

Abstract
Whole-cell patch-clamp recordings from freshly dissociated rat CA1 neurons revealed a large transient Na+ current (INa,T) and a smaller, inactivation-resistant persistent Na+ current (INa,P). Both currents could be blocked with TTX.
The average current densities of INa,T and INa,P in thirty cells were 111.0 ± 9.62 and 0.87 ± 0.13 pA pF−1, respectively.
Inhibiting oxidative phosphorylation by adding 5 mm sodium cyanide to the pipette solution significantly increased the amplitude of INa,P but had no significant effect on the amplitude of INa,T.
Exposing CA1 neurons to hypoxia for more than 7 min caused an increase in the amplitude of INa,P. There was also a delayed decrease in the amplitude of INa,T.
INa,P was more sensitive to the Na+ channel blockers TTX and lidocaine than INa,T. The IC50 for the effect of TTX on INa,P was 9.1 ± 1.2 nM whereas the IC50 for INa,T was 37.1 ± 1.2 nM, approximately 4-fold higher. Lidocaine (lignocaine; 1 μm) reduced INa,P to 0.24 ± 0.15 of control (n = 4) whereas INa,T was essentially unaffected (0.99 ± 0.11, n = 4).
These results show that INa,P is increased when oxidative metabolism is blocked in CA1 neurons. The persistent influx of Na+ through non-inactivating Na+ channels can be blocked by concentrations of Na+ channel blockers that do not affect INa,T.
Reduced oxygen supply to the brain results in a loss of ion homeostasis and energy depletion that leads to cell death. An early effect of hypoxia in neurons is an increase in intracellular sodium concentration ([Na+]i) which is followed by an elevation in intracellular calcium concentration ([Ca2+]i) (Friedman & Haddad, 1994a, b). In animal models of ischaemia, a low concentration of extracellular sodium ([Na+]o) and low concentrations of Na+ channel blockers (which do not affect normal action potential firing) have been shown to prevent the rise in [Ca2+]i and extracellular glutamate concentration, the decrease in [ATP]i and neuronal damage (Weber & Taylor, 1994; Fried, Amorim, Chambers, Cottrell & Kass, 1995; Taylor, Burke & Weber, 1995). It has been suggested, therefore, that an early increase in [Na+]i activates Na+-Ca2+ and glutamate exchangers in neurons in reverse mode leading to a rise in [Ca2+]i and glutamate release from cells (Stys, Waxman & Ransom, 1991; Taylor et al. 1995). The reason for the increase in [Na+]i has not been explained. Because the Na+ influx and depolarization recorded during hypoxia are sustained (Taylor, 1995), it has been thought that the classical transient Na+ channel current (INa,T) could not be responsible. However, a persistent, slowly inactivating, TTX-sensitive Na+ current has been recorded in neurons (Gilly & Armstrong, 1984; French & Gage, 1985; French, Sah, Buckett & Gage, 1990; Taylor, 1993; Crill, 1996) and it has been shown that hypoxia can cause an increase in a persistent sodium current (INa,P) in cardiac muscle (Ju, Saint & Gage, 1996). In normal healthy cells INa,P has been thought to function as a pacemaker current, contributing to synaptic potentials, enhancing rhythmicity and enabling repetitive firing of action potentials (Taylor, 1993). The aim of this study was to investigate the effect of inhibiting oxidative metabolism on INa,P in CA1 neurons and to assess the sensitivity of the current potentiated by hypoxia to TTX and lidocaine. We report here that inhibition of oxidative metabolism increases the persistent Na+ current in hippocampal neurons and propose that this is an early event during hypoxia that leads to Ca2+ accumulation and consequent damage to neurons. Some of these results have been presented previously in abstract form (Hammarström, Lim & Gage, 1996).
METHODS
Dissociation of CA1 neurons
Young adult rats (14-21 days old) were decapitated with a small animal guillotine (anaesthesia under CO2) and the brain quickly transferred to ice-cold artificial cerebrospinal fluid (ACSF) (124 mm NaCl, 26 mm NaH2CO3, 3 mm KCl, 1.3 mm MgSO4, 2.5 mm NaH2PO4 and 20 mm glucose) gassed with 95 % CO2-5 % O2 (BOC gases, Australia). Brain slices 500 μm thick were cut on a vibratome, in the presence of ice-cold ACSF equilibrated with 95 % CO2-5 % O2. The brain slices were enzymatically treated for 30 min with 200 units papain (Worthington Biochemicals), 1.1 mm cysteine (Sigma), 0.2 mm EDTA and 13.4 μm mercaptoethanol at 35°C, following the adaptation (French et al. 1990) of the procedure originally described by Kay & Wong, 1987. The CA1 region was subsequently removed and individual neurons obtained by careful mechanical trituration with a glass pipette. These methods have been approved by the Animal Experimentation Ethics Committee at the Australian National University (Ethics Approval No. J.NS.82.98).
Electrophysiological recording and data analysis
Tight gigaohm seal patch-clamp techniques using standard glass pipettes (GC150F-15, Clarke Electromedical Instruments) were used to record currents in whole-cell configuration from CA1 neurons. Neurons that were flat, swollen or grainy in appearance were not used. Pipettes were pulled on a List Medical vertical pipette puller (L/M-3P-A) and had resistances of 6-10 MΩ when filled with pipette solution. In order to obtain reproducible results, currents were not recorded for the first 10-15 min after achieving the whole-cell configuration.
Tetrodotoxin (TTX) (Boehringer Mannheim) and lidocaine (lignocaine; Sigma) were applied to whole cells through fine (200 μm i.d.) tubes carefully positioned to cause a rapid change in concentration of drugs close to the patched cell. Hypoxia was achieved by perfusing the patched cell through a fine stainless steel tube with a solution vigorously bubbled with 100 % N2 (BOC gases). This technique has already been used in our laboratory and produces a rapid decrease in oxygen tension around the cell (Ju et al. 1996).
The bath solution contained (mm): 135 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 5 CoCl2, 5 CsCl and 10 Tes adjusted to a pH of 7.4 with NaOH. The pipette solution contained (mm): 125 CsF, 5 NaF, 10 KCl, 10 Tes and 10 EGTA, pH 7.4. In experiments in which 5 mm sodium cyanide (BDH Chemicals, Australia) was added to the pipette solution the concentration of CsF in the pipette solution was reduced to 120 mm. In most instances 10 mm glucose was also included in the bath solution but its absence or presence did not appear to affect the Na+ currents (data not shown). Stocks of TTX and lidocaine were diluted in bath solution to the required concentration immediately prior to the experiment. All solutions were of similar osmolality (290 ± 10 mosmol kg−1).
Whole-cell currents were recorded with an Axopatch-1D amplifier (Axon Instruments) in response to voltage protocols imposed using an IBM-compatible PC and digital-to-analog interface. Currents were analysed using computer techniques and ‘in-house’ software (Ju, Saint & Gage, 1992; Ju et al. 1996). Subtraction of traces recorded before and after exposure to a high concentration of TTX (0.5 μm) was used throughout to isolate TTX-sensitive Na+ currents (Fig. 1).
Figure 1. Recording TTX-sensitive current.
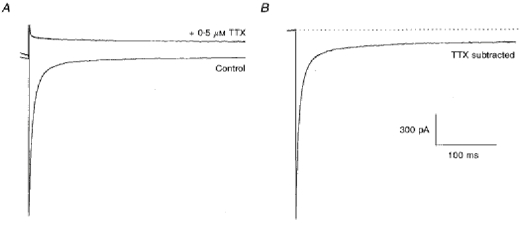
Currents shown were recorded with an electrode containing 5 mm sodium cyanide 15 min after the whole-cell seal was obtained. A, the two traces show currents recorded before (lower trace) and after exposure of the neuron to 0.5 μm TTX. The currents were generated by a voltage step to -30 mV from a holding potential of -100 mV. B, the TTX-sensitive current obtained by subtracting the two traces in A. The transient current is truncated to show the persistent current in more detail. The scale bars apply for both A and B. The dashed line denotes the subtracted current level before the depolarizing pulse (zero current).
The amplitude of INa,T was measured at its peak and the amplitude of INa,P was measured at the end of a 400 ms voltage pulse. These techniques have been described in more detail elsewhere (Ju et al. 1992, 1996).
All values are expressed as means ± 1 s.e.m. and the number of cells (n) in each group is given. Statistical analysis was performed using Student's two-tailed unpaired t test.
RESULTS
Control INa,P and INa,T
Sodium currents were recorded first in the absence and then in the presence of 0.5 μm TTX (Fig. 1A).
Subtraction of the traces revealed TTX-sensitive currents. An example of this procedure is shown in Fig. 1B. In this instance, the pipette contained 5 mm sodium cyanide. With this protocol, a transient and a persistent Na+ current could be seen free from contamination by other currents.
The current densities of INa,T and INa,P in control solution measured in thirty cells in this way were: peak INa,T, 111.0 ± 9.62 pA pF−1; INa,P, 0.87 ± 0.13 pA pF−1. That is, the persistent Na+ current was 0.7-1 % of the peak transient current amplitude. As has been reported previously (French et al. 1990; Alzheimer, Schwindt & Crill, 1993; Crill, 1996), INa,P activated at more negative potentials (V½ about -50 mV) than INa,T and did not show the same inactivation properties as INa,T. When the pre-pulse potential was altered between -150 and 0 mV, INa,P persisted at pre-pulse potentials that inactivated INa,T.
Effect of inhibiting oxidative metabolism with sodium cyanide
In order to block oxidative metabolism, neurons were exposed to 5 mm sodium cyanide through the pipette solution (Elliott, Smith & Allen, 1989; Ju et al. 1996). Solutions contained 10 mm glucose so that glycolysis was not impaired. With this treatment, the amplitudes of INa,P were much larger than the control amplitudes, as illustrated in Fig. 2A.
Figure 2. Inhibition of oxidative metabolism by hypoxia or cyanide increases persistent Na+ current.
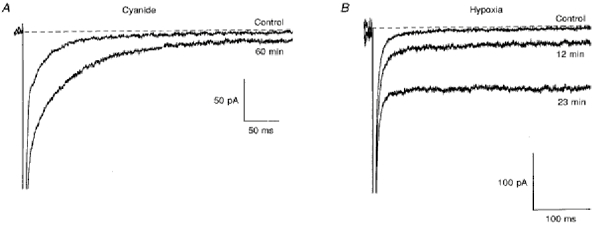
A, TTX-sensitive currents recorded from a neuron after 10 and 60 min of internal sodium cyanide exposure. Normally (in 12 out of 15 cells) the increase was large and stabilized within 10-15 min after the whole-cell seal was obtained. Currents shown were generated by a depolarizing step to -30 mV from a holding potential of -100 mV. B, TTX-sensitive currents recorded from a neuron before (top trace) and after exposure to hypoxic solution for 12 min (middle trace) and 23 min (lower trace). Currents were generated by a voltage step to -20 mV from a holding potential of -100 mV. The dashed line denotes the subtracted current level before the depolarizing pulse (zero current).
In ten out of fifteen cells in which the effect of cyanide was measured after the cell interior had been perfused with the drug for 10-15 min, the current density of INa,P recorded at the end of a 400 ms depolarizing pulse to -30 mV was 3.10 ± 0.71 pA pF−1 (n = 10), significantly greater than in the absence of cyanide (P < 0.0001). The current density of INa,T during exposure to cyanide was 119.70 ± 18.72 pA pF−1 (n = 10), not significantly different from control values (P > 0.05). Hence, cyanide caused an increase in the ratio of INa,P to INa,T to more than 2 % (0.022 ± 0.003, n = 10). In three out of the fifteen cells, a progressive time-dependent increase in the amplitude of INa,P was observed (in the other twelve cells the increase appeared to have stabilized 15-20 min after the whole-cell seal was obtained). One such cell is shown in Fig. 2A where the INa,P: INa,T ratio increased 3-fold (from 0.004 to 0.012) over a time period of 1 h.
Effects of hypoxia
In order to establish that the increase in INa,P caused by cyanide was due to inhibition of oxidative metabolism rather than a non-specific effect of the drug, the effect of reducing the O2 tension by perfusing a cell with solution that had been bubbled with 100 % N2 was examined in ten neurons. Exposure of a cell to a hypoxic solution clearly increased the amplitude of INa,P (Fig. 2B). In some neurons, a progressive increase in the amplitude of INa,P over time could be observed and currents recorded in one such neuron are shown in Fig. 2B. INa,P significantly increased from 1.21 ± 0.24 to 2.12 ± 0.34 pA pF−1 (n = 10, P < 0.05) during hypoxia. In contrast, there was generally a decrease in the amplitude of INa,T in neurons exposed to hypoxia in this way (0.87 ± 0.10, n = 9). The ratio of INa,P to INa,T increased 3-fold during hypoxia (from 0.014 ± 0.04 to 0.042 ± 0.018 of INa,T, n = 9).
An increase in the amplitude of INa,P during hypoxia was seen at all potentials, as can be seen in the current-voltage curves obtained from one neuron in Fig. 3A. Currents were recorded over a range of potentials before and after exposure to TTX and the amplitude of INa,P was measured from the subtracted current at the end of a 400 ms depolarizing pulse. Results shown in Fig. 3A were obtained before (•) and after 12-15 min of hypoxia (^). The lines through the data points were obtained from best fits to the conductance-voltage plots from the same cell (Fig. 3B) of the Boltzmann-type equation:
where Gmax is the maximum conductance, V the test potential, V′ the potential at which G was 50 % of Gmax and k a slope factor.
Figure 3. Current-voltage and conductance-voltage relationships before and after hypoxia.
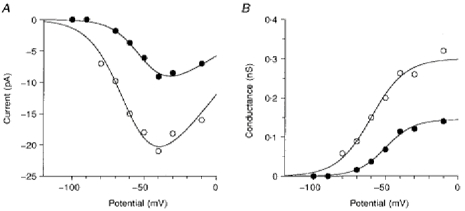
A, current-voltage relationship of the TTX-sensitive persistent Na+ current during normoxia (•) and after 12-15 min of hypoxia (^). The lines through the data points were obtained as described in the text. B, conductance-voltage relationship of the TTX-sensitive persistent Na+ current during normoxia (•) and after 12-15 min of hypoxia (^). The amplitude of INa,P was measured at the end of a 400 ms depolarizing pulse and divided by V - E0 to obtain the conductance (E0 is the potential at which I = 0). The lines through the data points were the best fits of the equation: G(V) = Gmax/(1 + exp((V′ - V)/k)). Further details are given in the text.
Before exposure to hypoxia, Gmax was 0.18 nS, V′ was -49 mV and k was 8.8. Following 12-15 min hypoxia, Gmax had increased to 0.33 nS, V′ was -60 mV and k was 11.8. It can be seen that INa,P is present at potentials near the resting membrane potential (around -70 mV) and reaches a peak at about -40 mV, a range that would be important in modulating the rate of action potential firing, or rhythmicity, in these cells.
Effects of Na+ channel blockers
TTX
The effects of a range of concentrations of TTX (0.5-500 nM) on Na+ current recorded with pipettes containing 5 mm sodium cyanide were examined. The persistent and transient sodium currents were evoked by a test pulse to -30 mV from a holding potential of -100 mV. Control responses were always established before and after exposure to solutions containing TTX. A solution containing 0.5 μm TTX was applied after each lower concentration of TTX so that the TTX-sensitive Na+ currents could be isolated by subtraction of the currents recorded in the presence of the high concentration of TTX. Currents analysed were obtained by subtraction of the currents remaining after exposure of the neuron to the supramaximal TTX concentration.
There was a noticeable difference in the sensitivity of INa,P and INa,T to TTX. At a concentration of 5 nM, TTX caused an obvious reduction in the amplitude of INa,P (Fig. 4A) whereas the peak amplitude of INa,T was not significantly affected.
Figure 4. Low concentrations of TTX and lidocaine block the persistent Na+ current more than the transient Na+ current.
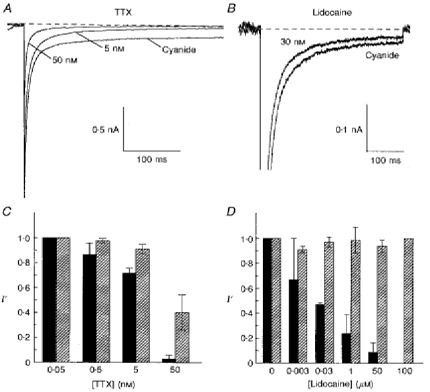
A, effects of 5 and 50 nM TTX on the persistent Na+ current generated by a voltage step from -100 to -30 mV 15-25 min after whole-cell seal was obtained. The pipette contained 5 mm cyanide. B, the persistent Na+ current after approximately 25 min of sodium cyanide exposure is depressed by 30 nM lidocaine. Currents were generated by a voltage pulse to -30 mV from a holding potential of -100 mV. C and D, the relationship between the concentration of TTX (C) or lidocaine (D) and depression of the normalized amplitudes (I′ = I/Icontrol) of the transient ( ) and persistent (▪) Na+ current. The bars show the average normalized amplitude of the current and the vertical lines denote 1 s.e.m.
) and persistent (▪) Na+ current. The bars show the average normalized amplitude of the current and the vertical lines denote 1 s.e.m.
In five cells exposed to 5 nM TTX, INa,P was reduced to 0.72 ± 0.03 of the control value (Fig. 4C). In the same cells, INa,T was 0.91 ± 0.03 of control which was significantly different from the effect of this concentration of TTX on INa,P (P < 0.005). In the presence of 50 nM TTX, INa,P was almost completely blocked while INa,T was reduced by about 50 % (Fig. 4A). In four cells exposed to 50 nM TTX, INa,P was 0.03 ± 0.03 of control whereas the peak INa,T was 0.40 ± 0.12 of control (Fig. 4C). In the presence of 0.5 μm TTX, both INa,P and INa,T were completely abolished (Fig. 1A). A least-squares fit of the Hill equation to the data gave an IC50 of 9.1 ± 1.2 nM for INa,P, which was significantly lower than the IC50 for INa,T, which was 37.1 ± 1.2 nM, approximately 4-fold higher (P < 0.001). A similar difference in the sensitivity of INa,T and INa,P to TTX was seen in neurons not exposed to cyanide (data not shown).
Lidocaine
The effects of lidocaine on the Na+ currents were examined in a similar manner. In three experiments in which a neuron was exposed to 3 nM lidocaine, the effects on both INa,P and INa,T were small (0.67 ± 0.33 and 0.91 ± 0.03 of control, respectively; Fig. 4D). At 30 nM, lidocaine caused a larger decrease in INa,P (Fig. 4B) while having little effect on INa,T. In four cells, 30 nM lidocaine reduced INa,P to 0.47 ± 0.01 of control; INa,T was 0.97 ± 0.04 of control, which was significantly different from the effect of this concentration on INa,P (P < 0.001). At 1 μm, lidocaine reduced INa,P further to 0.24 ± 0.15 whereas INa,T was 0.99 ± 0.11 of control (Fig. 4D, n = 4). Hence, INa,P responded to inhibition by lidocaine at concentrations that had little, if any, effect on INa,T.
DISCUSSION
These results show that inhibition of oxidative metabolism by sodium cyanide or hypoxia increases INa,P while having little effect on INa,T. It should be noted that, in these experiments, the intracellular [Na+] was kept constant so that any change in INa must have been due to a change in conductance rather than a change in driving force on sodium ions. The situation in cells in vivo would be different in that [Na+]i may change as extrusion mechanisms fail to cope with Na+ influx. Cyanide and hypoxia had similar effects, as has been described previously (Bickler & Hansen, 1994; Ju et al. 1996), indicating that they had a common mechanism of action.
Our results demonstrate the existence of a larger than normal persistent INa during hypoxia. An increase in INa,P would be expected to have a significant effect on neuronal function. Firstly, it would increase the excitability of neurons and disrupt the normal oscillations in membrane potential and rhythmic firing of action potentials (Alonzo & Llinás, 1989; Amitai, 1994). Similar effects have been observed when INa,P is increased with anemone toxin which significantly enhances bursting behaviour in rat neocortical pyramidal neurons (Mantegazza, Franceschetti & Avanzini, 1998). Secondly, an increase in a non-inactivating Na+ current is probably the primary cause of the increase in [Na+]i observed during hypoxia (Friedman & Haddad, 1994a) and may be involved both directly and indirectly in the sequence of events, including an increase in [Ca2+]i, that culminate in cell death caused by hypoxia. It has been shown that removing Na+ channel inactivation with agents such as veratridine during normoxia increases [Ca2+]i (Haigney, Lakatta, Stern & Silverman, 1994). Furthermore, neuronal damage caused by hypoxia or ischaemia can be alleviated by Na+ channel blockers such as TTX and lidocaine at concentrations so low that action potentials are not blocked. The rise in [Na+]i caused by an uncompensated increase in INa,P would reverse the Na+-Ca2+ exchanger and lead to an increase in [Ca2+]i and consequent cell damage. The depolarization caused by an increase in Na+ permeability could activate voltage-sensitive Ca2+ channels and this would tend to load cells with Ca2+ also. Thus, an increase in INa,P may be the primary change induced by hypoxia that eventually leads to cell death. Selective inhibition of the hypoxia-induced increase in neuronal INa,P by low concentrations of TTX and lidocaine provides an explanation for earlier reports that low concentrations of Na+ channel inhibitors prevent the [Ca2+]i increase and cell damage caused by hypoxia, while leaving normal action potential firing unchanged (Haigney et al. 1994; Fried et al. 1995; Taylor et al. 1995). Finally, the increase in [Na+]i would depress re-uptake of glutamate that is coupled to the [Na+] gradient. This, together with increased secretion of glutamate caused by the increase in [Ca2+]i, could explain the increase in extracellular glutamate concentration that is observed during hypoxia (Sanchez-Prieto & Gonzalez, 1988; Nicholls & Attwell, 1990).
The reason for the selective increase in INa,P during hypoxia is not clear. It has been postulated that the channels responsible for INa,P are the same as those that generate INa,T and that a small fraction of Na+ channels entering a non-inactivating mode generates INa,P (Alzheimer et al. 1993; Crill, 1996). Alternatively, the INa,P channels may be different from INa,T channels. For example, cloned Na+ type III channels have slower activation and particularly slower inactivation than type I and II Na+ channels but type III channels are expressed only at very early stages of development (Taylor, 1995). Furthermore, mutations in Na+ channel protein have been shown to cause a larger than normal INa,P (Rojas, 1996). Hence, the increase in the amplitude of INa,P may be due to increased probability of opening of ‘persistent Na+ channels’ or to an increased proportion of ‘normal’ Na+ channels (most probably of type I or maybe type II) switching to a non-inactivating state, possibly because of a change in phosphorylation mediated by protein kinase C.
Acknowledgments
A. K. M. Hammarström was a Postdoctoral Fellow supported by the National Heart Foundation of Australia and the Bruce Foundation (Anutech Pty Ltd). We are grateful to Michelle Lim for her help with dissociating cells.
References
- Alonzo A, Llinás RR. Subthreshold Na+-dependent theta-like rythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:1175–1177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- Alzheimer C, Schwindt PC, Crill WE. Modal gating of Na+ channels as a mechanism of persistent Na+ current in pyramidal neurons from rat and cat sensorimotor cortex. Journal of Neuroscience. 1993;13:660–673. doi: 10.1523/JNEUROSCI.13-02-00660.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amitai Y. Membrane potential oscillations underlying firing patterns in neocortical neurons. Neuroscience. 1994;63:151–161. doi: 10.1016/0306-4522(94)90013-2. 10.1016/0306-4522(94)90013-2. [DOI] [PubMed] [Google Scholar]
- Bickler PE, Hansen BM. Causes of calcium accumulation in rat cortical brain slices during hypoxia and ischaemia: role of ion channels and membrane damage. Brain Research. 1994;665:269–276. doi: 10.1016/0006-8993(94)91347-1. [DOI] [PubMed] [Google Scholar]
- Crill WE. Persistent sodium currents in mammalian central neurons. Annual Review of Physiology. 1996;58:349–362. doi: 10.1146/annurev.ph.58.030196.002025. [DOI] [PubMed] [Google Scholar]
- Elliott AC, Smith GL, Allen DG. Simultaneous measurements of action potential duration and intracellular ATP in isolated ferret hearts exposed to cyanide. Circulation Research. 1989;64:583–591. doi: 10.1161/01.res.64.3.583. [DOI] [PubMed] [Google Scholar]
- French CR, Gage PW. A threshold sodium current in pyramidal cells in rat hippocampus. Neuroscience Letters. 1985;56:289–293. doi: 10.1016/0304-3940(85)90257-5. 10.1016/0304-3940(85)90257-5. [DOI] [PubMed] [Google Scholar]
- French CR, Sah P, Buckett KJ, Gage PW. A voltage-dependent persistent sodium current in mammalian hippocampal neurons. Journal of General Physiology. 1990;95:1139–1157. doi: 10.1085/jgp.95.6.1139. 10.1085/jgp.95.6.1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fried E, Amorim P, Chambers G, Cottrell JE, Kass IS. The importance of sodium for anoxic transmission damage in rat hippocampal slices: mechanism of protection by lidocaine. The Journal of Physiology. 1995;489:557–565. doi: 10.1113/jphysiol.1995.sp021072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG. Anoxia induced an increase in intracellular sodium in rat cortical neurons in vitro. Brain Research. 1994a;663:329–334. doi: 10.1016/0006-8993(94)91281-5. 10.1016/0006-8993(94)91281-5. [DOI] [PubMed] [Google Scholar]
- Friedman JE, Haddad GG. Removal of extracellular sodium prevents anoxia-induced injury in freshly dissociated CA1 hippocampal neurons. Brain Research. 1994b;641:57–64. doi: 10.1016/0006-8993(94)91815-5. 10.1016/0006-8993(94)91815-5. [DOI] [PubMed] [Google Scholar]
- Gilly WF, Armstrong CM. Threshold channels - a novel type of sodium channel in squid giant axon. Nature. 1984;309:448–450. doi: 10.1038/309448a0. [DOI] [PubMed] [Google Scholar]
- Haigney MCP, Lakatta EG, Stern MD, Silverman HS. Sodium channel blockade reduces hypoxic sodium loading and sodium-dependent calcium loading. Circulation. 1994;90:391–399. doi: 10.1161/01.cir.90.1.391. [DOI] [PubMed] [Google Scholar]
- Hammarström AKM, Lim MSF, Gage PW. Effect of hypoxia and cyanide on the persistent sodium current of rat CA1 neurons. Proceedings of the Australian Physiological and Pharmacological Society. 1996;27:136P. abstract. [Google Scholar]
- Ju Y, Saint DA, Gage PW. Effects of lignocaine and quinidine on the persistent sodium current in rat ventricular myocytes. British Journal of Pharmacology. 1992;107:311–316. doi: 10.1111/j.1476-5381.1992.tb12743.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ju Y, Saint DA, Gage PW. Hypoxia increases persistent sodium current in rat ventricular myocytes. The Journal of Physiology. 1996;497:337–347. doi: 10.1113/jphysiol.1996.sp021772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay AR, Wong RK. Calcium current activation kinetics in isolated pyramidal neurones of the CA1 region of the mature guinea-pig hippocampus. The Journal of Physiology. 1987;392:603–616. doi: 10.1113/jphysiol.1987.sp016799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantegazza M, Franceschetti S, Avanzini G. Anemone toxin (ATX-II)-induced increase in persistent sodium current: effects on firing properties of rat neocortical pyramidal neurons. The Journal of Physiology. 1998;507:105–116. doi: 10.1111/j.1469-7793.1998.105bu.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D, Attwell D. The release and uptake of excitatory amino acids. Trends in Pharmacological Sciences. 1990;11:462–468. doi: 10.1016/0165-6147(90)90129-v. 10.1016/0165-6147(90)90129-V. [DOI] [PubMed] [Google Scholar]
- Rojas CV. Ion channels and human genetic diseases. News in Physiological Sciences. 1996;11:36–42. [Google Scholar]
- Sanchez-Prieto J, Gonzalez P. Occurrence of a large Ca2+-independent release of glutamate during anoxia in isolated nerve terminals (synaptosomes) Journal of Neurochemistry. 1988;50:1322–1324. doi: 10.1111/j.1471-4159.1988.tb10611.x. [DOI] [PubMed] [Google Scholar]
- Stys PK, Waxman SG, Ransom BR. Reverse operation of the Na+-Ca2+ exchanger mediates Ca2+ influx during anoxia in mammalian CNS white matter. Annals of the New York Academy of Sciences. 1991;639:328–332. doi: 10.1111/j.1749-6632.1991.tb17321.x. [DOI] [PubMed] [Google Scholar]
- Taylor CP. Na+ currents that fail to inactivate. Trends in Neurosciences. 1993;16:455–460. doi: 10.1016/0166-2236(93)90077-y. 10.1016/0166-2236(93)90077-Y. [DOI] [PubMed] [Google Scholar]
- Taylor CP. Na+ channels as targets for neuroprotective drugs. Trends in Pharmacological Sciences. 1995;16:309–316. doi: 10.1016/s0165-6147(00)89060-4. 10.1016/S0165-6147(00)89060-4. [DOI] [PubMed] [Google Scholar]
- Taylor CP, Burke SP, Weber ML. Hippocampal slices: glutamate overflow and cellular damage from ischemia are reduced by sodium-channel blockade. Journal of Neuroscience Methods. 1995;59:121–128. doi: 10.1016/0165-0270(94)00202-r. 10.1016/0165-0270(94)00202-R. [DOI] [PubMed] [Google Scholar]
- Weber ML, Taylor CP. Damage from oxygen and glucose deprivation in hippocampal slices is prevented by tetrodotoxin, lidocaine and phenytoin without blockade of action potentials. Brain Research. 1994;664:167–177. doi: 10.1016/0006-8993(94)91967-4. 10.1016/0006-8993(94)91967-4. [DOI] [PubMed] [Google Scholar]