

Abstract
The time course and voltage dependence of inactivation of KvLQT1 channels expressed in Xenopus oocytes were studied using two-microelectrode voltage-clamp techniques.
Tail current analysis was used to characterize the kinetics of channel inactivation and deactivation. The time constant for recovery from channel inactivation was voltage dependent and varied from 30 ± 2 ms at −90 mV to 36 ± 1 ms at −30 mV. The time constant for deactivation varied from 186 ± 21 to 986 ± 43 ms over the same voltage range.
Inactivation of KvLQT1 channels was incomplete, reducing fully activated current by 35 % at +40 mV. Inactivation of KvLQT1 channels was half-maximal at −18 ± 2 mV.
The onset of KvLQT1 channel inactivation during a single depolarization to +20 mV was exponential (τ = 130 ± 10 ms), and developed after a delay of ≈75 ms. In contrast, when inactivation was reinduced following transient recovery of channels to the open state(s), the onset of inactivation was immediate and 10 times faster. These findings suggest multiple open states, and a sequential gating model for KvLQT1 channel activation and inactivation (C1 ⇌ Cn ⇌ O1 ⇌ O2 ⇌ I).
Delayed rectifier K+ (IKs) channels formed by heteromultimeric coassembly of KvLQT1 and minimal K+ channel (minK) subunits did not inactivate. Thus, minK subunits eliminate, or greatly slow, the gating associated with channel inactivation when coassembled with KvLQT1.
KVLQT1 is a novel K+ channel gene discovered by positional cloning (Wang et al. 1996). Heterologous expression of KVLQT1 in oocytes and mammalian cells induced a delayed-rectifier K+ current that did not resemble any previously described cardiac current. This suggested that KvLQT1 might coassemble with other subunits to form a channel with biophysical properties similar to a known cardiac K+ current. When KvLQT1 subunits were coexpressed with minimal K+ channel (minK) subunits, the biophysical properties of the resulting current were identical to the cardiac slowly activating delayed-rectifier K+ current IKs (Barhanin, Lesage, Guillemare, Fink, Lazdunski & Romey, 1996; Sanguinetti et al. 1996). In contrast to KvLQT1 channel current, IKS activates very slowly, does not inactivate, and has a linear current-voltage (I-V) relationship.
The important physiological role of KvLQT1 is underscored by the recent finding that mutations in KVLQT1 are associated with two inherited human disorders. Autosomal dominant mutations in KVLQT1 cause long-QT syndrome (LQT) (Wang et al. 1996; Neyroud et al. 1997), a disorder of cardiac repolarization. The predicted functional consequences of mutations in KVLQT1 are a decrease in IKs, lengthened cardiac action potentials and a predisposition to ventricular tachyarrhythmias and sudden death. Autosomal recessive mutations in KVLQT1 cause Jervell and Lange-Nielsen syndrome (JLN), characterized by more severe LQT and bilateral deafness (Neyroud et al. 1997; Splawski, Timothy, Vincent, Atkinson & Keating, 1997). KVLQT1 is expressed in the stria vascularis of the mouse inner ear, and mutations in this gene may interfere with endolymph homeostasis and cause deafness in JLN (Neyroud et al. 1997).
KVLQT1 and minK are expressed together in the heart and stria vascularis. Thus, it is likely that in these tissues, KvLQT1 subunits coassemble with minK in a heteromultimeric complex to form IKs channels. KVLQT1 is also expressed in the pancreas, adrenal and thymus glands, lung, intestine and kidney (Sanguinetti et al. 1996; Yang, Levesque, Little, Conder, Shalaby & Blanar, 1997). The role of KvLQT1 in these tissues is unknown. In cells that express KVLQT1, but not minK, KvLQT1 subunits would probably coassemble to form homotetrameric channels. This may also occur in cells where selective gene expression results in a relative abundance of KvLQT1 compared with minK subunits.
In a previous study (Sanguinetti et al. 1996), we noted an unusual property of the current induced by heterologous expression of KVLQT1 in the absence of minK. Decay of KvLQT1 channel current after membrane repolarization was preceded by a transient increase, resulting in a hooked tail current. We hypothesized that this increase was caused by rapid recovery of channels from an inactivated state followed by a relatively slow time course of deactivation. In this study, we characterized the voltage dependence and kinetics of inactivation of KvLQT1 channels and show that IKs channels formed by coassembly of KvLQT1 and minK subunits do not inactivate. We conclude that minK eliminates or greatly slows inactivation gating of KvLQT1 when these subunits coassemble to form a heteromultimeric channel complex.
METHODS
Construction of a KVLQT1 expression plasmid and production of cRNA
A full-length KVLQT1 cDNA clone (Yang et al. 1997), isolated from a human cardiac cDNA library and subcloned into the pSP64 (Promega) plasmid expression vector, was provided by Dr Mark Keating (University of Utah). Before use in expression experiments, the KVLQT1 construct was characterized by restriction mapping and DNA sequence analyses. Complementary RNA (cRNA) for injection into oocytes was prepared with the mCAP RNA Capping Kit (Stratagene) following linearization of the expression construct with EcoRI. cRNA was quantified by UV spectroscopy, and quality was assessed by agarose gel electrophoresis.
Isolation of oocytes and injection of cRNA
Xenopus laevis frogs were anaesthetized by immersion in 0.2 % tricaine (Sigma) for 15-30 min. Ovarian lobes were digested with 2 mg ml−1 Type 1A collagenase (Sigma) in Ca2+-free ND96 solution (for composition, see below) for 1.5 h to remove follicle cells. Stage IV and V oocytes (Dumont, 1972) were injected with KVLQT1 cRNA (12 ng, 46 nl), then cultured in Barth's solution supplemented with 50 μg ml−1 gentamicin and 1 mM pyruvate at 18°C. Barth's solution contained (mM): 88 NaCl, 1 KCl, 0.4 CaCl2, 0.33 Ca(NO3)2, 1 MgSO4, 2.4 NaHCO3 and 10 Hepes; pH 7.4.
Two-microelectrode voltage clamp of oocytes
KvLQT1 channel currents were recorded 3-4 days after injection of oocytes with KVLQT1 cRNA. Oocytes were bathed in ND96 solution containing (mM): 96 NaCl, 2 KCl, 2 MgCl2, 0.1 CaCl2 and 5 Hepes; pH 7.6. CaCl2 was reduced to 0.1 mM to suppress endogenous Ca2+-activated Cl− current. Currents were recorded at room temperature (22-24°C) using standard two-microelectrode voltage-clamp techniques (Quick & Lester, 1994) and a Dagan TEV-200 amplifier (Dagan Corporation). Glass microelectrodes were filled with 3 M KCl and their tips broken to obtain a resistance of 0.5-1.5 MΩ. Voltage commands were generated using pCLAMP software (version 6.0.2, Axon Instruments), a personal computer and a TL-1 D/Ab interface (Axon Instruments). For 2 s voltage-clamp pulses, current signals were filtered at 0.5 kHz and digitized at 2 kHz. The oocyte membrane potential was held at −80 mV between test pulses.
Data analyses
The Chebyshev technique (pCLAMP 6.0.2) was used to fit exponential functions to current traces to determine the time constants and amplitudes for exponential functions. To determine the voltage dependence of channel activation, a single-exponential function was fitted to tail currents, extrapolated to the beginning of the repolarizing step (Fig. 2C), and this value (y) normalized to the maximum extrapolated tail current (ymax). The voltage dependence of KvLQT1 channel activation was determined by fitting the normalized extrapolated tail current (y/ymax) versus test potential (Vt) to a Boltzmann function:
Figure 2. Inactivation of KvLQT1 channels assessed by tail current analysis.
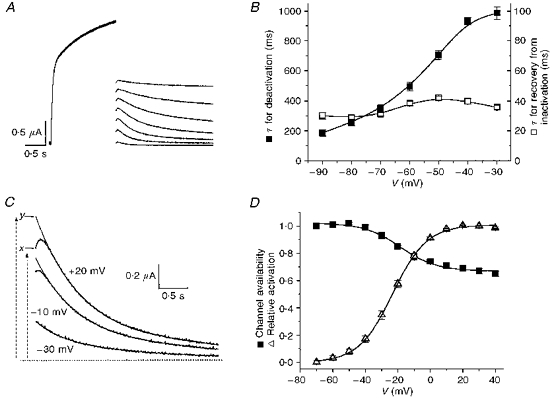
A, currents were activated with 1.6 s pulses to +20 mV; tail currents were elicited by hyperpolarizing pulses to voltages ranging from -90 to −30 mV. B, time constants of recovery from inactivation (for initial increase in current) and deactivation (slow decline) of KvLQT1 channel current (n = 16). C, tail currents measured at −70 mV after a 2 s pulse to -30, -10 and +20 mV. Tail currents were fitted with a single-exponential function between 0.2 and 1.0 s after repolarization. Initial current (x), and extrapolated (y) currents are indicated for the current measured after the pulse to +20 mV. D, voltage dependence of KvLQT1 channel inactivation. Initial tail current amplitude was divided by extrapolated amplitude to obtain the relative value (x/y) of channel availability (▪). Data were fitted with a Boltzmann function to determine the V0.5 (-18.3 ± 1.8 mV) and slope factor, k, (11.2 ± 1.7 mV) of the relationship (n = 18). The voltage dependence of activation (▵) is shown for comparison (V0.5 = -23 ± 0.4 mV; k = 10.6 ± 0.3 mV; n = 18).
where V0.5 is the voltage at which the current is half-activated, and k is the slope factor. The voltage dependence of inactivation was determined by fitting the ratio of extrapolated tail current (y) to the initial tail current (x;Fig. 2C) versus Vt to a Boltzmann function:
Data are expressed as means ± s.e.m. (n = number of oocytes).
RESULTS
Activation of KvLQT1 channel current
KvLQT1 channel current was elicited by membrane depolarization above −70 mV (Fig. 1A). The I-V relationship was determined by plotting current at the end of a 2 s pulse as a function of test potential. Currents were normalized relative to the magnitude of current at +60 mV for each oocyte. The mean values for eighteen oocytes are plotted in Fig. 1B. The slope conductance of the isochronal (2 s) I-V relationship decreased at progressively more positive potentials (Fig. 1B).
Figure 1. KvLQT1 channel current recorded in Xenopus oocytes.
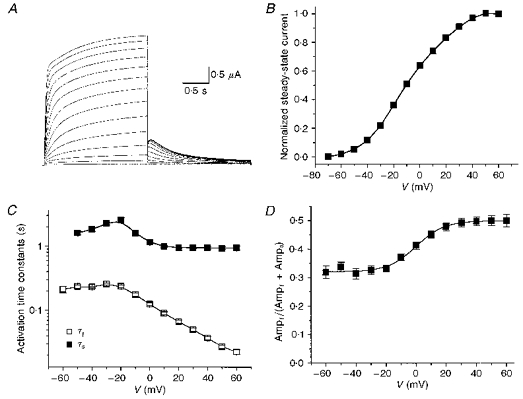
A, KvLQT1 channel current recorded during 2 s pulses from -70 to +40 mV. Deactivating tail currents were elicited by repolarization to −70 mV. B, normalized steady-state I-V relationship for KvLQT1 channel current. Current at the end of a 2 s pulse was normalized to current at +60 mV (n = 18). C, time constants for activation of KvLQT1 channel current, determined from two-exponential fits to the activating phase (n = 17). D, relative amplitude of the fast component of KvLQT1 channel activation (n = 17). Ampf and Amps are the amplitudes of the fast and slow components of activation, respectively.
The onset of current during a depolarizing step was best described by a two-exponential function after an initial, brief delay of a few milliseconds in duration. The fast (τf) and slow (τs) rates of activation increased as a function of membrane voltage between -20 and +60 mV (Fig. 1C). Activation was slowest at −20 mV (τf = 237 ± 13 ms, τs = 2.53 ± 0.25 s) and most rapid at +60 mV (τf = 23 ± 2 ms, τs = 937 ± 66 ms). The amplitudes of the fast (Ampf) and slow (Amps) components also varied with membrane potential. For example, the relative amplitude of the fast activation component (Ampf/[Ampf + Amps]) increased from 0.32 at −60 mV to 0.50 at +60 mV (Fig. 1D).
Voltage dependence of KvLQT1 channel inactivation
A possible cause of the non-linear I-V relationship was voltage- and time-dependent channel inactivation. After a 2 s depolarization to potentials > −30 mV, repolarization to −70 mV elicited a tail current that initially increased in amplitude (a hook), followed by a slow decay (Fig. 1A). Similar tail current hooks were previously described for the delayed-rectifier K+ current in cardiac atrial node cells (Shibasaki, 1987), and HERG channels expressed in oocytes (Sanguinetti, Jiang, Curran & Keating, 1995; Smith, Baukrowitz & Yellen, 1996; Spector, Curran, Zou, Keating & Sanguinetti, 1996), and were attributed to rapid recovery of channels from inactivation at a faster rate than deactivation. The rising and falling phases of KvLQT1 channel tail currents were also best described by a two-exponential function (Fig. 2A). The time constant for recovery from inactivation (initial increase in current) varied from 30 ± 2 ms at −90 mV to 42 ± 1 ms at −50 mV; the time constant for deactivation (slow decay) varied from 186 ± 21 ms at −90 mV to 986 ± 43 ms at −30 mV (Fig. 2B).
The voltage dependence of KvLQT1 channel inactivation was determined by tail current analysis. Two components of tail current were measured: initial tail current (x), and an extrapolated value (y) determined from a single-exponential fit of tail current beginning after the initial hook (Fig. 2C). Channel availability was estimated by the ratio x/y, equivalent to the fraction of current not inactivated during the prior test depolarization. The ratios were plotted as a function of the corresponding test pulse to determine the voltage dependence of inactivation (Fig. 2D). The data were well fitted by a Boltzmann function with a V0.5 of -18 ± 1.8 mV and a slope factor (k) of 11.2 ± 1.7 mV (n = 18). Channel availability reached a minimum value of 0.65 ± 0.02 at +40 mV. The V0.5 and k for channel availability were similar to the voltage dependence of activation (V0.5 = -23 ± 0.4 mV, k = 10.6 ± 0.3 mV; Fig. 2D).
Inactivation of KvLQT1 channels is delayed in onset
The rate of KvLQT1 channel inactivation was assessed by measuring relative inactivation (1 - x/y) as a function of pulse duration. KvLQT1 channels were activated at +20 mV and tail currents measured upon repolarization to −60 mV. For pulses > 100 ms, tail currents were hooked and the amplitude of the hook increased as a function of pulse duration (Fig. 3A). The onset of inactivation was best described with a single-exponential function after an initial delay of ∼75 ms (Fig. 3B). The average time constant for this function was 0.13 ± 0.01 s (n = 6).
Figure 3. The onset of KvLQT1 channel inactivation is delayed.
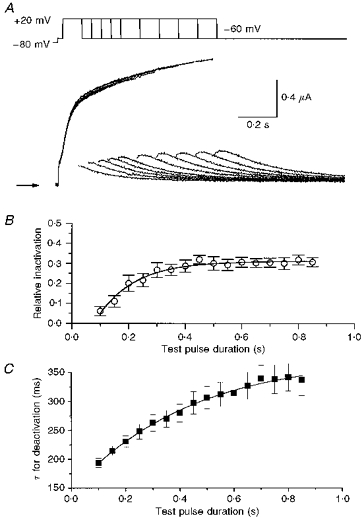
A, superimposed currents recorded during pulses of variable duration (0.1-1.6 s, in 0.1 s increments) to a potential of +20 mV, followed by a test pulse to −60 mV to elicit tail currents. Arrow indicates zero current. B, the fraction of inactivated channels (1 - x/y) was estimated by tail current analysis as described in Fig. 2C. Following an initial delay of ≈75 ms, inactivation proceeded monoexponentially (τ = 130 ± 10 ms; n = 6). C, time constants for decaying tail currents using pulse protocol in A are plotted against preceding pulse duration (n = 6).
The delayed onset of KvLQT1 channel inactivation suggests the presence of multiple open states that must be occupied before inactivation can occur. Such a sequential model of channel activation and inactivation predicts that deactivation rate would become slower as the duration of the preceding test pulse was lengthened. To test this possibility, we measured the rate of current deactivation for pulses to +20 mV that ranged in duration from 0.1 to 0.85 s. As predicted, the time constant for deactivation progressively increased as a function of the preceding test pulse duration (Fig. 3C). Because steady-state inactivation is achieved faster than steady-state activation, the time constant of deactivation continues to increase even after relative inactivation has saturated.
The rate of KvLQT1 channel inactivation is modulated by prior depolarization
To further test the sequential gating model and presence of multiple open states, a triple-pulse voltage-clamp protocol was used to record KvLQT1 channel current after transient removal of channel inactivation. Following a 2 s conditioning pulse to +40 mV to activate and inactive KvLQT1 channels, a brief hyperpolarizing interpulse was applied before a final test pulse to a variable potential (Fig. 4A). The duration (20 ms) and voltage (−130 mV) of the hyperpolarizing interpulse were chosen to permit the greatest amount of recovery from inactivation with the least amount of deactivation. After settling of the membrane capacitance transient (∼4 ms), the decay of current (reinduction of inactivation) during the test pulse was best fitted by a single-exponential function. The amplitude of instantaneous current was estimated by extrapolation of the fitted curve to the beginning of the test pulse. This measure assumes that inactivation is a first order process; however, this was not confirmed directly because the capacitance transient obscures the initial few milliseconds of inactivating current. The I-V relationship for instantaneous current determined in this manner was linear from -80 to +40 mV (Fig. 4B,n = 4). The time constant for inactivation during the third pulse varied from 14 ± 1 ms at +40 mV to 33 ± 1 ms at −30 mV (Fig. 4C). Unlike C-type inactivation (Lopez-Barneo, Hoshi, Heinemann & Aldrich, 1993), the rate of reinduction of inactivation was not dependent on [K+]o. The time constant for reinduction of inactivation was 16.8 ± 0.7 ms for 96 mM [K+]o, and 15.2 ± 0.8 ms for 2 mM [K+]o (n = 5, paired observations). The rapid decay of current during the reinduction of inactivation contrasts with the ∼75 ms delay and slow rate of inactivation (τ = 130 ms) determined by tail current analysis of single-step depolarizations to +20 mV (Fig. 3B). The rapid onset of inactivation using the triple-pulse protocol suggests that inactivated channels returned to the adjacent open state during the brief hyperpolarizing interpulse, and rapidly returned to the inactivated state during the test pulse.
Figure 4. Onset of KvLQT1 channel inactivation is rapid and occurs without a delay when measured using a triple-pulse protocol.
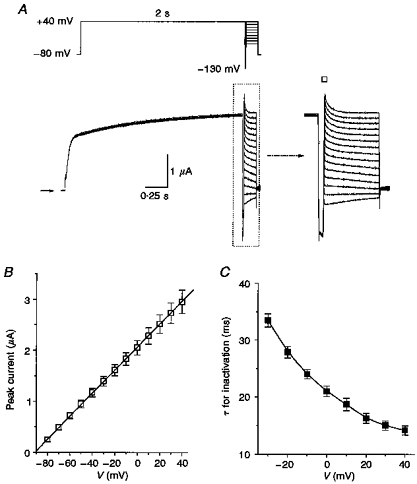
A, currents recorded during a pulse to +40 mV, followed by a 20 ms interpulse to −130 mV, before a third pulse to a variable potential (+40 to −80 mV, applied in 10 mV decrements). Arrow indicates zero current. Inset shows expanded view of currents delineated by dotted box. B, I-V relationship for fully activated KvLQT1 channel current determined by extrapolating currents to the beginning of the test pulse (denoted by □, inset in A). Mean data were fitted with a straight line by linear regression (n = 4). C, voltage dependence of inactivation time constants obtained from single-exponential fits to decaying current traces.
The triple-pulse protocol can also be used to estimate channel availability. The increased amplitude of instantaneous current during a test pulse to +45 mV relative to current during a 4 s conditioning pulse to +45 mV corresponds to a channel availabilty of 0.61 ± 0.04 (n = 5, data not shown). This is similar to channel availability estimated by tail current analysis after a single-step depolarization (0.65 ± 0.02 at +40 mV; Fig. 2D).
IKs channels formed by coassembly of KvLQT1 and minK do not inactivate
When KVLQT1 is coexpressed with minK in oocytes, the resulting current has properties nearly identical to cardiac IKs. The current activates very slowly, and the I-V relationship determined with 7.5 s pulses is linear (Sanguinetti et al. 1996). Oocytes were coinjected with KVLQT1 (6 ng cRNA per oocyte) and hminK (1 ng per oocyte) to determine if the resulting current retained the properties of inactivation characterized above for KvLQT1 channels.
Unlike KvLQT1 channel current, deactivation of IKs tail current was not preceded by an initial increase. In the example shown in Fig. 5, IKs was activated with 7.5 s pulses to +40 and +60 mV, and tail currents were measured at −50 mV. The sampling rate was increased from 130 to 500 Hz a few milliseconds before repolarization to increase the resolution of the initial tail current. Tail currents were not hooked and the decay of current was best described by a single exponential (Fig. 5B). Similar results were observed in fourteen other oocytes. The absence of hooks in tail currents indicates either that the channels did not inactivate during the preceding depolarization or that recovery from inactivation was too fast to be resolved from the capacity transient. It is more likely that IKs channels do not inactivate because the I-V relationship for IKs in oocytes is linear (Barhanin et al. 1996; Sanguinetti et al. 1996; Yang et al. 1997).
Figure 5. IKs does not inactivate.
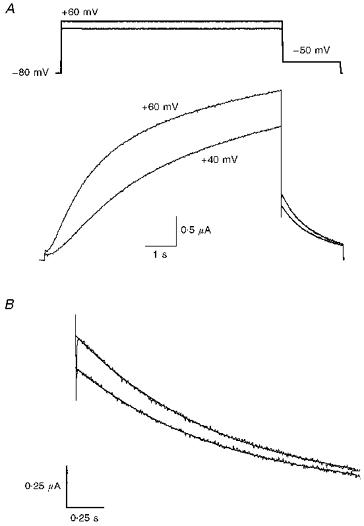
A, IKs (KvLQT1 + hminK) channels were activated by 7.5 s pulses to +40 and +60 mV. Tail currents were measured at −50 mV. B, expanded view of tail currents fitted with a single-exponential function. Deactivation was not preceded by an increase in current as observed for KvLQT1 alone.
DISCUSSION
In the heart, KvLQT1 subunits coassemble with minK to form IKs channels (Barhanin et al. 1996; Sanguinetti et al. 1996; Yang et al. 1997). The biophysical properties of KvLQT1 channels are altered by association with minK subunits. Current amplitude is increased severalfold and activation gating is markedly slowed compared with KvLQT1 homotetrameric channels. The single-channel amplitude of current through KvLQT1 channels has been reported to be decreased (Romey et al. 1997) or increased (Yang & Sigworth, 1998) by association with minK. In addition to these modulatory effects, our results indicate that minK also eliminates, or greatly slows, the gating process associated with inactivation of KvLQT1 channels.
Inactivation of KvLQT1 channels was suggested by the initial increase in tail current followed by a slow decline. We propose that the increase represents recovery of channels from inactivation (which develops during the preceding test pulse) at a rate much faster than deactivation. In contrast, IKs tail currents decayed monoexponentially, without a hook, indicating that IKs channels did not inactivate during the preceding depolarization. Channel inactivation was confirmed by the finding that rectification of the KvLQT1 channel I-V relationship could be eliminated using a triple-pulse protocol. As expected for a channel that does not inactivate, the I-V relationship for IKs (KvLQT1 + minK) channels is linear (Barhanin et al. 1996; Sanguinetti et al. 1996; Yang et al. 1997). The structural basis for the loss of inactivation of channels formed by the coassembly of KvLQT1 and minK is not known. Recently, Romey et al. (1997) used a yeast two-hybrid screen to demonstrate that the C-terminal region of minK can bind to the pore loop of KvLQT1. It was proposed that the minK C-terminus first occludes and then protrudes into the KvLQT1 tetrameric complex, resulting in ultraslow activation and an increased open channel probability of the IKs channel. Tai & Goldstein (1998) reported that residues of the minK transmembrane domain contribute to the selectivity filter of the KvLQT1-minK heteromultimeric complex. The physical association of minK residues with the pore region of KvLQT1 may prevent the conformational changes in KvLQT1 that would otherwise promote channel inactivation.
The rate of KvLQT1 channel inactivation was dependent on how current was activated. Based on tail current analysis, the onset of KvLQT1 channel inactivation during a single pulse to +20 mV occurred after a delay of about 75 ms, then developed exponentially with a time constant of 130 ms (Fig. 3). This contrasts with the immediate, and rapid, onset of inactivation (τ, ∼15 ms) for current measured during the third pulse of the triple pulse protocol (Fig. 4). These data suggest that reinduction of inactivation (following recovery of channels from an inactivated (I) state) may occur rapidly from an adjacent open channel state (O2), whereas inactivation during a single pulse requires transition of channels from a closed state (C) through at least two open states. The simplest state diagram to account for delayed inactivation and rapid reinduction of inactivation is shown below (Scheme 1).
Scheme 1.
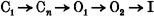
A slow transition from O1 to O2 could account for the delay in inactivation. If the reverse transition (O2 → O1) was also slow, then most of the channels would be in O2 after a brief hyperpolarizing interpulse, and would rapidly enter the I state (O2 → I) during a second depolarization. In support of the existence of at least two open states is the progressive increase in deactivation rate as function of preceding test pulse. Brief depolarizations would allow insufficient time for most channels to reach O2, thus upon repolarization, deactivation would proceed relatively rapidly. With longer pulses, channels would reach O2 and I, prolonging the time required for deactivation.
Inactivation of voltage-dependent K+ channels is usually considered to occur by either an N- or a C-type mechanism. N-type inactivation involves movement of the NH2-terminus to physically occlude the internal mouth of the open state of the channel (a tethered ball and chain mechanism; Hoshi, Zagotta & Aldrich, 1990). C-type inactivation is believed to require conformational changes near the external mouth of the pore which result in channel closure (Lopez-Barneo et al. 1993). The rate of inactivation is voltage independent for N- and C-type inactivation (Zagotta, Hoshi & Aldrich, 1989; Hoshi, Zagotta & Aldrich, 1991). In contrast, the rate of KvLQT1 channel inactivation varied from 14 ms at +40 mV to 33 ms at −30 mV. C-type inactivation is slowed by extracellular TEA (Choi, Aldrich & Yellen, 1991). KvLQT1 channel current was relatively insensitive to 96 mM extracellular TEA (∼40 % reduction, n = 6; data not shown), and therefore we could not test this criterion. The rate of C-type inactivation is also slowed by an increase in [K+]o (Lopez-Barneo et al. 1993), whereas changing [K+]o from 2 to 96 mM had no effect on the rate of KvLQT1 channel inactivation. Thus, KvLQT1 channels do not inactivate by a typical N- or C-type mechanism.
It is unknown if KvLQT1 forms homomultimeric channels in the heart, or if these subunits always coassemble with minK to form IKs channels. The presence of KvLQT1 channel current has not been reported in voltage-clamp studies of cardiac myocytes, but it would be difficult to detect because of overlap with ultrarapid (IKur), rapid (IKr) and slow (IKs) delayed rectifier K+ currents (Wang, Fermini & Nattel, 1993; Li, Fend, Yue, Carrier & Nattel,1996). If KvLQT1 homotetramers exist in cardiac myocytes, these channels would contribute to outward repolarizing current during the plateau phase of the action potential.
Acknowledgments
We are grateful to Heidi Orme and Monica Lin for technical assistance and Mark Keating and Igor Splawski for providing the KVLQT1 and hminK clones. We thank Dirk Synders for helpful comments. This work was supported by NIH NHBLI grants P50-HL52338 and R01-HL552336.
References
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium channel. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Choi KL, Aldrich RW, Yellen G. Tetraethylammonium blockade distinguishes two inactivation mechanisms in voltage-activated K+ channels. Proceedings of the National Academy of Sciences of the USA. 1991;88:5092–5095. doi: 10.1073/pnas.88.12.5092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumont JN. Oogenesis in Xenopus laevis (Daudin) Journal of Morphology. 1972;136:153–180. doi: 10.1002/jmor.1051360203. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 1990;250:533–538. doi: 10.1126/science.2122519. [DOI] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich RW. Two types of inactivation in Shaker K+ channels: effects of alterations in the carboxy-terminal region. Neuron. 1991;7:547–556. doi: 10.1016/0896-6273(91)90367-9. 10.1016/0896-6273(91)90367-9. [DOI] [PubMed] [Google Scholar]
- Li G-R, Feng J, Yue L, Carrier M, Nattel S. Evidence for two components of delayed rectifier K+ current in human ventricular myocytes. Circulation Research. 1996;78:689–696. doi: 10.1161/01.res.78.4.689. [DOI] [PubMed] [Google Scholar]
- Lopez-Barneo J, Hoshi T, Heinemann SH, Aldrich RW. Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors and Channels. 1993;1:61–71. [PubMed] [Google Scholar]
- Neyroud N, Tesson F, Denjoy I, Leibovici M, Donger C, Barhanin J, Faure S, Gary F, Coumel P, Petit C, Schwartz K, Guicheney P. A novel mutation in the potassium channel gene KVLQT1 causes the Jervell and Lange-Nielsen cardioauditory syndrome. Nature Genetics. 1997;15:186–189. doi: 10.1038/ng0297-186. 10.1038/ng0297-186. [DOI] [PubMed] [Google Scholar]
- Quick MW, Lester HA. Methods for expression of excitablility proteins in Xenopus oocytes. In: Narahashi T, editor. Methods in Neurosciences: Ion Channels of Excitable Cells. San Diego: Academic Press; 1994. pp. 261–279. [Google Scholar]
- Romey G, Attali B, Chouabe C, Abitbol I, Guillemare E, Barhanin J, Lazdunski M. Molecular mechanism and functional significance of the minK control of the KvLQT1 channel activity. Journal of Biological Chemistry. 1997;272:16713–16716. doi: 10.1074/jbc.272.27.16713. 10.1074/jbc.272.27.16713. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Shibasaki T. Conductance and kinetics of delayed rectifier potassium channels in nodal cells of the rabbit heart. The Journal of Physiology. 1987;387:227–250. doi: 10.1113/jphysiol.1987.sp016571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. Journal of General Physiology. 1996;107:611–619. doi: 10.1085/jgp.107.5.611. 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Vincent GM, Atkinson DL, Keating MT. Molecular basis of the long-QT syndrome associated with deafness. New England Journal of Medicine. 1997;336:1562–1567. doi: 10.1056/NEJM199705293362204. 10.1056/NEJM199705293362204. [DOI] [PubMed] [Google Scholar]
- Tai KK, Goldstein SAN. The conduction pore of a cardiac potassium channel. Nature. 1998;391:605–608. doi: 10.1038/35416. 10.1038/35416. [DOI] [PubMed] [Google Scholar]
- Wang Q, Curran ME, Splawski I, Burn TC, Millholland JM, VanRaay TJ, Shen J, Timothy KW, Vincent GM, de Jager T, Schwartz PJ, Towbin JA, Moss AJ, Atkinson DL, Landes GM, Connors TD, Keating MT. Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nature Genetics. 1996;12:17–23. doi: 10.1038/ng0196-17. 10.1038/ng0196-17. [DOI] [PubMed] [Google Scholar]
- Wang Z, Fermini B, Nattel S. Sustained depolarization-induced outward current in human atrial myocytes: evidence for a novel delayed rectifier K+ current similar to Kv1.5 cloned channel currents. Circulation Research. 1993;73:1061–1076. doi: 10.1161/01.res.73.6.1061. [DOI] [PubMed] [Google Scholar]
- Yang WP, Levesque PC, Little WA, Conder ML, Shalaby FY, Blanar MA. KvLQT1, a voltage-gated potassium channel responsible for human cardiac arrhythmias. Proceedings of the National Academy of Sciences of the USA. 1997;94:4017–4021. doi: 10.1073/pnas.94.8.4017. 10.1073/pnas.94.8.4017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Sigworth FJ. The conductance of KvLQT1 channels. Biophysical Journal. 1998;74:A19. [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Gating of single Shaker potassium channels in Drosophila muscle and in Xenopus oocytes injected with Shaker mRNA. Proceedings of the National Academy of Sciences of the USA. 1989;86:7243–7247. doi: 10.1073/pnas.86.18.7243. [DOI] [PMC free article] [PubMed] [Google Scholar]