HL 20558 from the National Heart, Lung and Blood Institute.
References
- Ackerman MJ, Clapham DE. Cardiac chloride channels. Trends in Cardiovascular Medicine. 1993;3:23–28. doi: 10.1016/1050-1738(93)90024-Z. 10.1016/1050-1738(93)90024-Z. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and diacyclycerol: two interacting second messengers. Annual Review of Biochemistry. 1987;56:159–193. doi: 10.1146/annurev.bi.56.070187.001111. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Inositol trisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Bertorello AM, Aperia A, Walaas SI, Nairn AC, Greengard P. Phosphorylation of the catalytic subunit of Na+, K+-ATPase inhibits the activity of the enzyme. Proceedings of the National Academy of Sciences of the USA. 1991;88:11359–11362. doi: 10.1073/pnas.88.24.11359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chibalin AV, Vasilets LA, Hennekes H, Pralong D, Geering K. Phosphorylation of Na, K-ATPase α-subunits in microsomes and in homogenates of Xenopus oocytes resulting from the stimulation of protein kinase A and protein kinase C. Journal of Biological Chemistry. 1992;267:22378–22384. [PubMed] [Google Scholar]
- Desilets M, Baumgarten CM. Isoproterenol directly stimulates the Na+-K+pump in isolated cardiac myocytes. American Journal of Physiology. 1986;251:H218–225. doi: 10.1152/ajpheart.1986.251.1.H218. [DOI] [PubMed] [Google Scholar]
- Ertl R, Jahnel U, Nawrath H, Carmeliet E, Vereecke J. Differential electrophysiologic and inotropic effects of phenylephrine in atrial and ventricular heart muscle preparations from rats. Naunyn-Schmiedeberg's Archives of Pharmacology. 1991;344:574–581. doi: 10.1007/BF00170655. [DOI] [PubMed] [Google Scholar]
- Falk RT, Cohen IS. Membrane current following activity in canine cardiac Purkinje fibers. Journal of General Physiology. 1984;83:771–799. doi: 10.1085/jgp.83.5.771. 10.1085/jgp.83.5.771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujii AM, Vatner SF. Sympathetic mechanisms regulating myocardial contractility in conscious animals. In: Fozzard HA, Haber E, Jennings RB, Katz AM, Morgan HE, editors. The Heart and Cardiovascular System. New York: Raven Press; 1986. pp. 1119–1132. [Google Scholar]
- Gadsby DC, Cranefield PF. Effects of electrogenic sodium extrusion on the membrane potential of cardiac Purkinje fibers. In: Paes De Carvalho A, Hoffman BF, Lieberman M, editors. Normal and Abnormal Conduction in the Heart. New York: Futura; 1980. pp. 225–247. [Google Scholar]
- Gadsby DC, Nakao M. Steady-state current-voltage relationship of the Na/K pump in guinea pig ventricular myocytes. Journal of General Physiology. 1989;94:511–537. doi: 10.1085/jgp.94.3.511. 10.1085/jgp.94.3.511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gambassi G, Spurgeon HA, Lakatta EG, Blank PS, Capogrossi MC. Different effects of α- and β-adrenergic stimulation on cytosolic pH and myofilament responsiveness to Ca2+ in cardiac myocytes. Circulation Research. 1992;71:870–882. doi: 10.1161/01.res.71.4.870. [DOI] [PubMed] [Google Scholar]
- Gao J, Cohen IS, Mathias RT, Baldo GJ. Regulation of the β-stimulation of the Na+-K+ pump current in guinea-pig ventricular myocytes by a cAMP-dependent PKA pathway. Journal of Physiology. 1994;477:373–380. doi: 10.1113/jphysiol.1994.sp020199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Isoprenaline, Ca2+ and the Na+-K+ pump in guinea-pig ventricular myocytes. Journal of Physiology. 1992;449:689–704. doi: 10.1113/jphysiol.1992.sp019109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Two functionally different Na/K pumps in cardiac ventricular myocytes. Journal of General Physiology. 1995;106:995–1030. doi: 10.1085/jgp.106.5.995. 10.1085/jgp.106.5.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Baldo GJ. Effects of acetylcholine on the Na+-K+ pump current in guinea-pig ventricular myocytes. Journal of Physiology. 1997a;501:527–535. doi: 10.1111/j.1469-7793.1997.527bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Shi J, Baldo GJ. The effects of β-stimulation on the Na+-K+ pump current-voltage relationship in guinea-pig ventricular myocytes. Journal of Physiology. 1996;494:697–708. doi: 10.1113/jphysiol.1996.sp021525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Mathias RT, Cohen IS, Sun X, Baldo GJ. Modulators of PKC affect Na/K pump current in guinea-pig ventricular myocytes. Biophysical Journal. 1997b;72:A51. [Google Scholar]
- Mosior M, McLaughlin S. Peptides that mimic the pseudosubstrate region of protein kinase C bind to acidic lipids in membranes. Biophysical Journal. 1991;60:149–159. doi: 10.1016/S0006-3495(91)82038-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakao M, Gadsby DC. [Na] and [K] dependence of the Na/K pump current-voltage relationship in guinea-pig ventricular myocytes. Journal of General Physiology. 1989;94:539–565. doi: 10.1085/jgp.94.3.539. 10.1085/jgp.94.3.539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nosek TM, Williams MF, Zeigler ST, Godt RE. Inositol trisphosphate enhances calcium release in skinned cardiac and skeletal muscle. American Journal of Physiology. 1986;250:C807–811. doi: 10.1152/ajpcell.1986.250.5.C807. [DOI] [PubMed] [Google Scholar]
- Okumura K, Kawai T, Hashimoto H, Ito T, Ogawa K, Satake T. Sustained diacylglycerol formation in norepinephrine stimulated rat heart is associated with α1-adrenergic receptors. Journal of Cardiovascular Pharmacology. 1988;11:651–656. doi: 10.1097/00005344-198806000-00004. [DOI] [PubMed] [Google Scholar]
- Scholz J, Schaefer B, Schmitz W, Scholtz H, Steinfath M, Lohse M, Schwabe U, Puurunen J. α1-Adrenoceptor-mediated positive inotropic effect and inositol trisphosphate increase in mammalian heart. Journal of Pharmacological Experimental Theory. 1988;245:327–335. [PubMed] [Google Scholar]
- Shah A, Cohen IS, Rosen MR. Stimulation of cardiac alpha receptors increases Na/K pump current and decreases gK via a pertussis toxin-sensitive pathway. Biophysical Journal. 1988;54:219–225. doi: 10.1016/S0006-3495(88)82950-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzic A, Anagnostopoulos T, Vogel SM. Opposite modulation of ouabain cardiotoxicity by hexamethyleneamiloride and phenylephrine. Naunyn-Schmiedeberg's Archives of Pharmacology. 1991;343:511–518. doi: 10.1007/BF00169554. [DOI] [PubMed] [Google Scholar]
- Tohse N, Hattori Y, Nakaya H, Kanno M. Effects of α-adrenoceptor stimulation on electrophysiological properties and mechanics in rat papillary muscle. General Pharmacology. 1987a;18:539–546. doi: 10.1016/0306-3623(87)90077-2. [DOI] [PubMed] [Google Scholar]
- Tohse N, Kameyama M, Irisawa H. Intracellular Ca2+ and protein kinase C modulate K+ current in guinea-pig heart cells. American Journal of Physiology. 1987b;253:H1321–1324. doi: 10.1152/ajpheart.1987.253.5.H1321. [DOI] [PubMed] [Google Scholar]
- Tohse N, Nakaya H, Kanno M. α1-adrenoceptor stimulation enhances the delayed rectifier K+ current of guinea pig ventricular cells through the activation of protein kinase. Circulation Research. 1992;71:1441–1446. doi: 10.1161/01.res.71.6.1441. [DOI] [PubMed] [Google Scholar]
- Vasilets LS, Schwarz W. Regulation of endogenous and expressed Na+/K+ pumps in Xenopus oocytes by membrane potential and stimulation of protein kinases. Journal of Membrane Biology. 1992;125:119–132. doi: 10.1007/BF00233352. [DOI] [PubMed] [Google Scholar]
- Walsh KB. Activation of a heart chloride conductance during stimulation of protein kinase C. Molecular Pharmacology. 1991;40:342–346. [PubMed] [Google Scholar]
- Wilde AAM, Kleber AG. Effect of norepinephrine and heart rate on intracellular sodium activity and membrane potential in beating guinea pig ventricular muscle. Circulation Research. 1991;68:1482–1489. doi: 10.1161/01.res.68.5.1482. [DOI] [PubMed] [Google Scholar]
- Williamson AP, Kennedy RH, Seifen E, Lindemann JP, Stimers JR. α1b-Adrenoceptor-mediated stimulation of Na-K pump current in adult rat ventricular myocytes. American Journal of Physiology. 1993;264:H1315–1318. doi: 10.1152/ajpheart.1993.264.4.H1315. [DOI] [PubMed] [Google Scholar]
- Zaza A, Kline RP, Rosen MR. Effects of α-adrenergic stimulation on intracellular sodium activity and automaticity in canine Purkinje fibers. Circulation Research. 1990;66:416–426. doi: 10.1161/01.res.66.2.416. [DOI] [PubMed] [Google Scholar]