

Abstract
To define the role of environmental oxygen in regulating postnatal maturation of the carotid body afferent pathway, light and electron microscopic methods were used to compare chemoafferent neurone survival and carotid body development in newborn rats reared from birth in normoxia (21 % O2) or chronic hyperoxia (60 % O2).
Four weeks of chronic hyperoxia resulted in a significant 41 % decrease in the number of unmyelinated axons in the carotid sinus nerve, compared with age-matched normoxic controls. In contrast, the number of myelinated axons was unaffected by hyperoxic exposure.
Chemoafferent neurones, located in the glossopharyngeal petrosal ganglion, already exhibited degenerative changes following 1 week of hyperoxia from birth, indicating that even a relatively short hyperoxic exposure was sufficient to derange normal chemoafferent development. In contrast, no such changes were observed in the vagal nodose ganglion, demonstrating that the effect of high oxygen levels was specific to sensory neurones in the carotid body afferent pathway. Moreover, petrosal ganglion neurones were sensitive to hyperoxic exposure only during the early postnatal period.
Chemoafferent degeneration in chronically hyperoxic animals was accompanied by marked hypoplasia of the carotid body. In view of previous findings from our laboratory that chemoafferent neurones require trophic support from the carotid body for survival after birth, we propose that chemoafferent degeneration following chronic hyperoxia is due specifically to the loss of target tissue in the carotid body.
Ventilatory chemoreflexes undergo profound maturational changes after birth. In the neonate, exposure to acute hypoxia produces a ‘biphasic’ response characterized by an initial increase, followed by a decrease in ventilation (Bureau, Foulon, Zinman & Begin, 1984; Eden & Hanson, 1987b). The relatively sustained increase in ventilation during hypoxia characteristic of adults does not appear until days after birth (Eden & Hanson, 1987b). Recent studies indicate that the transition from a neonatal to an adult hypoxic ventilatory response is not immutable, but rather is plastic and susceptible to modulation by environmental oxygen. Specifically, prolonged hypoxia from birth delays the onset of a ‘mature’ chemoreflex response to hypoxia (Eden & Hanson, 1987a; Hertzberg, Hellström, Holgert, Lagercrantz & Pequignot, 1992; Sladek, Parker, Grogaard & Sundell, 1993), whereas prolonged hyperoxia may accelerate it (Eden & Hanson, 1986; Hanson, Eden, Nijhuis & Moore, 1989). Interestingly, blunted ventilatory responses to acute hypoxia appear to be the long-term consequence of early exposure to either chronic hypoxia or chronic hyperoxia, and persist even after animals have been removed to a normoxic environment (Eden & Hanson, 1986; Hanson et al. 1989; Sladek et al. 1993; Ling, Olson, Vidruk & Mitchell, 1996, 1997b). Moreover, although hypoxic responsiveness is severely depressed in adult rats exposed to 60 % O2 during the first month of postnatal life, a similar exposure as adults does not alter hypoxic ventilatory reflexes (Ling et al. 1996, 1997b), indicating that chemoreflex development is particularly susceptible to modulation by environmental oxygen levels during the early postnatal period.
The mechanisms by which chemoreflex development is regulated by environmental oxygen are not known. Resetting of the peripheral chemoreceptors and/or persistence of central inhibitory influences have been proposed as contributing to the delay in chemoreflex development following chronic perinatal hypoxia (Eden & Hanson, 1987a). Chronic perinatal hyperoxia severely depresses responses to hypoxia recorded from the carotid sinus nerve of kittens and adult rats (Hanson et al. 1989; Vidruk, Olson, Ling & Mitchell, 1996), suggesting impairment of carotid body chemoreflex function. On the other hand, phrenic nerve responses to electrical stimulation of the carotid sinus nerve are nearly indistinguishable between adult rats reared in either normoxia or hyperoxia during the first postnatal month (Ling, Olson, Vidruk & Mitchell, 1997a), suggesting that early exposure to elevated oxygen levels does not impair central integration of carotid chemoreceptor afferent input. Based on these physiological studies, impairment of the carotid chemoreceptors per se has been suggested to underlie the deficits in hypoxic responsiveness seen in adult animals following exposure to chronic perinatal hyperoxia (Ling et al. 1997a). However, specific loci within the chemoafferent pathway at which environmental oxygen may influence chemoreflex development remain to be defined.
Previous studies in our laboratory demonstrated that chemosensory neurones that innervate the carotid body are immature at birth and subject to regulation by extrinsic factors, including trophic support derived from the carotid body (Hertzberg, Fan, Finley, Erickson & Katz, 1994). These cells, located in the glossopharyngeal petrosal ganglion, are the afferent link between the carotid chemoreceptors and the brainstem and thereby play a pivotal role in mediating peripheral chemoreflexes. Chemoafferent neurones, therefore, are one locus at which derangements of normal development could disrupt chemoreflex function. Consequently, the present study was undertaken to determine whether postnatal maturation of chemoafferent neurones is influenced by the level of environmental oxygen after birth.
METHODS
Chronic hyperoxia
Pregnant Sprague-Dawley rats (Zivic-Miller, Zelienople, PA, USA and Harlan Sprague-Dawley, Inc., Madison, WI, USA) were placed in individual cages 2 days prior to expected delivery, within large chambers supplied with room air (normoxic controls) or an oxygen-enriched atmosphere (hyperoxic animals; approximately 60 % O2; OM-11 O2 analyser, Beckman Instruments, Fullerton, CA, USA). After delivery the newborn animals were reared, with their mother, continuously from birth in normoxia or hyperoxia for 7 or 28 days. Gas flow was regulated to maintain a constant hyperoxic environment and to ensure that CO2 accumulation remained below 0.5 % (LB-2 CO2 analyser, Beckman). A separate group of juvenile rats was reared in normoxia or hyperoxia (60 % O2) for 1 week from postnatal day (P)21 to P28. In all experiments, food and water were provided ad libitum and fresh bedding was supplied as needed.
Histochemical and immunohistochemical procedures
At the end of all experiments, animals were deeply anaesthetized with saturated sodium pentobarbitone (36 mg, i.p.) and killed by perfusion through the heart with 4 % paraformaldehyde in 0.1 M sodium phosphate buffer (pH 7.4). Both carotid bifurcations, including the nodose-petrosal ganglion complex and the carotid body, were removed from each animal, infiltrated with 30 % sucrose in phosphate-buffered saline (PBS, pH 7.4), transferred to a 1 : 1 mixture of 30 % sucrose-PBS and Tissue Tek (Baxter Scientific, McGraw Park, IL, USA) for 24 h, embedded and frozen in Tissue Tek over dry ice, then stored at -80°C until use. Frozen sections (10 μm) were cut through the entire bifurcation and thaw-mounted onto gelatin-coated microscope slides. Every second or third section from the right ganglion complex was double immunostained using polyclonal anti-tyrosine hydroxylase (TH; Pel-Freez, Rogers, AR, USA) and monoclonal anti-neurofilament protein (NF200,160,68; Sigma) antibodies. Sections were: (1) air dried, rinsed in PBS for 15 min, soaked in dilution buffer (0.3 % Triton X-100, 20 g l−1 bovine serum albumin (Sigma) in phosphate buffer, pH 7.4) for 1 h and incubated overnight at room temperature (20-23°C) with anti-TH (1: 200) and anti-NF (1: 100) in dilution buffer; (2) washed in dilution buffer (2 × 30 min), incubated for 2 h in the dark at room temperature with secondary antibodies (goat anti-rabbit IgG-FITC, 1: 200, Boehringer Mannheim, and goat anti-mouse IgG-Rhodamine, 1: 200, Cappel Research Products, Organon Teknika, Durham, NC, USA) diluted in PBS-Triton containing 10 % goat serum and 10 % rat serum; (3) rinsed in dilution buffer (2 × 15 min) and PBS (15 min); (4) incubated with ρ-phenylenediamine (1 mg ml−1, Sigma) for 1 min, rinsed in PBS (2 × 5 min); then (5) coverslipped with glycerol gel.
Carotid body volume measurements
Serial sections of the carotid bifurcation immunostained for TH were used for carotid body volume measurements. In this material, the outer profile of the carotid body, as well as the TH-positive glomus cells within the organ, could be clearly delineated. The total volume of the carotid body and the volume occupied by glomus cells was determined in every second section using image analysis software (NIH Image, v. 1.55, NIH, USA). Values were calculated based on the cross-sectional areas of the entire carotid body or glomus cell compartment, section thickness, and the total number of sections containing the carotid body.
Neuronal cell counts
To determine the number of dopaminergic neurones in the petrosal or nodose ganglion, TH-immunostained neuronal profiles with a nucleus in the plane of section were counted separately for each ganglion. Neurofilament immunostaining of neurones and fibre tracts was used to distinguish the topographic limits of the two ganglia. Small intensely fluorescent (SIF) cells, distinguished by their small size, intense TH immunoreactivity and scant cytoplasm, were observed in both the nodose and petrosal ganglia of neonatal and juvenile animals, but were not included in the profile counts. The total number of TH-positive neurones was then estimated from the profile counts using the mean nuclear diameter derived from fifty individual measurements (Abercrombie, 1946).
Electron microscopy and axon counts
To estimate the number of axons in the carotid sinus nerve, a small section of the glossopharyngeal nerve, with the carotid sinus nerve attached, was carefully dissected and removed from animals reared in normoxia or hyperoxia for 28 days. The tissue was fixed for 1.5 h in 2 % glutaraldehyde in 0.1 M cacodylate buffer, rinsed in cacodylate buffer (3 × 20 min) then stored in buffer overnight at 4°C. Tissue was then post-fixed in 1 % osmium tetroxide (in 0.1 M cacodylate buffer) for 1 h, rinsed in distilled water (3 × 15 min), stained en bloc with 0.5 % uranyl acetate for 1 h in the dark, rinsed in distilled water (3 × 15 min), dehydrated through a graded series of acetone, infiltrated with Spurr resin (Electron Microscopy Sciences, Fort Washington, PA, USA) and polymerized for 24 h at 60°C. Cross-sections (80 nm) of the carotid sinus nerve were cut approximately 0.2-0.5 mm distal to its junction with the glossopharyngeal nerve using a Reichert Ultracut E ultra-microtome. Sections were counterstained with uranyl acetate and lead citrate, and examined using a JEOL 100CX transmission electron microscope. The total numbers of myelinated and unmyelinated axons were counted directly from photomontages prepared from electron photomicrographs (final magnification, × 20 400).
Statistical analyses
All values are reported as the mean ±s.e.m. Statistical analyses were performed using Student's unpaired t test. A P value of less than 0.05 was considered significant for all statistical tests.
RESULTS
Adult rats reared in hyperoxia (60 % O2) for 4 weeks from birth exhibit severely blunted ventilatory responses to hypoxia when tested at 3-5 months of age (Ling et al. 1996). To determine whether prolonged hyperoxia might alter hypoxic sensitivity by disrupting normal chemoafferent development, we compared the number of axons in the carotid sinus nerve from animals reared continuously for one month from birth in either normoxia (21 % O2) or chronic hyperoxia (60 % O2). The carotid sinus nerve contains the axons of chemosensory neurones located in the glossopharyngeal petrosal ganglion that convey afferent information from the carotid body chemoreceptors (for review see McDonald & Mitchell, 1975). In animals reared to postnatal day 28 in normoxia, the carotid sinus nerve contained a total of 764 ± 86 axons, and 40 ± 5 Schwann cell nuclei per nerve cross-section, values comparable to those previously reported by McDonald (1983) for adult rats. However, rearing animals in chronic hyperoxia resulted in a significant 41 % decrease in the number of unmyelinated carotid sinus nerve axons, compared with age-matched normoxic controls (Fig. 1; P0-P28 normoxia: 732 ± 85 vs. P0-P28 hyperoxia: 432 ± 54; P= 0.0247). In addition, we observed a significantly smaller number of Schwann cell nuclei in the nerve cross-sections from hyperoxic animals (Fig. 2; P0-P28 normoxia: 40 ± 5 vs. P0-P28 hyperoxia: 19 ± 3; P= 0.0114). There was no significant difference in the number of myelinated axons between normoxic and hyperoxic animals (P0-P28 normoxia: 32 ± 7 vs. P0-P28 hyperoxia: 39 ± 15; P= 0.6871).
Figure 1. Four-week exposure of newborn rats to chronic hyperoxia leads to loss of unmyelinated axons in the carotid sinus nerve.
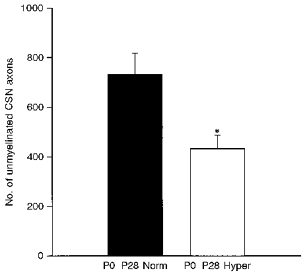
Animals were killed following continuous exposure to normoxia (▪, P0-P28 Norm, n= 4) or hyperoxia (□, P0-P28 Hyper, n= 4) from birth to 4 weeks of age. The number of axons in the carotid sinus nerve (CSN) was assessed by electron microscopic analysis of nerve cross-sections at a final magnification of × 20 400. Values are means +s.e.m.*P= 0.0247.
Figure 2. Electron photomicrographs of the carotid sinus nerve, cut in cross-section, from animals exposed to normoxia (A) or hyperoxia (B) from birth to 4 weeks of age.
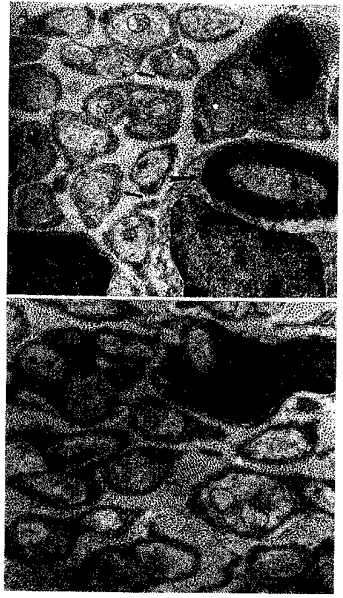
The morphology of axons within the carotid sinus nerve from a normoxic and hyperoxic animal is similar. Large arrow: myelinated axon. Small arrows: unmyelinated axons. SC, Schwann cell nucleus. Scale bar, 0.5 μm.
To begin defining early indices of chemoafferent degeneration, we examined expression of tyrosine hydroxylase (TH) immunoreactivity, a selective marker for a subset of petrosal ganglion neurones that innervate the carotid body (Katz & Black, 1986; Finley, Polak & Katz, 1992). These dopaminergic afferents project peripherally in the carotid sinus nerve and synapse directly on carotid body glomus cells (Finley et al. 1992). We found previously that TH expression in the petrosal ganglion decreases rapidly following disruption of the carotid sinus nerve between the petrosal ganglion and the carotid body (Katz & Black, 1986).
One week of hyperoxia from birth produced a 32 % decrease in the number of dopaminergic petrosal ganglion neurones, compared with normoxic controls (Fig. 3; P0-P7 normoxia: 334 ± 38 vs. P0-P7 hyperoxia: 226 ± 15; P= 0.0193). Thus, even a relatively short perinatal exposure to hyperoxia was sufficient to derange chemoafferent neurone development. In contrast, 1 week of hyperoxia had no effect on the number of nodose ganglion dopaminergic afferents, most of which do not innervate the carotid body, indicating that the effect of hyperoxia was specific to the carotid body chemosensory pathway (Fig. 4A; normoxia: 420 ± 18 vs. hyperoxia: 461 ± 26; P= 0.2097). Moreover, exposure of older animals to hyperoxia for 1 week, from postnatal days 21 to 28, had no effect on TH cell number in the petrosal ganglion (Fig. 4B; normoxia: 222 ± 18 vs. hyperoxia: 250 ± 16; P= 0.2552), demonstrating that the sensitivity of petrosal chemoafferents to hyperoxic exposure is restricted to the early postnatal period.
Figure 3. One-week exposure of newborn rats to hyperoxia deranges normal development of dopaminergic chemoafferent neurones in the petrosal ganglion.
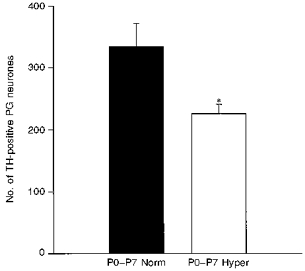
Animals were reared from birth to 1 week of age in normoxia (▪, P0-P7 Norm, n= 8) or hyperoxia (□, P0-P7 Hyper, n= 8). Hyperoxic exposure resulted in a 32 % decrease in the number of TH-immunoreactive chemoafferent neurones in the petrosal ganglion (PG) compared with normoxic controls. Values are means +s.e.m.*P= 0.0193.
Figure 4. The loss of dopaminergic afferent neurones associated with chronic hyperoxia is specific to the petrosal ganglion and restricted to early postnatal exposure.
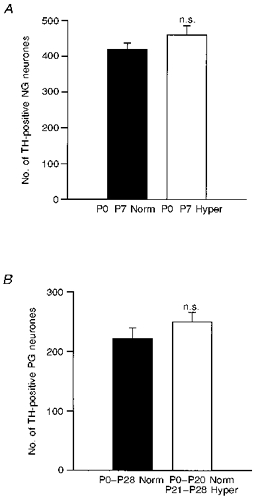
A, animals were reared in normoxia (▪, P0-P7 Norm, n= 8) or hyperoxia (□, P0-P7 Hyper, n= 8) for 1 week from birth. In contrast to the petrosal ganglion (Fig. 3), there was no significant difference in the number of dopaminergic neurones in the nodose ganglion (NG) between normoxic and hyperoxic animals. B, animals were reared continuously from birth in normoxia (▪, P0-P28 Norm, n= 11) or for 1 week in hyperoxia beginning at 3 weeks of age (□, P0-P20 Norm/P21-P28 Hyper, n= 13). In contrast to 1-week-old animals (Fig. 3), there was no significant difference in the number of dopaminergic neurones in the petrosal ganglion (PG) between normoxic and hyperoxic animals. Values are means +s.e.m.
Previous studies from our laboratory demonstrated that chemoafferent neurones require trophic support from the carotid body for survival after birth (Hertzberg et al. 1994). Thus, we hypothesized that chemoafferent degeneration following chronic hyperoxia might be secondary to a loss of target tissue in the carotid body. Since chemoafferent neurones already exhibited signs of degeneration (i.e. loss of TH expression) after 1 week of hyperoxia, we compared the volumes of the carotid body in animals reared in normoxia with those reared in hyperoxia for 1 week from birth (Fig. 5). Total carotid body volume decreased by 43 % in hyperoxic animals compared with normoxic controls (Fig. 6A; normoxia: (24.9 ± 2.8) × 106μm3vs. hyperoxia: (14.2 ± 1.6) × 106μm3; P= 0.0098). This change was paralleled by a 45 % decrease in the volume of the carotid body occupied by glomus cells (Fig. 6A; normoxia: (7.8 ± 0.8) × 106μm3vs. hyperoxia: (4.3 ± 0.6) × 106μm3; P= 0.0092). The ratio of glomic volume to total volume was the same in control and experimental animals (Fig. 6B; normoxia: 0.31 ± 0.01 vs. hyperoxia: 0.30 ± 0.02; P= 0.6667), indicating that the volume of both the glomic and non-glomic compartments within the carotid body decreased proportionately following 1 week of chronic hyperoxia.
Figure 5. Photomicrographs of sections through the carotid body from animals exposed to either normoxia (A) or hyperoxia (B) from birth to 1 week of age.
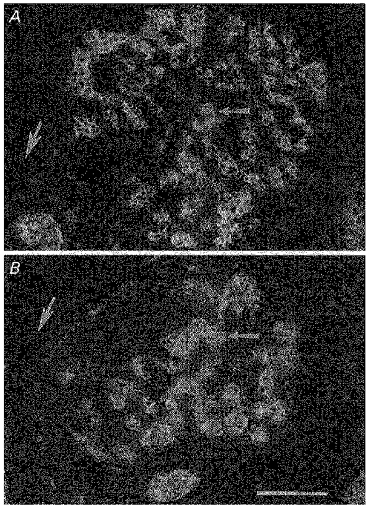
Sections were taken at the level of the carotid body artery (large arrows) and immunostained to detect tyrosine hydroxylase. Note that in these cross-sections, both the total area and the area occupied by glomus cells (small arrows) are markedly decreased in the carotid body from the chronically hyperoxic animal. Scale bar, 50 μm.
Figure 6. Exposure of newborn rats to chronic hyperoxia for 1 week from birth results in marked hypoplasia of the carotid body.
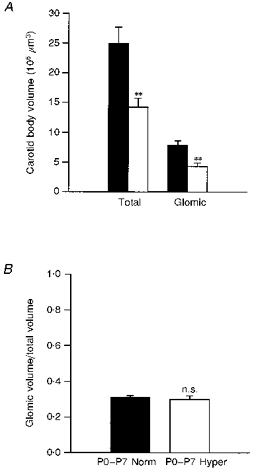
A, animals were killed at 1 week of age for morphometric analysis of carotid body volumes. Hyperoxic exposure (□, n= 5) resulted in a 43 % decrease in total volume (Total) and a 45 % decrease in the volume of the glomus cell compartment (Glomic) compared with normoxic controls (▪, n= 5). Values are means +s.e.m.**P < 0.01. B, the ratio of glomic to total volume was the same following 1 week of normoxia (▪, P0-P7 Norm) or hyperoxia (□, P0-P7 Hyper), indicating that both glomic and non-glomic compartments within the carotid body decreased proportionately following 1 week of chronic hyperoxia. P= 0.6667 (n.s.).
DISCUSSION
The present findings demonstrate that chronic exposure of newborn rats to elevated environmental oxygen results in the loss of chemoafferent axons projecting into the carotid sinus nerve. Moreover, evidence of early degenerative changes in chemoafferent cell bodies, i.e. loss of TH expression, after only 1 week of hyperoxia indicates that even a relatively brief elevation in environmental oxygen during the early postnatal period is sufficient to derange normal carotid body afferent development. In view of the fact that all petrosal neurones are postmitotic by embryonic day 15 (Altman & Bayer, 1984), at the beginning of the third trimester of gestation the axon loss we observed in hyperoxic animals can best be explained by degeneration of existing axons, rather than attenuation of chemoafferent neurone proliferation.
We hypothesize that chemoafferent degeneration in the chronically hyperoxic animals was related specifically to hypoplasia of the carotid body rather than a systemic effect of elevated tissue oxygen levels. Indeed, we have found previously that chemoafferent neurones depend on carotid body-derived trophic support for survival after birth (Hertzberg et al. 1994), indicating that the size of the chemoafferent population is related to target tissue availability, as in other developing sensory systems (Oppenheim, 1991). Although high levels of tissue oxygen can directly cause the death of various types of cells (Janssen, Van Houten, Borm & Mossman, 1993), the fact that chronic hyperoxia resulted in a loss of TH expression only in petrosal, and not in nodose neurones, argues against a non-selective cytotoxic mechanism. The petrosal and nodose ganglia both contain dopaminergic visceral sensory neurones; however, whereas virtually all dopaminergic petrosal afferents innervate the carotid body, most dopaminergic nodose afferents do not (Finley et al. 1992). Therefore afferent loss following chronic hyperoxia appears to be associated specifically with changes in the carotid body chemosensory pathway. In particular, we hypothesize that the loss of carotid body tissue leads to chemoafferent degeneration by decreasing the availability of target-derived trophic support. For example, we demonstrated previously that before birth, chemoafferent survival depends on brain-derived neurotrophic factor (BDNF), a member of the neurotrophin growth factor family that is expressed in the carotid body (Hertzberg et al. 1994; Erickson et al. 1996). More recent studies indicate that chronic hyperoxia decreases BDNF levels in the carotid body (D. M. Katz, unpublished observations), raising the possibility that loss of this factor contributes to the chemoafferent degeneration reported here.
There are several potential mechanisms by which chronic hyperoxia could lead to carotid body hypoplasia after birth. Normally, the carotid body increases in size during fetal and early postnatal development. For example, in the cat the volume of the carotid body increases by over 50 % between the late fetal and early postnatal period, with approximately proportional increases in the vascular and non-vascular compartments (Clarke, de Burgh Daly & Ead, 1990). In the rat, glomus cells occasionally have been reported to undergo mitosis during the late fetal and early postnatal period (Fishman & Schaffner, 1984; von Dalnok & Menßen, 1986). It is possible, therefore, that prolonged perinatal hyperoxia could decrease the size of the carotid body by directly attenuating cell proliferation, or inducing cell death. An alternative possibility is that the primary target of the hyperoxic insult is the carotid body vasculature, given that chronic hypoxia leads to carotid body hypertrophy in adult rats, due mainly to growth of blood vessels (Pequignot & Hellström, 1983; Pallot, 1986). It is possible, therefore, that hyperoxia inhibits angiogenesis in the carotid body leading secondarily to loss of glomus tissue. One candidate molecule that could mediate such an effect is vascular endothelial growth factor (VEGF), an angiogenic peptide that has recently been localized to the carotid body and whose expression is regulated by oxygen availability (Chen, Dinger, Jyung, Stensaas & Fidone, 1997).
Given that prolonged exposure of newborn rats to 60 % O2 results in marked chemoafferent neurone degeneration as well as carotid body hypoplasia, it is not surprising that such exposure leads to the blunting of hypoxic chemoreflexes (Ling et al. 1996, 1997b). Both impaired carotid body function (Ling et al. 1996, 1997b), and degeneration of chemoafferent inputs to the brainstem can be expected to result in attenuated chemoreflexes. However, electrical stimulation of the carotid sinus nerve is equally effective at increasing phrenic nerve output in normoxic and chronically hyperoxic animals, despite the blunting of hypoxic reflexes in the hyperoxic group (Ling et al. 1997a). This difference in the response to hypoxia vs. electrical stimulation in animals exposed to chronic hyperoxia from birth may indicate that the marked hypoplasia of the carotid body disrupts either functional connections between glomus cells and surviving chemoafferent fibres, or functioning of the glomus cells themselves, or both. Under such conditions electrical stimulation of the surviving subpopulation of chemoafferent fibres, but not physiological stimulation of the carotid body by hypoxia, could well be sufficient to produce a normal phrenic nerve response.
The fact that chemoafferent survival after birth is decreased by a chronic elevation in environmental oxygen may help to shed light on derangements in chemoreflex development observed in infants undergoing supplemental oxygen therapy. In particular, oxygen therapy has been linked to depressed hypoxic ventilatory drive, especially in a subset of low birth weight preterm infants (Katz-Salamon & Lagercrantz, 1994). Although it is unexpected that infants undergoing oxygen therapy would be subjected to the prolonged hyperoxaemia probably experienced by the chronically hyperoxic animals used in our study, we do not yet know the threshold level at which elevated arterial PO2 leads to a derangement of chemoafferent pathway development. Our findings demonstrate that carotid body hypoplasia and chemoafferent cell death are mechanisms by which increased oxygen availability could perturb normal chemoreflex maturation.
Note added in proof
Recent experiments from our laboratory (J. T. Erickson & D. M. Katz, unpublished observations) demonstrated that exposure of newborn rats to even transient, mild hyperoxia (1 week of 30 % O2) is sufficient to produce carotid body hypoplasia equivalent to that reported in the present study.
Acknowledgments
The authors wish to thank Hua Jun He, Roseann Brady and Candice Bartholomew for expert technical assistance and Dr Agnieszka Balkowiec and Teresa Brosenitsch for thoughtful reading of the manuscript. This work was supported by United States Public Health Service grants (HL-42131 and HL-25830, Project 4 to D. M. K. and HL-53319 to G. S. M.) and the Francis Families Foundation (J. T. E).
References
- Abercrombie M. Estimation of nuclear populations from microtome sections. Anatomical Record. 1946;94:239–247. doi: 10.1002/ar.1090940210. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the cranial nerve ganglia and related nuclei in the rat. Advances in Anatomy, Embryology and Cell Biology. 1984;74:1–87. doi: 10.1007/978-3-642-68479-1. [DOI] [PubMed] [Google Scholar]
- Bureau MA, Foulon P, Zinman R, Begin R. Diphasic ventilatory response to hypoxia in newborn lambs. Journal of Applied Physiology. 1984;56:84–90. doi: 10.1152/jappl.1984.56.1.84. [DOI] [PubMed] [Google Scholar]
- Chen J, Dinger B, Jyung R, Stensaas L, Fidone S. Upregulation of Vascular Endothelial Growth Factor (VEGF) and VEGF receptor (Flk-1) in chronically hypoxic rat carotid body. Society for Neuroscience Abstracts. 1997;27:433. [Google Scholar]
- Clarke JA, de Burgh Daly M, Ead HW. Comparison of the size of the vascular compartment of the carotid body of the fetal, neonatal and adult cat. Acta Anatomica. 1990;138:166–174. doi: 10.1159/000146934. [DOI] [PubMed] [Google Scholar]
- Eden GJ, Hanson MA. Effects of hyperoxia from birth on the carotid chemoreceptor and ventilatory responses of rats to acute hypoxia. The Journal of Physiology. 1986;374:24. P. [Google Scholar]
- Eden GJ, Hanson MA. Effects of chronic hypoxia from birth on the ventilatory response to acute hypoxia in the newborn rat. The Journal of Physiology. 1987a;392:11–19. doi: 10.1113/jphysiol.1987.sp016766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden GJ, Hanson MA. Maturation of the respiratory response to acute hypoxia in the newborn rat. The Journal of Physiology. 1987b;392:1–9. doi: 10.1113/jphysiol.1987.sp016765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erickson JT, Conover JC, Borday V, Champagnat J, Barbacid M, Yancopoulos G, Katz DM. Mice lacking brain-derived neurotrophic factor exhibit visceral sensory neuron losses distinct from mice lacking NT4 and display a severe developmental deficit in control of breathing. Journal of Neuroscience. 1996;16:5361–5371. doi: 10.1523/JNEUROSCI.16-17-05361.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finley JCW, Polak J, Katz DM. Transmitter diversity in carotid body afferent neurons: dopaminergic and peptidergic phenotypes. Neuroscience. 1992;51:973–987. doi: 10.1016/0306-4522(92)90534-9. 10.1016/0306-4522(92)90534-9. [DOI] [PubMed] [Google Scholar]
- Fishman MC, Schaffner AE. Carotid body cell culture and selective growth of glomus cells. American Journal of Physiology. 1984;246:C106–113. doi: 10.1152/ajpcell.1984.246.1.C106. [DOI] [PubMed] [Google Scholar]
- Hanson MA, Eden GJ, Nijhuis JG, Moore PJ. Peripheral chemoreceptors and other oxygen sensors in the fetus and newborn. In: Lahiri S, Forster RE, Davies RO, Pack AI, editors. Chemoreceptors and Reflexes in Breathing: Cellular and Molecular Aspects. New York: Oxford University Press; 1989. pp. 113–120. [Google Scholar]
- Hertzberg T, Fan G, Finley JCW, Erickson JT, Katz DM. BDNF supports mammalian chemoafferent neurons in vitro and following peripheral target removal in vivo. Developmental Biology. 1994;166:801–811. doi: 10.1006/dbio.1994.1358. 10.1006/dbio.1994.1358. [DOI] [PubMed] [Google Scholar]
- Hertzberg T, Hellström S, Holgert H, Lagercrantz H, Pequignot JM. Ventilatory response to hyperoxia in newborn rats born in hypoxia - possible relationship to carotid body dopamine. The Journal of Physiology. 1992;456:645–654. doi: 10.1113/jphysiol.1992.sp019358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen YMW, Van Houten B, Borm PJA, Mossman BT. Biology of disease. Cell and tissue responses to oxidative damage. Laboratory Investigation. 1993;69:261–274. [PubMed] [Google Scholar]
- Katz DM, Black IB. Expression and regulation of catecholaminergic traits in primary sensory neurons: relationship to target innervation in vivo. Journal of Neuroscience. 1986;6:983–989. doi: 10.1523/JNEUROSCI.06-04-00983.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz-Salamon M, Lagercrantz H. Hypoxic ventilatory defence in very preterm infants: attenuation after long term oxygen treatment. Archives of Disease in Childhood. 1994;70:F90–95. doi: 10.1136/fn.70.2.f90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH, Mitchell GS. Attenuation of the hypoxic ventilatory response in adult rats following one month of perinatal hyperoxia. The Journal of Physiology. 1996;495:561–571. doi: 10.1113/jphysiol.1996.sp021616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH, Mitchell GS. Integrated phrenic responses to carotid afferent stimulation in adult rats following perinatal hyperoxia. The Journal of Physiology. 1997a;500:787–796. doi: 10.1113/jphysiol.1997.sp022058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ling L, Olson EB, Jr, Vidruk EH, Mitchell GS. Phrenic responses to isocapnic hypoxia in adult rats following perinatal hyperoxia. Respiration Physiology. 1997b;109:107–116. doi: 10.1016/s0034-5687(97)00045-5. 10.1016/S0034-5687(97)00045-5. [DOI] [PubMed] [Google Scholar]
- McDonald DM. Morphology of the rat carotid sinus nerve. II. Number and size of axons. Journal of Neurocytology. 1983;12:373–392. doi: 10.1007/BF01159381. [DOI] [PubMed] [Google Scholar]
- McDonald DM, Mitchell RA. The innervation of glomus cells, ganglion cells and blood vessels in the rat carotid body: a quantitative ultrastructural analysis. Journal of Neurocytology. 1975;4:177–230. [Google Scholar]
- Oppenheim RW. Cell death during development of the nervous system. Annual Review of Neuroscience. 1991;14:453–501. doi: 10.1146/annurev.ne.14.030191.002321. 10.1146/annurev.ne.14.030191.002321. [DOI] [PubMed] [Google Scholar]
- Pallot DJ. The mammalian carotid body. Advances in Anatomy, Embryology and Cell Biology. 1986;102:1–89. doi: 10.1007/978-3-642-71857-1. [DOI] [PubMed] [Google Scholar]
- Pequignot J-M, Hellström S. Intact and sympathectomized carotid bodies of long-term hypoxic rats. A morphometric light microscopical study. Virchows Archives. 1983;400:235–243. doi: 10.1007/BF00612185. [DOI] [PubMed] [Google Scholar]
- Sladek M, Parker RA, Grogaard JB, Sundell HW. Long-lasting effect of prolonged hypoxemia after birth on the immediate ventilatory response to changes in arterial partial pressure of oxygen in young lambs. Pediatric Research. 1993;34:821–828. doi: 10.1203/00006450-199312000-00025. [DOI] [PubMed] [Google Scholar]
- Vidruk EH, Olson EB, Jr, Ling L, Mitchell GS. Carotid sinus nerve responses to hypoxia and cyanide are attenuated in adult rats following perinatal hyperoxia. Physiologist. 1996;39:190. [Google Scholar]
- von Dalnok GK, Menßen HD. A quantitative electron microscopic study of the effect of glucocorticoids in vivo on the early postnatal differentiation of paraneuronal cells in the carotid body and the adrenal medulla of the rat. Anatomy and Embryology. 1986;174:307–319. doi: 10.1007/BF00698781. [DOI] [PubMed] [Google Scholar]