

Abstract
Changes in cytosolic and mitochondrial [Ca2+] produced by brief trains of action potentials were measured in motor nerve terminals using a rapidly scanning confocal microscope. Cytosolic [Ca2+] was measured using ionophoretically injected Oregon Green BAPTA 5N (OG-5N). Mitochondrial [Ca2+] was measured using rhod-2, bath loaded as dihydrorhod-2.
In response to 100-250 stimuli at 25-100 Hz the average cytosolic [Ca2+] showed an initial rapid increase followed by a much slower rate of increase. Mitochondrial [Ca2+] showed no detectable increase during the first fifteen to twenty stimuli, but after this initial delay also showed an initially rapid rise followed by a slower rate of increase. The onset of the increase in mitochondrial [Ca2+] coincided with the slowing of the rate of rise of cytosolic [Ca2+]. The peak levels of cytosolic and mitochondrial [Ca2+] both increased with increasing frequencies of stimulation.
When stimulation terminated, the initial rate of decay of cytosolic [Ca2+] was much more rapid than that of mitochondrial [Ca2+].
After addition of carbonyl cyanide m-chlorophenyl hydrazone (CCCP, 1-2 μm) to dissipate the proton electrochemical gradient across the mitochondrial membrane, cytosolic [Ca2+] rose rapidly throughout the stimulus train, reaching levels much higher than normal. CCCP inhibited the increase in mitochondrial [Ca2+].
These results suggest that mitochondrial uptake of Ca2+ contributes importantly to buffering presynaptic cytosolic [Ca2+] during normal neuromuscular transmission.
Presynaptic terminals contain abundant mitochondria, consistent with the high energy demands associated with filling, releasing and recycling synaptic vesicles. Mitochondria can also take up Ca2+ via a uniporter, an energetically downhill process that utilizes the large, internally negative potential gradient created by oxidation-coupled H+ transport (reviewed in Gunter & Pfeiffer, 1990).
Studies using isolated mitochondria have reported half- maximal activation of uniporter transport at [Ca2+] ranging from ∼1 to 189 μm (Gunter & Pfeiffer, 1990), substantially higher than the resting values of 0.05-0.10 μm estimated for many excitable cells. Thus mitochondria have classically been viewed as a low-affinity uptake mechanism involved primarily in handling large Ca2+ loads. Consistent with this idea, the buffering of cytosolic [Ca2+] exceeding 500 nm produced by depolarizing pulses, high [K+], or flash photolysis of a photolabile Ca2+ chelator in neurohypophyseal nerve endings, sympathetic or hippocampal neuronal somata or adrenal chromaffin cells is greatly reduced by inhibitors of mitochondrial Ca2+ uptake (Steunkel, 1994; Friel & Tsien, 1994; Herrington, Park, Babcock & Hille, 1996; Xu, Naraghi, Kang & Neher, 1997; Sidky & Baimbridge, 1997). In addition, Tang & Zucker (1997) presented pharmacological evidence for significant mitochondrial Ca2+ uptake during prolonged tetanic stimulation (7-10 min at 20-33 Hz) of crayfish motor nerve terminals.
The goal of the present study was to determine whether mitochondria in presynaptic nerve terminals also help buffer the more modest Ca2+ loads associated with physiological patterns of stimulation. Indirect evidence for such a role includes reports that brief application of inhibitors of mitochondrial function to frog motor nerve terminals increases Ca2+-dependent evoked transmitter release (Alnaes & Rahamimoff, 1975; Zengel, Sosa, Poage & Mosier, 1994). More direct evidence includes demonstrations of significant mitochondrial Ca2+ uptake for cytosolic [Ca2+] elevations only 100-200 nm above rest in ATP-stimulated bovine endothelial cells (Fig. 4 of Rizzuto, Bastianutto, Brini, Murgia & Pozzan, 1994) and depolarized adrenal chromaffin cells (Fig. 5 in Babcock, Herrington, Goodwin, Park & Hille, 1997). Also, a recent study of isolated liver mitochondria presented evidence for a rapid uptake mode mediating limited mitochondrial uptake of Ca2+ at the onset of elevations of [Ca2+] as low as 200 nm (Sparagna, Gunter, Sheu & Gunter, 1995). Elevations of average cytosolic [Ca2+] in this range are achieved after brief (≤ 2 s) trains of action potentials at ≥ 20 Hz in crayfish and lizard motor nerve terminals (Ravin, Spira, Parnas & Parnas, 1997; David, Barrett & Barrett, 1997).
This study measured stimulation-induced changes in cytosolic and mitochondrial [Ca2+], either separately or simultaneously, in lizard motor nerve terminals (bouton diameter, 2-5 μm) filled with fluorescent indicator dyes. Mitochondrial Ca2+ uptake was detectable after relatively few stimuli (e.g. 25-50 at 50-100 Hz), when spatially averaged cytosolic [Ca2+] had increased by 50-300 nm. Inhibition of mitochondrial Ca2+ uptake by the protonophore CCCP resulted in much greater stimulation-induced elevations of cytosolic [Ca2+]. Thus mitochondrial Ca2+ uptake appears to contribute importantly to motor nerve terminal Ca2+ metabolism during normal activity.
METHODS
Experiments used external intercostal neuromuscular preparations dissected from lizards (Anolis sagrei), killed by decapitation and pithing following ether anaesthesia. The preparation was mounted in a chamber constructed on a thin (No. 1) glass coverslip and imaged with an inverted microscope using a × 40 water immersion or a × 60 oil immersion lens. The physiological saline contained (mm): 157 NaCl, 4 KCl, 2 CaCl2, 2 MgCl2 and 1 Hepes. Muscle contraction was blocked using carbachol (150-200 μm) or d-tubocurare (10 mg l−1). The motor nerve was stimulated by applying brief, suprathreshold depolarizing pulses (0.1-0.2 ms) via a suction electrode. Experiments were performed at room temperature (20-25°C).
Changes in cytosolic [Ca2+] were monitored using OG-5N (Kd, ∼60 μm) loaded ionophoretically via a microelectrode inserted into the motor axon (David et al. 1997). The Ca2+ indicator rhod-2 (Kd, ∼0.5 μm) was targeted into mitochondria by bath-loading the preparation with the acetoxymethylester (AM) form of dihydrorhod-2, freshly synthesized from rhod-2 AM using reduction by sodium borohydride (protocol from Molecular Probes). This indicator fluoresces only after it is oxidized to rhod-2, which occurs preferentially within mitochondria (Hajnóczky, Robb-Gaspers, Seitz & Thomas, 1995). Preparations were bathed in dihydrorhod-2 AM (5 μm, 1 h at 4°C followed by 30-60 min at 35°C, a modification of the technique of Trollinger, Cascio & Lemasters, 1997), and then washed with indicator-free medium for ≥ 3 h prior to the onset of imaging.
Stimulation-induced changes in indicator fluorescence were measured in terminals filled with only OG-5N (Figs 1A and 3A), with only rhod-2 (Figs 1C and 3B and C), or with both OG-5N and rhod-2 (Fig. 2). For co-imaging rhod-2 and OG-5N, both dyes were excited with 488 nm light and a dichroic mirror was used to separate the emitted light into a red component from rhod-2 (> 570 nm) and a green component from OG-5N (535 ± 20 nm). The confocal microscope (Odyssey XL, Noran Instruments, Middleton, WI, USA) was operated in dual photomultiplier mode, using a bandpass filter (535 ± 20 nm) to further filter the green light, and a long pass filter (> 590 nm) for the red light. Images were corrected for ‘cross-talk’ between OG-5N and rhod-2 signals as follows. After subtraction of background fluorescence, the total fluorescence recorded in the OG-5N (Ftot,OG) and rhod-2 (Ftot,rhod) channels is given by:
and
where FOG and Frhod are the true fluorescent signals of OG-5N and rhod-2, respectively, and αFtot,rhod and βFtot,OG indicate the extent to which the signal recorded for one dye is contaminated by cross-talk from the other dye. The myelinated portion of the axon (labelled a in Fig. 2A) contained OG-5N but no rhod-2, enabling estimation of β as Ftot,rhod/FOG. Extra-terminal regions (labelled e in Fig. 2A) were stained with rhod-2 but not OG-5N, enabling estimation of α as Ftot,OG/Frhod. α and β varied with both the relative dye concentrations and the gain settings used for recording, and hence were calculated separately for each terminal.
Figure 1. ΔF/F transients produced by stimulus trains of different frequencies in lizard external intercostal motor terminals filled with OG-5N (A-C) or rhod-2 (D-F).
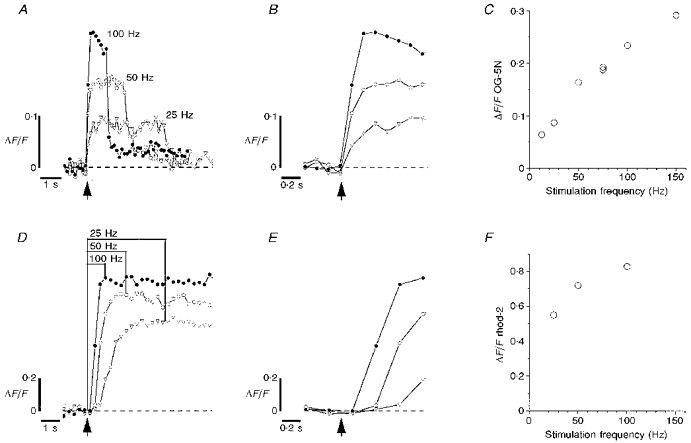
A and D, time courses of changes in ΔF/F in response to 100 stimuli at 25, 50 and 100 Hz. B and E,ΔF/F transients at the onset of stimulation plotted on a faster time scale. C and F, peak ΔF/F (averaged from the last 3 points collected during the train) as a function of stimulation frequency. In C, the order of administering the trains was (Hz): 50, 100, 25, 75 (two trains), 12.5, 150. For the 150 Hz point 200 rather than 100 stimuli were administered. OG-5N and rhod-2 images were collected at 7.50 and 3.75 s−1, respectively. Each plot in D was the mean of two identical stimulus trains (order of delivery: 50, 100, 25, 100, 25 and 50 Hz). The estimated change in cytosolic [Ca2+] (Δ[Ca2+]c) at the transition between fast and slow rates of rise of OG-5N fluorescence during 50 Hz stimulation was 200 nm, calculated as described in David et al. (1997) assuming Kd= 60 μm, resting [Ca2+]= 100 nm, and Fmax/Fmin= 50. The illustrated findings are representative of those obtained in 8 terminals filled with OG-5N and 5 terminals filled with rhod-2.
Figure 3. Effects of CCCP on cytosolic and mitochondrial ΔF/F transients.
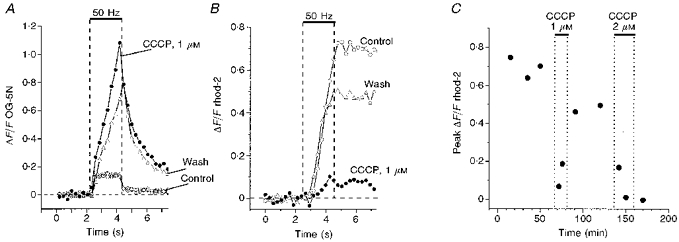
A and B,ΔF/F transients from terminals filled with OG-5N (A) or rhod-2 (B) in response to trains of 100 stimuli at 50 Hz (indicated by upper horizontal bar). ○, ΔF/F transients obtained under control conditions. A shows 3 superimposed trains separated by 15 min intervals, with a transition from fast to slow rates of rise at Δ[Ca2+]c of 150 nm. •, obtained 10 min (A) and 3 min (B) after adding 1 μm CCCP to the bath; Δ, obtained 30 min (A) and 48 min (B) after beginning CCCP washout. C, peak post-stimulation rhod-2 ΔF/F values in the terminal of B under control conditions, and following exposure to 1 μm CCCP, return to control medium, exposure to 2 μm CCCP, and return to control medium. Oligomycin (10 μg ml−1) was present during all but the first train. The trains plotted in B yielded the third, fourth and seventh points in C.
Figure 2. Pseudocolour fluorescence micrographs (A) and stimulation-induced ΔF/F transients (B and C) in terminals loaded with both OG-5N and rhod-2.
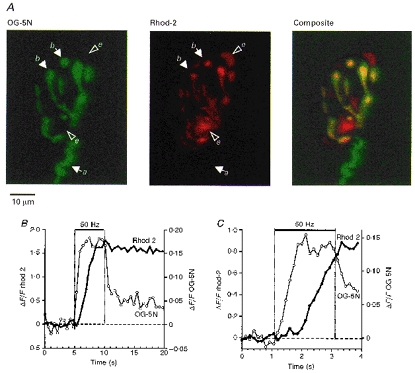
A, OG-5N (left, green) and rhod-2 (centre, red) fluorescence, and merged image (right). a, axon; b, boutons; and e, extra-terminal mitochondrial cluster. In the merged image regions containing both OG-5N and rhod-2 fluorescence appear yellow-orange. Each micrograph is the mean of 128 images. B and C,ΔF/F transients for rhod-2 (•, connected by thick line, left ordinate) and for OG-5N (○, thin line, right ordinate) produced by 50 Hz stimulation (indicated by upper horizontal bar). Transients in B came from the terminal in A, stimulated for 5 s and sampled at 1.875 s−1. For both dyes ΔF/F was averaged over the same area, defined by outlining all regions of the terminal that showed a stimulation-induced increase in OG-5N fluorescence (total of 4600 pixels, covering 167 μm2). Data in C, plotted on a faster time scale, came from a different terminal stimulated for 2 s and sampled at 7.5 s−1. Each plot in C is a mean of 3 trains (smoothed using a 3-point moving bin) repeated at 15 min intervals. In B and C the transition from fast to slow rates of rise of cytosolic [Ca2+] occurred at Δ[Ca2+]c of 180 and 150 nm, respectively. Ordinates in B and C were chosen to give peak ΔF/F transients of approximately the same amplitude, and are not intended to imply equality of the [Ca2+] within the mitochondrial and cytosolic compartments. OG-5N and rhod-2 images were corrected for cross-talk as described in Methods (for terminal in A and B α= 0.17, β= 0.79; for terminal in C α was negligible and β= 0.4). These findings are representative of those obtained in 3 terminals filled with both OG-5N and rhod-2.
Images were collected at rates ranging from 1.875 to 7.5 s−1 using an Indy workstation (Silicon Graphics) with Noran InterVision software, and stored and analysed as described in David et al. (1997). Data are plotted as ΔF/Frest (abbreviated as ΔF/F), where ΔF is the change in fluorescence and Frest is resting (pre-stimulation) fluorescence. For each terminal ΔF/F was averaged over regions of interest defined by drawing lines around all regions whose fluorescence increased in response to stimulation. In dual-imaging studies the regions of interest defined for OG-5N were also used to analyse rhod-2 fluorescence.
For the low-affinity dye OG-5N the plotted ΔF/F values were linearly related to changes in cytosolic [Ca2+], calculated as described in the legend of Fig. 1. ΔF/F measurements for rhod-2 were not converted into [Ca2+] estimates, but intramitochondrial rhod-2 is expected to be a reasonable indicator for increases in [Ca2+] within the mitochondrial matrix, since the estimated resting matrix [Ca2+] (≤ 0.2 μm) is less than the 0.5 μmKd of rhod-2, and matrix [Ca2+] is strongly buffered (estimated ratio of free to bound calcium of 0.0003; reviewed in Magnus & Keizer, 1997; see also Babcock et al. 1997). Preparations were checked for stability by administering the same train several times at 15 min intervals. Under control conditions ΔF/F transients were quite stable (see Fig. 3A), displaying little evidence of the ‘rundown’ often observed in internally dialysed cells. Terminals exhibiting abrupt increases in Frest were not analysed further.
Indicator dyes were purchased from Molecular Probes. All other reagents were from Sigma Chemical Co.
RESULTS
Figure 1 plots ΔF/F changes recorded in response to 100 action potentials evoked at different frequencies. Panels A-C came from a terminal filled with OG-5N to monitor changes in cytosolic [Ca2+], and panels D-F came from a separate terminal loaded with rhod-2 to monitor changes in mitochondrial [Ca2+]. At the onset of stimulation OG-5N fluorescence increased rapidly, at a rate corresponding to a Δ[Ca2+] of about 10-20 nm per impulse (Fig. 1A and B; see also David et al. 1997). Later in the train the rate of increase abruptly slowed to ∼0.1 nm per impulse or less (estimated from trains lasting 10-20 s, not shown). At 50 Hz the estimated increase in cytosolic [Ca2+]i at this transition averaged 186 nm (s.d. 30 nm, range 150-240 nm, n= 8 terminals). The peak ΔF/F after 100 stimuli increased with increasing stimulus frequency (12.5-150 Hz, Fig. 1C). The estimated increases in cytosolic [Ca2+] corresponding to these peak ΔF/F values were 70-370 nm.
In contrast, rhod-2 fluorescence showed no detectable increase during the interval corresponding to the first ten to fifteen stimuli in a train (Fig. 1D and E, n= 5 terminals). The absence of any significant increase in mitochondrial signal during the first ten stimuli was verified in line scans through single boutons sampled at 69 μs intervals (not shown). Also, other terminals stimulated with trains of ten action potentials at 50 Hz showed no significant increase in the mitochondrial signal during the train or for 80 s thereafter (not shown). After the initial delay rhod-2 fluorescence showed a rapid initial rise, followed by a much slower rate of rise evident in trains longer than those illustrated here. The mitochondrial signal attained following 100 stimuli increased with increasing stimulus frequency (Fig. 1F).
When stimulation stopped, cytosolic [Ca2+] showed a rapid initial decay followed by a slower decay (Fig. 1A). The initial decay of mitochondrial [Ca2+] was much slower (Fig. 1D); the sampling rates used here did not permit resolution of the complete time course of this decay, but in a preliminary experiment the mitochondrial signal retained 82 % of its maximal value 80 s after 100 stimuli at 50 Hz. Babcock et al. (1997) reported similar differences between the time courses of decay of cytosolic and mitochondrial [Ca2+]ΔF/F transients in adrenal chromaffin cells.
Similar differences between the time courses of stimulation- induced changes in cytosolic and mitochondrial [Ca2+] were observed in motor terminals in which both OG-5N and rhod-2 fluorescence were monitored simultaneously. Figure 2A shows an example of such a terminal, with green marking the OG-5N-filled cytosol, and red indicating rhod-2-filled mitochondrial clusters. In the merged image yellow indicates mitochondrial clusters within terminal boutons. Large structures outside the terminal cytoplasm that stained diffusely with rhod-2 (labelled e in Fig. 2A) probably represent mitochondrial clusters within the end- plate region of the muscle fibre; their fluorescence did not change during stimulation.
Figure 2B shows OG-5N and rhod-2 ΔF/F transients measured simultaneously in this terminal in response to a 5 s train at 50 Hz. The increase in the mitochondrial signal began with a delay following the onset of stimulation, at about the same time as the transition from the fast to the slow rate of rise of the cytoplasmic signal. This is best seen in Fig. 2C, which plots ΔF/F transients from another terminal in the same preparation sampled at a faster rate. As in Fig. 1, the initial decay of OG-5N fluorescence upon termination of stimulation was much more rapid than that of rhod-2 fluorescence.
The different kinetics of the OG-5N and rhod-2 ΔF/F transients were due to the differential localization of the dyes rather than simply to differences in dye kinetics. Preparations loaded with rhod-2 AM for only a brief time (labelling the cytosol, but not allowing sufficient time for mitochondrial localization) yielded ΔF/F transients with kinetics similar to those of ionophoretically injected indicators with similar Kd values (e.g. fluo-3, not shown).
To test further the hypothesis that mitochondrial Ca2+ uptake contributes importantly to controlling cytosolic [Ca2+] during brief stimulus trains, we applied the protonophore CCCP. Figure 3A shows that after addition of 1 μm CCCP, cytosolic [Ca2+] rose rapidly throughout the stimulus train, reaching a level severalfold higher than that measured under control conditions. CCCP also slowed the initial post-train decay of cytosolic [Ca2+]. Figure 3B and C illustrates the effects of 1-2 μm CCCP on mitochondrial ΔF/F transients in a different terminal. This experiment was done in the presence of oligomycin (10 μg ml−1), an inhibitor of F1, F0-ATP synthase, to minimize the component of ATP depletion associated with reverse action of this enzyme during CCCP application. CCCP greatly reduced the mitochondrial ΔF/F transient (Fig. 3B). The effects of 1 μm CCCP were partially reversible.
The finding that CCCP increased cytosolic ΔF/F transients but reduced mitochondrial ΔF/F transients supports the hypothesis that mitochondrial uptake makes an important contribution to the regulation of cytosolic [Ca2+] even during relatively brief stimulus trains. Several observations suggest that most of the effects of CCCP were due to reduction of the proton electrochemical potential across the mitochondrial membrane rather than to ATP depletion (see also Park, Herrington, Babcock & Hille, 1996). First, cytosolic ΔF/F transients eventually returned to baseline levels after brief exposure to CCCP (not shown), indicating that sufficient energy remained to power Ca2+ extrusion across the plasma membrane. Also, application of oligomycin alone had little effect on mitochondrial ΔF/F transients, and the effects of a low concentration of CCCP could be partially reversed by washing out CCCP when mitochondrial ATP synthesis was inhibited (Fig. 3C).
DISCUSSION
The experiments described here used action potential stimulation in patterns similar to those recorded during normal function (Carrier, 1989) under conditions associated with normal levels of Ca2+ entry. The intra-terminal environment was altered only by addition of indicator dyes. Under these nearly physiological conditions, mitochondria began to take up Ca2+ after only approximately twenty-five to fifty action potentials delivered at 25-100 Hz. This finding is consistent with the indirect evidence of Werth & Thayer 1994 for mitochondrial Ca2+ uptake following more than twenty-five action potentials at 10 Hz in the somata of dorsal root ganglion neurones. The rapid rise of mitochondrial [Ca2+] coincided with a slowing of the rate of rise of cytosolic [Ca2+], and the uncoupler CCCP increased cytosolic ΔF/F transients, and decreased mitochondrial ΔF/F transients. Thus mitochondrial uptake helps limit the increase in cytosolic [Ca2+] during physiological levels of activity in motor nerve terminals. Mitochondria also take up Ca2+ during physiological activity in rat cardiac myocytes (Miyata, Silverman, Sollott, Lakatta, Stern & Hansford, 1991; Trollinger et al. 1997). The marked slowing of the rate of rise of cytosolic [Ca2+] achieved in part by mitochondrial Ca2+ uptake may help maintain the responsiveness of the terminal to later stimuli in the train by preventing saturation of cytosolic Ca2+ buffers. Mitochondrial Ca2+ uptake also activates several dehydrogenases (reviewed in McCormack, Halestra & Denton, 1990; Gunter, Gunter, Sheu & Gavin, 1994), and thus may stimulate production of ATP needed to support Na+ and Ca2+ extrusion and rapid vesicle recycling.
Mitochondrial Ca2+ uptake was detectable for an increase in average cytosolic [Ca2+] of ≤ 200 nm, in agreement with the findings of Babcock et al. (1997) for depolarizing pulses applied to adrenal chromaffin cells. This spatially averaged [Ca2+] is probably a reasonably accurate estimate of the [Ca2+] around mitochondria, because electron micrographs show most mitochondria located near the centre of terminal boutons, or near non-secreting regions of the plasma membrane, rather than near the Ca2+ channels of the releasing surfaces. Mitochondrial Ca2+ uptake at such low cytosolic [Ca2+] might be mediated by the rapid uptake mode described by Sparagna et al. (1995). The rapid inactivation/saturation of this rapid uptake mode might make mitochondrial Ca2+ uptake more sensitive to pulsatile increases in [Ca2+] than to steady-state increases of comparable magnitude (see also Hajnóczky et al. 1995).
In our study mitochondrial [Ca2+] rose with a delay after cytosolic [Ca2+], and this delay decreased with increasing stimulation frequency. Babcock et al. (1997) found no such delay in adrenal chromaffin cells. Perhaps their application of intense (+10 mV, ≥ 50 ms) depolarizing pulses in elevated (10 mm) bath [Ca2+] produced increases in cytosolic [Ca2+] similar to those associated with a very high-frequency train of action potentials, in which case any delay between the increases in cytosolic and mitochondrial [Ca2+] might have been too brief to detect with the sampling intervals used (≥ 50 ms). Using electron-probe microanalysis after shock-freezing, Wendt-Gallitelli & Isenberg (1991) measured a delay between the increases in total cytosolic and mitochondrial calcium during the contraction cycle in ventricular myocytes. The mitochondrial model elaborated by Magnus & Keizer (1997) also predicts a delay between increases in cytosolic and mitochondrial [Ca2+]. This delay may result from the positive co-operativity of uniporter activation by extramitochondrial Ca2+ (Hill coefficient, ∼2.0; Gunter & Pfeiffer, 1990).
The stimulation-induced changes in cytosolic [Ca2+] measured here are not sufficient to predict changes in the magnitude of phasic evoked transmitter release. During continuous 50 Hz stimulation in physiological bath [Ca2+], cytosolic [Ca2+] remains above control levels, but evoked release usually declines below control levels (e.g. Glavinovic, 1979; Magleby, Pallotta & Terrar, 1981, for rat, mouse and frog neuromuscular junctions). One likely explanation for this discrepancy between cytosolic [Ca2+] and evoked release is that the pool of releasable vesicles becomes depleted at high release rates. Also, since evoked release is strongly influenced by Ca2+ influx near membrane release sites, the spatially averaged cytosolic [Ca2+] measured here might correlate better with asynchronous than with phasic evoked release (see e.g. Ravin et al. 1997).
Acknowledgments
This work was supported by US Public Health Service grant nos. NS12404 and NS12207, and by NCRR-BRS Shared Instrumentation Grant 1S10RR08368. We thank Ms Xiao-xi Zhou for help with preliminary experiments.
References
- Alnaes E, Rahamimoff R. On the role of mitochondria in transmitter release from motor nerve terminals. Journal of Physiology. 1975;248:285–306. doi: 10.1113/jphysiol.1975.sp010974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. Journal of Cell Biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrier DR. Ventilatory action of the hypaxial muscles of the lizard Iguana iguana: a function of slow muscle. Journal of Experimental Biology. 1989;143:435–457. doi: 10.1242/jeb.143.1.435. [DOI] [PubMed] [Google Scholar]
- David G, Barrett JN, Barrett EF. Stimulation-induced changes in [Ca2+] in lizard motor nerve terminals. Journal of Physiology. 1997;504:83–96. doi: 10.1111/j.1469-7793.1997.083bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive Ca2+ store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. Journal of Neuroscience. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glavinovic MI. Change of statistical parameters of transmitter release during various kinetic tests in unparalysed voltage-clamped rat diaphragm. Journal of Physiology. 1979;290:481–497. doi: 10.1113/jphysiol.1979.sp012785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu S-S, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. American Journal of Physiology. 1994;267:C313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. American Journal of Physiology. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Hajnóczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. 10.1016/S0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- McCormack JG, Halestra AP, Denton RM. Role of calcium ions in regulation of mammalian intramitochondrial metabolism. Physiological Reviews. 1990;70:391–425. doi: 10.1152/physrev.1990.70.2.391. [DOI] [PubMed] [Google Scholar]
- Magleby KL, Pallotta BS, Terrar DA. The effect of (+)-tubocurarine on neuromuscular transmission during repetitive stimulation in the rat, mouse, and frog. Journal of Physiology. 1981;312:97–113. doi: 10.1113/jphysiol.1981.sp013618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnus G, Keizer J. Minimal model of β-cell mitochondrial Ca2+ handling. American Journal of Physiology. 1997;273:C717–733. doi: 10.1152/ajpcell.1997.273.2.C717. [DOI] [PubMed] [Google Scholar]
- Miyata H, Silverman HS, Sollott SJ, Lakatta EG, Stern MD, Hansford RG. Measurement of mitochondrial free Ca2+ concentration in living single rat cardiac myocytes. American Journal of Physiology. 1991;261:H1123–1134. doi: 10.1152/ajpheart.1991.261.4.H1123. [DOI] [PubMed] [Google Scholar]
- Park YB, Herrington J, Babcock DF, Hille B. Ca2+ clearance mechanisms in isolated rat adrenal chromaffin cells. Journal of Physiology. 1996;492:329–346. doi: 10.1113/jphysiol.1996.sp021312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravin R, Spira ME, Parnas H, Parnas I. Simultaneous measurement of intracellular Ca2+ and asynchronous transmitter release from the same crayfish bouton. Journal of Physiology. 1997;501:251–262. doi: 10.1111/j.1469-7793.1997.tb00001.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. Journal of Cell Biology. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidky AO, Baimbridge KG. Calcium homeostatic mechanisms operating in cultured postnatal rat hippocampal neurones following flash photolysis of nitrophenyl-EGTA. Journal of Physiology. 1997;504:579–590. doi: 10.1111/j.1469-7793.1997.579bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparagna GC, Gunter KK, Sheu S-S, Gunter TE. Mitochondrial calcium uptake from physiological-type pulses of calcium. A description of the rapid uptake mode. Journal of Biological Chemistry. 1995;270:27510–27515. doi: 10.1074/jbc.270.46.27510. 10.1074/jbc.270.46.27510. [DOI] [PubMed] [Google Scholar]
- Stuenkel EL. Regulation of intracellular calcium and calcium buffering properties of rat isolated neurohypophysial nerve endings. Journal of Physiology. 1994;481:251–271. doi: 10.1113/jphysiol.1994.sp020436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y-G, Zucker RS. Mitochondrial involvement in post-tetanic potentiation of synaptic transmission. Neuron. 1997;18:483–491. doi: 10.1016/s0896-6273(00)81248-9. 10.1016/S0896-6273(00)81248-9. [DOI] [PubMed] [Google Scholar]
- Trollinger DR, Cascio WE, Lemasters JJ. Selective loading of rhod 2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochemical and Biophysical Research Communications. 1997;236:738–742. doi: 10.1006/bbrc.1997.7042. 10.1006/bbrc.1997.7042. [DOI] [PubMed] [Google Scholar]
- Wendt-Gallitelli MF, Isenberg G. Total and free myoplasmic calcium during a contraction cycle: X-ray microanalysis in guinea-pig ventricular myocytes. Journal of Physiology. 1991;435:349–372. doi: 10.1113/jphysiol.1991.sp018514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured dorsal root ganglion neurons. Journal of Neuroscience. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu T, Naraghi M, Kang H, Neher E. Kinetic studies of Ca2+ binding and Ca2+ clearance in the cytosol of adrenal chromaffin cells. Biophysical Journal. 1997;73:532–545. doi: 10.1016/S0006-3495(97)78091-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zengel JE, Sosa MA, Poage RE, Mosier DR. Role of intracellular Ca2+ in stimulation-induced increases in transmitter release at the frog neuromuscular junction. Journal of General Physiology. 1994;104:337–355. doi: 10.1085/jgp.104.2.337. 10.1085/jgp.104.2.337. [DOI] [PMC free article] [PubMed] [Google Scholar]