

Abstract
Missense mutations in the α-subunit of the human skeletal muscle sodium channel (hSkM1) have been detected in some heritable forms of myotonia. By recording Na+ currents from cells transfected with cDNA encoding either wild-type or mutant hSkM1, we characterized the functional consequences of two myotonia-associated mutations that lie at the cytoplasmic end of the sixth transmembrane segment in domain II (S804F) or domain III (V1293I).
Both mutations caused modest, but unequivocal, alterations in the voltage-dependent gating behaviour of hSkM1. For S804F, the abnormalities were limited to fast inactivation: the persistent Na+ current at the end of a 50 ms depolarization was increased 3-fold, the rate of inactivation from the open state was slowed 2-fold, and the voltage dependence of fast inactivation (h∞) was shifted by +3 mV. V1293I also disrupted fast inactivation, as evidenced by a 3-fold faster rate of recovery at hyperpolarized potentials (< = -70 mV). Activation was altered as well for V1293I: the voltage dependence was shifted by -6 mV (hyperpolarized).
Slow inactivation was not altered by S804F or V1293I.
We conclude that S804F and V1293I are not benign polymorphisms. Either mutation causes detectable alterations in channel gating and, in model simulations, the magnitude of the defects is sufficient to produce runs of myotonic discharges.
To date, nineteen distinct point mutations in the α-subunit of the adult human skeletal muscle sodium channel (hSkM1) have been identified in families with paramyotonia congenita (PMC), potassium-aggravated myotonia (PAM), and hyperkalaemic periodic paralysis (HyperPP) (for review, see Cannon, 1997). The functional consequences for many of these missense mutations have been studied by recording Na+ currents from either endogenously expressed channels in cultured myotubes, or from heterologously expressed channels. Mutations that segregate with myotonic phenotypes (PAM and PMC) partially disrupt fast inactivation of the channel (Chahine et al. 1994; Mitrovic et al. 1994, 1995; Yang et al. 1994; Hayward et al. 1996; Richmond et al. 1997). More pronounced defects in fast inactivation are often found for mutations that segregated with HyperPP in which the predominant symptom is episodic paralysis (Cannon et al. 1991, 1993). Disruption of slow inactivation occurs in the two most prevalent HyperPP mutations, T704M and M1592V (Cummins & Sigworth 1996; Hayward et al. 1997). In addition, a hyperpolarizing shift in the voltage dependence of channel activation has also been demonstrated for T704M (Cummins et al. 1993) and G1306E (Mitrovic et al. 1995). Based on these studies and others, it has been postulated that fast inactivation defects alone are sufficient to produce myotonia, while disruptions in slow inactivation increase the likelihood of paralysis (Hayward et al. 1997). One interpretation of these findings is that a positive correlation exists between the degree of aberrant channel behaviour and the severity of the clinical phenotype.
The functional consequences for two novel mutations associated with myotonic syndromes, S804F (McClatchey et al. 1992; Ricker et al. 1994) and V1293I (Koch et al. 1995) have not previously been studied. These missense mutations lie near the cytoplasmic face of sixth transmembrane segment (S6) of SkM1 in domains II (S804F) and III (V1293I). The S804F mutation has been reported in two families with PAM (McClatchey et al. 1992; Ricker et al. 1994). As is typical for PAM, affected individuals have myotonia that is aggravated by elevated serum potassium and responsive to acetazolamide. S804F is associated with a variant of PAM, called myotonia fluctuans, in which the stiffness is less severe than most forms of PAM and cold does not aggravate the myotonia (Ricker et al. 1994). The V1293I mutation has been found in three German families with PMC (Koch et al. 1995). Those affected have myotonia that is aggravated by cold and paradoxically worsens with repeated voluntary contractions. The V1293I variant is phenotypically milder than most forms of PMC, in that, weakness never occurs, even with extreme cooling (immersion in ice water).
We have characterized the functional consequences of the S804F and V1293I mutations by measuring Na+ currents from human embryonic kidney (HEK) cells transiently transfected with wild-type (WT) or mutant hSkM1. Here, we report that S804F and V1293I have mild alterations in both the kinetics of fast inactivation and the steady-state behaviour of the mutant channels. Model simulations demonstrate that these aberrant channel properties are sufficient to induce myotonia, but not paralysis.
METHODS
Mutagenesis and expression of sodium channels
The mutation V1293I was created in hSkM1 using the Altered Sites mutagenesis system (Promega). Briefly, single-strand DNA was rescued from pSELECT-hSkM1/D3 containing an 1817 bp Sph I-Hind III fragment (nucleotides 2235-4051), and used in mutagenesis reactions with a mutagenic primer: 5′-CTCAACCTCTTCATTGGCATCATCATCGATAACTTCAACAGCAGAAG-3′. The S804F mutation was created by a polymerase chain reaction-mediated mutagenesis procedure. Both mutations were subcloned into the mammalian expression plasmid pRc/CMV-hSkM1 and sequenced to verify the mutation and to exclude polymerase errors in the amplified region (S804F only). The human β1-subunit cDNA (McClatchey et al. 1993) was subcloned into the Eco RI site of the GW1-CMV expression vector (British Biotechnology, Oxford, UK).
A human embryonic kidney cell line, HEK 293t, transformed to express the large T antigen (provided by Dr B. Seed, Massachusetts General Hospital, Boston, MA, USA) was used as the expression system. HEK cells were maintained in DMEM media (Gibco) containing 45 g l−1 glucose, 25 mm Hepes, 2 mm L-glutamine, 3 mm taurine, 1 % penicillin-streptomycin (5000 units ml−1 and 5 mg ml−1, respectively) and 10 % fetal bovine serum. HEK cells were transiently cotransfected by the calcium phosphate method (Sambrook et al. 1989) with cDNA encoding either hSkM1 (1-2 μg per 22.6-mm dish) or a mutant α-subunit (1-6 μg), the human sodium channel β1 subunit (4-fold molar excess over the α-subunit DNA), and a reporter plasmid (0.25 μg) encoding the α-subunit of the human CD8 lymphocyte antigen. Transfection-positive cells were identified by attachment of CD8 antibody-coated beads (Dynal, Great Neck, NY, USA).
Whole-cell recording
All experiments were performed at room temperature (20-22°C). Sodium currents were measured in whole-cell mode with an Axopatch 200A amplifier (Axon Instruments). The amplifier output was filtered at 10 kHz and sampled at 50 kHz using a LM900 Laboratory Interface (Dagan Corporation, Minneapolis, MN, USA) controlled by a 486-based computer. A custom Axobasic (Axon Instruments) program was used for data acquisition. On-line leak subtraction was performed using linearly scaled responses to 30 mV depolarizations. Electrodes were constructed from borosilicate capillary tubes (1.65 mm o.d., VWR, West Chester, PA, USA) using a microprocessor-controlled puller (P-87, Sutter Instrument Co.). A coat of Sylgard (Dow Corning) was applied to the electrode shank to reduce capacitance before the tip was fired polished to a final diameter of 0.5-2 μm. The uncompensated series resistance was 0.5-1.0 MΩ, at least 85 % of which was compensated by the analog circuitry.
Several conditions were used to define acceptable Na+ currents. To minimize the contribution of endogenous Na+ currents in HEK cells (typically < 100 pA), current amplitudes were at least 1 nA upon a depolarization from -100 to -10 mV. Peak currents greater than 15 nA were excluded to minimize peak voltage errors due to series resistance. Only currents which displayed monoexponential decays were included to reduce modal-gating artifacts which were observed for both mutant and WT α-subunits.
The external solution contained (mm): 140 NaCl, 4 KCl, 10 Hepes, 2.5 glucose, 2 CaCl2 and 1 MgCl2 (pH 7.4). The internal solution contained (mm): 105 CsF, 35 NaCl, 10 EGTA and 10 Hepes (pH 7.4). Internal fluoride stabilized the seal and prolonged the life-time of the whole-cell recording. In HEK cells, fluoride induced a tetrodotoxin-insensitive outwardly rectifying current. Due to the slow onset (tens of milliseconds) and small peak amplitude (a few hundred picoamps) of this current, our Na+ current measurements were not contaminated, except for attempts to measure the small persistent Na+ current after a 50 ms depolarization. Therefore, we used a fluoride-free internal solution for experiments to assay steady-state currents, which contained (mm): 130 CsCl, 10 NaCl2, 10 Hepes, 5 EGTA and 2 MgCl2 (pH 7.4). To avoid a diminution in the Na+ current amplitude at depolarized potentials, a reversed Na+ gradient was used when measuring the kinetics of fast inactivation (Fig. 1). For the reversed gradient experiments, the external solution contained (mm): 150 choline chloride, 10 Hepes, 2 CaCl2 and 0.2 CdCl2 (pH 7.4); and the internal solution contained (mm): 130 NaCl, 10 Hepes and 10 EGTA (pH 7.4).
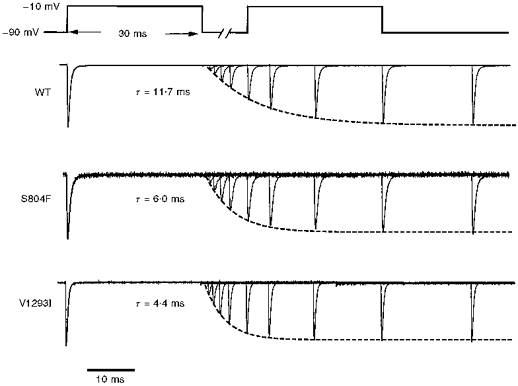
Figure 1. Fast inactivation from the open state was slowed for S804F channels.
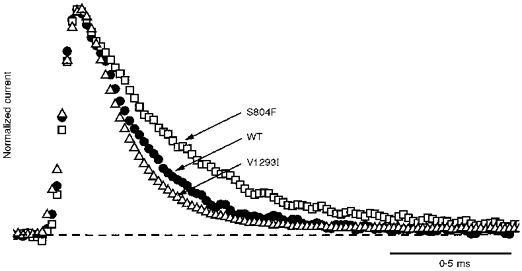
Sodium currents recorded in a reversed Na+ gradient were elicited at +50 mV from a holding potential of -100 mV, normalized by peak amplitude, and superimposed for comparison of macroscopic current fast inactivation. Peak amplitudes were 4.0 nA for WT, 3.8 nA for S804F and 12.0 nA for V1293I. The time constant, τh, was approximately 2-fold slower for S804F (0.42 ms) than for WT (0.24 ms), or V1293I (0.20 ms).
Data analysis
The data were analysed and displayed using a combination of computer programs: Axobasic, Excel (Microsoft Corporation), SigmaPlot (Jandel Scientific) and Origin (MicroCal). For statistical evaluation, the Student's t test was applied, and values for P < 0.05 were considered significant. All statistically derived values are given as means ± standard error of the mean.
The kinetics of fast inactivation were quantified from single exponential fits to the macroscopic current decay, and to the relaxation between closed and inactivated states as revealed by two-pulse protocols. The time constant of the decay, τh, was estimated by fitting the Na+ current, I, to a single exponential plus a constant term, I∞:
where A is the maximal amplitude and t is pulse duration. This same equation was used to estimate the time constant for the onset of fast inactivation from closed states. In this case, I represents the relative peak current elicited after a conditioning pulse of varying duration, t. To measure the time course of recovery, a 30 ms conditioning pulse to -10 mV was applied to fully fast inactivate the channels. The cell was then held at the recovery voltage, ranging from -120 to -70 mV, for a variable interval, and a test pulse to -10 mV was applied to assay the fraction of channels recovered. The relative peak current, I, as a function of recovery interval, t, was approximated by a single exponential using the equation:
where A is the maximal amplitude, td is a delay required to optimize a monoexponential fit, and τ is the time constant of recovery.
The steady-state voltage dependence was measured for fast and for slow inactivation by using either brief (300 ms) or long (60 s) conditioning pulses, respectively. The relative current availability, I/Ipeak, was fitted to a Boltzmann distribution with a non-zero pedestal current, Io, calculated as:
with V½ as the voltage for half-inactivation and k, the slope factor.
Na+ channel activation was characterized by the voltage dependence of the peak Na+ current and the time to half-peak. The peak conductance, G(V), was calculated using the relation:
where the reversal potential, Erev, was determined experimentally for each cell.
RESULTS
Expression of sodium channels was detectable for WT and both mutants within 24-48 h of transfection. The mean peak Na+ current, in response to a step from -100 to -10 mV, was not significantly different for WT (-5.1 ± 1.0 nA, n = 12), S804F (-4.2 ± 0.81 nA, n = 8) or V1293I (-5.3 ± 1.1 nA, n = 9).
Kinetics of fast inactivation
The kinetics of fast inactivation were characterized by measuring the time course and voltage dependence of fast inactivation entry and recovery. When macroscopic Na+ currents are recorded, the best estimate for the rate of inactivation from the open state is obtained by measuring the decay rate of the current elicited at strongly depolarized potentials (Aldrich et al. 1983). Figure 1 shows amplitude-normalized whole-cell Na+ currents for WT, S804F and V1293I recorded at +50 mV from a holding potential of -100 mV. The currents are outward because a reversed sodium gradient was used to prevent Na+ current reversal at depolarized potentials, where the normally small current amplitudes impede fitting of the decay phase. All subsequent experiments employed a conventional high external Na+ solution (see Methods). A 2-fold slower time course of current decay was observed for the S804F mutant compared with WT. At +50 mV, the time course of current decay for V1293I was not distinguishably different from WT. Inactivation time constants, τh, were obtained over the voltage range -30 to +80 mV by fitting the decay phase of the reverse-gradient Na+ current traces with a single exponential. The voltage dependence of τh is shown in Fig. 3 (squares). A defect in fast inactivation for the S804F mutant was discernible at voltages above 0 mV as a 2-fold slower τh. This slowing was also observed when a normal sodium gradient was used (data not shown). Conversely, at potentials greater than 10 mV, V1293I shared a similar time course for fast inactivation entry as WT. For milder depolarizations (-30 to 0 mV), however, the voltage dependence of τh for V1293I was left-shifted in comparison to WT.
Figure 3. S804F and V1293I caused distinct alterations in the kinetics of fast inactivation.
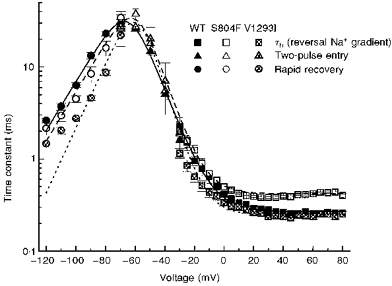
The voltage dependence of the equilibration time constants for fast inactivation is shown by combining data from two-pulse recovery (-120 to -70 mV), two-pulse entry (-70 to -30 mV), and single-pulse relaxation protocols. Smooth curves show the best fits for a two-state model with voltage-dependent forward (βh) and backward (αh) rates from available to fast-inactivated states, where 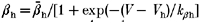 and
and 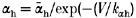 . Parameter values for WT channels were:
. Parameter values for WT channels were: 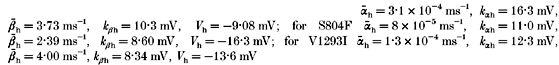
Two-pulse voltage protocols were used to measure the kinetics of recovery from fast inactivation and the kinetics of fast inactivation from closed states. Figure 2 shows typical recovery data at -90 mV, following a 30 ms conditioning pulse at -10 mV, for WT, S804F and V1293I channels. The recovery of the peak Na+ current was fitted well by a single exponential (dashed curves, Fig. 2). At -90 mV, recovery from fast inactivation was faster for the mutant channels than for WT (WT: 13.3 ± 1.2 ms, n = 12; S804F: 8.4 ± 1.6 ms, n = 5; V1293I: 4.6 ± 0.3 ms n = 6). The voltage dependence of the time constant for recovery from fast inactivation is shown in Fig. 3 (circles). Over the voltage range studied, -120 to -70 mV, recovery of the peak current was 3-fold faster for V1293I than for WT (n = 4-9, P < 0.005 for each voltage). Recovery for S804F was approximately 1.4 times faster than WT, from -100 to -80 mV. This small difference was statistically significant (-100 mV: n = 9, P < 0.003; -90 mV: n = 5 P < 0.03; -80 mV: n = 5, P < 0.02).
Figure 2. Recovery from fast inactivation was accelerated for both V1293I and S804F.
A two-pulse voltage protocol using a 30 ms conditioning pulse to -10 mV followed by a variable recovery interval at -90 mV was used to measure recovery from fast inactivation. V1293I currents recovered 3-fold faster than WT. S804F channels recovered 1.4-fold faster than WT. Current traces have been normalized in amplitude. Peak currents were -12.8 nA for WT, -9.4 nA for S804F and -13.3 nA for V1293I.
The kinetics of fast inactivation from closed states were measured over an intermediate range of voltages, -70 to -30 mV, by using a two-pulse protocol in which the conditioning pulse duration was varied from 0.1 to 80 ms. Peak Na+ current, as a function of conditioning pulse duration, was fitted by a single exponential, and the time constants are plotted versus voltage in Fig. 3 (triangles). The voltage dependence of the time constant for equilibration between closed and fast-inactivated states was not appreciably different for WT and either S804F or V1293I.
Steady-state fast inactivation
The voltage dependence of steady-state fast inactivation, h∞, was measured as the peak current elicited following 300 ms conditioning prepulses. Figure 4A (left) shows that the h∞ curve for each mutant was marginally right-shifted relative to WT. For S804F, the 3 mV depolarizing shift in the steady-state availability curve was significant when compared with WT (P < 0.05). There was no discernible difference in the slope parameter, k, between WT and S804F. While the h∞ curve for V1293I appeared shifted to the right of the WT curve, it was not significantly different in either the half-maximal voltage or slope when compared with WT. The average values for half-maximal voltage, V½, and slope, k, used for the Boltzmann fits in Fig. 4 are listed in Table 1.
Figure 4. Voltage dependence of fast inactivation and activation.
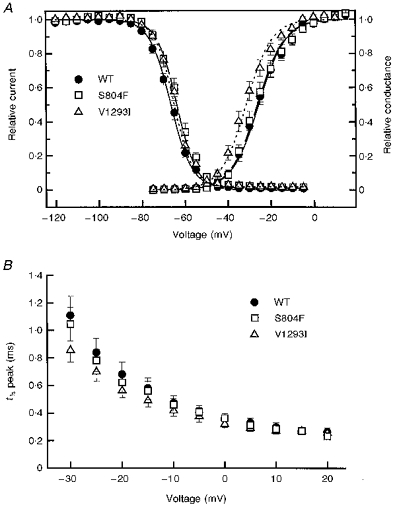
A, channel availability, the fraction not fast-inactivated, was measured after application of a 300 ms conditioning pulse. Fits of the availability data to a Boltzmann distribution (see Methods) showed a consistent, although statistically significant for only S804F (P < 0.05), depolarized shift: WT (n = 15): V½ = -66.7 ± 0.8 mV, k = 4.9 ± 0.2 mV; S804F (n = 9): V½ = -63.8 ± 1.2 mV, k = 4.6 ± 0.2 mV; V1293I (n = 11): V½ = -65.2 ± 0.6 mV, k = 4.3 ± 0.2 mV. The voltage dependence of activation was determined by applying a series of depolarizing steps from a holding potential of -100 mV. The voltage dependence of peak conductance G(V) was similar for S804F and WT: WT (n = 12): V½ = -25.7 ± 1.4 mV, k = 6.3 ± 0.3 mV and S804F (n = 8) V½ = -26.2 ± 1.6 mV, k = 6.3 ± 0.5 mV, but shifted in the hyperpolarized direction for V1293I (n = 9) V½ = -31.5 ± 1.6 mV, k = 6.0 ± 0.3 mV. B, the voltage dependence for the time to half-peak Na+ current was left-shifted for V1293I compared with WT and S804F.
Table 1.
Estimates for sodium current gating parameters at 20–22 °C
| Mutation | Activation, G(V) | Fast inactivation, h∞(V) | Slow inactivation, S∞(V) | ||||
|---|---|---|---|---|---|---|---|
| V½ (mV) | k (mV/e-fold) | V½ (mV) | k (mV/e-fold) | V½ (mV) | k (mV/e-fold) | Io | |
| WT | −25.7 ± 1.4 (12) | 6.3 ± 0.3 | −66.7 ± 0.8 (15) | 4.9 ± 0.2 | −63.3 ± 2.4 (5) | 13.8 ± 0.7 | 0.050 ± 0.002 |
| S804F | −26.2 ± 1.6 (8) | 6.3 ± 0.5 | −63.8 ± 1.2 (9)* | 4.6 ± 0.2 | −66.4 ± 2.5 (5) | 13.5 ± 0.3 | 0.058 ± 0.020 |
| V1293I | −31.5 ± 1.6 (9)* | 6.0 ± 0.3 | −65.2 ± 0.6 (11) | 4.3 ± 0.2 | −65.2 ± 1.4 (7) | 10.7 ± 0.7* | 0.078 ± 0.011 |
Values are means ±s.e.m., with number of experiments in parentheses.
Significant difference compared with WT (P < 0.05).
Activation
Sodium channel activation was characterized by measuring the voltage dependence for the time to half-peak and for the peak Na+ conductance. Peak conductance as a function of voltage, G(V), is shown in Fig. 4A (right) for each of the channels studied. Fits to these data by a Boltzmann distribution (smooth curves) are summarized in Table 1. There was a -6 mV (hyperpolarizing) shift in the peak conductance curve for V1293I (P < 0.025), whereas S804F was indistinguishable from WT. Figure 4A shows that the -6 mV shift in the G(V) curve for V1293I causes a substantial overlap, near -50 mV, where channels are activated appreciably and channel availability (h∞) is detectably greater than zero. This overlap predicts an anomalous persistent Na+ current for V1293I channels over the voltage range of -55 to -45 mV.
The relationship between Na+ current time to half-peak and voltage is shown in Fig. 4B. For test potentials from -30 to +20 mV, the V1293I time to half-peak is left-shifted (hyperpolarized) relative to WT or S804F. This finding supports the G(V) data, indicating that the V1293I mutation affected activation, whereas activation in the S804F mutation was unaltered.
For both S804F and V1293I, the reversal potential for the sodium conductance was not significantly different from WT (Table 1). Therefore, ion selectivity for Na+ over Cs+ was unaffected by either mutation.
Persistent Na+ current
The ratio of the steady-state current (Iss) during the last 5 ms of a 50 ms depolarization to the peak current (Ipeak), was measured over the voltage range of -20 to 0 mV for each of the channel types. This ratio gives an estimate of the fraction of channels which fail to fast inactivate at these voltages. The relative persistent Na+ current for S804F was significantly larger than WT (n = 8, P < 0.03). This persistent current was 0.47 ± 0.11 % for WT (n = 7), 1.5 ± 0.35 % for S804F (n = 8), and 0.75 ± 0.20 % for V1293I (n = 4). The 3-fold increase for S804F was measurable only when using chloride-based external solutions. With internal fluoride solutions, a tetrodotoxin-insensitive, outwardly rectifying current was often observed that activated over tens of milliseconds for depolarizations > 0 mV, which obscured the measurement of steady-state Na+ currents. The persistent current occurred at depolarized potentials, and therefore seems unlikely to be produced by a window current (i.e. the persistent current at -55 to -45 mV where the G(V) and h∞ curves have a non-zero overlap).
Slow inactivation
Steady-state slow inactivation, S∞, was assayed by a two-pulse protocol which employed a 60 s conditioning prepulse followed by a 20 ms step back to -100 mV, to allow recovery of fast, but not slow, inactivated channels (Hayward et al. 1997). The voltage dependence of the fraction of slow-inactivated channels was similar for WT, S804F and V1293I (Fig. 5). There was no significant difference in the half-maximal voltages or plateau value at +20 mV (Table 1). These data also show that even with prolonged depolarization lasting 60 s approximately 10 % of channels remain available. Thus slow inactivation is not capable of completely suppressing a persistent Na+ current arising from a defect in fast inactivation.
Figure 5. The voltage dependence of slow inactivation was similar for WT, V1293I and S804F.
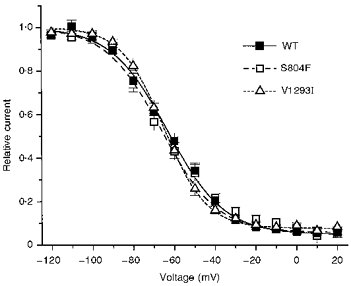
The steady-state availability for slow inactivation was measured after application of a 60 s conditioning pulse, as described in the text. Fits of the availability data to a Boltzmann distribution (I/Ipeak = (1 - Io/)/(1 + exp((V -V½)/k)) + Io) provided estimates of the voltage for half-inactivation, V½, steepness of the voltage-dependence, k, and pedestal current, Io: WT (n = 5): V½ = -63.3 ± 2.4 mV, k = 13.8 ± 0.7 mV, Io = 0.050 ± 0.002; S804F (n = 5): V½ = -66.4 ± 2.5 mV, k = 13.5 ± 0.3 mV, Io = 0.058 ± 0.020; V1293I (n = 7): V½ = -65.2 ± 1.4 mV, k = 10.7 ± 0.7 mV, Io = 0.078 ± 0.011.
DISCUSSION
The primary finding of these heterologous expression studies is that both S804F and V1293I cause detectable alterations in the voltage-dependent gating behaviour of hSkM1. This result supports the notion that these missense mutations are not benign polymorphisms. The alterations in fast inactivation and activation were modest, as is probably necessary for survival of affected individuals, but they were also unequivocal. Three lines of evidence suggest these gating defects are likely to be the direct cause of myotonia. First, similar defects have been reported for eleven other PMC/PAM-associated mutations in hSkM1 (reviewed in Cannon, 1997) which has led to the widely accepted consensus that mild disruption of fast inactivation or hyperpolarized shifts in activation give rise to myotonia. Second, heterologous expression studies are able to discriminate between normal and even mildly anomalous channel behaviour. Motivated by the failure to detect an abnormality in channel gating, we recently showed that a proposed mutation causing hyperkalaemic periodic paralysis and cardiac dysrhythmia, V781I, is actually a benign polymorphism (Green et al. 1997). Because the clinical symptoms are mild for patients carrying S804F or V1293I, compared with the myotonia produced by other missense mutations in hSkM1, it is noteworthy that channel behaviour was unambiguously altered by these mutations. Third, model simulations of muscle excitability (Cannon et al. 1993) have demonstrated that these modest functional defects are sufficient to produce the repetitive discharges of myotonia (see below).
Structure-function implications
The gating defects caused by S804F and V1293I are consistent with contemporary models of Na+ channel structure-function and extend our knowledge to new regions of the channel. Both mutations are predicted to lie near the cytoplasmic end of S6, in domain II for S804F and domain III for V1293I (George et al. 1992). These sites may reside near the intracellular mouth of the ion conducting pore, and consequently could affect fast inactivation mediated by docking of the interdomain III-IV loop (West et al. 1992) or any component of activation that involved rearrangement of the cytoplasmic face of the pore.
S804F is the less conservative of the two mutations, causing replacement of a small polar side chain by a hydrophobic aromatic ring. This mutation caused a mild destabilization of fast inactivation as evidenced by a 2-fold slowing of τh for voltages greater than +20 mV, a 3 mV depolarized shift in h∞, and a 3-fold increase in the persistent Na+ current at 50 ms. Similar effects have been reported for missense mutations at the cytoplasmic end of IVS6, M1592V (Cannon & Strittmatter 1993) and V1589M (Mitrovic et al. 1994). The short S4-S5 loop may play a similar role in forming the inner vestibule of the pore and a docking site for the III-IV loop. Mutations in IIS4-S5: T704M (Cannon & Strittmatter 1993; Yang et al. 1994); IIIS4-S5: I1160V (Richmond et al. 1997); and IVS4-S5: F1473S (Mitrovic et al. 1996) have all disrupted fast inactivation.
The V1293I mutation is biochemically a conservative substitution that increases the length of an aliphatic side chain by a methyl group. Fast inactivation was just barely perturbed. Recovery near -100 mV was accelerated 3-fold. The voltage dependence of activation was more dramatically altered, with a -6 mV (hyperpolarized) shift. Evidence for this shift was consistently found in three separate measures: the G(V) relation, τh(V) over the -35 to 0 mV range, and the time to half-peak. This is the third example of a mutation in hSkM1 associated with myotonia or periodic paralysis in which activation is altered, the other two being T704M (Cummins et al. 1993; Yang et al. 1994) and G1306E (Mitrovic et al. 1995; Hayward et al. 1996). Thus the original view promoted by us and others, that primarily defects of channel inactivation are responsible for the development of myotonia or paralysis, must be modified to include activation as well.
Neither S804F nor V1293I altered the voltage dependence of slow inactivation. Defects of slow inactivation cause a predilection for attacks of paralysis (Cummins & Sigworth 1996; Hayward et al. 1997) which does not occur in association with either S804F or V1293I. A coherent picture of which regions of the Na+ channel are critical for slow inactivation is not available. Missense mutations at the cytoplasmic ends of IIS5 (T704M) and IVS6 (M1592V) disrupt slow inactivation, whereas a mutation in IS6 (N434A in rat SkM1) enhances slow inactivation (Wang & Wang 1997). The two mutations studied herein provide pertinent data for further localization of slow inactivation.
Model predictions of altered excitability: the functional consequences of S804F and V1293I
To test whether the observed alterations in gating for S804F and V1293I are sufficient to cause myotonia, we simulated these abnormalities in channel behaviour in our computer-based model of muscle excitability. The model consists of two electrically coupled compartments, to simulate the sarcolemmal and T-tubular membranes (Cannon et al. 1993; Hayward et al. 1996). Defects in gating for S804F or V1293I channels were simulated by adjusting three features in the model. To simulate the persistent current, fast inactivation was completely disabled for a small fraction, f, of the Na+ conductance. The steady-state availability and kinetics of fast inactivation were altered by shifting the midpoint of the h∞ relation, and by altering the voltage dependence of τh(V). Finally, the midpoint of the activation state variable, m, was shifted to simulate the G(V) data. Slow inactivation was simulated by incorporating a new state variable, s(V, t), analogous to h(V, t) for fast inactivation (Hayward et al. 1997). The modified expression used to compute the Na+ current was:
Computed responses to injection of depolarizing current were used to characterize the excitability of the simulated muscle fibre. Figure 6 shows that with wild-type parameter values, a 100 ms current pulse results in a single action potential, and the membrane repolarizes at the end of the stimulus. To simulate the heterozygous state of diseased muscle, 50 % of the Na+ conductance retained wild-type characteristics, and the remainder was modelled with modified gating parameters.
Figure 6. Simulated responses to current injection for muscle containing wild-type or a mixture of wild-type and mutant Na+ channels.
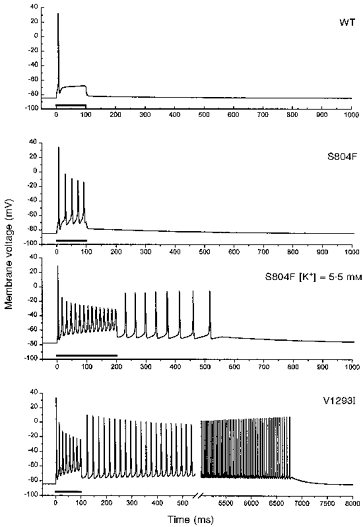
The S804F defect enhances the excitability of the simulated cell, but does not produce self-sustained myotonic discharges unless the extracellular [K+] is raised to 5.5 mm. The gating alterations for V1293I, especially the -6 mV shift in activation, cause a sustained 6.5 s burst of myotonic discharges. Parameter values for wild-type channels were as defined in Hayward and colleagues (1996). Gating defects were simulated by the Boltzmann fits to h∞(V) shown in Fig. 4, approximating the kinetics of fast inactivation by 1/(αh+βh) as in Fig. 3, and shifting the voltage dependence of activation m(V, t). Bars below traces indicate the application of a 25 mA cm−2 current injection.
The S804F defects were incorporated by shifting h∞(V) by +3 mV (cf. Fig. 4), removing fast inactivation for a small fraction (f = 0.015) of the conductance, and fitting the kinetics of fast inactivation with modified parameters for the αh(V, t) and βh(V, t) rate relations, as shown by the smooth curves in Fig. 3. With these modest changes alone, the simulated fibre had enhanced excitability, as evidenced by the repetitive discharges during the 100 ms current pulse (Fig. 6). However, the model did not produce self-sustained trains of action potentials, as occurs during myotonic runs. These after-discharges are triggered by activity-dependent accumulation of K+ in the T-tubular space (Adrian & Bryant, 1974; Cannon et al. 1993). At the end of the 100 ms stimulus, the T-tubular [K+] had risen to 6.6 mm. Simulations with a concentration jump in T-tubular [K+] showed that 10.2 mm was required to elicit after-discharges. As a provocative manoeuvre, the extracellular [K+] (bulk and equilibrated T-tubular) was increased from 4.0 to 5.5 mm and the stimulus interval was increased to 200 ms. These changes were sufficient to cause myotonic after-discharges in a simulated fibre with 50 % S804F-type channels (Fig. 6). For the simulated fibre, the raised extracellular [K+] depolarized the resting potential from -84.6 to -77.9 mV, reduced the action potential threshold for T-tubular [K+] to 9 mm, and resulted in a T-tubular [K+] of 10.2 mm by the end of the 200 ms stimulus. This dependence on raised extracellular [K+] is reminiscent of the exacerbation of clinical myotonia in PAM by K+ loading. For a model cell composed of wild-type Na+ channels, these alterations in external [K+] and stimulus duration did not change the excitability - only a single action potential was elicited.
The gating changes observed for V1293I channels were simulated by shifting the voltage dependence of activation by -6 mV (cf. Fig. 4), fitting the kinetics of fast inactivation with modified αh(V, t) and βh(V, t) rate relations (cf. Fig. 3), shifting h∞(V) rightward by 1.5 mV, and removing fast inactivation for f = 0.007. In the simulated fibre, the V1293I defects caused pronounced myotonia wherein a single 100 ms current pulse elicited a train of discharges that lasted for 6.5 s (Fig. 6). The threshold T-tubular [K+] to elicit after-discharges was much lower when the altered fraction of the Na+ conductance behaved like V1293I (5.5 mm) rather than like S804F (10.2 mm). By varying each of the parameters for V1293I independently, we conclude that, of all the gating changes, the -6 mV shift in activation has the strongest influence on the development of myotonic discharges.
The gating defects for V1293I and S804F did not result in a stable, anomalously depolarized resting potential, regardless of variations in the current stimulus or the value of extracellular or T-tubular [K+], within the physiological range. Thus neither mutation caused gating defects sufficient to produce the model equivalent of depolarization-induced flaccid paralysis.
Acknowledgments
We thank Adriana Pechanova for assistance with tissue culture and plasmid preparation. This work was supported by the NIH (GM15605 to D. S. G., RO1-NS32387 to A. L. G., RO1-42703 to S. C. C.), the Freudenberger Fund (D. S. G.), and the Klingenstein Foundation (S. C. C.). A. L. G. is a Lucille P. Markey Scholar.
References
- Adrian RH, Bryant SH. On the repetitive discharge in myotonic muscle fibres. The Journal of Physiology. 1974;235:103–131. doi: 10.1113/jphysiol.1974.sp010620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aldrich RW, Corey DP, Stevens CF. A reinterpretation of mammalian sodium channel gating based on single channel recording. Nature. 1983;306:436–441. doi: 10.1038/306436a0. [DOI] [PubMed] [Google Scholar]
- Cannon SC. From mutation to myotonia in sodium channel disorders. Neuromuscular Disorders. 1997;7:241–249. doi: 10.1016/s0960-8966(97)00430-6. [DOI] [PubMed] [Google Scholar]
- Cannon SC, Brown RH, Jr, Corey DP. A sodium channel defect in hyperkalemic periodic paralysis: potassium-induced failure of inactivation. Neuron. 1991;6:619–626. doi: 10.1016/0896-6273(91)90064-7. [DOI] [PubMed] [Google Scholar]
- Cannon SC, Brown RH, Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophysical Journal. 1993;65:270–288. doi: 10.1016/S0006-3495(93)81045-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannon SC, Strittmatter SM. Functional expression of sodium channel mutations identified in families with periodic paralysis. Neuron. 1993;10:317–326. doi: 10.1016/0896-6273(93)90321-h. [DOI] [PubMed] [Google Scholar]
- Chahine M, George AL, Jr, Zhou M, Ji S, Sun W, Barchi RL, Horn R. Sodium channel mutations in paramyotonia congenita uncouple inactivation from activation. Neuron. 1994;12:281–294. doi: 10.1016/0896-6273(94)90271-2. [DOI] [PubMed] [Google Scholar]
- Cummins TR, Sigworth FJ. Impaired slow inactivation in mutant sodium channels. Biophysical Journal. 1996;71:227–236. doi: 10.1016/S0006-3495(96)79219-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummins TR, Zhou J, Sigworth FJ, Ukomadu C, Stephan M, Ptacek LJ, Agnew WS. Functional consequences of a Na+ channel mutation causing hyperkalemic periodic paralysis. Neuron. 1993;10:667–678. doi: 10.1016/0896-6273(93)90168-q. [DOI] [PubMed] [Google Scholar]
- George AL, Jr, Komisarof J, Kallen RG, Barchi RL. Primary structure of the adult human skeletal muscle voltage-dependent sodium channel. Annals of Neurology. 1992;31:131–137. doi: 10.1002/ana.410310203. [DOI] [PubMed] [Google Scholar]
- Green DS, Hayward LJ, George AL, Cannon SC. A proposed mutation, Val781Ile, associated with hyperkalemic periodic paralysis and cardiac dysrhythmia is a benign polymorphism. Annals of Neurology. 1997;42:253–256. doi: 10.1002/ana.410420219. [DOI] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Inactivation defects caused by myotonia-associated mutations in the sodium channel III-IV linker. Journal of General Physiology. 1996;107:559–576. doi: 10.1085/jgp.107.5.559. 10.1085/jgp.107.5.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward LJ, Brown RH, Jr, Cannon SC. Slow inactivation differs among mutant Na channels associated with myotonia and periodic paralysis. Biophysical Journal. 1997;72:1204–1219. doi: 10.1016/S0006-3495(97)78768-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MC, Baumbach K, George AL, Ricker K. Paramyotonia congenita without paralysis on exposure to cold: a novel mutation in the SCN4A gene (Val1293Ile) NeuroReport. 1995;6:2001–2004. doi: 10.1097/00001756-199510010-00012. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, Cannon SC, Slaugenhaupt SA, Gusella JF. The cloning and expression of a sodium channel β1-subunit cDNA from human brain. Human Molecular Genetics. 1993;2:745–749. doi: 10.1093/hmg/2.6.745. [DOI] [PubMed] [Google Scholar]
- McClatchey AI, McKenna-Yasek D, Cros D, Worthen HG, Kuncl RW, DeSilva SM, Cornblath DR, Gusella JF, Brown RH., Jr Novel mutations in families with unusual and variable disorders of the skeletal muscle sodium channel. Nature Genetics. 1992;2:148–152. doi: 10.1038/ng1092-148. 10.1038/ng1092-148. [DOI] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Heine R, Wagner S, Pika U, Hartlaub U, Zhou M, Lerche H, Fahlke C, Lehmann-Horn F. K(+)-aggravated myotonia: destabilization of the inactivated state of the human muscle Na+ channel by the V1589M mutation. The Journal of Physiology. 1994;478:395–402. doi: 10.1113/jphysiol.1994.sp020260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, George AL, Jr, Lerche H, Wagner S, Fahlke C, Lehmann-Horn F. Different effects on gating of three myotonia-causing mutations in the inactivation gate of the human muscle sodium channel. The Journal of Physiology. 1995;487:107–114. doi: 10.1113/jphysiol.1995.sp020864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitrovic N, Lerche H, Heine R, Fleischhauer R, Pika-Hartlaub U, Hartlaub U, George AL, Lehmann-Horn F. Role in fast inactivation of conserved amino acids in the IV/S4-S5 loop of the human muscle Na+ channel. Neuroscience Letters. 1996;214:9–12. doi: 10.1016/0304-3940(96)12866-4. [DOI] [PubMed] [Google Scholar]
- Richmond JE, Featherstone DE, Ruben PC. Human Na+ channel fast and slow inactivation in paramyotonia congenita mutants expressed in Xenopus laevis. The Journal of Physiology. 1997;499:589–600. doi: 10.1113/jphysiol.1997.sp021952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricker K, III Moxley RT, Heine R, Lehmann-Horn F. Myotonia fluctuans. A third type of muscle sodium channel disease. Archives of Neurology. 1994;51:1095–1102. doi: 10.1001/archneur.1994.00540230033009. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T. Molecular Cloning: A Laboratory Manual. 2. Cold Spring Harbour, NY, USA: Cold Spring Harbour Laboratory; 1989. [Google Scholar]
- Wang S, Wang GK. A mutation in segment I-S6 alters slow inactivation of sodium channels. Biophysical Journal. 1997;72:1633–1640. doi: 10.1016/S0006-3495(97)78809-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West JW, Patton DE, Scheuer T, Wang Y, Goldin AL, Catterall WA. A cluster of hydrophobic amino acid residues required for fast Na+-channel inactivation. Proceedings of the National Academy of Sciences of the USA. 1992;89:10910–10914. doi: 10.1073/pnas.89.22.10910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N, Ji S, Zhou M, Ptacek LJ, Barchi RL, Horn R, George AL., Jr Sodium channel mutations in paramyotonia congenita exhibit similar biophysical phenotypes in vitro. Proceedings of the National Academy of Sciences of the USA. 1994;91:12785–12789. doi: 10.1073/pnas.91.26.12785. [DOI] [PMC free article] [PubMed] [Google Scholar]