

Abstract
Mechanisms of Ca2+ sensitization of force production by noradrenaline were investigated by measuring contractile responses, intracellular Ca2+ concentration ([Ca2+]i) and phosphorylation of the myosin light chain (MLC) in intact and α-toxin-permeabilized rat mesenteric small arteries.
The effects of noradrenaline were investigated at constant membrane potential by comparing fully depolarized intact arteries in the absence and presence of noradrenaline. Contractile responses to K-PSS (125 mM K+) and NA-K-PSS (K-PSS + 10 μM noradrenaline) were titrated to 30 and 75 %, respectively, of control force, by adjusting extracellular Ca2+ ([Ca2+]o). At both force levels, [Ca2+]i was substantially lower with NA-K-PSS than with K-PSS. With K-PSS, the proportion of MLC phosphorylated (≈30 %) was similar at 30 and 75 % of control force; with NA-K-PSS, MLC phosphorylation was greater at the higher force level (40 vs. 34 %).
In α-toxin-permeabilized arteries, the force response to 1 μM Ca2+ was increased by 10 μM noradrenaline, and MLC phosphorylation was increased from 35 to 45 %. The protein kinase C (PKC) inhibitor calphostin C (100 nM) abolished the noradrenaline-induced increase in MLC phosphorylation and contractile response, without affecting the contraction in response to Ca2+. Treatment with ATPγS in the presence of the MLC kinase inhibitor ML-9 increased the sensitivity to Ca2+ and abolished the response to noradrenaline.
The present results show that in rat mesenteric small arteries noradrenaline-induced Ca2+ sensitization is associated with an increased proportion of phosphorylated MLC. The results are consistent with a decreased MLC phosphatase activity mediated through PKC. Furthermore, while MLC phosphorylation is a requirement for force production, the results show that other factors are also involved in force regulation.
Activation by noradrenaline of α1-adrenoceptors in vascular smooth muscle cells induces contraction in part through a rise in cytosolic Ca2+ (for review see Somlyo & Somlyo, 1994), which in rat mesenteric small arteries is determined mainly by the membrane potential (Nilsson, Jensen & Mulvany, 1994). In addition, noradrenaline is able to increase contraction at a constant level of cytoplasmic Ca2+, both in intact and in Staphylococcus aureusα-toxin- or β-escin-permeabilized vessels (Kitazawa, Kobayashi, Horiuti, Somlyo & Somlyo, 1989; Jensen, Mulvany & Aalkjaer, 1992; Jensen, 1996). These studies also showed that the action of Ca2+-sensitizing agonists is coupled to GTP-binding proteins, since they are potentiated by GTP, inhibited by GDPβS and mimicked by the non-hydrolysable GTP analogue GTPγS.
At the level of the contractile filaments, the major direct controller of contraction is thought to be the degree of phosphorylation of the 20 kDa myosin light chain (MLC), which is determined by the balance between the activity of the Ca2+-calmodulin-dependent MLC kinase and the MLC phosphatase (Somlyo et al. 1989). The Ca2+-sensitizing effects of noradrenaline may be mediated through inhibition of the MLC phosphatase (Somlyo et al. 1989; reviewed in Somlyo & Somlyo, 1994). The link between receptor activation and inhibition of the MLC phosphatase is now believed to involve the small GTP-binding protein, RhoA (Hirata et al. 1992; Uehata et al. 1997; Gong, Fujihara, Somlyo & Somlyo, 1997). Other Ca2+-sensitizing mechanisms operating independently of MLC phosphorylation may include phosphorylation of caldesmon by mitogen-activated protein kinase or calponin by protein kinase C (reviewed in Horowitz, Menice, Laporte & Morgan, 1996), although a direct role for caldesmon and calponin in the regulation of contraction by agonists remains to be shown in situ.
In the present study, we have analysed the role of MLC phosphorylation in noradrenaline-induced Ca2+ sensitization in rat mesenteric arteries, and the mechanisms controlling MLC phosphorylation. Both intact and permeabilized arteries were used. In intact arteries, we compared responses to full depolarization (125 mM K+, K-PSS) with responses to K-PSS plus 10 μM noradrenaline (NA-K-PSS). This protocol allowed assessment of the effects of noradrenaline which were independent of the changes in membrane potential seen with noradrenaline alone (Nilsson et al. 1994). [Ca2+]i was varied by varying [Ca2+]o. In permeabilized arteries, we investigated the possible role of protein kinase C (PKC) using the PKC inhibitor calphostin C, and the possible role of MLC phosphatase by irreversibly inhibiting the phosphatase by treatment with ATPγS in the presence of the MLC kinase inhibitor ML-9 (Trinkle-Mulcahy, Ichikawa, Hartshorne, Siegman & Butler, 1995). The results show that in rat mesenteric small arteries MLC phosphorylation is a requirement for force production and for noradrenaline-induced Ca2+ sensitization. However, the results also show that other factors are involved in force regulation.
METHODS
Male Wistar rats (12–16 weeks old) were killed by CO2 inhalation. Segments (2–3 mm long) of mesenteric arteries were isolated and mounted as previously described (Mulvany & Halpern, 1977). The arteries were dissected free from connective tissue and mounted on wires in an isometric myograph (JP Trading, Denmark). The internal circumference of the artery was set to that for which the active tension was maximal (Mulvany & Halpern, 1977). The internal diameter for the mounted arteries was about 250 μm.
Experiments on intact arteries
Arteries were equilibrated in physiological saline solution (PSS) buffered with bicarbonate (37°C) and oxygenated with 95 % O2-5 % CO2 (saline solutions are described in the ‘Solutions’ section, below). Before the experimental protocol, the arteries were stimulated once for 5 min with K-PSS and twice for 5 min with NA-K-PSS to check the viability. For Ca2+ measurements, the myograph was placed on the stage of an inverted microscope with optics for epifluorescence as previously described (Jensen et al. 1992). The artery was illuminated with 340 and 380 nm light, and emitted light was passed through filters (500–530 nm, < 720 nm) and detected by a photomultiplier. Data from light emission were captured at 10 Hz and computer processed (Felix program, Photon Technology International, Monmouth Junction, NJ, USA).
The artery was loaded with fluorescent indicator by incubating in a loading solution (see ‘Solutions’ section below) twice for 30 min at 37°C. Intracellular Ca2+ concentration was calculated from:
where Kd= 211 nM for Ca2+ bound to fura-2 (Peng, Jensen, Nilsson & Aalkjær, 1998), β is the ratio of emission at 380 nM at maximum and minimum Ca2+ levels, R is the ratio between emission at 340 nM illumination and emission at 380 nM illumination, Rmax and Rmin are values of R at maximum and minimum Ca2+ levels. Rmin and Rmax were determined in each vessel at the end of the experiment as described previously (Jensen et al. 1992). Background fluorescent signal in the preparation, found by quenching fura-2 with 25 mM Mn2+ at the end of the experiment, was subtracted from the measured emission levels.
At the start of experiments, arteries were stimulated with NA-K-PSS for 5 min to obtain the control response, F1. Force was then titrated to 30 and 75 % of F1 by adjusting [Ca2+]o (in the range from 20 μM to 2.5 mM), the [Ca2+]o needed in each case being estimated from the response to one test stimulation, where the [Ca2+]o was a prediction of that required. Before each stimulation, the intracellular Ca2+ stores were depleted by 2 min consecutive treatments with 10 μM noradrenaline and 0.1 mM EGTA in Ca2+-free PSS, repeated twice and followed by a 2 min incubation in Ca2+-free PSS with 1 μM phentolamine if stimulation with K-PSS followed.
Experiments on permeabilized arteries
Arteries were permeabilized with Staphylococcus aureusα-toxin as described by Jensen (1996). Arteries were mounted in the myograph and kept at room temperature (22°C) throughout the experiment. Before permeabilization, the arteries were first stimulated once with K-PSS, and then the medium was changed to relaxing solution (see ‘Solutions’ section below) oxygenated with 100 % O2. Permeabilization was obtained by incubation with α-toxin for 20–25 min in 10 μl relaxing solution containing 1 μM free Ca2+, permeabilization being monitored by the contraction in response to Ca2+ entering the cells. The permeabilized artery was stimulated three times for 10 min with 1 μM Ca2+ in relaxing solution, with 20 min between stimulations, before the experimental protocol was initiated. All solutions contained 2 μM GTP, unless otherwise indicated. In some experiments, calphostin C was included; here the myograph was illuminated in order to enhance its inhibitor action on PKC (Bruns et al. 1991).
Treatment with ATPγS to irreversibly phosphorylate MLC phosphatase, using the reversible inhibitor ML-9 to protect the MLC from phosphorylation, was performed as described by Trinkle-Mulcahy et al. (1995). The artery was pre-incubated for 5 min with relaxing solution containing 300 μM ML-9 and for 10 min in a solution containing 1 mM ATPγS and 300 μM ML-9. The artery was washed twice in relaxing solution and then kept in relaxing solution. Thiophosphorylated proteins were identified by labelling with [35S]ATPγS. A segment of artery approximately 3 mm long was incubated in a solution containing [35S]ATPγS (3 mCi ml−1) in relaxing solution containing 100 μM ATPγS and 300 μM ML-9. The artery was dispersed in a glass-glass homogenizer and proteins were precipitated in 10 % trichloroacetic acid. The precipitate was washed twice with acetone and dissolved in SDS sample buffer (see ‘Solutions’ section below), and loaded on 4–20 % SDS-PAGE gradient gels. The fixed gel was treated with a scintillation enhancer (Ampliphy, Amersham, UK) and exposed to X-ray (Kodak, X-Omat) films.
Measurements of MLC phosphorylation
For determination of MLC phosphorylation, two arteries (3 mm long) were pooled. Both arteries were mounted in a double myograph and, at the moment of the given contractile condition, the arteries were frozen by changing the medium in the tissue chamber to melting acetone containing 10 mM dithiothreitol (DTT) (about −75°C). The arteries were transferred to a test-tube containing 600 μl precooled acetone, 10 % (w/v) trichloroacetic acid, 10 mM DTT, and stored at −70°C. The artery was dispersed in the storage solution by using a small glass-glass homogenizer, and proteins were precipitated by centrifugation (3000 g, 15 min). The precipitate was washed three times with acetone containing 10 mM DTT (4°C), and dissolved in urea-glycerol gel electrophoresis sample buffer (see ‘Solutions’ section below) (Persechini, Kamm & Stull, 1986). The sample was vortexed for 3 h and undissolved material was removed by centrifugation (6000 g, 2.5 min).
Phosphorylated and non-phosphorylated MLCs were separated by using urea-glycerol gel electrophoresis followed by blotting to a polyvinylidene difluoride (PVDF) membrane and detection of MLC by an antibody-based technique (Persechini et al. 1986). The monoclonal antibody MY-21 (Sigma) was used as primary antibody and goat anti-mouse IgM conjugated with peroxidase (Sigma) was used as secondary antibody. MLC-containing bands on the blot were visualized by using an enhanced chemiluminescence detection system (Amersham) with Hyperfilm-ECL (Amersham). Quantification of the MLC content in each band was done by densitometric scanning using a scanner equipped for transillumination (Hewlett Packard ScanJet llcx/T) and the software ImageQuant (Molecular Dynamics, Sunnyvale, CA, USA) for data analysis. All sample bands were normalized to an arbitrary standard curve, made from a dilution series of a batch homogenate from arteries, which then was run on a gel parallel to the samples and blotted to the same membrane as the samples. Exposure time was adjusted to ensure that measurements were made within the dynamic range of the film.
Solutions
PSS used for the intact arteries contained (mM): 119 NaCl, 4.7 KCl, 1.18 KH2PO4, 1.17 MgSO4, 25 NaHCO3, 5.5 glucose, with 26 μM EDTA (pH 7.4); CaCl2 was 2.5 mM, unless otherwise indicated. K-PSS was the same as PSS but with 119 mM NaCl replaced by 119 mM KCl. NA-K-PSS consisted of K-PSS with 10 μM noradrenaline.
Relaxing solution used for the permeabilized arteries contained (mM): 2 EGTA, 130 potassium methane sulphonate, 4 MgCl2, 20 Tris maleate, 4 Na2ATP, 10 creatine phosphate, with 1 mg ml−1 creatine phosphokinase. pH was adjusted to 7.1 with KOH. Activation was achieved by addition of CaCl2 as described by Jensen (1996) to give the [Ca2+] indicated.
The loading solution for fura-2 contained: 10 μM fura-2 AM, 0.5 % (v/v) dimethylsulphoxide, 0.02 % (w/v) Pluronic F-127 and 0.1 % (v/v) Cremophor EL.
The SDS sample buffer contained: 62.5 mM Tris-HCl (pH = 6.8), 2 % (w/v) SDS, 10 % (v/v) glycerol, 50 mM DTT, 0.001 % (w/v) Bromophenol Blue.
Urea-glycerol gel electrophoresis sample buffer contained: 20 mM Tris, 23 mM glycine (pH = 8.6), 8 M urea, 10 mM DTT, 0.005 % (w/v) Bromophenol Blue.
Chemicals
Staphylococcus aureusα-toxin was from Gibco. Na2ATP, Na2GTP, Li3GDPβS, GTPγS, ATPγS and creatine phosphokinase were from Böhringer Mannheim. [35S]ATPγS (specific activity > 1000 Ci mmol−1) was from Amersham. EGTA was from Fluka Chemie (Buchs, Switzerland). Calphostin C was from Calbiochem. Molecular mass marker for gel electrophoresis was from Bio-Rad (cat. no. 161–0318, Hercules, CA, USA). All other chemicals were analytical quality and were purchased from Sigma and Merck.
Statistics
Data are expressed as means ±s.e.m. Student's t test was used, with paired or unpaired testing as indicated. One-way ANOVA were used for multiple testing, and the Fisher's LSD test was used for post hoc analysis. P < 0.05 was considered significant.
RESULTS
Noradrenaline-induced Ca2+ sensitization of force response in intact arteries
The Ca2+-sensitizing effect of noradrenaline was assessed in fully depolarized arteries, by comparing responses to K-PSS and NA-K-PSS. Any effects of noradrenaline were thus not due to depolarization, and the ability of noradrenaline to increase Ca2+ sensitivity under these conditions is demonstrated in Fig. 1. Arteries were activated with either K-PSS or NA-K-PSS in increasing [Ca2+]o. For a given [Ca2+]o, the force response to NA-K-PSS was greater than that for K-PSS. Simultaneously measured [Ca2+]i showed that there was a fixed relation between [Ca2+]i and [Ca2+]o regardless of whether K-PSS or NA-K-PSS was used.
Figure 1. Force and [Ca2+]i measured simultaneously in response to [Ca2+]o.
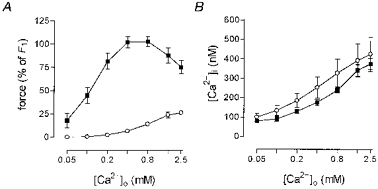
A, force relative to control force, F1, in response to cumulative increasing [Ca2+]o in K-PSS (^, 125 mM K+) and NA-K-PSS (▪, K-PSS plus 10 μM noradrenaline) in intact rat mesenteric small arteries. B, from the same experiments; simultaneous measurements of [Ca2+]i using fura-2 plotted against [Ca2+]o. The [Ca2+]i recorded is the mean concentration in the last 20 s of a stimulation period of 3 min. Error bars indicate s.e.m. (n = 4).
Time course of noradrenaline-induced Ca2+ sensitization of force response, and relation to MLC phosphorylation in intact arteries
Arteries were activated for 20 min while [Ca2+]i was measured simultaneously (Fig. 2); in other experiments, arteries were frozen at 2, 5 or 20 min for determination of MLC phosphorylation (Fig. 3). As in Fig. 1, the contractile response was in all cases greater with NA-K-PSS than in K-PSS. The Ca2+ traces were qualitatively similar in arteries stimulated in the absence or presence of noradrenaline, except that the initial peak was higher and a small elevation of [Ca2+]i (∼10 % at all three time points) was seen with NA-K-PSS stimulation compared with arteries stimulated by K-PSS (Fig. 2). MLC phosphorylation was also greater for the NA-K-PSS-stimulated arteries at 2 and 5 min, but was similar to the K-PSS-stimulated arteries at 20 min (Fig. 3). All subsequent experiments with intact arteries were performed using 5 min stimulations, at which time the force and noradrenaline-induced Ca2+ sensitization had reached a peak.
Figure 2. Time course of the Ca2+-sensitizing effect of noradrenaline investigated by simultaneous measurements of [Ca2+]i and contractile response in intact rat mesenteric small arteries.
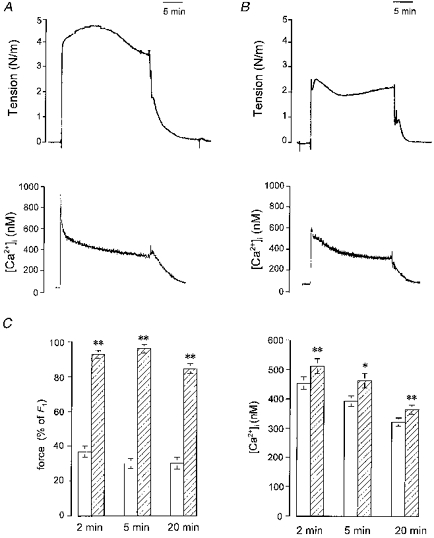
A and B, original traces showing contractile responses (upper) and [Ca2+]i (lower) measured in arteries mounted in a myograph under isometric conditions. In A, the artery was stimulated with K-PSS for 20 min. In B, the artery was stimulated with NA-K-PSS. C, readings at 2, 5 and 20 min of simultaneously measured contractile responses (left panel, measured relative to control force, F1) and [Ca2+]i (right panel). Arteries were stimulated with K-PSS (open bars) or with NA-K-PSS (hatched bars). Error bars indicate s.e.m. (n = 7). *P < 0.05, **P < 0.01, using a Student's paired t test to test the difference between responses to K-PSS and NA-K-PSS at each time point.
Figure 3. Time course of the Ca2+-sensitizing effect of noradrenaline investigated by simultaneously measured MLC phosphorylation and contractile response in intact rat mesenteric small arteries.
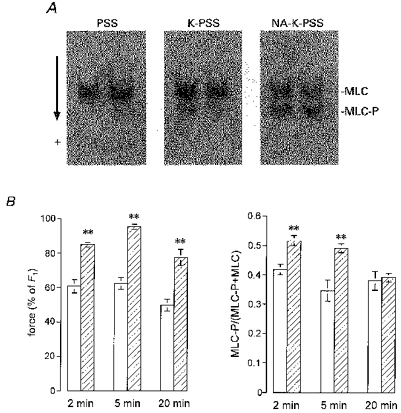
A, Western blot of homogenates of arteries which had been mounted in an isometric myograph and quick-frozen under the conditions indicated, i.e. relaxation conditions (PSS), after stimulation with K-PSS for 5 min, and after stimulation with NA-K-PSS for 5 min. Arterial proteins were extracted, separated by urea-glycerol gel electrophoresis, blotted onto a PVDF membrane, labelled with MLC antibodies and detected by a chemiluminescence detection system. Each lane contains proteins from two 3 mm segments mounted in the same double myograph. The upper band is the unphosphorylated MLC and the lower band is the monophosphorylated MLC. Each panel represents double determinations. The arrow indicates the direction of migration. B, developed force (left) measured relative to control force, F1, and the fraction of phosphorylated MLC (right) of arteries frozen 2, 5 and 20 min after start of stimulation with K-PSS (125 mM K+) (open bars) or NA-K-PSS (hatched bars). Error bars indicate s.e.m. (n = 4–6). **P < 0.01, using a Student's unpaired t test to test the difference between responses to K-PSS and NA-K-PSS at each time point.
Correlation between MLC phosphorylation and the contractile response in intact arteries
Figure 4 shows a series of experiments where the contractile response to Ca2+-depleted depolarized arteries was titrated to 30 or 75 % of the control response to NA-K-PSS (F1; see Methods), both in the presence and absence of noradrenaline, by appropriate adjustment of [Ca2+]o. The [Ca2+]o (and thus the [Ca2+]i; see Fig. 1B) needed to produce both force levels was less with NA-K-PSS stimulation than with K-PSS stimulation (Fig. 4A), again confirming the Ca2+-sensitizing effect of noradrenaline. The MLC phosphorylation was about 5 % in resting arteries and 29–40 % in arteries contracted to 30 and 75 % control force levels, both with K-PSS and NA-K-PSS activation, despite the differences in [Ca2+]o (and thus [Ca2+]i) (Fig. 4B). With 2.5 mM [Ca2+]o in the presence of noradrenaline, which gives maximum force, MLC phosphorylation rose to approximately 55 %. Plotting the force and MLC phosphorylation data of Fig. 4A and B against each other shows (Fig. 4C) that the relation between force and MLC phosphorylation was rather similar, regardless of whether NA-K-PSS or K-PSS had been used as the activator, even though the presence of NA did shift the relation slightly to the right, particularly at 75 % of control force. As can also be seen from Fig. 4C, the relation between contractile response and MLC phosphorylation was steep, especially for the K-PSS-stimulated arteries, where there was no significant difference in MLC phosphorylation at the two force levels. In contrast, with NA-K-PSS activation, MLC phosphorylation was slightly increased at the higher force level (34 vs. 40 %, P < 0.01, Student's paired t test). These results indicate that MLC phosphorylation is a requirement for force production, and to this extent force production is related more directly to MLC phosphorylation than to [Ca2+]o (or to [Ca2+]i). However, the steepness of the MLC phosphorylation-force relation raises questions concerning the degree to which MLC phosphorylation is the only regulator of tone, as discussed below.
Figure 4. Effect of noradrenaline on the relation between MLC phosphorylation and force in intact rat mesenteric small arteries.
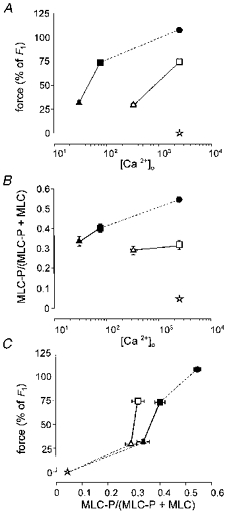
Arteries were depleted for Ca2+ in intracellular stores and stimulated with K-PSS (open symbols) or NA-K-PSS (filled symbols) with variable [Ca2+]o as indicated. The upper graphs show, as functions of [Ca2+]o (μM), force development (A) and phosphorylation (B) determined after 5 min of stimulation, where [Ca2+]o was adjusted to give either 30 % (triangles) or 75 % (squares) force relative to control force, F1 (see Methods). C, same data plotting force versus MLC phosphorylation.  , non-stimulated arteries (held in PSS) (n = 2); •, arteries stimulated for 5 min with NA-K-PSS, both with high [Ca2+]o (2·5 mM) (n = 2). Continuous lines connect paired data (n = 12), where the arteries were mounted on two double myographs run in parallel. Dashed lines indicate tentative curves connecting paired K-PSS or NA-K-PSS data with minimum and maximum values. Error bars indicate s.e.m. where this exceeds the size of the symbol.
, non-stimulated arteries (held in PSS) (n = 2); •, arteries stimulated for 5 min with NA-K-PSS, both with high [Ca2+]o (2·5 mM) (n = 2). Continuous lines connect paired data (n = 12), where the arteries were mounted on two double myographs run in parallel. Dashed lines indicate tentative curves connecting paired K-PSS or NA-K-PSS data with minimum and maximum values. Error bars indicate s.e.m. where this exceeds the size of the symbol.
GTP, in micromolar concentrations, was required to obtain repeatable responses to Ca2+ and noradrenaline in α-toxin-permeabilized arteries
In small arteries permeabilized with α-toxin, the responses to Ca2+, and the sensitizing effects of NA, were determined in repetitive stimulations. As shown in Fig. 5A (upper panel), in the absence of GTP there was initially a concentration-dependent response to Ca2+, and noradrenaline caused a potentiation of the response to 1 μM Ca2+. If the stimulation was repeated, the response to Ca2+ decreased, as did the potentiating effect of noradrenaline (Fig. 5A, upper panel). However, if 6 μM GTP was present in the Ca2+ solutions, there was no diminution of the response either to Ca2+ or to noradrenaline (Fig. 5A, lower panel). This ability of GTP to maintain the response to Ca2+ and to noradrenaline was also seen in part at concentrations of 2 and 4 μM (Fig. 5B). Moreover, none of the concentrations affected the initial responses to Ca2+ and noradrenaline. Further experiments (n = 3) showed that 100 μM GDPβS, which inhibits GTP-binding proteins, markedly inhibited the response to 1 μM Ca2+ (Fig. 6).
Figure 5. Force responses of permeabilized rat mesenteric small arteries to the permeabilization procedure and to subsequent application of Ca2+.
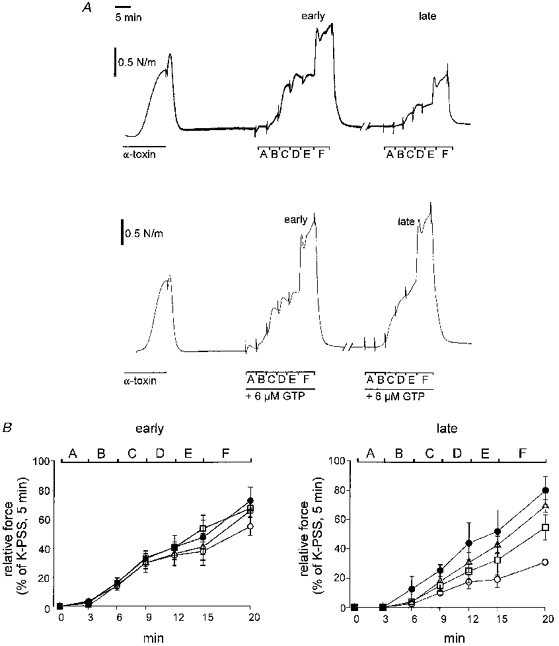
A, traces show the response of an artery to permeabilization with α-toxin in the presence of 1 μM Ca2+, then to cumulative applications of Ca2+, and then a repeat of the latter 80 min later. The permeabilization was performed (left traces) with 1000 U ml−1α-toxin in the presence of 1 μM Ca2+ until the force appeared to be close to its maximum. In centre and right traces, Ca2+ was applied with increasing concentrations at intervals of 3 min: A, 115 nM; B, 317 nM; C: 573 nM; D, 737 nM; E, 955 nM. In F, 10 μM noradrenaline was added in addition to 955 nM Ca2+ for 5 min. During the break in the trace (80 min), the artery was stimulated twice with 955 nM Ca2. In the lower trace, 6 μM GTP was present during the time indicated by the bar. In the upper trace, no exogenous GTP was present in the bath at any time. The left panel in B (early) shows Ca2+ response curves made just after the permeabilization in the presence of different GTP concentrations: 0 μM (^), 2 μM (□), 4 μM (▵), 6 μM (•). The right panel (late) shows the second Ca2+ response curves, as described in A, repeated with the respective GTP concentrations (identified as in left panel). Error bars indicate s.e.m.
Figure 6. Effect of GDPβS on the contractile response to 1 μM Ca2+ in permeabilized rat mesenteric small arteries.
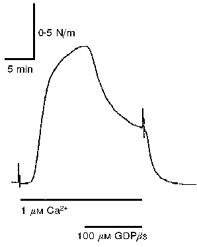
Trace shows 100 μM GDPβS added in addition to stimulation with 1 μM Ca2+ 20 min after permeabilization. No exogenous GTP was present. Trace is representative of 3 experiments.
Noradrenaline increased the Ca2+ sensitivity of the contractile response in α-toxin-permeabilized arteries
The effect of noradrenaline on a contraction in response to 1 μM Ca2+ was assayed. As in the experiment shown in Fig. 5, addition of noradrenaline caused an increase in tension which consisted of an initial peak followed by a slower developing phase over the following 30 min (Fig. 7A, right). MLC phosphorylation was measured in arteries (n = 4) after 10 min and in other arteries after a further 30 min of stimulation with Ca2+ in the absence or presence of noradrenaline (Fig. 7 and Table 1). No significant change in MLC phosphorylation was seen during the further 30 min stimulation with Ca2+ alone, but an increase was seen in the presence of noradrenaline. In other experiments, the protein kinase C inhibitor calphostin C was included in the solutions. Calphostin C (100 nM) abolished the noradrenaline-induced increase in phosphorylation and contractile response, without having inhibitory effects on responses to Ca2+ alone (Table 1).
Figure 7. Inhibition of noradrenaline-induced Ca2+ sensitization in permeabilized rat mesenteric small arteries with calphostin C.
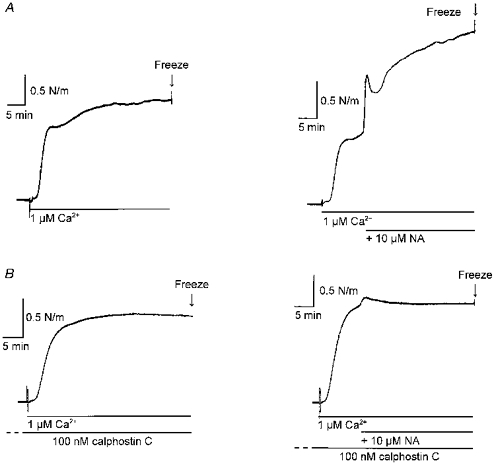
A, traces show 40 min stimulation with 1 μM Ca2+ in a permeabilized artery (left), and similar stimulation with 1 μM Ca2+, but with 10 μM noradrenaline (NA) added at 10 min (right). B, as A but in the presence of 100 nM calphostin C (after 1 h pre-incubation).
Table 1.
Inhibition of noradrenaline-induced Ca2+ sensitization in permeabilized rat mesenteric small arteries with calphostin C
| Control | + 100 nM calphostin C | |||||
|---|---|---|---|---|---|---|
| Ca-10 | Ca-40 | NA-40 | Ca-10 | Ca-40 | NA-40 | |
| MLC phosphorylation | 0.337 ± 0.020 a | 0.354 ± 0.018 b | 0.446 ± 0.015 a,b | 0.367 ± 0.005 | 0.367 ± 0.017 | 0.389 ± 0.022 |
| Force | 80.5 ± 9.0 c | 106.8 ± 22.3 d | 167.8 ± 17.7 c,d | 81.0 ± 9.2 | 135.4 ± 13.9 | 113.7 ± 17.0 |
MLC phosphorylation and the simultaneously measured force production in permeabilized arteries stimulated with 1 μM Ca2+ for 10 min (Ca-10), for 40 min (Ca-40), or for 40 min with noradrenaline added at 10 min (NA-40). Protocol illustrated in Fig. 7. MLC phosphorylation is calculated from the equation MLC-P/(MLC-P + MLC) where MLC-P is phosphorylated MLC. Force is given as a percentage of the initial response to 1 μM Ca2+. Paired differences were tested with one-way ANOVA, and Fisher's LSD test was used for post hoc analysis (mean ±s.e.m., n = 4). Values with the same superscripts are significantly different
P < 0.01
P < 0.05
Evidence for noradrenaline signal transduction through the MLC phosphatase
Following the technique described by Trinkle-Mulcahy et al. (1995) using rabbit portal vein, we treated permeabilized small arteries with ATPγS in the presence of the MLC kinase inhibitor ML-9. The aim was to thiophosphorylate the myosin light chain phosphatase without thiophosphorylating MLC. Following this treatment, the contractile response to 1 μM Ca2+ increased substantially (n = 5), as indicated in Fig. 8A.
Figure 8. Thiophosphorylation in the presence of ML-9 increases the response of rat mesenteric small arteries to Ca2+.
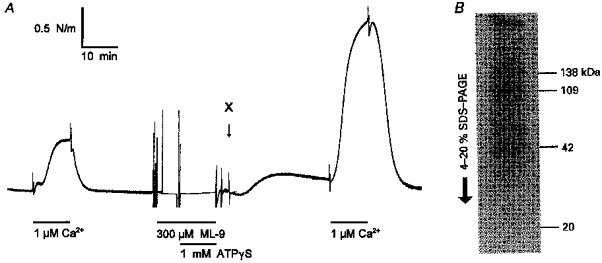
A, permeabilized arteries were treated with ML-9 and ATPγS as described by Trinkle-Mulcahy et al. (1995). The increased Ca2+ sensitivity following this treatment is shown by a comparison of the stimulations with 1 μM Ca2+ before and after treatment. B, detection of proteins labelled with [35S]ATPγS (see Methods) in the presence of ML-9 and absence of Ca2+. Following the protocol shown in A, but with 50 μM GTPγS as well as [35S]ATPγS, arteries (3 mm long) were frozen at the point labelled ‘X’, and total protein was extracted, separated by gel electrophoresis (4–20 % SDS-PAGE) and prepared for autoradiography. The arrow indicates direction of migration. The molecular masses of the three major bands and the expected position of MLC (20 kDa) is shown on the right. The gel is typical of three experiments.
In order to estimate the pattern for thiophosphorylation by ATPγS, we labelled arteries with [35S]ATPγS, and proteins were extracted and separated by gradient SDS-PAGE (Fig. 8B). Three protein bands with apparent molecular masses of 138, 109 and 42 kDa were labelled. In the presence of Ca2+ and absence of ML-9, a 20 kDa band was also labelled (not shown). Furthermore, following treatment, noradrenaline failed to increase the response to 0.15 μM Ca2+ (Fig. 9), consistent with the possibility that noradrenaline causes sensitization through inhibition of the MLC phosphatase, as discussed below.
Figure 9. Thiophosphorylation in the presence of ML-9 inhibits noradrenaline-induced Ca2+ sensitization of rat mesenteric small arteries.
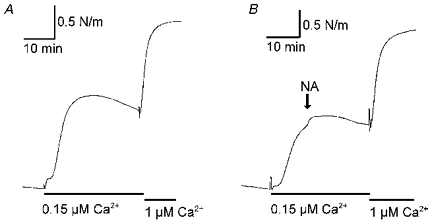
Traces are from two arteries treated with ATPγS-ML-9 and stimulated with 0.15 μM Ca2+ followed by stimulation with 1 μM Ca2+, giving near-maximal contraction. A, control. B, noradrenaline was added on top of stimulation with 0.15 μM Ca2+ as indicated; note the small size of the response to noradrenaline (NA). The traces are representative for four experiments. In some of these (as here), ATP was omitted from the relaxing solution during thiophosphorylation.
DISCUSSION
The main results of this investigation can be summarized as follows. First, noradrenaline-induced Ca2+ sensitization in depolarized rat mesenteric small arteries was associated with increased phosphorylation of the 20 kDa myosin light chain (MLC). Second, calphostin C, an inhibitor of protein kinase C, eliminated the noradrenaline-induced Ca2+ sensitization. Third, conditions which were designed to inhibit MLC phosphatase also eliminated the noradrenaline-induced Ca2+ sensitization. Fourth, especially in the absence of noradrenaline, changes in force were associated with only minor changes in MLC phosphorylation, indicating that other Ca2+-dependent mechanisms may also play a role in the control of activation.
Activation mechanisms
Our finding that GTP was necessary in the bathing solution to obtain repeatable responses to both Ca2+ and noradrenaline in permeabilized preparations (Fig. 5) indicates that GTP is slowly lost from the cytoplasm upon permeabilization. This finding is in agreement with previous findings concerning the influence of GTP on agonist-induced Ca2+ sensitivity (Kitazawa et al. 1989). A requirement for GTP to maintain Ca2+ sensitivity in the absence of noradrenaline has also been noted previously (Akata, 1996). It was of interest that the concentration of GTP required (2 μM) was similar to the Km value reported for the high affinity GTPase activity of GTP-binding proteins in caudal artery membranes (in the range from 0.24 to 1 μM, Weber & MacLeod, 1996). The concentration of GTP required was, however, two orders of magnitude less than the concentrations of free GTP which have been reported in smooth muscle (Coburn, Azim, Fillers & Baron, 1993), and it is therefore unlikely, as was pointed out by these authors, that changing levels of GTP in vivo could be a limiting or regulatory factor for contraction. Further support for the importance of GTP in the response to Ca2+ comes from our finding that this response was inhibited by GDPβS. Thus our findings support the view (for review see Somlyo & Somlyo, 1994) that GTP proteins play a key role in mediating activation of both Ca2+-dependent activation and noradrenaline-induced Ca2+ sensitization.
Relations between force production and MLC phosphorylation
Ca2+, calmodulin-dependent activation of myosin light chain kinase, and subsequent phosphorylation of the MLC, is well established as a major pathway of smooth muscle actomyosin interaction (reviewed by Somlyo & Somlyo, 1994), where specifically it is phosphorylation of serine-19 on the 20 kDa myosin light chain that is involved. For example, correlations between vascular tone and MLC phosphorylation have been found in portal vein and pulmonary and femoral arteries (Kitazawa, Gaylinn, Denney & Somlyo, 1991), and in cremaster arterioles (Zou, Ratz & Hill, 1995). In contrast, a discrepancy between MLC phosphorylation and force was noted by Gerthoffer (1986) in canine tracheal muscle: in that study, MLC phosphorylation remained high when the force of agonist-activated preparations was reduced by stepwise reduction of [Ca2+]o. Thus, although in general MLC phosphorylation is an important determinant of actomyosin interaction, conditions appear to exist where other processes are also involved.
The present study supports the view that in rat mesenteric small arteries, MLC phosphorylation is a requirement for contraction, as in other smooth muscle preparations. Force production was associated with MLC phosphorylation (e.g. Fig. 3), an apparent threshold of about 25 % MLC phosphorylation being required for even small force production (Fig. 4). On the other hand, two discrepancies were observed, suggesting that processes different from MLC phosphorylation also play a role.
First, prolonged (20 min) activation with NA-K-PSS produced a maintained force response, yet both MLC phosphorylation and [Ca2+]i declined (Figs 2 and 3). This suggests the development of a latch state (for review see Murphy, 1994). On the other hand, 20 min exposure to K-PSS did not seem to be associated with a latch development, which contrasts with the behaviour of other preparations where high potassium solution did induce latch (for review see Murphy, 1994). These results suggest that our activations may not have been of long enough duration to produce the phenomenon, but that noradrenaline is able to accelerate the process. This needs to be investigated further.
The second discrepancy between MLC phosphorylation and force production is seen in the steep, apparently vertical, relation between these parameters in the experiments where the force in K-PSS-activated preparations was titrated to either 30 or 75 % of maximum force by adjusting [Ca2+]o (Fig. 4). In NA-K-PSS-activated preparations, the relation was less steep, but even here the difference between the two force levels was associated with only a small change in MLC phosphorylation (from 34 to 40 %). Steep relations between force production and MLC phosphorylation have also been noted by Rembold (1992) (where production of 50 % of maximum force was associated with an increase in MLC phosphorylation from about 8 to 15 %). This suggests that processes auxiliary to MLC phosphorylation are also involved, at least in the case of K-PSS activation. Probably these auxiliary processes are Ca2+ dependent, since to increase the force level from 30 to 75 % of control force required a 10-fold increase in [Ca2+]o (and thus an expected 3-fold increase in [Ca2+]i; Fig. 1). Possible Ca2+-sensitive substrates are the thin-filament proteins caldesmon and calponin, both of which may be phosphorylated at high levels of Ca2+ and thus remove the inhibitory action of these proteins on actomyosin activation (see Horowitz et al. 1996 for review).
The basis for the apparent threshold of 25 % MLC phosphorylation for force production suggested by Fig. 4 is not clear. In swine carotid artery (see Rembold (1992) for review) the threshold is about 8 % MLC phosphorylation. It cannot be excluded that the Ca2+-depletion protocol which we used enhanced the inhibition of contraction by mechanisms auxiliary to MLC phosphorylation (Gerthoffer, 1986), which could only be overcome by relatively high MLC phosphorylation levels. Our data are, however, generally consistent with other reports indicating that maximum contraction can be obtained with 40–50 % MLC phosphorylation (e.g. Rembold, 1992). The data are also consistent with the finding that thiophosphorylated smooth muscle myosin heads can produce substantial mechanical activity, even when paired with an unphosphorylated partner (Harris, Stromski, Hayes & Warshaw, 1995). The highest degree of MLC phosphorylation which we noted (∼55 %) was not a maximum value, since a value in excess of 75 % has been observed in intact unstretched arteries kept at room temperature and stimulated for 20 s with NA-K-PSS (C. L. Buus, unpublished observations). This indicates that phosphorylated myosin molecules do not prevent phosphorylation of their partners (Harris et al. 1995). Nevertheless, the fact that relatively low levels of MLC phosphorylation are able to produce high levels of force may be related to an ability of phosphorylated myosin molecules to operate co-operatively with unphosphorylated partners (Vyas, Mooers, Narayan, Witherell, Siegman & Butler, 1992).
Ca2+ sensitization
The ability of agonist activation of smooth muscle to cause increased force production at constant [Ca2+]i is a phenomenon common to most smooth muscle preparations (for review see Somlyo & Somlyo, 1994). As in the present study (Figs 1 and 3), this is associated with an ability to increase the level of MLC phosphorylation independently of a rise in [Ca2+]i (Kitazawa et al. 1991; see review by Rembold, 1992). The degree of MLC phosphorylation is determined by a balance between MLC kinase and MLC phosphatase activity. Since MLC kinase activity is determined primarily by [Ca2+]i (reviewed in Gallagher, Herring & Stull, 1997), particular attention has been paid to the possibility that noradrenaline may affect regulation of MLC phosphatase (Somlyo et al. 1989; Kitazawa et al. 1991). This indeed appears to be the case, as has been demonstrated by Trinkle-Mulcahy et al. (1995) in rabbit vascular smooth muscle, where inhibition of the MLC phosphatase by thiophosphorylation caused Ca2+ sensitization. This may also have been the case in our experiments using the same protocol (Fig. 8), and further support for this interpretation comes from our finding that under these conditions, noradrenaline was not able to cause Ca2+ sensitization (Fig. 9).
The MLC phosphatase in vascular smooth muscle consists of one catalytic subunit (PP1c) and two non-catalytic subunits (M110 and M21) (Chen et al. 1994), where phosphorylation of the M110 subunit (molecular mass 110 kDa) inhibits the phosphatase activity (Trinkle-Mulcahy et al. 1995; Ichikawa, Ito & Hartshorne, 1996). Available evidence suggests that inhibition of MLC phosphatase may be mediated through arachidonic acid (Gong et al. 1992), diacylglycerol (Gailly, Gong, Somlyo & Somlyo, 1997), or activation of PKC (Masuo, Reardon, Ikebe & Kitazawa, 1994; Ikebe & Brozovich, 1996). The latter possibility is supported by the finding that inhibition of the phosphatase in chicken gizzard muscle is achieved by phosphorylation of the larger non-catalytic subunit at a site which appears to be a substrate for PKC (Ichikawa et al. 1996).
We found that calphostin C (100 nM), which inhibits PKC with half-maximal inhibitory concentration of ∼50 nM by acting on the regulatory domain of PKC (Kobayashi, Nakano, Morimoto & Tamaoki, 1989), was able to completely inhibit the ability of noradrenaline, at constant [Ca2+]i, to increase force and MLC phosphorylation (Fig. 7). In contrast, calphostin C had no significant effect on the response to calcium in the absence of noradrenaline, either in force or in phosphorylation. The inhibiting effect on the noradrenaline response thus cannot be explained by any enhancing effect by calphostin C itself precluding any further enhancement by noradrenaline, but appears rather due to a specific inhibition of the increase in phosphorylation caused by noradrenaline. This is consistent with reports from permeabilized preparations on inhibition of phenylephrine contraction in isolated cells by PKC pseudosubstrate inhibitor (Collins, Walsh & Morgan, 1992; Brozovich, 1995) and in rabbit mesenteric arteries by the PKC inhibitor Ro 31–8220 (Parsons, Sumner & Garland, 1996). On the other hand, downregulation of PKC by phorbol esters did not inhibit Ca2+ sensitization by phenylephrine (Jensen, Gong, Somlyo & Somlyo, 1996). Another discrepancy is that while calphostin C is thought to act mainly on the typical PKC isoforms, Gailly et al. (1997) found that it is the atypical PKC isoforms which are responsible for agonist-induced Ca2+ sensitization. These discrepancies need to be clarified.
The findings described above can be interpreted as indicating a role for PKC in agonist-induced inhibition of the MLC phosphatase. Another means by which PKC can cause Ca2+ sensitization in smooth muscle is thought to be by a PKC-mediated phosphorylation of calponin and caldesmon (see review by Horowitz et al. 1996), as described above. However, in the intact arteries the phosphorylation-force relation was shifted slightly to the right by noradrenaline (Fig. 4C), and not to the left as expected if agonists, acting via PKC on caldesmon and calponin, increase sensitivity of force to MLC phosphorylation.
Recently, the role of Rho-associated protein kinase, activated by RhoA in the Ca2+-sensitization process has been examined (Hirata et al. 1992; Kimura et al. 1996; Gong et al. 1997), and it was shown that Rho-associated protein kinase was able to phosphorylate MLC phosphatase causing its inactivation (Kimura et al. 1996). The importance of Rho-associated protein kinase has been convincingly supported by the demonstration that a pyridine derivative (Y-27632), which preferentially inhibited phenylephrine-induced contraction (IC50= 0.7 μM) in rabbit aorta compared with contraction in response to potassium (IC50 > 30 μM), is a specific inhibitor of Rho-associated protein kinase (Uehata et al. 1997). RhoA is activated through a heterotrimeric GTP-binding protein, and during this process is translocated to the membrane (Gong et al. 1997), although the link between agonist activation and RhoA activation is not established. Our present results, showing that the noradrenaline-induced Ca2+ sensitization of force and MLC phosphorylation was inhibited by calphostin C, suggest that if RhoA is involved in this process then either it is activated by PKC, or else that calphostin C can inhibit RhoA more directly (as, for example, is found in permeabilized U937 promonocytes, where calphostin C inhibited translocation of RhoA from cytosol to membrane (Dubyak & Kertesy, 1997)).
Experimental conditions
In the intact preparations, most experiments were performed 5 min after activation, at which time force had reached a stable level (Fig. 2). In the permeabilized arteries, activation periods of more than 20 min were needed to obtain stable contractions, as has been found previously in rat (Jensen, 1996) and rabbit mesenteric artery preparations (Moreland, Nishimura, van Breemen, Ahn & Moreland, 1992). However, it should be noted that the experiments with permeabilized preparations were performed at 22°C, while the experiments with intact preparations were performed at 37°C. It is likely that the temperature difference is largely responsible for the different time courses. The similarity between the free Ca2+ concentrations required to provide activation in the permeabilized preparations (Fig. 5) and the measured [Ca2+]i during contraction in the intact preparations (Fig. 1) suggests that the conditions for muscle contraction in the two preparations were comparable.
Conclusion
The present results indicate that noradrenaline-induced Ca2+ sensitization is associated with an increased proportion of phosphorylated MLC in rat mesenteric small arteries. The Ca2+ sensitization as regards both force development and MLC phosphorylation is inhibited by calphostin C, suggesting that Ca2+ sensitization involves PKC. Furthermore, conditions designed to inhibit MLC phosphatase-produced Ca2+ sensitization also inhibited further Ca2+ sensitization. These findings support the concept that Ca2+ sensitization is mediated at least in part through inhibition of MLC phosphatase. However, the steep relation between MLC phosphorylation and force production suggests that mechanisms auxiliary to MLC phosphorylation are also involved in force regulation.
Acknowledgments
We thank Karen Skjødt for technical assistance. The work has been supported by the Danish Medical Research Council and the Danish Heart Foundation.
References
- Akata T. Possible involvement of guanosine-5′-triphosphate in calcium-activation of contractile proteins in vascular smooth muscle. Journal of Vascular Research. 1996;33(suppl. 2):30. (abstract) [Google Scholar]
- Brozovich FV. PKC regulates agonist-induced force enhancement in single alpha-toxin-permeabilized vascular smooth muscle cells. American Journal of Physiology. 1995;268:C1202–1206. doi: 10.1152/ajpcell.1995.268.5.C1202. [DOI] [PubMed] [Google Scholar]
- Bruns RF, Miller FD, Merriman RL, Howbert JJ, Heath WF, Kobayashi E, Takahashi I, Tamaoki T, Nakano H. Inhibition of protein kinase C by calphostin C is light-dependent. Biochemical and Biophysical Research Communications. 1991;176:288–293. doi: 10.1016/0006-291x(91)90922-t. [DOI] [PubMed] [Google Scholar]
- Chen YH, Chen MX, Alessi DR, Campbell DG, Shanahan C, Cohen P, Cohen PTW. Molecular cloning of cDNA encoding the 110 kDa and 21 kDa regulatory subunits of smooth muscle protein phosphatase 1 M. FEBS Letters. 1994;356:51–55. doi: 10.1016/0014-5793(94)01231-8. [DOI] [PubMed] [Google Scholar]
- Coburn RF, Azim S, Fillers WS, Baron CB. Smooth muscle guanine nucleotides and receptor-effector coupling following inhibition of oxidative energy production. American Journal of Physiology. 1993;264:L1–6. doi: 10.1152/ajplung.1993.264.1.L1. [DOI] [PubMed] [Google Scholar]
- Collins EM, Walsh MP, Morgan KG. Contraction of single vascular smooth muscle cells by phenylephrine at constant [Ca2+]i. American Journal of Physiology. 1992;262:H754–762. doi: 10.1152/ajpheart.1992.262.3.H754. [DOI] [PubMed] [Google Scholar]
- Dubyak GR, Kertesy SB. Inhibition of GTP-gamma-S-dependent phospholipase D and Rho membrane association by calphostin is independent of protein kinase C catalytic activity. Archives of Biochemistry and Biophysics. 1997;341:129–139. doi: 10.1006/abbi.1997.9946. 10.1006/abbi.1997.9946. [DOI] [PubMed] [Google Scholar]
- Gailly P, Gong MC, Somlyo AV, Somlyo AP. Possible role of atypical protein kinase C activated by arachidonic acid in Ca2+ sensitization of rabbit smooth muscle. The Journal of Physiology. 1997;500:95–109. doi: 10.1113/jphysiol.1997.sp022002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallagher PJ, Herring BP, Stull JT. Myosin light chain kinases. Journal of Muscle Research and Cell Motility. 1997;18:1–16. doi: 10.1023/a:1018616814417. 10.1023/A:1018616814417. [DOI] [PubMed] [Google Scholar]
- Gerthoffer WT. Calcium dependence of myosin phosphorylation and airway smooth muscle contraction and relaxation. American Journal of Physiology. 1986;250:C597–604. doi: 10.1152/ajpcell.1986.250.4.C597. [DOI] [PubMed] [Google Scholar]
- Gong MC, Fuglsang A, Alessi D, Kobayashi S, Cohen P, Somlyo AV, Somlyo AP. Arachidonic acid inhibits myosin light chain phosphatase and sensitizes smooth muscle to calcium. Journal of Biological Chemistry. 1992;267:21492–21498. [PubMed] [Google Scholar]
- Gong MC, Fujihara H, Somlyo AV, Somlyo AP. Translocation of rhoA associated with Ca2+ sensitization of smooth muscle. Journal of Biological Chemistry. 1997;272:10704–10709. doi: 10.1074/jbc.272.16.10704. 10.1074/jbc.272.16.10704. [DOI] [PubMed] [Google Scholar]
- Harris DE, Stromski CJ, Hayes E, Warshaw DM. Thiophosphorylation independently activates each head of smooth muscle myosin in vitro. American Journal of Physiology. 1995;269:C1160–1166. doi: 10.1152/ajpcell.1995.269.5.C1160. [DOI] [PubMed] [Google Scholar]
- Hirata K, Kikuchi A, Sasaki T, Kuroda S, Kaibuchi K, Matsuura Y, Seki H, Saida K, Takai Y. Involvement of rho p21 in the GTP-enhanced calcium ion sensitivity of smooth muscle contraction. Journal of Biological Chemistry. 1992;267:8719–8722. [PubMed] [Google Scholar]
- Horowitz A, Menice CB, Laporte R, Morgan KG. Mechanisms of smooth muscle contraction. Physiological Reviews. 1996;76:967–1003. doi: 10.1152/physrev.1996.76.4.967. [DOI] [PubMed] [Google Scholar]
- Ichikawa K, Ito M, Hartshorne DJ. Phosphorylation of the large subunit of myosin phosphatase and inhibition of phosphatase activity. Journal of Biological Chemistry. 1996;271:4733–4740. doi: 10.1074/jbc.271.9.4733. 10.1074/jbc.271.9.4733. [DOI] [PubMed] [Google Scholar]
- Ikebe M, Brozovich FV. Protein kinase C increases force and slows relaxation in smooth muscle: Evidence for regulation of the myosin light chain phosphatase. Biochemical and Biophysical Research Communications. 1996;225:370–376. doi: 10.1006/bbrc.1996.1182. 10.1006/bbrc.1996.1182. [DOI] [PubMed] [Google Scholar]
- Jensen PE. Calphostin C-sensitive enhancements of force by lysophosphatidylinositol and diacylglycerols in mesenteric arteries from the rat. British Journal of Pharmacology. 1996;119:15–22. doi: 10.1111/j.1476-5381.1996.tb15671.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PE, Gong MC, Somlyo AV, Somlyo AP. Separate upstream and convergent downstream pathways of G-protein- and phorbol ester-mediated Ca2+ sensitization of myosin light chain phosphorylation in smooth muscle. Biochemical Journal. 1996;318:469–475. doi: 10.1042/bj3180469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen PE, Mulvany MJ, Aalkjaer C. Endogenous and exogenous agonist-induced changes in the coupling between [Ca2+]i and force in rat resistance arteries. Pflügers Archiv. 1992;420:536–543. doi: 10.1007/BF00374630. [DOI] [PubMed] [Google Scholar]
- Kimura K, Ito M, Amano M, Chihara K, Fukata Y, Nakafuku M, Yamamori B, Feng J, Nakano T, Okawa K, Iwamatsu A, Kaibuchi K. Regulation of myosin phosphatase by Rho and Rho-associated kinase (Rho-kinase) Science. 1996;273:245–248. doi: 10.1126/science.273.5272.245. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Gaylinn BD, Denney GH, Somlyo AP. G-protein-mediated Ca2+ sensitization of smooth muscle contraction through myosin light chain phosphorylation. Journal of Biological Chemistry. 1991;266:1708–1715. [PubMed] [Google Scholar]
- Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle. Role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ Journal of Biological Chemistry. 1989;264:5339–5342. [PubMed] [Google Scholar]
- Kobayashi E, Nakano H, Morimoto M, Tamaoki T. Calphostin C (UCN-1028C), a novel microbial compound, is a highly potent and specific inhibitor of protein kinase C. Biochemical and Biophysical Research Communications. 1989;159:548–553. doi: 10.1016/0006-291x(89)90028-4. [DOI] [PubMed] [Google Scholar]
- Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca2+-sensitizing effect of protein kinase C on vascular smooth muscle: Inhibition of myosin light chain phosphatase. Journal of General Physiology. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreland S, Nishimura J, van Breemen C, Ahn HY, Moreland RS. Transient myosin phosphorylation at constant Ca2+ during agonist activation of permeabilized arteries. American Journal of Physiology. 1992;263:C540–544. doi: 10.1152/ajpcell.1992.263.2.C540. [DOI] [PubMed] [Google Scholar]
- Mulvany MJ, Halpern W. Contractile properties of small arterial resistance vessels in spontaneously hypertensive and normotensive rats. Circulation Research. 1977;41:19–26. doi: 10.1161/01.res.41.1.19. [DOI] [PubMed] [Google Scholar]
- Murphy RA. What is special about smooth muscle? The significance of covalent crossbridge regulation. FASEB Journal. 1994;8:311–318. doi: 10.1096/fasebj.8.3.8143937. [DOI] [PubMed] [Google Scholar]
- Nilsson H, Jensen PE, Mulvany MJ. Minor role for direct adrenoceptor-mediated calcium entry in rat mesenteric small arteries. Journal of Vascular Research. 1994;31:314–321. doi: 10.1159/000159059. [DOI] [PubMed] [Google Scholar]
- Parsons SJ, Sumner MJ, Garland CJ. Phospholipase A2 and protein kinase C contribute to myofilament sensitization to 5-HT in the rabbit mesenteric artery. The Journal of Physiology. 1996;491:447–453. doi: 10.1113/jphysiol.1996.sp021228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng H-L, Jensen PE, Nilsson H, Aalkjaer C. Effect of acidosis on tension and [Ca2+]i in rat cerebral arteries - is there a role for the membrane potential? American Journal of Physiology. 1998;274:H655–662. doi: 10.1152/ajpheart.1998.274.2.H655. [DOI] [PubMed] [Google Scholar]
- Persechini A, Kamm KE, Stull JT. Different phosphorylated forms of myosin in contracting tracheal smooth muscle. Journal of Biological Chemistry. 1986;261:6293–6299. [PubMed] [Google Scholar]
- Rembold CM. Regulation of contraction and relaxation in arterial smooth muscle. Hypertension. 1992;20:129–137. doi: 10.1161/01.hyp.20.2.129. [DOI] [PubMed] [Google Scholar]
- Somlyo AP, Kitazawa T, Himpens B, Matthijs G, Houriuti K, Kobayashi S, Goldman YE, Somlyo AV. Modulation of Ca2+-sensitivity and of contraction in smooth muscle: a major role of protein phosphatases? Advances in Protein Phosphatases. 1989;5:181–195. [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Trinkle-Mulcahy L, Ichikawa K, Hartshorne DJ, Siegman MJ, Butler TM. Thiophosphorylation of the 130-kDa subunit is associated with a decreased activity of myosin light chain phosphatase in α-toxin-permeabilized smooth muscle. Journal of Biological Chemistry. 1995;270:18191–18194. doi: 10.1074/jbc.270.31.18191. [DOI] [PubMed] [Google Scholar]
- Uehata M, Ishizaki T, Satoh H, Ono T, Kawahara T, Morishita T, Tamakawa H, Yamagami K, Inui J, Maekawa M, Narumiya S. Calcium sensitization of smooth muscle mediated by a Rho-assosiated protein kinase. Nature. 1997;389:990–994. doi: 10.1038/40187. [DOI] [PubMed] [Google Scholar]
- Vyas TB, Mooers SU, Narayan SR, Witherell JC, Siegman MJ, Butler TM. Cooperative activation of myosin by light chain phosphorylation in permeabilized smooth muscle. American Journal of Physiology. 1992;263:C210–219. doi: 10.1152/ajpcell.1992.263.1.C210. [DOI] [PubMed] [Google Scholar]
- Weber LP, Macleod KM. Noradrenaline stimulation of high-affinity GTPase activity in membranes from rat aorta and caudal artery. Biochemical Pharmacology. 1996;52:677–684. doi: 10.1016/0006-2952(96)00344-9. [DOI] [PubMed] [Google Scholar]
- Zou H, Ratz PH, Hill MA. Role of myosin phosphorylation and [Ca2+]i in myogenic reactivity and arteriolar tone. American Journal of Physiology. 1995;269:H1590–1596. doi: 10.1152/ajpheart.1995.269.5.H1590. [DOI] [PubMed] [Google Scholar]