

Abstract
The action of philanthotoxin 343 (PhTX) on rat homomeric GluR6(Q) recombinant glutamate receptor channels was analysed using concentration-jump techniques and outside-out patches from HEK 293 cells. Both onset and recovery from block by external PhTX were dependent on the presence of agonist, indicating that channels must open for PhTX to bind and that channel closure can trap PhTX.
Block by external PhTX developed with double-exponential kinetics. The rate of onset of the fast component of block showed an exponential increase per 27 mV hyperpolarization over the range -40 to -100 mV. The rate of onset of the slow component of block showed a non-linear concentration dependence indicating a rate-limiting step in the blocking mechanism.
The extent of block by 1 μM external PhTX was maximal at -40 mV and did not increase with further hyperpolarization; the rate of recovery from block by external PhTX increased 6-fold on hyperpolarization from -40 to -100 mV suggesting that PhTX permeates at negative membrane potentials.
Apparent Kd values for block by external PhTX estimated from dose-inhibition experiments decreased 300-fold on hyperpolarization from +40 mV (Kd, 19.6 μM) to -40 mV (Kd, 69 nM); there was little further increase in affinity with hyperpolarization to -80 mV (Kd, 56 nM), consistent with permeation of PhTX at negative membrane potentials.
Block by internal PhTX showed complex kinetics and voltage dependence. Analysis with voltage ramps from -120 to +120 mV indicated a Kd at 0 mV of 20 μM, decreasing e-fold per 16 mV depolarization. However, at +90 mV the extent of block by 1 and 10 μM internal PhTX (73 % and 95 %, respectively) reached a maximum and did not increase with further depolarization.
Voltage-jump analysis of block by 100 μM internal PhTX revealed partial trapping. With 100 ms jumps from -100 to -40 mV, onset and recovery from block were complete within 5 ms. With jumps of longer duration the extent of block increased, with a time constant of 8.1 s, reaching 84 % at 30 s. On repolarization to -100 mV, recovery from block showed fast and slow components.
The amplitude of the slow component of block by internal PhTX showed a biphasic voltage dependence, first increasing then decreasing with progressive depolarization. Maximum block was obtained at 0 mV.
Our results suggest that PhTX acts as an open channel blocker; however, provided that the toxin remains bound to the channel, an allosteric mechanism destabilizes the open state, inducing channel closing and trapping PhTX. Strong depolarization for internal PhTX, or strong hyperpolarization for external PhTX, forces the toxin to permeate before it triggers entry into closed blocked states.
Glutamate receptor ion channels in both vertebrates and invertebrates are blocked by nanomolar concentrations of the polyamine toxins present in spider and wasp venoms. Although a large number of synthetic analogues of these toxins have been developed, allowing considerable insight into the structure-activity relationships required for their potent activity (Anis et al. 1990; Usherwood & Blagbrough, 1991), their blocking action is not understood in any detail. The main mechanism by which polyamine toxins act on glutamate receptors appears to be use- and voltage-dependent ion channel block (Herlitze et al. 1993), but there have been reports of weak competitive antagonism of NMDA receptors (Donevan & Rogawski, 1996), and polyamine-like potentiation of NMDA- and kainate-activated receptors (Ragsdale et al. 1989; Brackley, Goodnow, Nakanishi, Sudan & Usherwood, 1990; Donevan & Rogawski, 1996).
The common structural motif of these toxins is a polyamine chain with an amide-linked aromatic amino acid at one end. The toxin molecules are typically 2.0-4.0 nm in length, making it likely that they interact with multiple amino acid residues within the ion channel pore. Photoaffinity labelling of nicotinic receptors (Choi, Kalivretenos, Usherwood & Nakanishi, 1995), as well as structure-activity studies for glutamate receptors using analogues with modifications in either the polyamine chain or aromatic head of the toxin molecule (Anis et al. 1990; Bruce et al. 1990), support a model of action in which the linear polyamine chain penetrates and occludes the ion channel pore while the distal aromatic moiety interacts with amino acid residues situated above the narrowest region of the pore.
The cloning of a large family of glutamate receptors (GluRs) has now allowed examination of the molecular determinants required for high-potency inhibition by polyamine toxins (Seeburg, 1993; Hollmann & Heinemann, 1994). Studies on recombinant non-NMDA receptors expressed in Xenopus oocytes have shown that an amino acid residue which regulates Ca2+ permeability also controls block by polyamine toxins, since both require a glutamine residue at the mRNA-editing (Q/R) site in the M2 segment (Hume, Dingledine & Heinemann, 1991; Burnashev, Monyer, Seeburg & Sakmann, 1992; Blaschke, Keller, Rivosecchi, Hollmann, Heinemann & Konnerth, 1993; Brackley, Bell, Choi, Nakanishi & Usherwood, 1993; Herlitze et al. 1993); an asparagine residue at the corresponding site in the M2 segments of NMDA receptor subunits is required for high-affinity block by argiotoxin (Raditsch et al. 1993). For non-NMDA receptor channels, mRNA editing at the Q/R site also controls ‘intrinsic’ rectification (Verdoorn, Burnashev, Monyer, Seeburg & Sakmann, 1991). The subsequent finding that such ‘intrinsic’ rectification is due to voltage-dependent block by cytoplasmic polyamines, principally spermine and spermidine (Bowie & Mayer, 1995; Donevan & Rogawski, 1995; Isa, Iino, Itazawa & Ozawa, 1995; Kamboj, Swanson & Cull-Candy, 1995; Koh, Burnashev & Jonas, 1995), suggests that the molecular determinants for block by internal polyamines and externally applied polyamine toxins are likely to overlap. Although spermine is known to permeate mammalian non-NMDA receptors (Bowie & Mayer, 1995; Bähring, Bowie, Benveniste & Mayer, 1997), due to their much larger size polyamine toxins would not be expected to pass easily through glutamate receptor channels, raising the possibility that block by internal and external toxins could occur via separate mechanisms.
In prior studies on block of recombinant non-NMDA receptor channels by polyamine toxins, experiments were performed using two-electrode voltage clamp in Xenopus oocyte expression systems, where slow drug application prevented any detailed kinetic analysis; in addition, inward rectification due to ion channel block by endogenous cytoplasmic polyamines seriously interfered with analysis of the voltage dependence of toxin block (Brackley et al. 1993; Herlitze et al. 1993; Washburn & Dingledine, 1996). In the present study these limitations were overcome using fast-perfusion techniques and outside-out membrane patches to study block of GluR6(Q) glutamate receptors in the absence of endogenous polyamines. For these experiments we used philanthotoxin 343 (PhTX), a synthetic analogue of a conjugated polyamine present in the venom of the wasp Philanthus triangulum (Eldefrawi et al. 1988). Our results show that PhTX acts initially as an open channel blocker; however, provided that the toxin remains bound to the channel for sufficient time, an allosteric mechanism destabilizes the open state, inducing channel closing and trapping PhTX. We suggest that in the presence of agonist, glutamate receptor channels can close with PhTX bound, and that this represents an important component of the blocking mechanism for external PhTX. In addition, we show that PhTX is weakly permeable, and that this produces partial trapping, limiting the extent of block at high membrane electric field strengths, since strong depolarization for internal PhTX, or strong hyperpolarization for external PhTX, destabilize the binding of PhTX, forcing dissociation and permeation of toxin before it triggers entry into closed blocked states.
METHODS
Transfection and recording techniques
Homomeric glutamate receptor channels assembled from GluR6(Q) subunits were transiently expressed in HEK 293 cells as described previously (Bowie & Mayer, 1995; Bähring et al. 1997). Cotransfection of glutamate receptor cDNA (150 ng per 35 mm dish) with cDNA for the S65T mutant of green fluorescent protein (GFP), both gifts from Dr P. Seeburg (Department of Molecular Neurobiology, Max-Planck-Institute for Medical Research, Heidelberg), allowed the identification of transfected cells during experiments. A high ratio of GFP to GluR6 (1:1) was used to lower expression of glutamate receptor channels, since we found that with high levels of expression the collapse of transmembrane ionic concentration gradients can distort electrophysiological analysis.
All recordings were done in the outside-out patch configuration with equal activities of Na+ (150 mM) on either side of the membrane. The external solution contained (mM): NaCl, 150; CaCl2, 0.1; MgCl2, 0.1; and Hepes, 5; 0.01 mg ml−1 Phenol Red; pH 7.3; 295 mosmol l−1. Only 100 μM Ca2+ and 100 μM Mg2+ were added to the external solution to minimize divalent cation block; where specifically noted, Ca2+ and Mg2+ concentrations were increased to 200 and 800 μM, respectively, to improve patch stability; block by PhTX appeared not to be altered at these divalent cation concentrations. Recording electrodes had a tip resistance of 2-5 MΩ when filled with a solution containing (mM): NaCl, 110; NaF, 10; CaCl2, 0.5; Hepes, 5; Na4BAPTA, 5; and Na2ATP, 10; pH 7.2; 295 mosmol l−1. Except for analysis of the action of internal PhTX, ATP was included in the internal solution to chelate residual endogenous polyamines; as described previously, this completely abolished inward rectification (Bähring et al. 1997).
Solutions were applied with a stepper motor-based fast-perfusion system which allowed solution exchange in less than 10 ms (Vyklicky, Benveniste & Mayer, 1990). In order to reduce desensitization, patches were treated with 0.3 mg ml−1 concanavalin A (Sigma, Type IV) for 1 min prior to recording. GluR6(Q) responses were evoked by 50 μM domoic acid (Tocris Cookson). Philanthotoxin 343 trifluoroacetate was obtained from RBI and for simplicity is referred to as PhTX. During experiments, patches were held at 0 mV except during acquisition of data. This increased the stability of recordings and allowed analysis of responses to repeated applications of PhTX to the same patch. Recovery from block by prior applications of PhTX was obtained by repeated voltage jumps (300 ms duration, 1 Hz, to +60 mV) from a holding potential of 0 mV in the constant presence of domoate. All experiments were conducted at room temperature (22-25°C).
Data acquisition and analysis
Responses were recorded from outside-out patches voltage clamped using an Axopatch 200 amplifier (Axon Instruments), filtered at a frequency between 0.5 and 10 kHz with an 8-pole Bessel filter, and amplified as required. Signals were digitized with sampling periods of 50 μs to 20 ms and stored on a Power Macintosh 7600/132 computer using an ITC16 interface under the control of the data acquisition and analysis program Synapse (Synergy Research Inc., Silver Spring, MD, USA).
To calculate apparent dissociation constants for PhTX block at different holding potentials, a one binding site model was fitted to dose-inhibition plots:
| (1) |
where Imax is the control current amplitude measured before application of PhTX, [B] the PhTX concentration, and Kd the apparent dissociation constant for PhTX at any given membrane potential.
To characterize the voltage dependence of block by PhTX we used a Boltzmann function modified to account for the observed incompleteness of block at strongly hyperpolarized or depolarized potentials:
| (2) |
where Gmax is the fully unblocked conductance, G the conductance at a membrane potential Vm, and Gmin the minimum conductance at strongly hyperpolarized membrane potentials; V½ is the potential at which block is half-maximal, and k is the slope factor. For comparison with prior work, responses were also fitted with a Woodhull model for an impermeable ion channel blocker (Woodhull, 1973). For external PhTX, the Kd for block as a function of membrane potential is given by: Kd = Kd(0)exp(VmzδF/RT), where Kd(0) is the dissociation constant at 0 mV; z is the valence of PhTX, which has been determined by NMR to be +3 at physiological pH (Jaroszewski, Matzen, Frolund & Krogsgaard-Larsen, 1996); and δ represents the fraction of the membrane electric field measured from the outer surface experienced by PhTX at its binding site within the channel. F, R and T have their usual meanings. The appropriate equations were used for block by internal PhTX.
The kinetics of onset and recovery from PhTX block were usually best described by a double-exponential function. To allow comparison of the recovery kinetics at different holding potentials we calculated weighted recovery time constants:
where W1 and W2 represent the relative percentage contributions of the respective time constants to the total current decay. Results are presented as means ± s.e.m. from analysis of pooled data, unless otherwise stated. Data analysis was performed with Synapse and Kaleidagraph (Synergy Software, Reading, PA, USA). Statistical tests were performed by variance analysis using the program GraphPad InStat (GraphPad Software, San Diego, CA, USA).
RESULTS
Use dependence of PhTX block revealed by concentration-jump experiments
At a membrane potential of -60 mV, PhTX applied to outside-out patches at a concentration of 1 μM caused substantial (94.5 ± 0.5 %, mean ± s.d.; n = 4) block of GluR6(Q) responses activated by 50 μM domoate (Fig. 1A). Block by PhTX developed rapidly, reaching equilibrium within 6 s. In order to test whether the blocking action of PhTX (1 μM) required activation of ion channel gating by agonist, PhTX was applied for 8 s prior to a concentration-jump application of domoate in the continued presence of PhTX. With this protocol, the application of agonist caused an inward current transient with a peak amplitude that reached 96.6 ± 7.0 % (mean ± s.d.; n = 4) of the current evoked by domoate in the same patch in the absence of PhTX (Fig. 1B). The current then decayed to a steady-state level of 7.1 ± 2.7 % (mean ± s.d.; n = 4) of the peak response due to onset of block by PhTX, similar to the response observed when PhTX was applied in the presence of domoate (Fig. 1C). Identical results were obtained when 1 μM PhTX was applied for 60 s prior to application of domoate (n = 3). A consistent trend for the peak amplitude of the response to domoate applied in the constant presence of PhTX to be slightly below 100 % of control is likely to result from block by PhTX which develops during the onset of the response to agonist (the 10-90 % rise time of currents activated by 50 μM domoate in the absence of PhTX was 44.9 ± 2.9 ms; n = 4). Indeed, when exponential fits to the onset of block by PhTX were extrapolated to the mid-point of the rising phase of the response to domoate, the peak amplitude increased to 107 ± 12 % of control, suggesting essentially no block by external PhTX when applied to closed channels. The results of our experiments thus exclude an inhibitory action of PhTX in the absence of agonist, indicating that block of GluR6(Q) channels by external PhTX is use dependent. This is typical for the action of toxins that need to access the channel pore in order to exert their blocking action, and is a consistent finding for the action of polyamine toxins on non-NMDA receptors (Brackley et al. 1993; Herlitze et al. 1993; Iino, Koike, Isa & Ozawa, 1996; Washburn & Dingledine, 1996). In the simplest case such an open channel block mechanism would follow a first-order bimolecular relationship reflecting binding of the toxin to a site within the channel. In this case the kinetics of onset of block should be single exponential. The fast drug application system used in our experiments allowed a detailed analysis of the onset kinetics for PhTX block. They were best described by a double-exponential function (Fig. 1A and B), which suggests that after channel opening more than one step is involved in the blocking reaction.
Figure 1. Block by external PhTX requires activation by agonist.
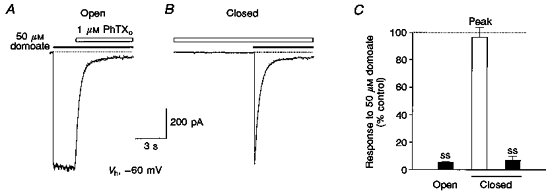
A, rapid onset of block by 1 μM external PhTX (PhTXo, open bar) applied to an outside-out patch after channels have opened in response to 50 μM domoate (filled bar). B, in the same patch PhTX failed to produce block of the response to domoate when applied to closed channels prior to the application of agonist, but rapidly attenuated the inward current generated by domoate as soon as channels opened in response to a concentration-jump application of agonist. Both responses were recorded at -60 mV. Dotted lines indicate the holding current prior to application of agonist. Curves fitted to the onset of block by PhTX represent double-exponential functions of time constants: τfast = 370, τslow = 790 ms (36 %, A) and τfast = 360, τslow = 890 ms (13 %, B). C, mean values from 4 experiments for block by 1 μM PhTX plotted relative to the amplitude of control responses to 50 μM domoate. The large amplitude of peak responses to domoate after prior application of PhTX in the absence of agonist (97 ± 7 %) indicates minimal closed channel block; steady-state (ss) current during block by 1 μM PhTX is similar when toxin is applied prior to or after application of agonist.
Concentration dependence of external PhTX blocking kinetics
A simple bimolecular reaction scheme for open channel block predicts that the association rate increases linearly with toxin concentration. Based on the assumption that one of the two observed components of block represents such an open channel blocking reaction, we studied the concentration dependence of the rate of block by external PhTX. For this purpose, PhTX was applied at five different concentrations (0.3, 1, 3, 10 and 30 μM) to individual patches at a holding potential of -60 mV (n = 4). The rate of onset of block increased with concentration of PhTX and was best described by a double-exponential function (Fig. 2A). For 0.3, 1, 3 and 30 μM PhTX we calculated mean values for τfast, the time constant of the fast component of block, of 1148 ± 211, 553 ± 42, 174 ± 19 and 26 ± 2 ms, respectively; for τslow, the time constant of the slow component of block, the values were 3.4 ± 0.5 s, 1.8 ± 0.3 s, 896 ± 108 ms and 211 ± 21 ms, respectively (n = 4). The relative amplitude of the slow component was 55 ± 9.4 % for 0.3 μM, 28.7 ± 7.9 % for 1 μM, 20.8 ± 2.4 % for 3 μM and only 8.9 ± 1.9 % for 30 μM PhTX.
Figure 2. Fast and slow components of block by external PhTX.
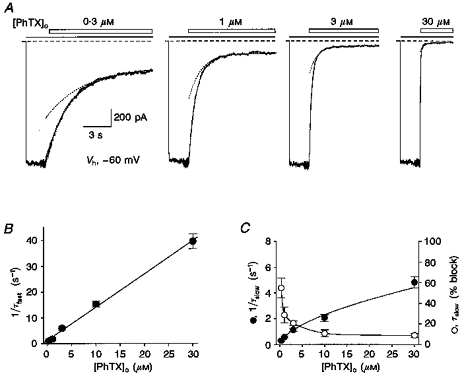
A, responses from the same patch to 4 concentrations of PhTX (open bars) applied during responses to 50 μM domoate (filled bars). Dashed lines represent the holding current at -60 mV prior to application of agonist. The external solution contained 200 and 800 μM external Ca2+ and Mg2+, respectively, to improve patch stability. The apparent increase in noise preceding the response to PhTX is due to a change in sample clock frequency. Lines drawn through the data points are double-exponential functions fitted to the onset of block by external PhTX. Dotted lines show the exponential for the slow component of onset of block. The time constants were: 0.3 μM PhTX, τfast = 1.68 s, τslow = 4.12 s (53 %); 1 μM PhTX, τfast = 0.58 s, τslow = 1.36 s (43 %); 3 μM PhTX, τfast = 207 ms, τslow = 810 ms (26 %); 30 μM PhTX, τfast = 32 ms, τslow = 380 ms (9 %). B, rates for the fast component of onset of block (1/τfast) plotted against PhTX concentration; the continuous line shows a linear fit to mean values from 4 patches which gives an estimate for the association rate constant for PhTX at -60 mV of 1.3 × 106 M−1 s−1. C, plot of PhTX concentration versus rate of onset of the slow component of block (1/τslow, •) and the percentage of the onset of block accounted for by the slow component (○) clearly demonstrates non-linear concentration dependence and suggests saturation at high concentrations of PhTX (lines connecting the data points have no significance).
The calculated rates for onset of block, 1/τfast and 1/τslow, when plotted against PhTX concentration show the expected linear relationship for a bimolecular interaction for 1/τfast, while for 1/τslow the rate of onset of block deviated from linearity (Fig. 2B and C). For example, a 10-fold increase in PhTX concentration, from 3 to 30 μM, caused only a 4-fold increase in 1/τslow, from 1.16 to 4.84 s−1. In contrast, the plot of 1/τfastversus PhTX concentration was linear with a slope of 1.29 × 106 M−1 s−1 at -60 mV (Fig. 2B).
These results suggest that the fast component of the action of PhTX is likely to represent an open channel blocking reaction. The non-linear concentration dependence of the slow component, however, indicates a rate-limiting step in the blocking reaction, possibly a conformational change in the channel protein due to the binding of PhTX to an allosteric site. Although it is possible that PhTX could act at some extracellular allosteric site which becomes accessible only after binding of agonist, based on subsequent experiments which revealed a strong voltage dependence of PhTX action we favoured another hypothesis for the interpretation of our results: that the rate-limiting step which causes saturation of 1/τslow with increasing PhTX concentration is the closing of GluR6(Q) channels after PhTX has bound to a site within the ion channel pore. Channel closure with PhTX bound would be expected to cause trapping of the blocker with the consequence that channels would have to reopen in order to unblock. If the unbinding of PhTX occurs slowly enough this would result in an accumulation of receptors in the closed blocked state. At non-saturating PhTX concentrations, the rate of accumulation in the closed blocked state would depend on the occupancy of the open blocked state, and hence increase with concentration of PhTX. However, at higher PhTX concentrations, the closing rate would become rate limiting. In the absence of PhTX, the decay of responses to domoate following removal of agonist was best fitted by the sum of two exponentials which reveal ‘fast’ (1/τ1 = 0.44 s−1) and slow rates (1/τ2 = 0.08 s−1) of deactivation. Since the rate for the slow component of block by 30 μM PhTX at -60 mV (1/τslow), 4.84 s−1 (Fig. 2C), is 10 times faster than the rate of even the ‘fast’ component of deactivation, our results suggest that the closing rate constant must increase at least 10-fold when PhTX is bound to the channel.
The relative amplitude of the slow component of block decreased with increasing PhTX concentration (Fig. 2C). This suggests that due to the faster on-rate for open channel block at high concentrations of PhTX this mechanism accounts for most of the observed blocking kinetics, whereas at low concentrations of PhTX virtually all of the decay will be due to the accumulation of channels in a closed blocked state. However, although at high concentrations of PhTX channels are likely to first block due to binding of toxin to open channels, subsequent transitions to the closed blocked state would be expected to underlie prolonged recovery from block by PhTX as described below.
Trapping suggests that GluR6(Q) channels can close with PhTX bound
To establish that GluR6(Q) channels are indeed able to close with PhTX bound, which would be expected to cause trapping of the blocker, we studied the agonist dependence of recovery from block. In these experiments strong block of domoate-activated responses was induced by the application of 10 μM PhTX at -60 mV for 10 s. When PhTX was removed in the continued presence of domoate, recovery from block developed with slow kinetics, reaching 75.2 ± 4.4 % (n = 6) of control after 60 s (Fig. 3A). The recovery kinetics could be described by a double-exponential function with τ1 = 6.5 ± 1.2 s and τ2 = 85.0 ± 18.6 s (60.2 ± 5.0 % of the total decay; n = 6). Extrapolation of exponential fits suggested that full recovery from PhTX block at -60 mV required more than 8 min; such slow recovery from block was difficult to study experimentally due to patch instability, and normally patches were depolarized to 0 mV before full recovery was obtained (see Methods).
Figure 3. Recovery from block by PhTX requires activation by agonist.
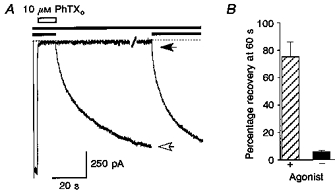
A, slow recovery from block of responses to 50 μM domoate (filled bars) by 10 μM external PhTX (open bar) at -60 mV. Two responses from the same patch are shown superimposed; one was recorded with agonist applied continuously following removal of PhTX, the second was recorded with a 60 s wash with control solution following termination of the application of PhTX before a second application of agonist. The open arrow indicates 80 % recovery from block at 60 s when PhTX was removed in the presence of agonist; the filled arrow recorded after a 60 s wash with control solution indicates the amplitude of the ‘instantaneous’ component of the response to domoate due to activation of a small fraction of unblocked channels, which is followed by a slow increase in current due to recovery from block by PhTX. B summarizes results from 6 patches (means ± s.d.) analysed using the same protocol, and indicates that recovery from block by PhTX is greatly enhanced by the application of agonist.
To test whether PhTX could dissociate in the absence of agonist, domoate was removed after the onset of block by PhTX but 20 s prior to the removal of toxin to allow deactivation to proceed in the presence of blocker (Fig. 3A). The subsequent removal of PhTX was followed by a 60 s period of superfusion with control solution. Such experiments showed that recovery from block was greatly reduced in the absence of agonist, as indicated by only a small amplitude ‘instantaneous’ current (5.8 ± 0.5 % of control; n = 6) when domoate was reapplied 60 s after removal of toxin (Fig. 3B). Notably, no additional recovery was achieved when the membrane potential was depolarized to 0 mV or +40 mV during the 60 s period of superfusion with control solution (not shown). Although our results suggest that PhTX gets trapped in closed GluR6(Q) channels, they do not exclude the possibility that in the absence of agonist the toxin dissociates at a rate too slow to be detected in our experiments; assuming that PhTX can dissociate in the absence of agonist, 6 % recovery at 60 s gives a recovery time constant of 16 min, much too slow to be tested easily by patch clamp analysis.
Taken together, our experiments show that both onset and recovery from block of GluR6(Q) channels by PhTX are use dependent. These results suggest that the blocking mechanism follows an open channel blocking scheme with the existence of at least one toxin-bound closed state. This is summarized in Scheme 1, which represents an extension of the classical sequential model of ion channel block (Neher, 1983):
Scheme 1.

where A represents the agonist, and the binding of two agonist molecules leads from the closed state (R) to the open state of the receptor channel complex (R *). The rate constants for binding and unbinding of agonist are represented by k+ and k-, respectively, and β and α are the rate constants for channel opening and closing, respectively. The association rate constant, kon, and the dissociation rate constant, koff, for the blocker B define the open channel blocking reaction, and the rate constants for channel gating in the presence of blocker are given by β’ and α’. Provided that PhTX alters the opening or closing rate constants to lower the maximum open probability (a similar result would be obtained if PhTX-blocked channels have a lower affinity for agonist), the binding of PhTX eventually leads to a high level of occupancy of closed blocked states (Lingle, 1983; Blanpied, Boeckman, Aizenman & Johnson, 1997).
The above model is likely to be oversimplified since double-exponential deactivation kinetics observed following removal of domoate in the absence of PhTX suggest at least two open states, although other mechanisms such as reopening of desensitized channels could underlie the biexponential rate of decay. Trapping of PhTX might then be possible from only one or from both open blocked states. The recovery kinetics from PhTX block after trapping (Fig. 3A) were also best described by a double-exponential function when agonist was reapplied following a 60 s wash with control solution (τ1 = 4.5 ± 0.8 s; τ2 = 40.8 ± 9.7 s, 63.2 ± 4.2 % of the total decay; n = 6), and were slightly faster than when PhTX was removed in the presence of agonist without a 60 s wash with control solution (τ1 = 6.5 ± 1.2 s; τ2 = 85.0 ± 18.6 s, 60.2 ± 5.0 % of the total decay). The trend for slower recovery when PhTX was removed in the presence of domoate, without a 60 s wash in control solution, might result from accumulation of toxin in an unstirred layer at the membrane surface in addition to the specific interaction of the toxin with the ion channel pore.
Voltage dependence of block by external PhTX
For an open channel block mechanism in which PhTX binds to a site within the membrane electric field, the rate constants for binding and dissociation of external PhTX would show an inverse voltage dependence such that membrane hyperpolarization leads to an increase in kon and a decrease in koff (Scheme 1). Due to the resulting higher occupancy of the open blocked state A2R *B, the probability for channels to enter the closed blocked state A2RB and hence the amount of equilibrium block should also increase with hyperpolarization.
In order to estimate the voltage dependence of PhTX binding, we studied in detail the kinetics of block by external PhTX over a wide range of membrane potentials. This was possible because we were able to completely abolish rectification due to endogenous cytoplasmic polyamines by adding ATP to the internal solution (see Methods). We chose a concentration of 1 μM PhTX, which caused strong but incomplete block at -60 mV (Fig. 1). Both the rate of onset and the amount of block by PhTX were measured at holding potentials between +40 mV and -100 mV (Fig. 4A). The block observed at +40 mV developed with a mean time constant of 32.4 ± 8.8 s (n = 3). At +20 and -20 mV the onset of block by 1 μM PhTX was also well fitted by single-exponential functions with time constants of 14.7 ± 1.5 s (n = 5) and 3.7 ± 0.3 s (n = 7), respectively (Fig. 4A). Apparently, at these membrane potentials the major component of block reflects accumulation of receptors in a closed blocked state, similar to the findings with lower PhTX concentrations at -60 mV (Fig. 2). However, at more negative membrane potentials a faster component of block became resolvable, and, as expected, the rate of onset of block increased with further hyperpolarization. At -40 mV the time constant of the fast component of block, τfast, was 1.3 ± 0.1 s (n = 4); at -100 mV block developed with much more rapid kinetics, τfast = 142 ± 13 ms (n = 5). As shown in Fig. 4B, τfast changed as an exponential function of membrane potential, indicating an e-fold enhancement of the rate of binding of PhTX to its blocking site per 27.1 mV hyperpolarization. Assuming that PhTX has a charge of +3 (Jaroszewski et al. 1996), this suggests that the energy barrier which PhTX crosses to reach its binding site senses 1/3 of the membrane electric field.
Figure 4. Rate of onset of block by external PhTX increases with hyperpolarization.
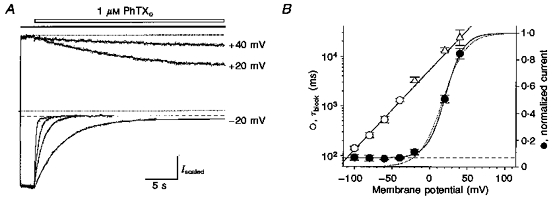
A, block by 1 μM PhTX (open bar) of responses to 50 μM domoate (filled bar) at holding potentials from -100 to +40 mV. The amplitudes of control responses prior to the onset of block are scaled to the response at -100 mV; the upper and lower dotted lines represent the unblocked current at +40 mV and the holding current prior to application of agonist, respectively; the dashed line indicates saturation of block for responses at -100, -60 and -40 mV. Lines through the data points show double-exponential functions fitted to the onset of block by PhTX, except for responses at +40, +20 and -20 mV, which were well fitted by single-exponential functions; to calculate the extent of block at equilibrium at these potentials, fits were extended until they reached steady state. B, time constants for the fast component of onset of block, τfast (○), with 1 μM PhTX plotted versus membrane potential (pooled data from 3 to 8 patches; ▵ indicates single exponential fits at +40, +20 and -20 mV); the time constant of onset of block decreased e-fold per 27.1 mV hyperpolarization (the line through the data points was fitted by least squares). Equilibrium block (•, measured relative to control) was well fitted by a modified Boltzmann function (V½, 21 mV; k,12 mV), accounting for the incompleteness of block at negative membrane potentials (dashed line). The dotted line indicates a Woodhull fit assuming complete block, which gave Kd(0) of 308 nM, zδ = 1.7.
In contrast to the observed increase in on-rate for open channel block, the expected corresponding increase in the amount of equilibrium block with hyperpolarization was not observed. Instead, the block induced by 1 μM PhTX showed saturation at membrane potentials negative to -40 mV rather than increasing with hyperpolarization as expected for an open channel blocking mechanism (Fig. 4A). The amount of equilibrium block plotted against membrane potential is shown in Fig. 4B. At -60 mV, 1 μM PhTX caused 94.4 ± 1.2 % block (mean ± s.d.; n = 8), which was not significantly different from the amount of block at -100 mV, 93.1 ± 2.1 %, mean ± s.d. (n = 5). Indeed, although the difference in the amount of block at -40 and -100 mV failed to reach significance, there was a clear trend for the block to show slight relief on hyperpolarization from -60 to -100 mV (Fig. 4A and B). This finding is unexpected for an open channel blocker of molecular weight 435, but suggests that PhTX may permeate GluR6(Q) channels to give relief from block at membrane potentials negative to -40 mV.
Fitting the Woodhull model for the action of an impermeable blocker to the data points for equilibrium block by PhTX clearly revealed the weaker than expected block by PhTX on hyperpolarization negative to -40 mV (Fig. 4B). Data points at more positive potentials were reasonably well fitted assuming a Kd at 0 mV (Kd(0)) of 308 nM. A zδ value of 1.71 suggests that if PhTX is fully ionized in the lumen of the channel (z = +3) the binding site for externally applied PhTX senses 56 % of the membrane electric field. A modified Boltzmann function which accounts for the incompleteness of block at negative membrane potentials (see Methods) yielded a membrane potential for half-maximal block (V½) of 21 mV and a slope factor (k) of 12 mV.
PhTX affinity fails to increase at hyperpolarized membrane potentials
Woodhull analysis of the voltage dependence of GluR6(Q) responses in the presence of 1 μM external PhTX suggests that the affinity for block fails to increase with hyperpolarization beyond about -40 mV (Fig. 4B). To test this directly we determined the voltage dependence of the Kd for PhTX from dose-inhibition analysis at steady-state block for different membrane potentials. For this purpose, PhTX at concentrations between 10 nM and 10 μM was applied by concentration jump in the presence of 50 μM domoate to outside-out patches at membrane potentials between +40 mV and -80 mV (Fig. 5A). As expected, the kinetics of onset of block by 10 and 100 nM PhTX at both -40 and -80 mV could be well described by a single-exponential function (Fig. 5A), whereas at higher concentrations both fast and slow components of block were clearly distinguishable. Calculation of kon from the linear concentration dependence of 1/τfast (not shown) yielded values of 0.67 × 106 M−1 s−1 for -40 mV and 3.72 × 106 M−1 s−1 for -80 mV. Combining these results with the value obtained for -60 mV (Fig. 2B) suggested an e-fold enhancement of the rate of binding of PhTX per 23.3 mV hyperpolarization, which is in good agreement with the voltage dependence of τfast shown in Fig. 4B
Figure 5. Anomalous voltage dependence for block by external PhTX revealed by dose-response analysis.
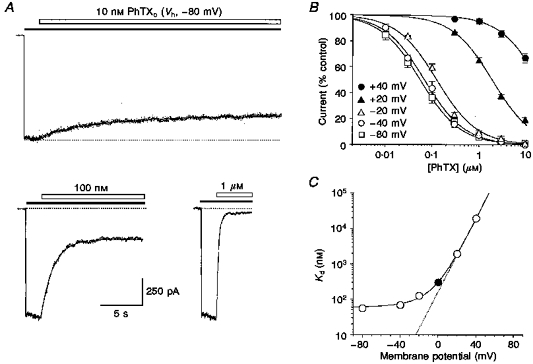
A, block of responses to 50 μM domoate (filled bars) at -80 mV by 10 nM to 1 μM PhTX (open bars) as indicated; the upper and lower dotted lines represent the unblocked current at -80 mV and the holding current, respectively. B, PhTX dose-inhibition plots at holding potentials between +40 mV and -80 mV (means ± s.d., pooled data from 5 to 13 patches) fitted by a single-binding site model to estimate PhTX affinity as a function of membrane potential. The Kd values were: +40 mV, 19.6 μM; +20 mV, 1.9 μM; -20 mV, 126 nM; -40 mV, 69 nM; -80 mV, 56 nM. C, when plotted on a logarithmic scale, Kd values at negative membrane potentials deviate from the expected exponential increase predicted by extrapolation of Kd values at +40 and +20 mV (dotted line). • indicates the Kd(0) of 308 nM obtained from Woodhull analysis of block by 1 μM PhTX (Fig. 4).
Current amplitudes during equilibrium block relative to control were plotted against PhTX concentration, and pooled data fitted with a single-binding site model (see Methods) to yield apparent Kd values for chosen membrane potentials (Fig. 5B). These experiments did not reveal polyamine-like potentiation at low concentrations of PhTX (Ragsdale et al. 1989; Brackley et al. 1990). The dose-inhibition analysis amply confirmed the unusual voltage dependence observed for block by 1 μM PhTX (Fig. 4). Although hyperpolarizing the membrane potential from +40 mV to -40 mV resulted in a 300-fold decrease in Kd from 19.6 μM to 69 nM, further hyperpolarization to -80 mV (Kd, 56 nM) caused only a 1.2-fold increase in affinity. The deviation from an exponential increase in affinity for PhTX block with membrane hyperpolarization is illustrated in Fig. 5C, which shows Kd values, including Kd(0) from a Woodhull analysis of block by 1 μM PhTX (Fig. 4), plotted against membrane potential. The voltage dependence of Kd clearly deviated from the expected exponential relationship at membrane potentials more negative than about 0 mV, and at strongly hyperpolarized potentials the affinity for PhTX block saturates at around 60 nM. Taken together, these results indicate that on hyperpolarization from -40 to -100 mV, the experimentally observed incomplete block of GluR6(Q) channels by PhTX (see Fig. 4) is due to a lack of increase in affinity for PhTX at negative membrane potentials.
Since the rate of onset of block changed as an exponential function of membrane potential over the same membrane potential range that equilibrium block by PhTX shows saturation (cf. Figs 4 and 5), it was probable that an increase in the kinetics of unblock was responsible for the lack of increase in affinity at negative membrane potentials. In Scheme 1, unblock is determined only by koff, the rate constant for dissociation of PhTX from its binding site to the extracellular solution, which would be expected to decrease with hyperpolarization. This view might, however, be oversimplified and it is possible that at negative membrane potentials PhTX can both permeate to the intracellular side as well as dissociate to the extracellular solution.
Recovery from block by external PhTX is enhanced by hyperpolarization
Since the rate of dissociation of PhTX to the external side and permeation to the inside of the membrane would be expected to show an inverse dependence on membrane potential, we studied the voltage dependence of recovery from block in order to test our hypothesis that PhTX is able to permeate GluR6(Q) channels, thereby producing relief from block at negative potentials. For this purpose currents induced by 50 μM domoate at -80 mV were rapidly blocked by the application of 10 μM PhTX. Simultaneous with the removal of PhTX in the continued presence of domoate, the membrane potential was stepped to different values between +40 mV and -100 mV for 30 s. During this time interval the kinetics of recovery from PhTX block were monitored. A subsequent voltage jump back to -80 mV allowed measurement of the amount of recovery that had occurred during the 30 s period at the test potential (Fig. 6A). The observed relaxations due to recovery from block were best fitted by a double-exponential function; however, to simplify comparison of the recovery kinetics at different holding potentials a weighted recovery time constant, τ'rec, was calculated which accounted for the relative contributions of both τ1 and τ2 to the total decay (see Methods). Our experimental results showed that recovery from PhTX block was accelerated by hyperpolarization and as a consequence the amount of recovery obtained after the 30 s test period was larger at more negative membrane potentials (Fig. 6B). At -100 mV, recovery was complete after 30 s (τ'rec = 5.6 ± 0.6 s; n = 4), whereas at -20 mV recovery reached only 43.8 ± 2.3 % (τ'rec = 59.8 ± 9.3 s; n = 5).
Figure 6. Recovery from block by external PhTX is accelerated by hyperpolarization.
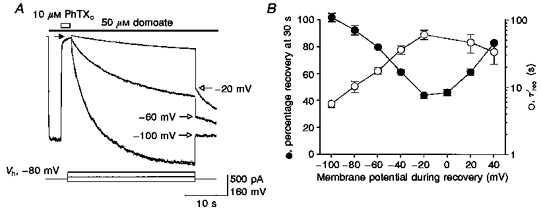
A, block of responses to 50 μM domoate (filled bar) by 10 μM PhTX (open bar) at -80 mV, with recovery from block measured at -20, -60 and -100 mV (see voltage protocol below current traces). Open arrows indicate the initial amplitude of responses at -80 mV after 30 s recovery from block at the indicated membrane potentials. Lines fitted to the recovery responses represent double-exponential functions of time constants: -100 mV, τfast = 2.9 s, τslow = 11.2 s (53 %); -60 mV, τfast = 4.6 s, τslow = 24.7 s (72 %); -20 mV, τfast = 14.4 s, τslow = 56.6 s (72 %). B, recovery from block measured after 30 s (•) at the indicated membrane potentials (pooled data from 5 patches) shows biphasic voltage dependence. Weighted time constants for recovery from block (○) reveal a striking increase in rate of unblock at hyperpolarized membrane potentials.
The voltage dependence of recovery from PhTX block was biphasic (Fig. 6B), as would be generated by two recovery mechanisms with opposite voltage dependence. The slowest kinetics resulting in the smallest amount of recovery were measured close to the reversal potential. These findings strongly support the idea that PhTX is a permeant blocker of GluR6(Q) channels, and hence the gating scheme, Scheme 1, described previously requires, in addition to koff, another reaction pathway defined by the rate constant kesc which defines the permeation of PhTX to the intracellular solution. Scheme 2 represents such a modification of the reaction pathway from open blocked to unblocked states in order to account for the permeation of PhTX:
Scheme 2.
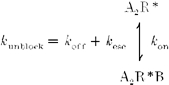
where at any given membrane potential koff and kesc sum to give the unblocking rate constant, kunblock. While koff decreases with hyperpolarization, kesc must increase at more negative membrane potentials to produce relief from block. At positive membrane potentials kesc would be expected to play a negligible role, with the observed enhancement of recovery solely due to a voltage-dependent increase in koff.
Low-affinity block and permeation by internal PhTX
The reaction diagram shown in Scheme 2 does not take into account channel block from the inside by PhTX molecules that have permeated the channel, and which has been postulated as a component of the mechanism of block by externally applied polyamine toxins (Usherwood & Blagbrough, 1989; Iino et al. 1996). To address this, the affinity and voltage dependence for block by internal PhTX were measured directly by adding PhTX to the internal solution (Fig. 7). Analysis of conductance-voltage relationships using voltage ramps for block by 1, 10, 100 and 1000 μM internal PhTX with a Woodhull model for an impermeable blocker, where φ is the electrical distance to the toxin binding site measured from the inside of the membrane, fitted well the observed responses for all concentrations of PhTX except 1 and 10 μM, for which block was incomplete at strongly positive membrane potentials (Fig. 7). The Kd(0) and zφ values were 27.7 ± 2.0 μM and 1.82 ± 0.01, respectively (n = 5), for 1000 μM internal PhTX, and 19.1 ± 2.7 μM and 1.89 ± 0.04, respectively, (n = 3) for 100 μM internal PhTX. At these concentrations, similar to results from previous experiments, we observed no obvious relief from block on depolarization from +50 to +100 mV (Bähring et al. 1997). However, for 1 and 10 μM internal PhTX, block was clearly incomplete at strongly positive membrane potentials. These results suggest that with an internal PhTX concentration of 100 μM and higher, the on-rate for toxin block greatly exceeds the permeation rate, and it is only at lower concentrations of PhTX that incomplete block is observable on strong depolarization. Fitting the Woodhull model to the data obtained with these low concentrations over a limited voltage range (Fig. 7B) yielded Kd(0) and zφ values for 10 μM internal PhTX of 22.3 ± 3.4 μM and 1.67 ± 0.05, respectively (n = 8); for 1 μM internal PhTX the values were 10.5 ± 1.9 μM and 1.41 ± 0.10, respectively (n = 4). The weak concentration dependence of Kd(0) could arise from several mechanisms, including a decrease in ionic activity at high concentrations of PhTX, competition between PhTX molecules for entry into the channel, or multiple blocking mechanisms with different kinetics since, to maintain equilibrium block during voltage ramps, the ramp rate was 6-fold slower for 1 μM PhTX than for higher concentrations. Despite this, a Kd(0) of 20 μM indicates a 60-fold lower affinity for block by internal than external PhTX (Fig. 4), indicating that the high potency for block by external PhTX does not require access to a binding site on the internal face of the membrane.
Figure 7. Low-affinity block by internal PhTX.
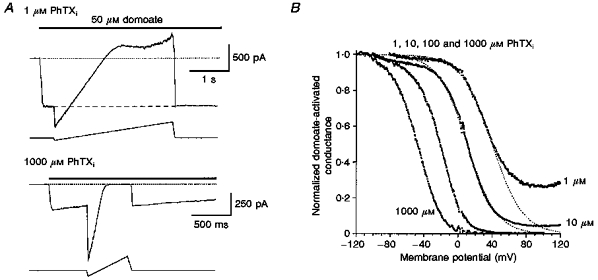
A, currents recorded in response to 50 μM domoate during voltage ramp protocols; the pipette solution contained PhTX at the indicated concentrations (internal PhTX, PhTXi) and no ATP. To maintain equilibrium block during changes in membrane potential, a slower ramp rate was required with 1 μM internal PhTX (-80 to +120 mV, 0.07 V s−1; upper panel) than with 1000 μM internal PhTX (-120 to +100 mV, 0.44 V s−1; lower panel). Holding potential, -60 mV. Traces are leak subtracted. Dotted lines represent zero current. Note the slow decline in the response to domoate at -60 mV observed with 1000 μM but not 1 μM PhTX. B, G-V curves for block by internal PhTX, normalized to the response recorded at the most negative membrane potential tested, for pooled data from 4 (1 μM), 8 (10 μM), 3 (100 μM) and 5 (1000 #181;M) patches, show a leftward shift with increasing PhTX concentrations (mean V½ values from Boltzman analysis are +41.5, +11.4, -22.6 and -50.4 mV, with slope factors of 18.4, 15.2, 13.5 and 14.0 mV−1 for 1, 10, 100 and 1000 μM PhTX, respectively). At positive potentials complete block was observed for both 1000 μM and 100 μM internal PhTX, whereas for 1 and 10 μM PhTX maximum block reached an asymptote and failed to increase with further depolarization. Dotted lines show the G-V relationships expected for a non-permeant blocker and represent fits of the Woodhull model of ion channel block to data points more negative than +40 mV for 10 μM PhTX and +30 mV for 1 μM PhTX.
Partial trapping by internal PhTX suggests accumulation in a stable blocked state
Of interest, with 1000 μM internal PhTX we observed a slow decay of the response to domoate at -60 mV even though patches were treated with concanavalin A to attenuate desensitization (Fig. 7A, lower panel). Such a decrease in response amplitude, which was not observed with lower internal PhTX concentrations, is reminiscent of the slow accumulation of GluR6(Q) channels in a stable blocked state observed with external PhTX. In order to test the hypothesis that internally applied PhTX can cause both low-affinity open channel block with rapid onset and recovery and a slowly developing more persistent block, we performed experiments in which block of domoate responses by 100 μM internal PhTX was induced by voltage steps from -100 mV to more depolarized potentials. Figure 8A shows an experiment in which steps to -40 mV of different duration were applied. Depolarizing voltage steps of < 100 ms duration caused 26.3 ± 2.0 % block at -40 mV (n = 7), calculated assuming a linear I-V relationship and a reversal potential of 0 mV, as found in previous experiments (Bähring et al. 1997). The amount of block was in excellent agreement with that measured at -40 mV from voltage ramp analysis with 100 μM internal PhTX (25.7 ± 2.9 % block; n = 3). During individual voltage steps of longer duration, the amount of block at -40 mV showed a slow increase, such that after 30 s block increased to 84.4 ± 0.9 % (n = 7). Fitting a single-exponential function to the observed slow current decay during a 30 s step (not shown) yielded a time constant of 7.9 ± 0.4 s (n = 7).
Figure 8. Tail current analysis of partial trapping by internal PhTX.
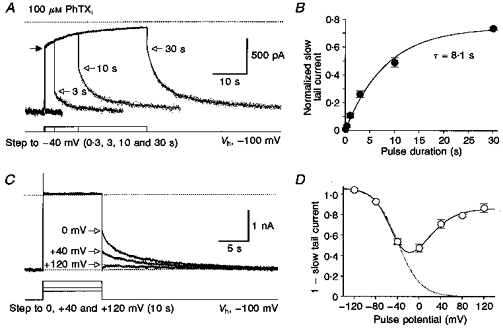
A, voltage steps from -100 to -40 mV for different durations reveal slow block by 100 μM internal PhTX of responses to 50 μM domoate. The voltage protocol is shown below a series of superimposed current traces recorded from the same patch. The dotted line represents zero current. The filled arrow on the left indicates the current amplitude at -40 mV predicted by analysis of ramp I-V relationships (Fig. 7). Lines through the data points are double-exponential functions fitted to the slow relaxations which followed the instantaneous increase in current on return to -100 mV (open arrows). The time constants of unblock, τfast and τslow, were (respectively): 2.2 s and 7.9 s (45 %) for 3 s steps to -40 mV; 2.3 s and 11.7 s (46 %) for 10 s steps; and 2.4 s and 12.1 s (47 %) for 30 s steps. B, the amplitude of the slow relaxation relative to the control response at -100 mV plotted against test pulse duration for observations from 6 to 7 patches per data point. Fitting an exponential function yielded a time constant of 8.1 s for the development of the slow component of block by internal PhTX. C, responses for voltage steps 10 s in duration from -100 mV to different test potentials as indicated; the upper dotted line shows zero current; because traces were not leak subtracted there was a small outward current at +120 mV which was the same value as that recorded during voltage ramps in the absence of agonist. Note that the amplitude of the slow component of recovery from block (open arrows) decreases with depolarization. D, biphasic voltage dependence for the slow component of block by 100 μM internal PhTX revealed by analysis of tail currents at -100 mV (mean values from 3 to 9 patches per data point). The amplitude of the slow component of the tail current at -100 mV was estimated by fitting exponentials to responses like those shown in C and extrapolating these to the time at which the membrane potential was returned from the test potential to -100 mV.
When the voltage step was terminated and the membrane potential returned to -100 mV an instantaneous increase in current amplitude was observed which represented both an increase in driving force and fast unblock of GluR6(Q) channels in the open blocked state. For voltage steps of longer than 100 ms duration, fast unblock was followed by a slow current relaxation. The amplitude of this slow relaxation relative to the control current at -100 mV must represent the fraction of channels that have accumulated in a stable blocked state. The size of this fraction was dependent on the step duration (Fig. 8B) and amounted to 74 ± 1 % block (n = 7) after 30 s. Exponential analysis of the increase in amplitude of the slow tail current following repolarization from -40 to -100 mV resulted in a time constant of 8.1 s, in excellent agreement with the time constant obtained from fitting the current decay at -40 mV during the depolarizing voltage step (Fig. 8B). The kinetics of the slow component of recovery from block at -100 mV could be described by a double-exponential function (Fig. 8A) with a weighted recovery time constant, τ'rec, of 5.4 ± 0.5 s (n = 9 patches tested with a step duration of 10 s), identical to the value obtained by analysis of the recovery kinetics at -100 mV following the removal of external PhTX (Fig. 6). This suggests that the observed current relaxation represents recovery from the same blocked state(s) independent of whether PhTX gained access to its binding site from the internal or external solution.
To study the voltage dependence of the slow component of block by 100 μM internal PhTX, voltage steps of 10 s duration to different potentials were evoked from a holding potential of -100 mV (Fig. 8C). As expected from our voltage ramp experiments, no obvious relief from block was observed at positive potentials, most probably because the permeation rate of PhTX is low and the on-rate for open channel block high. Unexpectedly, we found that the fraction of channels that entered the stable blocked state decreased with depolarization beyond 0 mV (Fig. 8C and D). Following termination of voltage steps to +80 and +120 mV a transient inward current was frequently observed at -100 mV which was followed by a fast decay and subsequent slow recovery from block to the control amplitude measured before the step (Fig. 8C). The fast decay possibly results from a reduced driving force due to ionic accumulation on the internal side, or from onset of block by PhTX molecules that have accumulated in or near the outer vestibule of the channel. To avoid an underestimation of the fraction of trapped channels in these cases, we calculated the amount of block from the extension of a single-exponential fit to the slow current relaxation (Fig. 8C). Notably, steps to -120 mV caused additional slow unblock, and slow onset of block was observed on return to -100 mV (not shown), indicating that even at -100 mV a small fraction of channels is in the stable blocked state. Since in our analysis of block by internal PhTX data have been normalized to the values obtained at -100 mV, the additional unblock resulted in a value of > 1 for the fraction of channels in the non-trapped state after 10 s at -120 mV (Fig. 8D). As shown in Fig. 8D, the voltage dependence of cumulative block observed after depolarizing voltage steps in the presence of 100 μM internal PhTX could be described by the sum of two Boltzmann functions with opposite voltage dependence. The reduced block at positive membrane potentials suggests that, despite a high occupancy by PhTX, a long dwell time is necessary for trapping to occur and that an increase in the permeation rate for PhTX which occurs during strong depolarization destabilizes the binding of toxin.
DISCUSSION
In the present study we examined the mechanisms by which philanthotoxin blocks GluR6(Q) subtype glutamate receptor channels. Three major conclusions can be drawn from our experiments: (1) a major component of the blocking mechanism for externally applied PhTX, especially at nanomolar concentrations of toxin, results from trapping of PhTX due to channel closure via an allosteric mechanism; (2) PhTX can permeate GluR6(Q) channels, causing relief from block at negative membrane potentials for external PhTX, with opposite voltage dependence for onset and relief from block for internal PhTX; (3) although PhTX permeates GluR6(Q) channels, its mechanism of action from the external solution does not involve binding to a high-affinity site preferentially accessible from the internal face of the membrane.
Trapping ion channel block by PhTX
Using fast-perfusion techniques we were able to resolve two components of block by external PhTX: a fast component, which we believe represents an open channel blocking reaction because it is linearly dependent on PhTX concentration, and a slow component, which shows a saturating concentration dependence indicative of an allosteric mechanism. A single-channel analysis of the block of crayfish muscle glutamate receptors by argiotoxin (ATX) also clearly revealed two components of block by polyamine toxins (Antonov, Dudel, Franke & Hatt, 1989). Sequential open channel block was detectable as brief interruptions of the open state, but this was resolvable only at micromolar concentrations of toxin, reflecting the need for a high forward rate for entry into the open blocked state. At nanomolar concentrations of ATX a higher affinity blocking mechanism was recorded as a gradual loss of single channel activity. This was interpreted as binding of ATX to agonist-bound closed states (Antonov et al. 1989). In our experiments we observed no inhibitory action of PhTX on closed channels (Fig. 1), while recovery from block was largely prevented in the absence of agonist (Fig. 6), suggesting that for GluR6 the allosteric component of block may represent the accumulation of channels in a closed blocked state which is accessible only from the open blocked state. Accumulation in the closed blocked state occurs when channels close with blocker bound to them and the blocking molecule gets trapped in the channel. Trapping of blocker molecules in closed channels was first suggested by Lingle (1983) to account for the cumulative block of nicotinic ACh receptor channels by different cholinergic antagonists and for the agonist dependence of recovery from block. A similar mechanism was suggested to explain agonist-dependent recovery from PhTX block of responses to AMPA receptor agonists in rat brainstem neurons and oocytes expressing glutamate receptors generated by injection of rat brain mRNA (Jones, Anis & Lodge, 1990; Brackley et al. 1993), but a kinetic analysis has not been undertaken previously.
Blanpied et al. (1997) have studied in detail trapping ion channel block by amantadine and memantine for native and recombinant NMDA receptors, and have performed simulations based on a model similar to Scheme 1. Their experimental finding that only a fraction of channels seemed to trap the blocker (‘partial trapping’) could be reproduced by assuming that when bound to the channel, memantine influenced both agonist affinity and channel gating. We performed simulations using their model which revealed that incomplete block at equilibrium (Fig. 4), and partial trapping (Fig. 8), occur with higher probability when the dissociation rate constant for blocker is increased, for example by permeation. Prior analysis of block by internal and external spermine with a three-barrier two-site rate theory model (Bähring et al. 1997) suggests a mechanism by which block by internal PhTX could occur via two mechanisms with strikingly different kinetics. Low-affinity block recorded using voltage ramps and depolarizing jumps of < 100 ms duration could reflect a low affinity for binding to the inner site, with binding of PhTX to the external site required for inducing an allosteric transition to closed blocked states. A high central barrier would limit the rate at which internal PhTX moved to the outer site, producing a component of block with slow onset as was observed with voltage jumps of 1-30 s duration. For block by external PhTX, the rate of occupancy of the outer site would be much faster and determined by the external barrier height. As noted earlier, residence time on the external site would regulate the rate of transitions to the closed blocked state, with permeation of PhTX allowing channels to avoid entry into the closed blocked state.
Permeation of PhTX for glutamate receptor channels
Although prior studies with two-electrode voltage clamp of GluRs expressed in Xenopus oocytes revealed an increase in block by external PhTX and ATX on membrane potential hyperpolarization (Brackley et al. 1993; Herlitze et al. 1993; Washburn & Dingledine, 1996), strong rectification due to channel block by endogenous cytoplasmic polyamines prevented any detailed analysis. In the present study this was circumvented by recording from outside-out patches with ATP present in the internal solution to chelate residual polyamines. As shown by an analysis of the voltage dependence of the Kd for external PhTX, a large reduction in efficiency of block develops with very modest hyperpolarization and is apparent at membrane potentials of only -20 to -40 mV (Fig. 5C). Voltage-jump experiments with internal PhTX reveal a similar marked reduction of entry into a stable blocked state with depolarization to only 0 or +40 mV (Fig. 8).
We propose that the paradoxical weak voltage dependence for block by PhTX results from permeation of toxin. Our results indicate that entry into the more stable closed blocked state, which we suggest underlies the high affinity of PhTX for GluR channels, occurs at a slow rate and only when PhTX remains bound to the channel for a sufficiently long period to trigger a change in conformation to the closed blocked state. Although for block by high concentrations of internal PhTX there is no detectable current flow on depolarization to +120 mV, entry into the closed blocked state is greatly reduced with strong depolarization (Fig. 8). We propose that this occurs because, with strong depolarization, block by PhTX results from a rapid equilibrium between open and open blocked channels, with relief from block due to permeation of PhTX compensated by a high rate of binding of new toxin molecules. When the permeation rate is lower (e.g. at -40 mV) PhTX remains bound to the channel for a sufficiently long period to allow a high probability of entry into the closed blocked state. A prior analysis of block of crayfish muscle glutamate receptor channels by external ATX revealed an analogous relief from block with strong hyperpolarization over the range -120 to -240 mV (Antonov et al. 1989). Although in this study a model was considered in which ATX binds directly to closed channels, in part due to interactions of ‘a phenolic (negative) group’ with the crayfish muscle glutamate receptor, with relief from block at negative potentials due to ‘destabilization of the negative binding site’, NMR analysis of ATX (Raditsch, Geyer, Kalbitzer, Jahn, Ruppersberg & Witzemann, 1996) and PhTX (Jaroszewski et al. 1996) indicates that the OH groups of the phenol group in these toxins have pKa values much too high to allow deprotonation at physiological pH. It seems more likely that ATX block for crayfish muscle glutamate receptor channels also occurs via an open channel block mechanism which leads to formation of a stable closed blocked state, and that relief from block at strongly negative potentials results from permeation of ATX similar to the mechanism proposed here for PhTX.
Although permeation of PhTX at first seemed surprising given a molecular weight of 435, this is not unreasonable given that the dimension of the narrowest cross-section of the PhTX molecule derived from CPK models (0.75 nm) is equal to the diameter of the narrowest constriction of the ion channel pore for GluR6(Q) (Burnashev, Villarroel & Sakmann, 1996). The pore diameter of AMPA receptors measures 0.78 nm (Burnashev et al. 1996) and it seems reasonable to assume that permeation of polyamine toxins is possible in these channels as well. On first inspection, permeation through mammalian GluRs of even larger polyamine toxins such as ATX (molecular weight, 636) seems even less likely. However, ATX is also only 0.75 nm wide in narrowest cross-section, making it probable that ATX will also permeate AMPA and kainate receptor channels. In contrast, for NMDA receptors the pore is only 0.55 nm wide at its narrowest constriction (Villarroel, Burnashev & Sakmann, 1995), making it extremely unlikely that PhTX or ATX would be permeable, consistent with experimental measurements for ATX block of NMDA receptors (Ruppersberg, von Kitzing & Schoepfer, 1994; Raditsch et al. 1996). However, mutant NMDA receptors generated by simultaneous substitution of glycine for asparagine at the N and the N + 1 sites in the NR1 and NR2A subunits, respectively, form a channel with an estimated pore diameter of 0.87 nm (Wollmuth, Kuner, Seeburg & Sakmann, 1996). Such glycine-substituted NMDA receptor channels show pronounced relief from block on hyperpolarization for N1-dansyl-spermine which, due to its bulky naphthalene ring, is not able to permeate wild-type NMDA receptor channels (Chao et al. 1997).
Comparison of AMPA and kainate receptor channel block by PhTX
The IC50 for extracellular PhTX block of GluR6(Q) responses, 60 nM at -60 mV, indicates 10- to 50-fold higher affinity than previously reported for block of homomeric recombinant AMPA receptors assembled from the flop splice variants of either GluR1 or GluR3 (Brackley et al. 1993; Washburn & Dingledine, 1996). Possible explanations for the lower affinity block of AMPA receptor responses could include genuine subunit-dependent differences in affinity for PhTX; inhibition by cytoplasmic polyamines of block by external PhTX, since prior experiments were performed using Xenopus oocyte two-electrode voltage clamp (Brackley et al. 1993; Washburn & Dingledine, 1996); or differences in the functional properties of receptors expressed in Xenopus oocytes and mammalian fibroblasts, e.g. Sivilotti, McNeil, Lewis, Nassar, Schoepfer & Colquhoun, 1997. We performed preliminary experiments to address these issues using outside-out patches from HEK 293 cells transfected with the flip splice variant of GluR-A (GluR1). In the presence of cyclothiazide, which blocks desensitization for AMPA receptor flip splice variants (Partin, Patneau & Mayer, 1994), 30 nM PhTX caused almost complete block of responses to glutamate at -60 mV, an effect even stronger than observed for GluR6, indicating a much higher potency for AMPA receptor block than previously reported (Brackley et al. 1993; Washburn & Dingledine, 1996). The effect of PhTX was not weakened when 50 μM spermine and 150 μM spermidine were added simultaneously to the internal solution to mimic whole-cell conditions, nor when the experiment was repeated with two-electrode voltage clamp in oocytes injected with cRNA for GluR-Aflip (authors’ unpublished observations). These results raise the possibility that the previously observed lower potency for PhTX block of AMPA-type glutamate receptors in Xenopus oocytes occurred because kainate and not glutamate was used as an agonist. It is also possible that flip splice variants of AMPA receptor subunits are more sensitive to block by PhTX than the corresponding flop splice variants used in prior experiments, or that block of desensitization by cyclothiazide increases the binding of PhTX. Although further experiments are required to address these possibilities, it is well known that kainate acts as a partial agonist at AMPA receptors (Sommer et al. 1990; Patneau & Mayer, 1991; Patneau, Vyklicky & Mayer, 1993). Such partial agonism, if due to a reduced maximum open probability, might limit the accessibility of the open channel to PhTX; in addition, activation of subconductance states by kainate might interfere with block by PhTX if the toxin has different affinity for the main and substates of AMPA receptor channels. Our results show that under the appropriate conditions PhTX can bind to both AMPA and kainate receptors with very high affinity, indicating that PhTX and related polyamine toxins are likely to become increasingly useful tools for characterizing the physiological role of native AMPA receptors which lack GluR-B subunits.
Acknowledgments
We thank Dr Peter Seeburg for the gift of plasmids, Drs D. Bowie and K. Swartz for comments on the manuscript, and Ms C. Glasser for preparing cDNAs.
References
- Anis N, Sherby S, Goodnow R, Jr, Niwa M, Konno K, Kallimopoulos T, Bukownik R, Nakanishi K, Usherwood P, Eldefrawi A, Eldefrawi M. Structure-activity relationships of philanthotoxin analogs and polyamines on N-methyl-D-aspartate and nicotinic acetylcholine receptors. Journal of Pharmacology and Experimental Therapeutics. 1990;254:764–773. [PubMed] [Google Scholar]
- Antonov SM, Dudel J, Franke C, Hatt H. Argiopine blocks glutamate-activated single-channel currents on crayfish muscle by two mechanisms. The Journal of Physiology. 1989;419:569–587. doi: 10.1113/jphysiol.1989.sp017887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bähring R, Bowie D, Benveniste M, Mayer ML. Permeation and block of rat GluR6 glutamate receptor channels by internal and external polyamines. The Journal of Physiology. 1997;502:575–589. doi: 10.1111/j.1469-7793.1997.575bj.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman FA, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. Journal of Neurophysiology. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- Blaschke M, Keller BU, Rivosecchi R, Hollmann M, Heinemann S, Konnerth A. A single amino acid determines the subunit-specific spider toxin block of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionate/kainate receptor channels. Proceedings of the National Academy of Sciences of the USA. 1993;90:6528–6532. doi: 10.1073/pnas.90.14.6528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowie D, Mayer ML. Inward rectification of both AMPA and kainate subtype glutamate receptors generated by polyamine-mediated ion channel block. Neuron. 1995;15:453–462. doi: 10.1016/0896-6273(95)90049-7. [DOI] [PubMed] [Google Scholar]
- Brackley P, Goodnow R, Jr, Nakanishi K, Sudan HL, Usherwood PN. Spermine and philanthotoxin potentiate excitatory amino acid responses of Xenopus oocytes injected with rat and chick brain RNA. Neuroscience Letters. 1990;114:51–56. doi: 10.1016/0304-3940(90)90427-b. [DOI] [PubMed] [Google Scholar]
- Brackley PT, Bell DR, Choi SK, Nakanishi K, Usherwood PN. Selective antagonism of native and cloned kainate and NMDA receptors by polyamine-containing toxins. Journal of Pharmacology and Experimental Therapeutics. 1993;266:1573–1580. [PubMed] [Google Scholar]
- Bruce M, Bukownik R, Eldefrawi AT, Eldefrawi ME, Goodnow R, Jr, Kallimopoulos T, Konno K, Nakanishi K, Niwa M, Usherwood PN. Structure-activity relationships of analogues of the wasp toxin philanthotoxin: non-competitive antagonists of quisqualate receptors. Toxicon. 1990;28:1333–1346. doi: 10.1016/0041-0101(90)90098-r. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Monyer H, Seeburg PH, Sakmann B. Divalent ion permeability of AMPA receptor channels is dominated by the edited form of a single subunit. Neuron. 1992;8:189–198. doi: 10.1016/0896-6273(92)90120-3. [DOI] [PubMed] [Google Scholar]
- Burnashev N, Villarroel A, Sakmann B. Dimensions and ion selectivity of recombinant AMPA and kainate receptor channels and their dependence on Q/R site residues. The Journal of Physiology. 1996;496:165–173. doi: 10.1113/jphysiol.1996.sp021674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao J, Seiler N, Renault J, Kashiwagi K, Masuko T, Igarashi K, Williams K. N1-dansyl-spermine and N1-(n-octanesulfonyl)-spermine, novel glutamate receptor antagonists: block and permeation of N-methyl-D-aspartate receptors. Molecular Pharmacology. 1997;51:861–871. doi: 10.1124/mol.51.5.861. [DOI] [PubMed] [Google Scholar]
- Choi SK, Kalivretenos AG, Usherwood PNR, Nakanishi K. Labeling studies of photolabile philanthotoxins with nicotinic acetylcholine receptors: mode of interaction between toxin and receptor. Chemistry and Biology. 1995;2:23–32. doi: 10.1016/1074-5521(95)90077-2. [DOI] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. Intracellular polyamines mediate inward rectification of Ca2+-permeable α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid receptors. Proceedings of the National Academy of Sciences of the USA. 1995;92:9298–9302. doi: 10.1073/pnas.92.20.9298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donevan SD, Rogawski MA. Multiple actions of arylalkylamine arthropod toxins on the N-methyl-D-aspartate receptor. Neuroscience. 1996;70:361–375. doi: 10.1016/0306-4522(95)00342-8. 10.1016/0306-4522(95)00342-8. [DOI] [PubMed] [Google Scholar]
- Eldefrawi AT, Eldefrawi ME, Konno K, Mansour NA, Nakanishi K, Oltz E, Usherwood PN. Structure and synthesis of a potent glutamate receptor antagonist in wasp venom. Proceedings of the National Academy of Sciences of the USA. 1988;85:4910–4913. doi: 10.1073/pnas.85.13.4910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herlitze S, Raditsch M, Ruppersberg JP, Jahn W, Monyer H, Schoepfer R, Witzemann V. Argiotoxin detects molecular differences in AMPA receptor channels. Neuron. 1993;10:1131–1140. doi: 10.1016/0896-6273(93)90061-u. 10.1016/0896-6273(93)90061-U. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- Hume RI, Dingledine R, Heinemann SF. Identification of a site in glutamate receptor subunits that controls calcium permeability. Science. 1991;253:1028–1031. doi: 10.1126/science.1653450. [DOI] [PubMed] [Google Scholar]
- Iino M, Koike M, Isa T, Ozawa S. Voltage-dependent blockage of Ca2+-permeable AMPA receptors by joro spider toxin in cultured rat hippocampal neurones. The Journal of Physiology. 1996;496:431–437. doi: 10.1113/jphysiol.1996.sp021696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isa T, Iino M, Itazawa S, Ozawa S. Spermine mediates inward rectification of Ca(2+)-permeable AMPA receptor channels. NeuroReport. 1995;6:2045–2048. doi: 10.1097/00001756-199510010-00022. [DOI] [PubMed] [Google Scholar]
- Jaroszewski JW, Matzen L, Frolund B, Krogsgaard-Larsen P. Neuroactive polyamine wasp toxins: nuclear magnetic resonance spectroscopic analysis of the protolytic properties of philanthotoxin-343. Journal of Medicinal Chemistry. 1996;39:515–521. doi: 10.1021/jm950488s. 10.1021/jm950488s. [DOI] [PubMed] [Google Scholar]
- Jones MG, Anis NA, Lodge D. Philanthotoxin blocks quisqualate-, AMPA- and kainate-, but not NMDA-, induced excitation of rat brainstem neurones in vivo. British Journal of Pharmacology. 1990;101:968–970. doi: 10.1111/j.1476-5381.1990.tb14189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamboj SK, Swanson GT, Cull-Candy SG. Intracellular spermine confers rectification on rat calcium-permeable AMPA and kainate receptors. The Journal of Physiology. 1995;486:297–303. doi: 10.1113/jphysiol.1995.sp020812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koh DS, Burnashev N, Jonas P. Block of native Ca2+-permeable AMPA receptors in rat brain by intracellular polyamines generates double rectification. The Journal of Physiology. 1995;486:305–312. doi: 10.1113/jphysiol.1995.sp020813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingle C. Different types of blockade of crustacean acetylcholine-induced currents. The Journal of Physiology. 1983;339:419–437. doi: 10.1113/jphysiol.1983.sp014724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E. The charge carried by single channel currents of rat cultured muscle cells in the presence of local anaesthetics. The Journal of Physiology. 1983;339:663–678. doi: 10.1113/jphysiol.1983.sp014741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Partin KM, Patneau DK, Mayer ML. Cyclothiazide differentially modulates desensitization of AMPA receptor splice variants. Molecular Pharmacology. 1994;46:129–138. [PubMed] [Google Scholar]
- Patneau DK, Mayer ML. Kinetic analysis of interactions between kainate and AMPA: evidence for activation of a single receptor in mouse hippocampal neurons. Neuron. 1991;6:785–798. doi: 10.1016/0896-6273(91)90175-y. 10.1016/0896-6273(91)90175-Y. [DOI] [PubMed] [Google Scholar]
- Patneau DK, Vyklicky L, Mayer ML. Hippocampal neurons exhibit cyclothiazide-sensitive rapidly desensitizing responses to kainate. Journal of Neuroscience. 1993;13:3496–3509. doi: 10.1523/JNEUROSCI.13-08-03496.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raditsch M, Geyer M, Kalbitzer HR, Jahn W, Ruppersberg JP, Witzemann V. Polyamine spider toxins and mammalian N-methyl-D-aspartate receptors. Structural basis for channel blocking and binding of argiotoxin636. European Journal of Biochemistry. 1996;240:416–426. doi: 10.1111/j.1432-1033.1996.0416h.x. 10.1111/j.1432-1033.1996.0416h.x. [DOI] [PubMed] [Google Scholar]
- Raditsch M, Ruppersberg JP, Kuner T, Günther W, Schoepfer R, Seeburg PH, Jahn W, Witzemann V. Subunit-specific block of cloned NMDA receptors by argiotoxin636. FEBS Letters. 1993;324:63–66. doi: 10.1016/0014-5793(93)81533-6. 10.1016/0014-5793(93)81533-6. [DOI] [PubMed] [Google Scholar]
- Ragsdale D, Gant DB, Anis NA, Eldefrawi AT, Eldefrawi ME, Konno K, Miledi R. Inhibition of rat brain glutamate receptors by philanthotoxin. Journal of Pharmacology and Experimental Therapeutics. 1989;251:156–163. [PubMed] [Google Scholar]
- Ruppersberg JP, Kitzing von E, Schoepfer R. The mechanism of magnesium block of NMDA receptors. Seminars in the Neurosciences. 1994;6:87–96. 10.1006/smns.1994.1012. [Google Scholar]
- Seeburg P. The molecular biology of mammalian glutamate receptor channels. Trends in Neurosciences. 1993;16:359–365. doi: 10.1016/0166-2236(93)90093-2. 10.1016/0166-2236(93)90093-2. [DOI] [PubMed] [Google Scholar]
- Sivilotti LG, McNeil DK, Lewis TM, Nassar MA, Schoepfer R, Colquhoun D. Recombinant nicotinic receptors, expressed in Xenopus oocytes, do not resemble native rat sympathetic ganglion receptors in single-channel behaviour. The Journal of Physiology. 1997;500:123–138. doi: 10.1113/jphysiol.1997.sp022004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommer B, Keinänen K, Verdoorn TA, Wisden W, Burnashev N, Herb A, Köhler M, Takagi T, Sakmann B, Seeburg PH. Flip and flop: a cell-specific functional switch in glutamate-operated channels of the CNS. Science. 1990;249:1580–1585. doi: 10.1126/science.1699275. [DOI] [PubMed] [Google Scholar]
- Usherwood PNR, Blagbrough IS. Antagonism of insect muscle glutamate receptors – with particular reference to arthropod toxins. In: Narahashi T, Chambers JE, editors. Insecticide Action: From Molecule to Organism. New York: Plenum Press; 1989. pp. 13–31. [Google Scholar]
- Usherwood PNR, Blagbrough IS. Spider toxins affecting glutamate receptors: polyamines in therapeutic neurochemistry. Pharmacology and Therapeutics. 1991;52:245–268. doi: 10.1016/0163-7258(91)90012-b. 10.1016/0163-7258(91)90012-B. [DOI] [PubMed] [Google Scholar]
- Verdoorn TA, Burnashev N, Monyer H, Seeburg PH, Sakmann B. Structural determinants of ion flow through recombinant glutamate receptor channels. Science. 1991;252:1715–1718. doi: 10.1126/science.1710829. [DOI] [PubMed] [Google Scholar]
- Villarroel A, Burnashev N, Sakmann B. Dimensions of the narrow portion of a recombinant NMDA receptor channel. Biophysical Journal. 1995;68:866–875. doi: 10.1016/S0006-3495(95)80263-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vyklicky L, Benveniste M, Mayer ML. Modulation of N-methyl-D-aspartic acid receptor desensitization by glycine in mouse cultured hippocampal neurones. The Journal of Physiology. 1990;428:313–331. doi: 10.1113/jphysiol.1990.sp018214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Washburn MS, Dingledine R. Block of alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors by polyamines and polyamine toxins. Journal of Pharmacology and Experimental Therapeutics. 1996;278:669–678. [PubMed] [Google Scholar]
- Wollmuth LP, Kuner T, Seeburg PH, Sakmann B. Differential contribution of the NR1- and NR2A-subunits to the selectivity filter of recombinant NMDA receptor channels. The Journal of Physiology. 1996;491:779–797. doi: 10.1113/jphysiol.1996.sp021257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodhull AM. Ionic blockage of sodium channels in nerve. Journal of General Physiology. 1973;61:687–708. doi: 10.1085/jgp.61.6.687. 10.1085/jgp.61.6.687. [DOI] [PMC free article] [PubMed] [Google Scholar]