

Abstract
Properties of the endothelium-dependent hyperpolarization evoked by acetylcholine (ACh) in smooth muscle of the guinea-pig coronary artery were investigated using conventional microelectrode techniques.
ACh hyperpolarized the membrane in an endothelium-dependent manner. The hyperpolarization comprised two components: an initial and a slow hyperpolarization. The former appeared during application of ACh, while the latter occurred after withdrawal of ACh.
Indomethacin and diclofenac, inhibitors of the enzyme cyclo-oxygenase, blocked only the slow hyperpolarization, indicating that this potential was produced by endothelial prostanoids.
Clotrimazole and SKF 525a, known inhibitors of the enzyme cytochrome P450, inhibited both the initial and the slow hyperpolarizations, suggesting that these chemicals acted as non-selective inhibitors of arachidonic acid metabolism. Inhibition of the lipoxygenase pathway of arachidonic acid metabolism by nordihydroguaiaretic acid had no effect on either component of the hyperpolarization.
The slow hyperpolarization was inhibited by 4-aminopyridine (4-AP; 10−4–10−3 M) and glibenclamide (10−6 M). The initial hyperpolarization was greatly inhibited by charybdotoxin (CTX; 5 × 10−8 M) and partially inhibited by apamin (10−7 M), but was not inhibited by glibenclamide (10−5 M). Ba2+ (10−4 M) depolarized the membrane and increased the amplitude of both components of the ACh-induced hyperpolarization.
Hyperpolarizations produced by Y-26763, a K+ channel opener, were inhibited by glibenclamide, but not by 4-AP.
The results indicate that the slow hyperpolarization is produced by endothelial prostanoids through activation of 4-AP-sensitive K+ channels (possibly delayed rectifier type). The initial hyperpolarization is produced mainly through activation of CTX-sensitive K+ channels (possibly Ca2+-sensitive type).
In vascular smooth muscle, many types of agonist hyperpolarize the membrane in an endothelium-dependent manner. The mediators of such responses are endothelium-derived relaxing factor (EDRF; Tare, Parkington, Coleman, Neild & Dusting, 1990), endothelium-derived hyperpolarizing factor (EDHF; Chen, Suzuki & Weston, 1988; Chen & Suzuki, 1989) or prostanoids (Parkington, Tare, Tonta & Coleman, 1993). The acetylcholine (ACh)-induced hyperpolarization is accompanied by an increase in ionic conductance of the membrane, thereby suggesting an increased conductance of potassium ions (Komori & Suzuki, 1987; Suzuki & Chen, 1990; Hashitani & Suzuki, 1997). An involvement of K+ channels in the endothelium-dependent hyperpolarization is also supported by evidence that the hyperpolarization is not generated in high [K+]o solution (Chen & Suzuki, 1989; Garland, Plane, Kemp & Cocks, 1995), or after K+ channels have been blocked with tetrabutylammonium, a non-selective K+ channel inhibitor (Nagao & Vanhoutte, 1992).
Many types of K+ channel have been identified in smooth muscle cells (Nelson & Quayle, 1995) and the involvement of different types of K+ channel in individual vasodilator responses has been estimated from the effects of drugs which selectively inhibit certain K+ channels. EDRF may be nitric oxide (NO) or a NO-containing substance metabolized from L-arginine (Moncada, Palmer & Higgs, 1991). EDRF hyperpolarizes the membrane in the uterine artery of the guinea-pig (Tare et al. 1990) possibly by increasing the open probability of the high conductance Ca2+-sensitive K+ channel, since NO or NO-containing substances activate this type of K+ channel (Robertson, Schubert, Hescheler & Nelson, 1993; Bolotina, Najibi, Palacino, Pagano & Cohen, 1994). EDRF may also activate ATP-sensitive K+ channels in smooth muscle of the rabbit cerebral artery (Standen, Quayle, Davies, Brayden, Huang & Nelson, 1989) and mesenteric artery (Murphy & Brayden, 1995a). EDHF may be a metabolite of arachidonic acid derived from cytochrome P450-dependent mono-oxygenase activity in the porcine coronary artery (Hecker, Bara, Bauersachs & Busse, 1994; Campbell, Gebremedhin, Pratt & Harder, 1996) and rat mesenteric artery (Chen & Cheung, 1996), since chemicals which inhibit cytochrome P450 inhibit the generation of endothelium-dependent hyperpolarization. The products of the activation of cytochrome P450 which evoke membrane hyperpolarization are epoxyeicosatrienoic acids (EETs) and their regioisomers (Fitzpatrick & Murphy, 1988). These eicosanoids activate Ca2+-sensitive K+ channels in porcine coronary artery (Hu & Kim, 1993; Campbell et al. 1996). Selective blockade of Ca2+-sensitive K+ channels with charybdotoxin (CTX) abolishes the ACh-induced hyperpolarization, suggesting that EETs may be the mediators of such responses (Chen & Cheung, 1997; Petersson, Zygmunt & Högestätt, 1997). However, in the guinea-pig carotid artery (Corriu, Félétou, Canet & Vanhoutte, 1996) and rat mesenteric artery (Vanheel & Van de Voorde, 1997; Fukao, Hattori, Kanno, Sakuma & Kitabatake, 1997), inhibitors of cytochrome P450 may not have specific inhibitory effects on the EDHF-induced hyperpolarization. In the rabbit mesenteric artery, the EDHF-induced hyperpolarization is inhibited by apamin, suggesting that the small conductance Ca2+-sensitive K+ channel is involved (Murphy & Brayden, 1995b). The ACh-induced hyperpolarizations generated in the rat mesenteric artery (Chen & Cheung, 1997) and in the submucosal arterioles of guinea-pig (Hashitani & Suzuki, 1997) are abolished by simultaneous application of CTX and apamin, suggesting that two types of Ca2+-sensitive K+ channel mediate the hyperpolarization. Prostacyclin may be the main prostanoid derived from the endothelium (Gryglewski, Botting & Vane, 1991), and this prostanoid is known to hyperpolarize arterial smooth muscle (Parkington et al. 1993) by activating glibenclamide-sensitive K+ channels (possibly the ATP-sensitive K+ channel) in the guinea-pig coronary artery (Parkington, Tonta, Coleman & Tare, 1995) and the rabbit mesenteric artery (Murphy & Brayden, 1995b).
We have investigated the properties of the endothelium-dependent hyperpolarization in the smooth muscle of the guinea-pig coronary artery. In this artery, the hyperpolarization, in response to ACh, consists of three components produced by EDHF, EDRF and prostanoids (Parkington et al. 1993). As the type of K+ channel activated by these endothelial factors may differ, an attempt was made to separate the hyperpolarization produced by individual factors using drugs which inhibit different types of K+ channel. Experiments were first carried out to examine the substances which contributed to each component of the ACh-induced hyperpolarization, using inhibitors of metabolic pathways of arachidonic acid or NO. The effects of several types of drugs which inhibit K+ channels were then tested on the ACh-induced hyperpolarization. The results suggest that, in the guinea-pig coronary artery, the ACh-induced hyperpolarization is produced by EDHF and prostanoids which mainly activate CTX- and 4-aminopyridine (4-AP)-sensitive types of K+ channel, respectively.
METHODS
Albino guinea-pigs of either sex (200–250 g) were exsanguinated by decapitation after being stunned by a blow to the head or after CO2 anaesthesia. The proximal part of the main coronary artery extending to the left ventricle was dissected from the heart and cleaned by removal of surrounding connective and cardiac tissue. The recording chamber (capacity of about 0.5 ml, 8 mm width, 8 mm depth, 30 mm length) was made from Lucite plate with a silicone rubber plate fixed at the bottom. The artery was mounted on the rubber plate using small pins, and superfused with warmed (35.5°C) Krebs solution at a constant flow rate of about 3 ml min−1. Endothelial cells were removed either by rubbing the internal surface of the vessel or by passing distilled water through it. In the former case, the vessels were opened by vertical cutting and the internal surface was rubbed with a moistened cotton ball. Each method resulted in a depolarization of the membrane by about 10 mV. The successful removal of effective endothelial cells was confirmed by the absence of the typical hyperpolarization response to ACh.
Membrane potentials were recorded using a conventional microelectrode technique, using glass capillary microelectrodes filled with 3 M KCl. Smooth muscle cells were impaled from the adventitial side. The criteria for good penetrations were: (1) a sharp drop of the potential in the negative direction by about 50 mV, and (2) an ability to maintain the potential at a stable level for over 3 min. Usually, a stable intracellular potential could be monitored for 1–2 h.
The ionic composition of the Krebs solution was as follows (mM): Na+, 137.4; K+, 5.9; Ca2+, 2.5; Mg2+, 1.2; HCO3−, 15.5; H2PO4−, 1.2; Cl−, 134; and glucose, 11.5. The solution was aerated with O2 containing 5% CO2, and the pH of the solution was maintained at 7.2–7.3.
Drugs used were acetylcholine chloride, 4-AP, apamin, atropine sulphate, clotrimazole, 4-diphenylacetoxy-N-methylpiperidine methiodide (4-DAMP), glibenclamide and indomethacin (Sigma); Nω-nitro-L-arginine (nitroarginine) and CTX (Peptide Institute, Osaka, Japan); diclofenac sodium salt and N,N-diethylaminoethyl-2,2-diphenylvalerate hydrochloride (SKF 525a) (Research Biochemicals International, Natick, MA, USA); nordihydroguaiaretic acid (NDGA; Biomolecular Research Laboratories, Plymouth Meeting, PA, USA); and Y-26763 (Yoshitomi Pharmaceutical Ind., Osaka, Japan). Drugs were dissolved in distilled water (4-AP, apamin, CTX, diclofenac, 4-DAMP, nitroarginine, SKF 525a), dimethyl sulphoxide (clotrimazole, glibenclamide, Y-26763), ethanol (NDGA) or 5 mM Na2CO3 solution (indomethacin), and added to the Krebs solution to obtain the desired concentration. The final concentration of these solvents in the Krebs solution did not exceed 1:1000, and no alteration of the pH of the solution was detected following these procedures.
Measured values were expressed as means ± standard deviation (s.d.). Statistical significance was tested using Student's paired and unpaired t tests, and probabilities of less than 5% were considered significant.
RESULTS
Hyperpolarization produced by ACh
In cells of the guinea-pig coronary artery with intact endothelium, the resting membrane potential was stable, ranging between −45 and −55 mV (Table 1). Application of 10−6 M ACh for 1 min evoked a membrane hyperpolarization which comprised two components: a large hyperpolarization, which had an amplitude of about 20 mV and lasted about 1.5 min (initial hyperpolarization), followed by a sustained hyperpolarization which appeared at the withdrawal of ACh and which had an amplitude of about 5 mV and a duration of about 5 min (slow hyperpolarization; Fig. 1A). In twenty-five arteries studied, the amplitude of the initial hyperpolarization ranged between 10 and 25 mV, while the slow hyperpolarization ranged between 1 and 8 mV (Table 1) and lasted between 4 and 10 min (mean, 6.5 ± 2.0 min). Impairment of the endothelial cells by perfusion with distilled water reduced the membrane potential to between −35 and −45 mV (mean, −40.2 ± 2.8 mV; n = 8; P < 0.05). In endothelium-denuded arteries, ACh depolarized the membrane by 1–5 mV in five preparations (mean value, 3.3 ± 1.2 mV; Fig. 1B) but hyperpolarized the membrane by 1–3 mV in three preparations (mean value, 1.5 ± 1.0 mV; Fig. 1C). The results indicate that the ACh-induced hyperpolarization is an endothelium-dependent phenomenon.
Table 1.
Membrane potential and ACh-induced hyperpolarization in the guinea-pig coronary artery
| ACh-induced hyperpolarization (mV) | |||
|---|---|---|---|
| Condition | Membrane potential (mV) | Initial | Slow |
| Control (resting potential) | -51.5 ± 2.6 (31) | 20.5 ± 3.1 (31) | 4.9 ± 1.5 (31) |
| Indomethacin (5 × 10−6 M) | -50.5 ± 2.5 (10) | 20.1 ± 4.0 (10) | 0 (10) * |
| Diclofenac (10−6 M) | -51.5 ± 3.5 (11) | 19.5 ± 3.3 (11) | 0 (11) * |
| Nitroarginine (10−5 M) | -51.1 ± 3.8 (12) | 19.1 ± 4.0 (12) | 5.9 ± 1.8 (12) |
| Nitroarginine (10−5 M) + indomethacin (5 × 10−6 M) | -50.8 ± 2.8 (8) | 20.2 ± 3.8 (8) | 0 (8) * |
| SKF 525a (3 × 10−5 M) | -48.0 ± 4.8 (7)* | 2.5 ± 1.2 (10)* | 0.6 ± 0.5 (10)* |
| SKF 525a (10−4 M) | -43.7 ± 2.5 (10)* | 0.7 ± 0.7 (10)* | 0 (10)* |
| Clotrimazole (10−5 M) | -33.5 ± 1.7 (6)* | 2.5 ± 1.2 (6)* | 0.5 ± 0.1 (6)* |
| NDGA (10−5 M) | -49.3 ± 3.5 (8) | 21.5 ± 2.8 (8) | 5.2 ± 1.7 (8) |
| CTX (5 × 10−8 M) | -44.9 ± 2.5 (10)* | 2.5 ± 1.5 (10)* | 3.8 ± 1.5 (10)* |
| Apamin (10−7 M) | -48.8 ± 3.5 (7) | 16.5 ± 3.5 (7)* | 5.0 ± 2.0 (7) |
| Ba2+ (10−4 M) | -46.5 ± 3.0 (6)* | 22.2 ± 2.0 (6)* | 6.7 ± 1.5 (6)* |
| Ba2+ (10−3 M) | -38.5 ± 3.3 (7)* | 16.3 ± 2.1 (7)* | 1.7 ± 0.5 (7)* |
| Glibenclamide (10−6 M) | -43.3 ± 3.2 (12)* | 23.5 ± 2.5 (12)* | 0 (11)* |
Membrane potentials were measured after addition of individual drugs for over 5 min. ACh (10−6 M) was then applied for 1 min, and the peak amplitude of the initial and slow components of the hyperpolarization was measured from the potential before application of ACh. Values are means ±s.d. (number of observations).
P < 0.05, compared with control.
Figure 1. ACh-induced hyperpolarization and endothelial cells in the guinea-pig coronary artery.
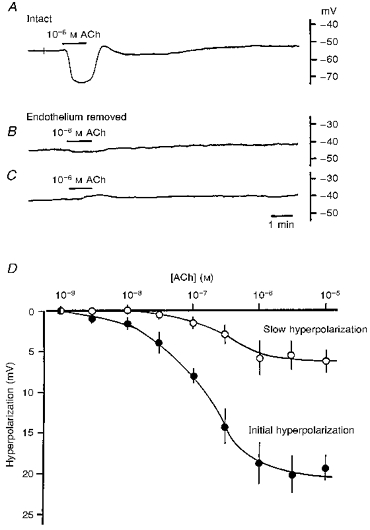
ACh (10−6 M) was applied for 1 min as indicated by the horizontal bars. Endothelial cells were either intact (A) or damaged with distilled water (B and C). Each response was recorded from a different vessel. D, relationship between the concentration of ACh and the amplitude of the hyperpolarization. ACh (10−9-10−5 M) was applied for 1 min and the peak amplitudes of the initial (•) and slow (^) hyperpolarizations were measured. Means ±s.d. (n = 5–18 observations) obtained from 5 arteries.
The concentration-response relationship for the effects of ACh on the membrane potential (Fig. 1D) shows that the threshold concentrations of ACh required for generation of the initial and slow hyperpolarizations were 10−8 and 10−7 M, respectively. The amplitude of both the initial and slow hyperpolarizations increased in a concentration-dependent manner to reach maximum values of about 20 and 5 mV at 10−6 and 10−5 M ACh, respectively. In the presence of atropine (10−7 M) or 4-DAMP (10−8 M), ACh produced no detectable response (n = 2 for atropine, n = 3 for 4-DAMP; data not shown). Neither atropine nor 4-DAMP affected the resting membrane potential. The actions of atropine, a non-selective muscarinic receptor antagonist, and 4-DAMP, an M3 receptor-selective antagonist (Eglen, Hegde & Watson, 1996), suggested that the responses were mediated by activation of muscarinic M3 subtype receptors.
Effects of inhibitors of enzymes involved in the metabolism of NO or arachidonic acid
The effects of inhibitors of cyclo-oxygenase (indomethacin or diclofenac) or of NO synthase (nitroarginine) on the ACh-induced hyperpolarization were investigated. None of these inhibitors had any significant effect on the resting membrane potential (Table 1). Indomethacin (5 × 10−6 M) inhibited the slow hyperpolarization with no significant alteration of the initial hyperpolarization (Fig. 2A and B and Table 1). Additional application of nitroarginine (10−5 M) produced no detectable alteration of the ACh-induced response (Fig. 2C). In different preparations, nitroarginine did not alter either component of the ACh-induced hyperpolarization (Fig. 2D and E and Table 1) while indomethacin (5 × 10−6 M), added to the nitroarginine-containing solution, again inhibited the slow hyperpolarization, with no significant alteration of the initial hyperpolarization (Fig. 2F and Table 1). In the presence of both nitroarginine and indomethacin, withdrawal of ACh produced a transient depolarization which was often accompanied by spike generation (Fig. 2F). In different experiments, the effects of diclofenac (10−6 M) on the ACh-induced hyperpolarization were found to be identical to those of indomethacin (Table 1).
Figure 2. Indomethacin, but not nitroarginine, inhibits the slow component of the ACh-induced hyperpolarization in the guinea-pig coronary artery.
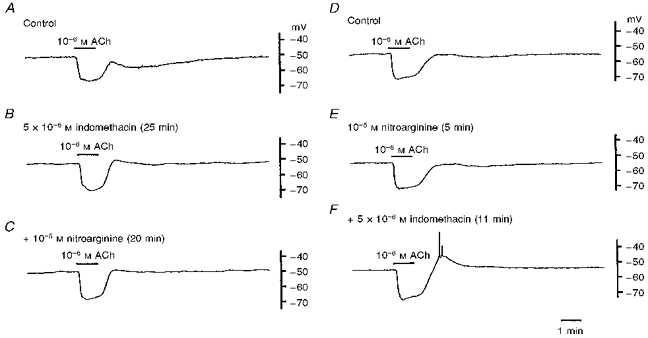
ACh (10−6 M) was applied for 1 min before (A; Control) and after application of 5 × 10−6 M indomethacin for 25 min (B), and indomethacin and 10−5 M nitroarginine together for 20 min (C), or before (D; Control) and after application of 10−5 M nitroarginine for 5 min (E) and nitroarginine and 5 × 10−6 M indomethacin together for 11 min (F). Traces shown in A-C and D-F were recorded from different single cells.
Figure 3 shows the effects of clotrimazole and SKF 525a, inhibitors of the enzyme cytochrome P450, on the ACh-induced hyperpolarization. Clotrimazole (10−5 M) inhibited the ACh-induced hyperpolarization, with depolarization of the membrane (Fig. 3A–C and Table 1). Inhibition of the two components of the hyperpolarization developed slowly and required over 60 min to reach a steady level. The initial hyperpolarization was altered to a transient form with a reduced amplitude and the slow hyperpolarization was abolished (Fig. 3C). Recovery from inhibition by clotrimazole was not the same for both components of the hyperpolarization; the slow hyperpolarization fully recovered after washout of clotrimazole whereas the initial hyperpolarization did not (Fig. 3D).
Figure 3. Cytochrome P450 inhibitors and ACh-induced hyperpolarization.
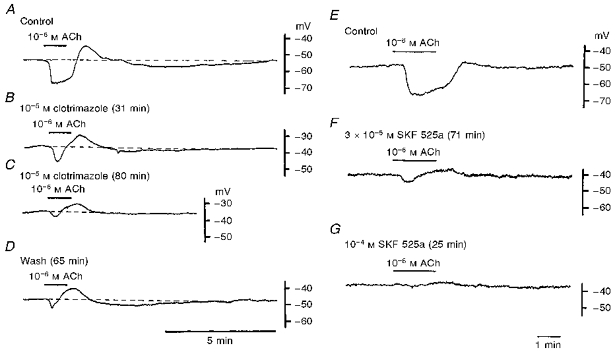
A-D, ACh (10−6 M) was applied for 1 min in the absence (A; Control) and presence (31 min, B; 80 min, C) of 10−5 M clotrimazole, and 65 min after removal of clotrimazole from the superfusate (D). E-G, ACh was applied for 1 min before (E; Control) and after application of SKF 525a (3 × 10−5 M for 71 min, F; 10−4 M for 25 min, G). Traces shown in A-D and E-G were recorded from single cells in different vessels.
SKF 525a was also a potent inhibitor of the ACh-induced hyperpolarization. In the presence of 3 × 10−5 M SKF 525a, for over 30 min, the initial hyperpolarization was changed to a transient form with a reduced amplitude of about 15% of control, and the slow hyperpolarization was abolished (Fig. 3E and F and Table 1). When the concentration of SKF 525a was increased to 10−4 M, the membrane was depolarized by about 8 mV and both components of the ACh-induced hyperpolarization were inhibited (Fig. 3G and Table 1). The inhibition by SKF 525a of both components of the ACh-induced hyperpolarization was irreversible, for up to 2 h after withdrawal of the inhibitor (n = 3 tissues).
Application of NDGA (10−5 M), a lipoxygenase inhibitor, for up to 2 h, had no effect on either the ACh-induced hyperpolarization or the membrane potential (Table 1), suggesting that metabolites produced through the lipoxygenase pathway were not involved in the generation of the ACh-induced response.
Effects of inhibitors of K+ channels on the ACh-induced hyperpolarization in guinea-pig coronary artery
The drugs tested were CTX which blocks high conductance Ca2+-sensitive K+ channels, apamin which blocks small conductance Ca2+-sensitive K+ channels, 4-AP which blocks voltage-dependent K+ channels, Ba2+ which blocks inward rectifier K+ channels and glibenclamide which blocks ATP-sensitive K+ channels (Nelson & Quayle, 1995).
CTX (5 × 10−8 M) depolarized the membrane by about 7 mV and reduced the amplitude of the ACh-induced initial and slow hyperpolarizations to a similar value (Fig. 4A–C and Table 1). Recovery of the hyperpolarization was incomplete: about 60% recovery was achieved after washout of CTX for over 70 min (Fig. 4D). Apamin (10−7 M) reduced the amplitude of the initial hyperpolarization, with no alteration of either the membrane potential or the slow hyperpolarization (Fig. 4E and F and Table 1). The inhibitory actions of apamin were fully reversible after washout for 30 min (data not shown).
Figure 4. CTX and apamin inhibit the ACh-induced hyperpolarization in the guinea-pig coronary artery.
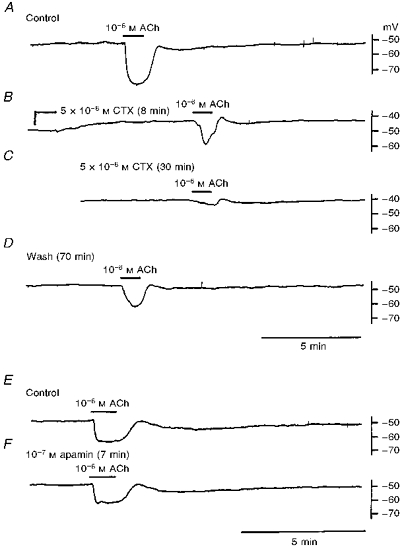
ACh (10−6 M) was applied for 1 min before (A; Control), during (8 min, B; 30 min, C) application of 5 × 10−8 M CTX (applied at the point indicated in B), and after removal of CTX for 70 min (D), or before (E; Control) and during (F) application of 10−7 M apamin for 7 min. Traces in A-D and E and F were recorded from single cells in different tissues.
Glibenclamide (10−6 M) depolarized the membrane by about 8 mV, inhibited the slow hyperpolarization, but enhanced the initial hyperpolarization (Fig. 5A and B and Table 1). The effects of two concentrations of Ba2+ (10−4 and 10−3 M) were tested on the ACh-induced hyperpolarization; 10−4 M Ba2+ depolarized the membrane by about 5 mV and increased the amplitude of both the initial and slow hyperpolarizations (Fig. 5C and D and Table 1), while 10−3 M Ba2+ depolarized the membrane by about 13 mV and reduced the amplitude of both the initial and slow hyperpolarizations (Fig. 5E and Table 1).
Figure 5. Effects of glibenclamide and Ba2+ on the ACh-induced hyperpolarization in the guinea-pig coronary artery.
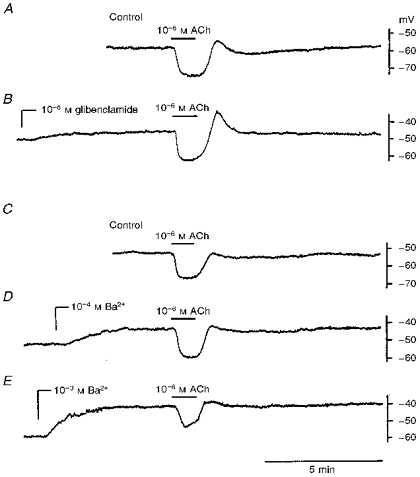
ACh (10−6 M) was applied for 1 min before (A and C; Control) and after application (at the points indicated) of 10−6 M glibenclamide (B), 10−4 M Ba2+ (D), and 10−3 M Ba2+ (E). Traces in A-B and C-E were recorded from different single cells in different vessels.
Figure 6 shows the effects of 4-AP on the ACh-induced hyperpolarization. Low concentrations (2 × 10−4 M) of 4-AP inhibited the slow hyperpolarization with no alteration of the initial hyperpolarization (Fig. 6A and B); high concentrations (10−3 M) of 4-AP inhibited both components (Fig. 6C). These inhibitory effects of 4-AP were partially reversible (Fig. 6D). The concentration-response relationship of the effects of 4-AP on the ACh-induced hyperpolarization shows that the slow hyperpolarization was inhibited at concentrations greater than 10−4 M (Fig. 6E; ▵), while the initial hyperpolarization was inhibited at a concentration of 10−3 M (Fig. 6E; •) with no significant alteration of the resting membrane potential (Fig. 6E; ^).
Figure 6. 4-AP selectively inhibits the ACh-induced hyperpolarization in the guinea-pig coronary artery.
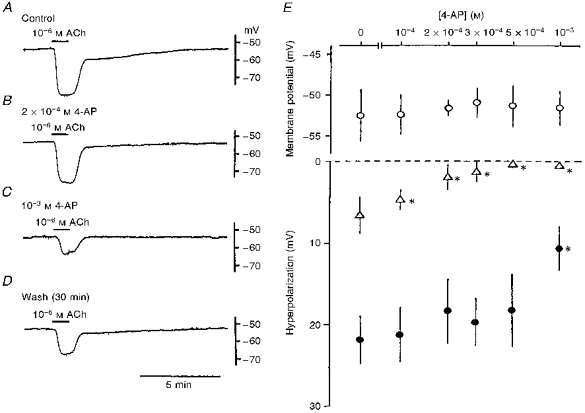
ACh (10−6 M) was applied for 1 min before (A; Control) and during application of 4-AP (2 × 10−4 M, B; 10−3 M, C), and after washout of 4-AP for 30 min (D). Traces in A-D were recorded from the same cell. In E, the membrane potential (^), and the amplitudes of the slow (▵) and initial (•) hyperpolarizations are shown as a function of the concentration of 4-AP (0 and 10−4–10−3 M). *P < 0.05, compared with control (0 M 4-AP).
These results indicate that the K+ channel involved in the generation of the slow hyperpolarization is inhibited by both glibenclamide and 4-AP. These two K+ channel blockers inhibit different types of K+ channel: glibenclamide inhibits ATP-sensitive K+ channels and 4-AP inhibits voltage-dependent K+ channels (Nelson & Quayle, 1995). Attempts were made to compare the effects of glibenclamide and 4-AP on the hyperpolarization produced by Y-26763, a K+ channel opener (Itoh, Ito, Shafiq & Suzuki, 1994), in the guinea-pig coronary artery. The hyperpolarization produced by 10−7 M Y-26763 (7.3 ± 1.9 mV; n = 7) was inhibited by 10−6 M glibenclamide (0.2 ± 0.2 mV; n = 8; P < 0.01) but not by 5 × 10−4 M 4-AP (7.1 ± 2.1 mV; n = 6; P > 0.1). Thus, the results indicate that the K+ channels activated by Y-26763 (possibly the ATP-sensitive type) are inhibited by glibenclamide but not by 4-AP.
DISCUSSION
The present experiments confirm the observation reported by Parkington et al. (1993) that smooth muscle of the guinea-pig coronary artery responds to ACh with two components of hyperpolarization. These ACh-induced responses were endothelium dependent and were significantly inhibited or converted to a depolarizing response after damage to the endothelial cells. The damage to the endothelial cells was accompanied by a depolarization of the smooth muscle membrane, possibly due to the simultaneous impairment of some of the smooth muscle cells during the procedure. In this artery, the ACh-induced hyperpolarization reportedly consists of three components: an initial hyperpolarization which is generated by EDRF and EDHF and a slow hyperpolarization generated by prostanoids (Parkington et al. 1993). The present experiments confirmed that the slow hyperpolarization is indeed produced by endothelial prostanoids metabolized through activation of cyclooxygenase, since this component of the response was selectively inhibited when the activity of the enzyme was blocked by indomethacin or diclofenac. The component of the ACh-induced initial hyperpolarization produced by EDRF was detected by elevating the perfusion pressure of the vessel (Parkington et al. 1993). The present experiments were carried out using a vessel with no perfusion pressure, and this may account for the absence of any nitroarginine-sensitive component in the ACh-induced hyperpolarization.
The endothelium-dependent hyperpolarization produced after inhibition of the production of prostanoids and NO may be generated mainly by EDHF (Suzuki, Chen, Yamamoto & Miwa, 1993; Garland et al. 1995), that is, ACh hyperpolarizes smooth muscle membrane by both EDHF and prostanoids in the guinea-pig coronary artery. EDHF may be a metabolite of arachidonic acid which is produced through the epoxygenase pathway which involves the enzyme cytochrome P450 mono-oxygenase (Hecker et al. 1994). An involvement of a cytochrome P450-related pathway in the biosynthesis of EDHF is suggested from the inhibition of the endothelium-dependent hyperpolarization by inhibitors of this enzyme such as SKF 525a or clotrimazole (Campbell et al. 1996; Chen & Cheung, 1996). Furthermore, activation of this pathway by β-naphthoflavone enhances the production of EDHF (Popp, Bauersachs, Hecker, Fleming & Busse, 1996). However, this is not the case for all blood vessels. In the guinea-pig carotid artery (Corriu et al. 1996) or rat mesenteric artery (Vanheel & Van de Voorde, 1997), the ACh-induced hyperpolarization is not inhibited by inhibitors of cytochrome P450. Chemicals which inhibit the activity of cytochrome P450 are reported to have additional actions, such as the inhibitory action of SKF 525a on Ca2+ movements in endothelial cells (Graier, Simecek & Sturek, 1995) or the blockade by clotrimazole of K+ channels in vascular smooth muscle (Edwards, Zygmunt, Högestätt & Weston, 1996). In the present experiments, SKF 525a and clotrimazole each inhibited both components of the ACh-induced hyperpolarization, indicating that these chemicals also inhibit the activity of both cyclo-oxygenase and epoxygenase. Alternatively, these chemicals may have non-selective inhibitory actions on the metabolism of arachidonic acid, possibly indirectly through inhibition of Ca2+ movements. Thus, the present experiments could not confirm that EETs are indeed the mediators of the endothelium-dependent hyperpolarization in the guinea-pig coronary artery. NDGA, an inhibitor of lipoxygenase, had no effect on the ACh-induced hyperpolarization, indicating that arachidonic acid metabolites produced through the lipoxygenase pathway may not be EDHF.
The type of K+ channel activated in the ACh-induced hyperpolarization was deduced from the effects of different types of K+ channel inhibitor. CTX was highly selective in inhibiting the initial hyperpolarization, suggesting that a high conductance Ca2+-sensitive K+ channel is involved. In addition to CTX, apamin and high concentrations of Ba2+ and 4-AP were effective in blocking the initial hyperpolarization. Apamin inhibited the initial hyperpolarization only, with no alteration of the slow hyperpolarization, indicating that some of the small conductance Ca2+-sensitive K+ channels are involved in the generation of this potential. Complete inhibition of the ACh-induced hyperpolarization by simultaneous application of apamin and CTX, but not by CTX alone, was also observed in the mesenteric artery of rat (Chen & Cheung, 1997) and the submucosal arterioles of guinea-pig (Hashitani & Suzuki, 1997). The inhibition of the initial hyperpolarization by high concentrations of Ba2+ and 4-AP may be due to the non-selective actions, as suggested in other tissues (Nelson & Quayle, 1995).
The slow hyperpolarization was effectively inhibited by glibenclamide and 4-AP in the guinea-pig coronary artery. Glibenclamide inhibits ATP-sensitive K+ channels while 4-AP inhibits voltage-dependent K+ channels (Nelson & Quayle, 1995), suggesting that these two different types of K+ channel are involved in the generation of the slow hyperpolarization. In the guinea-pig coronary artery, however, the hyperpolarization produced by Y-26763, which activates ATP-sensitive K+ channels (Itoh et al. 1994), could be inhibited by glibenclamide but not by 4-AP, indicating that 4-AP does not have the ability to inhibit ATP-sensitive K+ channels. Some K+ channel inhibitors have actions other than channel inhibition. In particular, in the case of glibenclamide, inhibitory actions on responses produced by several kinds of prostanoids are also known (Cocks, King & Angus, 1990; Zhang, Stockbridge, Weir, Krueger & Cook, 1991; Zhang, Weir, Stockbridge, Doi & Cook, 1992). The slow hyperpolarization may be produced by prostanoids, since this potential was inhibited after blockade of the activity of cyclo-oxygenase. Thus, we speculate that the actions of glibenclamide on the slow hyperpolarization are probably due to inhibition of the actions of prostanoids rather than inhibition of the ATP-sensitive K+-channels, and that this potential is produced mainly by activation of 4-AP-sensitive K+ channels. The K+ channels inhibited by 4-AP in arterial smooth muscles are mainly voltage dependent (Nelson & Quayle, 1995), although in vein, the cromakalim-activated K+ channels are inhibited by 4-AP at very low concentrations (ID50= 0.2 mM; Beech & Bolton, 1989).
Inhibition of both components of the ACh-induced hyperpolarization occurred in the presence of CTX or high concentrations of Ba2+. The release or production of endothelium-derived vasodilators requires an increase in endothelial [Ca2+] (Chen & Suzuki, 1990; Moncada et al. 1991), and such could be brought about by the hyperpolarization due to the absence of any voltage-sensitive Ca2+ channels in the membrane (Adams, 1994). The increase in cytosolic [Ca2+] may hyperpolarize the endothelial membrane by activation of Ca2+-sensitive K+ channels (Busse, Fichtner, Luckhoff & Kohlhardt, 1988; Adams, 1994). CTX is also effective in inhibiting the Ca2+-sensitive K+ channel in endothelial cells and inhibits the hyperpolarization (Rusko, Tanzi, van Breemen & Adams, 1992). Thus, some components of the ACh-induced hyperpolarization inhibited by CTX or Ba2+ may be related to the reduced production of endothelial vasodilators indirectly through inhibition of the endothelial hyperpolarization.
The response of the smooth muscle membrane of the coronary artery to K+ channel inhibitors differed: the membrane was depolarized by CTX, glibenclamide and Ba2+, but not by apamin or 4-AP. These observations suggest that, at rest, current flowing through high conductance Ca2+-sensitive K+ channels, ATP-sensitive K+ channels and inward rectifier K+ channels each contribute to the membrane potential. The small conductance Ca2+-sensitive K+ channels and voltage-dependent K+ channels do not appear to be active. The resting membrane potential was not changed following inhibition of the production of prostanoids by indomethacin or diclofenac, indicating that the depolarization of the membrane by glibenclamide is mainly due to inhibition of ATP-sensitive K+ channels, and not to inhibition of the actions of prostanoids. At rest, the continuous release of EDRF is deduced from the actions of inhibitors of NO synthase (Moncada et al. 1991). Therefore, possible inhibition of the actions of endothelial NO by glibenclamide (Murphy & Brayden, 1995a) or CTX (Robertson et al. 1993; Bolotina et al. 1994) is also considered as a mechanism of depolarization produced by K+ channel inhibitors, although significant depolarization was not elicited by inhibiting the production of NO with nitroarginine in the guinea-pig coronary artery.
We conclude that, in the guinea-pig coronary artery, ACh releases EDHF and prostanoids from the endothelial cells, to produce an initial and then a slow hyperpolarization, respectively. EDHF stimulates CTX-sensitive (high conductance Ca2+-sensitive) K+ channels while prostanoids stimulate 4-AP-sensitive (voltage-dependent) K+ channels. The results obtained could not confirm that any arachidonic acid metabolites were involved in the generation of the initial hyperpolarization.
Acknowledgments
This work was partly supported by the Grant-in-Aid for Scientific research from the Ministry of Culture and Education, Japan, to H. S. (nos 06670122 and 09470011). Y-26763 was a gift from Yoshitomi Pharmaceutical Ind., Ltd (Japan). The authors are grateful to Dr M. Bramich for critical reading of the manuscript.
References
- Adams DJ. Ionic channels in vascular endothelial cells. Trends in Cardiovascular Medicine. 1994;4:18–26. doi: 10.1016/1050-1738(94)90021-3. 10.1016/1050-1738(94)90021-3. [DOI] [PubMed] [Google Scholar]
- Beech DJ, Bolton TB. Properties of the cromakalim-induced potassium conductance in smooth muscle cells isolated from the rabbit portal vein. British Journal of Pharmacology. 1989;98:851–864. doi: 10.1111/j.1476-5381.1989.tb14614.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolotina VM, Najibi S, Palacino JJ, Pagano PJ, Cohen RA. Nitric oxide directly activates calcium-dependent potassium channels in vascular smooth muscle. Nature. 1994;368:850–853. doi: 10.1038/368850a0. [DOI] [PubMed] [Google Scholar]
- Busse R, Fichtner H, Luckhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cells. American Journal of Physiology. 1988;255:H965–969. doi: 10.1152/ajpheart.1988.255.4.H965. [DOI] [PubMed] [Google Scholar]
- Campbell WG, Gebremedhin D, Pratt PF, Harder DR. Identification of epoxyeicosatrienoic acids as endothelium-derived hyperpolarizing factors. Circulation Research. 1996;78:415–423. doi: 10.1161/01.res.78.3.415. [DOI] [PubMed] [Google Scholar]
- Chen G, Cheung DW. Modulation of endothelium-dependent hyperpolarization and relaxation to acetylcholine in rat mesenteric artery by cytochrome P450 enzyme activity. Circulation Research. 1996;79:827–833. doi: 10.1161/01.res.79.4.827. [DOI] [PubMed] [Google Scholar]
- Chen G, Cheung DW. Effect of K+-channel blockers on ACh-induced hyperpolarization and relaxation in mesenteric arteries. American Journal of Physiology. 1997;272:H2306–2312. doi: 10.1152/ajpheart.1997.272.5.H2306. [DOI] [PubMed] [Google Scholar]
- Chen G, Suzuki H. Some electrical properties of the endothelium-dependent hyperpolarization recorded from rat arterial smooth muscle cells. The Journal of Physiology. 1989;410:91–106. doi: 10.1113/jphysiol.1989.sp017522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Suzuki H. Calcium dependency of the endothelium-dependent hyperpolarization in smooth muscle cells of the rabbit carotid artery. The Journal of Physiology. 1990;421:521–534. doi: 10.1113/jphysiol.1990.sp017959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Suzuki H, Weston AH. Acetylcholine releases endothelium derived hyperpolarizing factor and EDRF from rat blood vessels. British Journal of Pharmacology. 1988;95:1165–1174. doi: 10.1111/j.1476-5381.1988.tb11752.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cocks TM, King SJ, Angus JA. Glibenclamide is a competitive antagonist of the thromboxane A2 receptor in dog coronary artery in vitro. British Journal of Pharmacology. 1990;100:375–378. doi: 10.1111/j.1476-5381.1990.tb15812.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corriu C, Félétou M, Canet E, Vanhoutte PM. Inhibitors of the cytochrome P450-mono-oxygenase and endothelium-dependent hyperpolarizations in the guinea-pig isolated carotid artery. British Journal of Pharmacology. 1996;117:607–610. doi: 10.1111/j.1476-5381.1996.tb15233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards G, Zygmunt PM, Högestätt ED, Weston AH. Effects of cytochtome P450 inhibitors on potassium currents and mechanical activity in rat portal vein. British Journal of Pharmacology. 1996;119:691–701. doi: 10.1111/j.1476-5381.1996.tb15728.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eglen RM, Hegde SS, Watson N. Muscarinic receptor subtype and smooth muscle function. Pharmacological Reviews. 1996;48:531–565. [PubMed] [Google Scholar]
- Fitzpatrik FA, Murphy RC. Cytochrome P-450 metabolism of arachidonic acid: formation and biological actions of ‘epoxygenase’-derived eicosanoids. Pharmacological Reviews. 1988;40:229–241. [PubMed] [Google Scholar]
- Fukao M, Hattori Y, Kanno M, Sakuma I, Kitabatake A. Evidence against a role of cytochrome P450-dependent arachidonic acid metabolites in endothelium-dependent hyperpolarization by acetylcholine in rat isolated mesenteric artery. British Journal of Pharmacology. 1997;120:439–446. doi: 10.1038/sj.bjp.0700932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garland CJ, Plane F, Kemp BK, Cocks TM. Endothelium-dependent hyperpolarization: a role in the control of vascular tone. Trends in Pharmacological Sciences. 1995;16:23–30. doi: 10.1016/s0165-6147(00)88969-5. 10.1016/S0165-6147(00)88969-5. [DOI] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of Ca2+ entry in human and bovine endothelial cells. The Journal of Physiology. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gryglewski RJ, Botting RM, Vane JR. Prostacyclin: from discovery to clinical application. In: Rubanyi GM, editor. Cardiovascular Significance of Endothelium-Derived Vasoactive Factors. Mount Kisco, NY, USA: Futura Publishing; 1991. pp. 3–37. [Google Scholar]
- Hashitani H, Suzuki H. K+ channels which contribute to the acetylcholine-induced hyperpolarization in smooth muscle of the guinea-pig submucosal arteriole. The Journal of Physiology. 1997;501:319–329. doi: 10.1111/j.1469-7793.1997.319bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hecker M, Bara A, Bauersachs J, Busse R. Characterization of endothelium-dependent hyperpolarizing factor as a cytochrome P450-derived arachidonic acid metabolite in mammals. The Journal of Physiology. 1994;481:407–414. doi: 10.1113/jphysiol.1994.sp020449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu S, Kim HS. Activation of K+ channels in vascular smooth muscles by cytochrome P450 metabolites of arachidonic acid. European Journal of Pharmacology. 1993;230:215–221. doi: 10.1016/0014-2999(93)90805-r. 10.1016/0014-2999(93)90805-R. [DOI] [PubMed] [Google Scholar]
- Itoh T, Ito S, Shafiq J, Suzuki H. Effects of a newly synthesized K+ channel opener, Y-26763, on noradrenaline-induced Ca2+ mobilization in smooth muscle of the rabbit mesenteric artery. British Journal of Pharmacology. 1994;111:165–172. doi: 10.1111/j.1476-5381.1994.tb14039.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komori K, Suzuki H. Electrical responses of smooth muscle cells during cholinergic vasodilatation in the rabbit saphenous artery. Circulation Research. 1987;61:586–593. doi: 10.1161/01.res.61.4.586. [DOI] [PubMed] [Google Scholar]
- Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: Physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- Murphy ME, Brayden JE. Nitric oxide hyperpolarizes rabbit mesenteric arteries via ATP-sensitive potassium channels. The Journal of Physiology. 1995a;486:47–58. doi: 10.1113/jphysiol.1995.sp020789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy ME, Brayden JE. Apamin-sensitive K+ channels mediate an endothelium-dependent hyperpolarization in rabbit mesenteric arteries. The Journal of Physiology. 1995b;489:723–734. doi: 10.1113/jphysiol.1995.sp021086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagao T, Vanhoutte PM. Hyperpolarization as a mechanism for endothelium-dependent relaxation in the porcine coronary artery. The Journal of Physiology. 1992;445:355–367. doi: 10.1113/jphysiol.1992.sp018928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Quayle JM. Physiological roles and properties of potassium channels in arterial smooth muscle. American Journal of Physiology. 1995;268:C799–822. doi: 10.1152/ajpcell.1995.268.4.C799. [DOI] [PubMed] [Google Scholar]
- Parkington HC, Tare M, Tonta MA, Coleman HA. Stretch revealed three components in the hyperpolarization of guinea-pig coronary artery in response to acetylcholine. The Journal of Physiology. 1993;465:459–476. doi: 10.1113/jphysiol.1993.sp019687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parkington HC, Tonta MA, Coleman HA, Tare M. Role of membrane potential in endothelium-dependent relaxation of guinea-pig coronary arterial smooth muscle. The Journal of Physiology. 1995;484:469–480. doi: 10.1113/jphysiol.1995.sp020679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersson J, Zygmunt PM, Högestätt ED. Characterization of the potassium channels involved in EDHF-mediated hyperpolarization in cerebral arteries. British Journal of Pharmacology. 1997;120:1344–1350. doi: 10.1038/sj.bjp.0701032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp R, Bauersachs J, Hecker M, Fleming I, Busse R. A transferable, β-naphthoflavone-inducible, hyperpolarizing factor is synthesized by native and cultured porcine coronary endothelial cells. The Journal of Physiology. 1996;497:699–709. doi: 10.1113/jphysiol.1996.sp021801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson BE, Schubert J, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca2+-activated K+ channels in cerebral artery smooth muscle cells. American Journal of Physiology. 1993;265:C299–303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- Rusko J, Tanzi F, Van Breemen C, Adams DJ. Calcium-activated potassium channels in native endothelial cells from rabbit aorta: conductance, Ca2+ sensitivity and block. The Journal of Physiology. 1992;455:601–621. doi: 10.1113/jphysiol.1992.sp019318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Standen NB, Quayle JM, Davies NW, Brayden JE, Huang Y, Nelson MT. Hyperpolarizing vasodilators activate ATP-sensitive K+ channels in arterial smooth muscle. Science. 1989;245:177–180. doi: 10.1126/science.2501869. [DOI] [PubMed] [Google Scholar]
- Suzuki H, Chen G. Endothelium-dependent hyperpolarizing factor (EDHF): an endogenous potassium-channel activator. News in Physiological Sciences. 1990;5:212–215. [Google Scholar]
- Suzuki H, Chen G, Yamamoto Y, Miwa K. Nitroarginine-sensitive and insensitive components of the endothelium-dependent relaxation in the guinea-pig carotid artery. Japanese The Journal of Physiology. 1993;42:335–347. doi: 10.2170/jjphysiol.42.335. [DOI] [PubMed] [Google Scholar]
- Tare M, Parkington HC, Coleman HA, Neild TO, Dusting GJ. Hyperpolarization and relaxation of arterial smooth muscle caused by nitric oxide derived from the endothelium. Nature. 1990;346:69–71. doi: 10.1038/346069a0. 10.1038/346069a0. [DOI] [PubMed] [Google Scholar]
- Vanheel B, Van de Voorde J. Evidence against the involvement of cytochrome P450 metabolites in endothelium-dependent hyperpolarization of the rat main pulmonary artery. The Journal of Physiology. 1997;501:331–341. doi: 10.1111/j.1469-7793.1997.331bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Stockbridge N, Weir B, Krueger C, Cook D. Glibenclamide relaxes vascular smooth muscle constriction produced by prostaglandin F2α. European Journal of Pharmacology. 1991;195:27–35. doi: 10.1016/0014-2999(91)90378-4. 10.1016/0014-2999(91)90378-4. [DOI] [PubMed] [Google Scholar]
- Zhang H, Weir B, Stockbridge N, Doi M, Cook D. Glibenclamide inhibits the contractile responses of canine middle cerebral artery to eicosanoids and oxyhemoglobin. Cerebrovascular Diseases. 1992;2:51–57. [Google Scholar]