

Abstract
In order to assess ionic mechanisms mediating renal afferent arteriolar myogenic constriction, experiments were performed using isolated perfused hydronephrotic rat kidneys.
Increasing pressure progressively constricted the afferent arteriole (-0·26 ± 0·02 % mmHg−1, n= 21, r= 0·97). Gadolinium (10 μM), a mechanosensitive cation channel blocker, abolished this myogenic constriction. However, high potassium media (30 mm) constricted the afferent arteriole in the presence of gadolinium.
Lowering extracellular sodium concentration gradually attenuated afferent arteriolar myogenic constriction. In the perfusate containing 50 mm sodium, the myogenic response was arrested.
Afferent arteriolar myogenic constriction was prevented in calcium-free perfusate or by the L-type calcium channel blocker diltiazem (10 μM).
Our present findings provide evidence that increasing pressure gates mechanosensitive cation channels on the afferent arteriole, thereby eliciting membrane depolarization and activating voltage-dependent calcium channels.
Bayliss (1902) initially reported the arterial myogenic response which may participate in the autoregulation of capillary blood flow and pressure. In particular, kidneys exhibit a remarkable capacity to maintain a constant glomerular blood flow and pressure in the face of marked variations in blood pressure (Navar et al. 1996). In progressive renal diseases, renal autoregulatory capacity is compromised, often in the presence of systemic hypertension, resulting in the elevation of glomerular capillary pressure, which leads to further glomerular injury (Takenaka et al. 1992; Griffin et al. 1995). Furthermore, renal failure presents serious medical and socio-economical problems (Rettig & Levinsky, 1991). Investigations into the mechanisms mediating renal autoregulation may provide a framework for developing therapeutic strategies against various renal diseases.
Previous studies demonstrated that a stronger myogenic response was observed in arterioles than in arteries (D'Angelo & Meninger, 1994). Indeed, although the elevation of renal arterial pressure constricts all segments of preglomerular vasculature, the afferent arteriole plays the most important role in autoregulatory adjustments of renal vascular resistance (Carmines et al. 1990). We have previously demonstrated that in addition to tubuloglomerular feedback, an intact afferent arteriolar myogenic response, mediated by the activation of voltage-dependent calcium channels, is required for efficient autoregulation of glomerular blood flow (Takenaka et al. 1994). Increasing transmural pressure elicits gradual membrane depolarization in renal vasculature (Harder et al. 1987). Although chloride channels underlie myogenic responses in cerebral arteries (Nelson et al. 1997), recent studies indicate that calcium-activated potassium or chloride channels are not involved in afferent arteriolar myogenic constriction (Loutzenhiser & Parker, 1994; Takenaka et al. 1996a). Thus, the ionic mechanisms mediating alterations of membrane potential by pressure in the afferent arteriole are still not clear.
In order to assess the relationship between pressure-induced channel activation and afferent arteriolar constrictor responses, we examined the involvement of mechanosensitive cation channels (Yang & Sachs, 1989) in myogenic responses, using isolated perfused hydronephrotic kidneys, which enable direct visualization of renal microvascular behaviour free from influences of tubuloglomerular feedback (Takenaka et al. 1996a). Our present findings provide evidence that the elevation of pressure gates mechanosensitive cation channels on the afferent arteriole, thereby eliciting membrane depolarization and consequently activating voltage-dependent calcium channels.
Part of the data in this manuscript was presented at the 29th annual meeting of the American Society of Nephrology in New Orleans, LA, USA (Takenaka et al. 1996b), and Experimental Biology 98 in San Francisco, CA, USA (Takenaka et al. 1998).
METHODS
Adult male Sprague-Dawley rats (Charles River Japan, Atsugi, Kanagawa, Japan) had free access to food and water. Animals were anaesthetized with ether (Showa Chemicals, Tokyo, Japan), and the right ureter was ligated via a small abdominal incision, using sterile techniques. Anaesthesia was administered to the animals by inhalation of ether in a glass chamber, and maintained by nasal inhalation in an air-conditioned room. The depth of anaesthesia was assessed by loss of corneal reflex. The abdomen was then closed and the animals were allowed to recover. After 8-12 weeks from the surgery (Marin-Grez et al. 1986), the rats were again anaesthetized with ether, and the right renal artery was cannulated by introducing a perfusion cannula across the aorta through a superior mesenteric artery. Perfusion with warm, oxygenated physiological saline solution (PSS, pH 7.4) was initiated during this cannulation procedure. PSS consisted of (mm): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 5 Hepes and 5 glucose. The kidney was then excised and placed on the stage of an inverted microscope (model T041, Olympus), which accommodated a heat table equipped with a thin glass viewing port at the bottom. After the kidney was removed, the rat was exsanguinated under anaesthesia.
The kidney was provided with perfusate from a pressurized chamber. The chamber pressure was maintained by the inflow of warm, hydrated, oxygenated gas, which exited through an adjustable back-pressure regulator (model 10BP, Fairchild Industrial Products, Winston Salem, NC, USA). Perfusion pressure, which was measured at the level of the renal artery, could be varied arbitrarily by altering the rate of gas exit, and was maintained at 80 mmHg except during pressure protocols. The kidneys were allowed to equilibrate in perfusate for at least 30 min before initiating experimental protocols. Myogenic responses were obtained by stepwise increases in perfusion pressure. Renal arterial pressure was kept constant for at least 2 min before further alterations were made (Takenaka et al. 1996a). Video images of renal microvessels were obtained using a CCD video camera (model ICD-42AC, Ikegami, Tokyo, Japan) and recorded by a videocassette recorder (model EVO-9850, Sony). To determine the vessel diameter, the video recording was transmitted to an IBM-AT computer equipped with a display board (model TARGA+, Truevision, Indianapolis, IN, USA). Vessel diameters were estimated by an automated program custom designed to determine the mean distance between parallel vessel walls. Arteriolar diameters were obtained during the plateau of the response. A segment of afferent arteriole near the interlobular artery (Takenaka et al. 1994), approximately 10 μm in length, was scanned at 2-5 s intervals.
In the first series of studies, the effects of gadolinium on afferent arteriolar myogenic constriction were assessed. Gadolinium was selected as a pharmacological probe because it potently blocks mechanosensitive cation channels, but seems to be cell impermeant (Yang & Sachs, 1989). Initially, basal afferent arteriolar responses to pressure changes were observed. Then, the kidneys (n= 6) were exposed to increasing doses of gadolinium (1-10 μM). Pressure changes were made at each concentration of gadolinium. Finally, in the presence of 10 μM gadolinium, the potassium concentration of perfusate was isosmotically increased to 30 mm by adding potassium-rich solution, in which KCl was substituted for NaCl (Loutzenhiser et al. 1989). In complementary studies (4 kidneys), effects of increasing doses of gadolinium (1 μM to 1 mm) on afferent arteriolar constriction by KCl-induced depolarization were studied.
In the second series of experiments (6 kidneys), the effects of isosmotic lowering of extracellular sodium concentration on afferent arteriolar myogenic constriction were examined. After basal myogenic responses were observed, sodium concentration was decreased to 100, 70 and 50 mm by adding sodium-free media, in which NaCl was replaced with N-methyl-D-glucamine chloride. Pressure challenge was done at each sodium concentration. Finally, the influence of high potassium media (30 mm) was assessed. In separate studies, the effects of the cation channel inhibitors tetrodotoxin (1 and 3 μM) and streptomycin (10 and 100 μM) (4 kidneys for each) on afferent arteriolar myogenic constriction were examined (Oka, 1995; Hamill & McBride, 1996).
In the third group, the kidneys (n= 5) were perfused with calcium-free media containing 2 mm EGTA (Hishikawa et al. 1994) after basal pressure responses had been obtained. Subsequently, pressure challenge was performed. Then, the perfusate was returned to normal PSS and pressure responses were again observed. In additional experiments (4 kidneys), influences of diltiazem (10 μM) on afferent arteriolar constriction during myogenic stimulation and KCl-induced depolarization were evaluated (Takenaka et al. 1996a).
All experimental procedures described above were approved by our institutional committees. Gadolinium chloride was obtained from Aldrich, and the other chemicals were purchased from Sigma. Data are expressed as means ±s.e.m. Statistical differences were checked with regression analysis or analysis of variance followed by Neumann-Kuels test. P < 0.05 was considered significant.
RESULTS
As shown in Fig. 1A, increasing perfusion pressure elicited progressive afferent arteriolar constriction. Gadolinium inhibited the afferent arteriolar myogenic constriction in a dose-dependent manner. Significant inhibition of afferent arteriolar myogenic constriction was observed with 3 μM gadolinium (Fig. 1C). At 10 μM, gadolinium abolished myogenic constriction (Fig. 1D). Mean data from seven afferent arterioles are described below. In the control condition, increasing pressure from 80 to 120 mmHg constricted afferent arterioles by 13 ± 2 % (from 20.5 ± 0.9 to 17.5 ± 1.0 μm, n= 7, P < 0.01). Elevating pressure to 160 mmHg elicited further afferent arteriolar constriction by 20 ± 2 % (to 15.8 ± 0.9 μm, P < 0.01). Although the addition of 1 μM gadolinium did not alter afferent arteriolar myogenic responsiveness, 3 μM gadolinium exerted an attenuative effect (P < 0.05vs. respective controls at each pressure). Thus, in the presence of gadolinium (3 μM), increasing pressure from 80 to 160 mmHg decreased afferent arteriolar diameter by only 11 ± 2 % (from 20.1 ± 1.0 to 17.3 ± 0.9 μm, P < 0.01). At 10 μM, gadolinium abolished myogenic constriction (20.1 ± 1.0 μm at 80 mmHg vs. 19.5 ± 0.8 μm at 160 mmHg), providing pharmacological evidence that mechanosensitive cation channels are involved in arteriolar myogenic response. Furthermore, in the presence of gadolinium (10 μM), the isosmotic increase in potassium concentration of the medium to 30 mm, which exclusively activates voltage-dependent calcium channels (Loutzenhiser et al. 1989), constricted afferent arterioles by 41 ± 4 % (from 20.0 ± 0.9 to 11.8 ± 0.7 μm, P < 0.01).
Figure 1. Representative tracings illustrating the effects of gadolinium (1-10 μM) on myogenic constriction of single afferent arterioles.
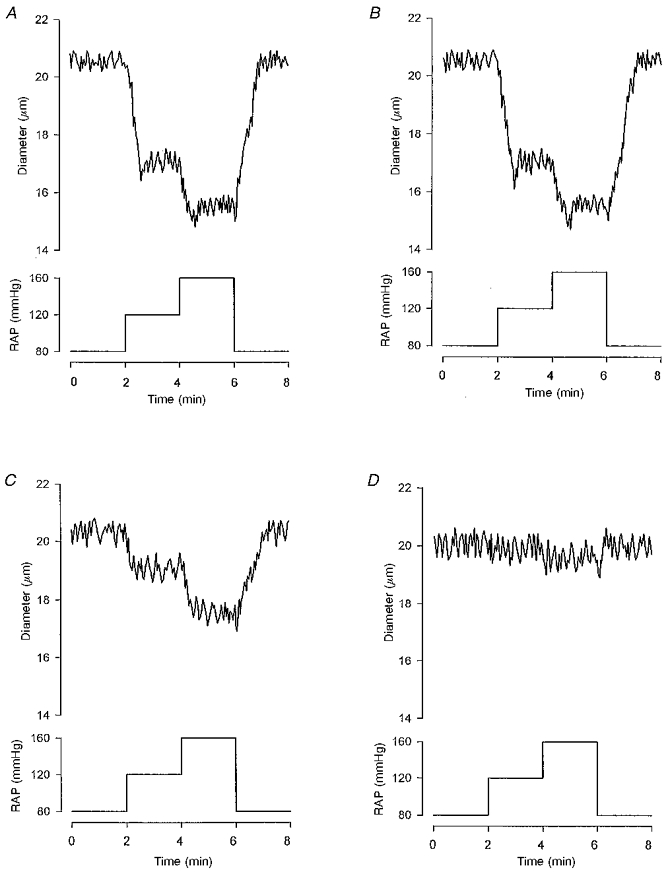
In the control state (A), the elevation of renal arterial pressure (RAP) from 80 to 120 mmHg decreased afferent arteriolar diameter from 20.5 to 17.1 μm. Further raising of the pressure to 160 mmHg constricted the afferent arteriole to a diameter of 15.6 μm. Although gadolinium did not alter myogenic responsiveness at 1 μM (B), 3 μM gadolinium attenuated myogenic constriction (C). In the presence of 3 μM gadolinium, increasing pressure from 80 to 160 mmHg considerably reduced afferent arteriolar diameter, from 20.1 to 17.6 μm. Afferent arteriolar myogenic constriction was abolished by 10 μM gadolinium (D).
Since gadolinium may interact with voltage-dependent calcium channels (Lansman, 1990; Davis et al. 1992), which are required for intact myogenic constriction (Takenaka et al. 1994), the effects of increasing doses of gadolinium on KCl-induced afferent arteriolar constriction were examined, and the data are summarized in Fig. 2. High potassium (30 mm) solutions constricted afferent arterioles by 43 ± 3 % (from 18.9 ± 0.6 to 11.3 ± 1.0 μm, n= 5, P < 0.01). Although 1 mm gadolinium reversed KCl-induced afferent arteriolar constriction, to a diameter of 18.6 ± 0.8 μm (n.s. vs. control), 10 μM gadolinium did not modify KCl-induced decrements in afferent arteriolar diameter, arguing against interactions between gadolinium (up to 10 μM) and voltage-dependent calcium channels in the renal microcirculation.
Figure 2. Influences of gadolinium (Gd3+) on afferent arteriolar constriction by KCl (30 mm)-induced depolarization.
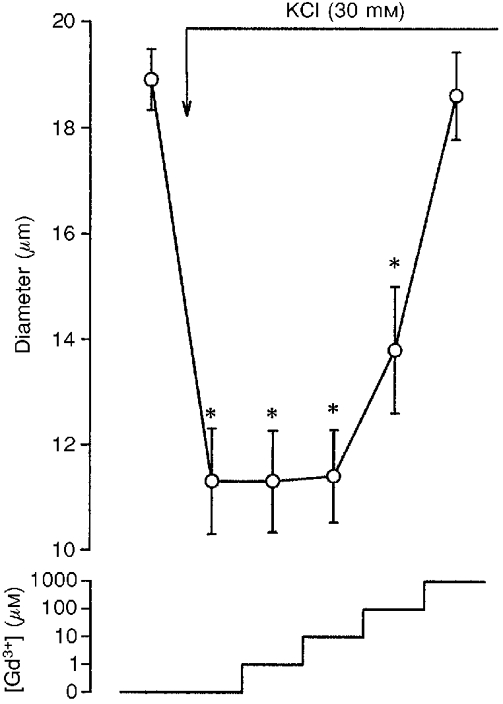
* Significant difference from basal value.
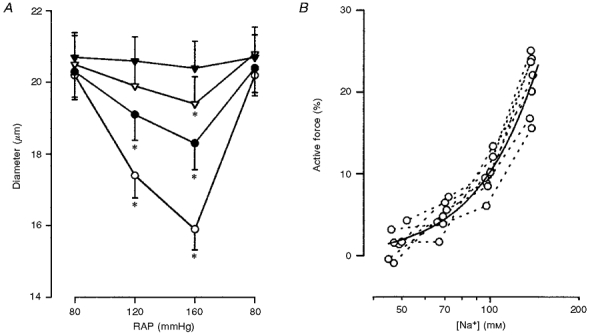
The effects of isosmotic decreases in extracellular sodium concentration, which reduce the electrochemical gradient for sodium, on the afferent arteriolar myogenic response were assessed (Fig. 3A). In the basal state, afferent arteriolar myogenic constriction was well preserved. The afferent arteriolar myogenic responsiveness was significantly diminished with 100 mm sodium. Thus, in low (100 mm) sodium media, the elevation of pressure from 80 to 120 mmHg reduced afferent arteriolar diameter by only 6 ± 1 % (from 20.3 ± 0.7 to 19.0 ± 0.7 μm, n= 7, P < 0.05). A further increase in pressure to 160 mmHg constricted afferent arterioles by 10 ± 2 % (to 18.3 ± 0.7 μm, P < 0.01). At 70 mm sodium, the elevation of pressure from 80 to 120 mmHg failed to constrict afferent arterioles (from 20.5 ± 0.8 to 20.0 ± 0.7 μm). In the perfusate containing 50 mm sodium, afferent arteriolar myogenic constriction was abolished (20.8 ± 0.7 μm at 80 mmHg vs. 20.4 ± 0.7 μm at 160 mmHg). However, in low sodium solutions (50 mm), increasing potassium concentration to 30 mm constricted afferent arterioles by 38 ± 5 % (from 20.7 ± 0.6 to 12.8 ± 0.5 μm, P < 0.01).
Figure 3. Effect of decreases in extracellular sodium concentration on arteriolar myogenic constriction and active force development.
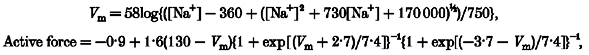 |
Figure 4 demonstrates the influences of cation channel blockers on afferent arteriolar myogenic responses. As depicted in Fig. 4A, in the presence of tetrodotoxin (1 μM), the elevation of pressure from 80 to 160 mmHg decreased afferent arteriolar diameter by 21 ± 3 % (n= 5, P < 0.01). When the kidneys were treated with 3 μM tetrodotoxin, raising pressure from 80 to 160 mmHg elicited a 19 ± 3 % reduction in afferent arteriolar diameter. Thus, tetrodotoxin, a voltage-dependent sodium channel blocker, failed to alter afferent arteriolar myogenic responsiveness. Figure 4B illustrates the effects of streptomycin, a putative mechanosensitive cation channel blocker, on afferent arteriolar myogenic constriction. In contrast to tetrodotoxin, streptomycin manifested a dose-dependent inhibition of myogenic constriction. Thus, at 10 μM, streptomycin attenuated the magnitude of decrements in afferent arteriolar diameter induced by the elevation of pressure from 80 to 160 mmHg (20 ± 2 vs. 9 ± 1 %, n= 6, P < 0.05). Furthermore, at 100 μM, streptomycin blocked afferent arteriolar myogenic constriction. Finally, in the presence of 100 μM streptomycin, an isosmotic increase in potassium concentration to 30 mm constricted afferent arterioles from a diameter of 21.1 ± 1.0 to 13.5 ± 0.8 μm (P < 0.01).
Figure 4. Effects of cation channel inhibitors on afferent arteriolar constriction.
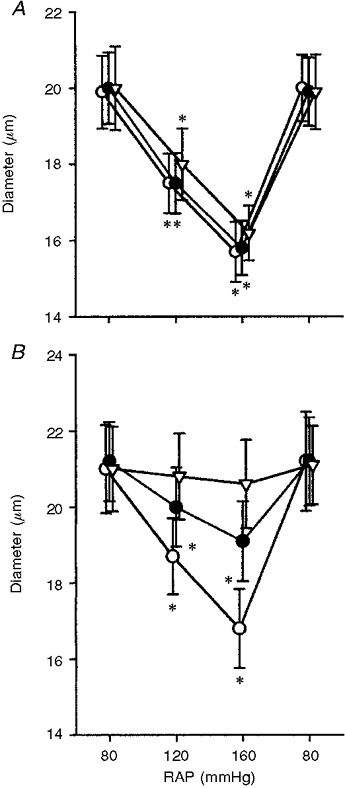
A, influence of tetrodotoxin on afferent arteriolar constriction by increasing renal arterial pressure. ○, • and ▿ represent values obtained during the control condition and in the presence of 1 and 3 μM tetrodotoxin, respectively. * Significantly different from respective basal value at 80 mmHg. B, effects of streptomycin on afferent arteriolar constriction by increasing RAP. ○, • and ▿ represent values obtained during the control condition, and in the presence of 10 and 100 μM streptomycin, respectively. * Significantly different from respective basal value at 80 mmHg.
Since mechanosensitive cation channels carry calcium ions, the electrochemical gradient for calcium was physically reversed by calcium-free solutions. As shown in Fig. 5A, afferent arteriolar myogenic responsiveness was well preserved in PSS. When calcium-free medium was applied, afferent arteriolar myogenic constriction was blocked (21.6 ± 1.0 μm at 80 mmHg vs. 22.0 ± 1.1 μm at 160 mmHg, n= 6). Following the exchange of calcium-free perfusate for PSS, increasing pressure from 80 to 160 mmHg decreased afferent arteriolar diameter by 19 ± 2 % (from 21.4 ± 1.0 to 17.7 ± 0.9 μm, P < 0.01), serving as time controls. Since voltage-dependent calcium channels are present on the afferent arteriole (Loutzenhiser et al. 1989), a separate series of experiments was performed to assess the effects of the calcium antagonist diltiazem (10 μM) on afferent arteriolar myogenic constriction (Fig. 5B). This dose of diltiazem was chosen because previous results showed that it did not deplete intracellular calcium stores under our experimental conditions (Takenaka et al. 1996a). Afferent arteriolar myogenic constriction was inhibited by diltiazem (20.2 ± 1.1 μm at 80 mmHg vs. 19.5 ± 1.2 μm at 160 mmHg, n= 5), suggesting that the magnitude of the increase in cytosolic calcium by calcium influx through mechanosensitive cation channels is insufficient to induce pressure-induced vasoconstriction. In addition, diltiazem (10 μM) prohibited afferent arteriolar constriction by KCl-induced depolarization (to 19.7 ± 1.0 μm).
Figure 5. Role of calcium in afferent arteriolar myogenic constriction.
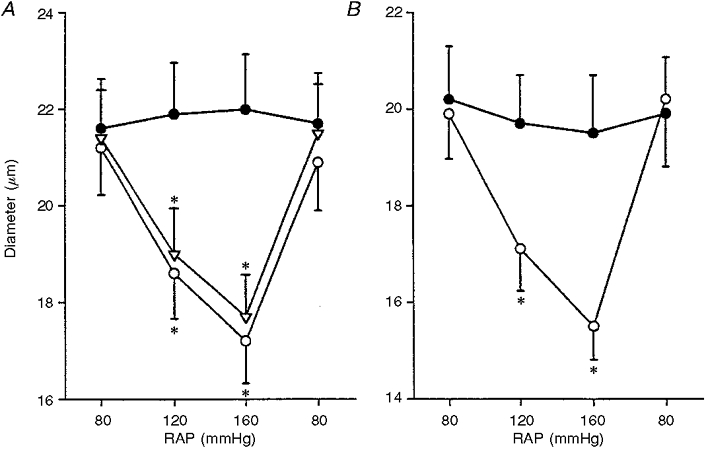
A, effects of calcium-free solution on afferent arteriolar myogenic constriction. ○, • and ▿ represent values obtained with normal PSS, calcium-free solution and re-application of PSS, respectively. * Significant difference from respective basal value at 80 mmHg. B, impact of diltiazem on afferent arteriolar myogenic responsiveness. ○ and • represent control condition and presence of diltiazem (10 μM), respectively. * Significantly different from basal value at 80 mmHg.
DISCUSSION
Cells respond to mechanical stimuli (Morris, 1990). For example, plant roots respond to gravity. Stretch of atrial muscle stimulates the secretion of natriuretic peptide (Laine et al. 1994). These responses are inhibited by gadolinium, a potent mechanosensitive cation channel blocker (Yang & Sachs, 1989). Increasing pressure raises the tension of the vascular wall (Bayliss, 1902). Mechanosensitive cation channels could participate in initiating this myogenic response. Since mechanosensitive cation channels conduct, rather non-specifically, mono- and divalent cations including potassium, sodium and calcium (Yang & Sachs, 1989; Davis et al. 1992), the reversal potential for mechanosensitive cation channels on vascular smooth muscle cells in PSS was reported as -15 mV (Davis et al. 1992), which is substantially higher than the resting potential. The increase in open probability of mechanosensitive cation channels on arteriolar myocytes by pressure should elicit substantial membrane depolarization. Our present results show that gadolinium blocks myogenic constriction in afferent arterioles, and further provide evidence that mechanosensitive cation channels mediate alterations in the membrane potential of afferent arterioles during myogenic activation.
This notion is supported by the present demonstration that low sodium solutions attenuate afferent arteriolar myogenic constriction. In addition, afferent arteriolar myogenic constriction was prevented by streptomycin, another mechanosensitive cation channel blocker (Hamill & McBride, 1996), but not by tetrodotoxin, a specific inhibitor of voltage-dependent sodium channels (Oka, 1995). Collectively, these results suggest that sodium conductance through mechanosensitive cation channels constitutes an important determinant of afferent arteriolar myogenic responsiveness. Membrane potential (Vm) was estimated according to the Goldman-Hodgkin-Katz constant field equation:
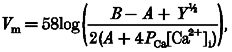 |
where:
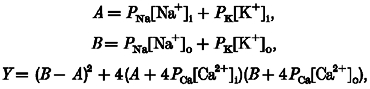 |
P represents the permeability of each ion, and i and o indicate intracellular and extracellular, respectively (Sperelakis, 1979). Because arteriolar tension is proportional to both pressure and diameter (Laplace's law), active force development (F) induced by elevating perfusion pressure (P) relates to the difference between purely passive (non-distensive) and given arteriolar diameters (D):
The diameter at 80 mmHg was taken as a passive one, because increasing pressure from 80 to 160 mmHg did not alter afferent arteriolar diameter in calcium-free media (Fig. 5A). Since calcium antagonists block afferent arteriolar myogenic constriction (Fig. 5B), active force depends on calcium entry through voltage-dependent calcium channels, which can be calculated from the product of the difference between membrane potential and calcium equilibrium potential (ECa - Vm), the availability of calcium channels to open (1 + exp[(Vm - Vi)/k]}−1, where Vi is the voltage at which half the calcium channels should be inactivated), open probability of calcium channels (1 + exp[(Vo - Vm)/k]}−1, where Vo is the voltage at which the open probability of the calcium channels is 50 %), and a factor related to single channel conductance and the number of calcium channels. As shown in Fig. 3B, our data are in agreement with this premise (Lee & Tsien, 1983; McDonald et al. 1986).
We have not, however, extended our investigations to single-channel responses to pressure-utilizing patch clamp methods, because the patch itself might generate artifacts such as sucking effects on cell membrane, possibly giving misleading results (Morris & Horn, 1991; Sokabe et al. 1991). Furthermore, since the afferent arteriole responds very actively to variations in pressure, non-invasive methods including video microscopy are uniquely suited for the investigation of afferent arteriolar behaviour (Casellas & Moore, 1993; Takenaka et al. 1994). Finally, patch clamp would produce morphological changes in arterioles, making both an intact arteriolar response to pressure and accurate measurement of diameter impractical.
In cerebral arteries, endothelial cells could produce vasoconstrictors in response to pressure alterations, mediating pressure-induced constriction (D'Angelo & Meninger, 1994). Indeed, endothelial cells possess mechanosensitive cation channels which respond to the variations in pressure (Lansman et al. 1987; Sokabe et al. 1991). The possibility remains that calcium entry through mechanosensitive cation channels into endothelial cells may prompt the release of vasoconstrictors. However, Nelson et al. (1997) have recently demonstrated the presence of myogenic tone in cerebral arteries without endotheLium. Similarly, previous data indicate that endothelial removal does not inhibit renal autoregulatory responses (Liu et al. 1989; Navar et al. 1996). Furthermore, afferent arteriolar myogenic constriction was preserved after immunological removal of endothelial cells (Juncos et al. 1995). The present results also argue against a mediatory role of the endothelium in afferent arteriolar myogenic constriction. Although lowering extracellular sodium may alter the membrane potential of endothelial cells, the lack of voltage-dependent calcium channels on these cells (Demirel et al. 1993) precludes membrane potential-dependent changes in cytosolic calcium and release of vasoconstrictor (Fig. 3B). Collectively, these data suggest that afferent arteriolar myocytes possess all the components (from the sensor to the effector) that are essential for myogenic constriction.
We previously showed that the stimulation of phospholipase C with intracellular calcium mobilization plays a crucial role in arterial myogenic response (Hishikawa et al. 1994). However, in the present study, we have demonstrated that afferent arteriolar myogenic constriction is inactivated in calcium-free media. Furthermore, our preliminary results suggest that 200 μM 2-nitro-4-carboxyphenyl-N,N-diphenyl-carbamate, an inhibitor of phospholipase C (Takenaka et al. 1997), does not alter afferent arteriolar myogenic constriction, suggesting a small role of calcium mobilization in arteriolar myogenic responses. Our findings indicate that the calcium equilibrium potential or calcium entry itself plays a crucial role in afferent arteriolar myogenic constriction. On vascular smooth muscle cell membrane, the density of voltage-dependent calcium channels is much higher than that of mechanosensitive cation channels (Yang & Sachs, 1989; Nelson et al. 1990). The present data indicate that afferent arteriolar myogenic constriction is inhibited by 10 μM diltiazem, a calcium antagonist (Takenaka et al. 1996a). Our results are consistent with those cited above (Yang & Sachs, 1989; Nelson et al. 1990), and suggest that although increased calcium conductance through mechanosensitive cation channels could contribute to the membrane depolarization process, calcium entry through voltage-dependent calcium channels is a requisite for active afferent arteriolar constriction during myogenic stimulation.
In summary, our present data provide the first evidence that increasing pressure gates mechanosensitive cation channels on afferent arterioles, thereby eliciting membrane depolarization and consequent activation of voltage-dependent calcium channels.
Acknowledgments
The authors thank Drs Keiji Fujiwara, Hiroyuki Sasamura and Konosuke Konishi for their help during performance of experiments and preparation of the manuscript. This study was partly supported by grant from the Ministry of Health and Welfare, Japan.
References
- Bayliss WN. On the local reactions of the arterial wall to changes of internal pressure. The Journal of Physiology. 1902;28:220–231. doi: 10.1113/jphysiol.1902.sp000911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmines PK, Inscho EW, Gensure RC. Arterial pressure effects on preglomerular microvasculature of juxtamedullary nephrons. American Journal of Physiology. 1990;258:F94–102. doi: 10.1152/ajprenal.1990.258.1.F94. [DOI] [PubMed] [Google Scholar]
- Casellas D, Moore LC. Autoregulation of intravascular pressure in preglomerular juxtamedullary vessels. American Journal of Physiology. 1993;264:F315–321. doi: 10.1152/ajprenal.1993.264.2.F315. [DOI] [PubMed] [Google Scholar]
- D'Angelo G, Meninger GA. Transduction mechanisms involved in the regulation of myogenic activity. Hypertension. 1994;23:1096–1105. doi: 10.1161/01.hyp.23.6.1096. [DOI] [PubMed] [Google Scholar]
- Davis MJ, Donovitz JA, Hook JD. Stretch-activated single-channel and whole cell currents in vascular smooth muscle cells. American Journal of Physiology. 1992;262:C1083–1088. doi: 10.1152/ajpcell.1992.262.4.C1083. [DOI] [PubMed] [Google Scholar]
- Demirel E, Laskey RE, Purkerson S, van Breemen C. Passive calcium leak in cultured porcine aortic endothelial cells. Biochemical and Biophysical Research Communications. 1993;191:1197–1203. doi: 10.1006/bbrc.1993.1344. 10.1006/bbrc.1993.1344. [DOI] [PubMed] [Google Scholar]
- Griffin KA, Picken M, Bidani AK. Deleterious effects of calcium channel blockade on pressure transmission and glomerular injury in rat remnant kidneys. Journal of Clinical Investigation. 1995;96:793–800. doi: 10.1172/JCI118125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, McBride DW. The pharmacology of mechanogated membrane ion channels. Pharmacological Reviews. 1996;48:231–252. [PubMed] [Google Scholar]
- Harder DR, Gilbert R, Lombard JH. Vascular muscle cell depolarization and activation in renal arteries on elevation in transmural pressure. American Journal of Physiology. 1987;253:F778–781. doi: 10.1152/ajprenal.1987.253.4.F778. [DOI] [PubMed] [Google Scholar]
- Hishikawa R, Nakaki T, Marumo T, Hayashi M, Suzuki H, Kato R, Saruta T. Pressure promotes DNA synthesis in cultured rat vascular smooth muscle cells. Journal of Clinical Investigation. 1994;93:1975–1980. doi: 10.1172/JCI117189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juncos LA, Garvin J, Carretero OA, Ito S. Flow modulates myogenic responses in isolated microperfused rabbit afferent arterioles via endothelium-derived nitric oxide. Journal of Clinical Investigation. 1995;95:2741–2748. doi: 10.1172/JCI117977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine M, Arjamaa O, Vuolteenaho O, Ruskoaho H, Weckstorm M. Block of stretch-activated atrial natriuretic peptide secretion by gadolinium in isolated rat atrium. The Journal of Physiology. 1994;480:553–561. doi: 10.1113/jphysiol.1994.sp020383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansman JB. Blockade of current through single calcium channels by trivalent lanthanide cations. Journal of General Physiology. 1990;95:679–696. doi: 10.1085/jgp.95.4.679. 10.1085/jgp.95.4.679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lansman JB, Hallam TJ, Rink TJ. Single stretch- activated ion channels in vascular endothelial cells as mechanotransducers. Nature. 1987;325:811–813. doi: 10.1038/325811a0. 10.1038/325811a0. [DOI] [PubMed] [Google Scholar]
- Lee KS, Tsien RW. Mechanism of calcium channel blockade by verapamil, D600, diltiazem and nitrendipine in single dialyzed heart cells. Nature. 1983;302:790–794. doi: 10.1038/302790a0. [DOI] [PubMed] [Google Scholar]
- Liu Y, Harder DR, Lombard JH. Myogenic activation of canine small renal arteries after nonchemical removal of endothelium. American Journal of Physiology. 1994;267:H302–307. doi: 10.1152/ajpheart.1994.267.1.H302. [DOI] [PubMed] [Google Scholar]
- Loutzenhiser R, Hayashi K, Epstein M. Divergent effects of KCl-induced depolarization on afferent and efferent arterioles. American Journal of Physiology. 1989;257:F561–564. doi: 10.1152/ajprenal.1989.257.4.F561. [DOI] [PubMed] [Google Scholar]
- Louzenhiser R, Parker M. Hypoxia inhibits myogenic reactivity of renal afferent arterioles by activating ATP-sensitive K channels. Circulation Research. 1994;74:861–869. doi: 10.1161/01.res.74.5.861. [DOI] [PubMed] [Google Scholar]
- McDonald TF, Cavalie A, Trautwein W, Pelzer D. Voltage-dependent properties of macroscopic and elementary calcium currents in guinea pig ventricular myocytes. Pflügers Archiv. 1986;406:437–448. doi: 10.1007/BF00583365. [DOI] [PubMed] [Google Scholar]
- Marin-Grez M, Fleming J, Steinhausen M. Atrial natriuretic peptide causes pre-glomerular vasodilation and post- glomerular vasoconstriction in rat kidney. Nature. 1986;324:473–476. doi: 10.1038/324473a0. [DOI] [PubMed] [Google Scholar]
- Morris CE. Mechanosensitive ion channels. Journal of Membrane Biology. 1990;113:93–107. doi: 10.1007/BF01872883. [DOI] [PubMed] [Google Scholar]
- Morris CE, Horn R. Failure to elicit neuronal macroscopic mechanosensitive currents anticipated by single-channel studies. Science. 1991;251:1246–1249. doi: 10.1126/science.1706535. [DOI] [PubMed] [Google Scholar]
- Navar LG, Inscho EW, Majid DSA, Imig JD, Harrison-Bernard LM, Mitchell KD. Paracrine regulation of the renal microcirculation. Physiological Reviews. 1996;76:425–536. doi: 10.1152/physrev.1996.76.2.425. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. The Journal of Physiology. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, III, Standen NB. Calcium channels, potassium channels and voltage dependence of arterial smooth muscle tone. American Journal of Physiology. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Oka Y. Tetrodotoxin-resistant persistent Na+ current underlying pacemaker potentials of fish gonadotropin-releasing hormone neurons. The Journal of Physiology. 1995;482:1–6. doi: 10.1113/jphysiol.1995.sp020494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rettig RA, Levinsky NG. Kidney Failure and the Federal Government. Washington DC: National Academy Press; 1991. [PubMed] [Google Scholar]
- Sokabe M, Sachs F, Jing Z. Quantitative video microscopy of patch-clamped membrane: stress, strain, capacitance and stretch channel activation. Biophysical Journal. 1991;59:722–728. doi: 10.1016/S0006-3495(91)82285-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sperelakis N. Origin of the cardiac resting potential. In: Berne RM, editor. Handbook of Physiology, section 2, The Cardiovascular System. Vol. 1. Baltimore, MA, USA: Williams & Wilkins Co.; 1979. pp. 187–267. [Google Scholar]
- Takenaka T, Forster H, De Micheli A, Epstein M. Impaired myogenic responsiveness in renal microvessels of Dahl salt-sensitive rats. Circulation Research. 1992;71:471–480. doi: 10.1161/01.res.71.2.471. [DOI] [PubMed] [Google Scholar]
- Takenaka T, Harrison-Bernard LM, Inscho EW, Carmines PK, Navar LG. Autoregulation of afferent arteriolar blood flow in juxtamedullary nephrons. American Journal of Physiology. 1994;267:F879–887. doi: 10.1152/ajprenal.1994.267.5.F879. [DOI] [PubMed] [Google Scholar]
- Takenaka T, Kanno Y, Kitamura Y, Hayashi K, Suzuki H, Saruta T. Role of chloride channels in afferent arteriolar constriction. Kidney International. 1996a;50:864–872. doi: 10.1038/ki.1996.386. [DOI] [PubMed] [Google Scholar]
- Takenaka T, Kitamura Y, Hayashi K, Suzuki H, Saruta T. A non-selective cation channel blocker inhibits afferent arteriolar constriction by pressure. Journal of the American Society of Nephrology. 1996b;7:1589. [Google Scholar]
- Takenaka T, Suzuki H, Fujiwara K, Kanno Y, Ohno Y, Hayashi K, Nagahama T, Saruta T. Cellular mechanisms mediating rat renal microvascular constriction by angiotensin II. Journal of Clinical Investigation. 1997;100:2107–2114. doi: 10.1172/JCI119745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takenaka T, Suzuki H, Okada H, Hayashi K, Kanno Y, Saruta T. Mechanosensitive cation channels mediate renal afferent arteriolar myogenic constriction. FASEB Journal. 1998;12:A997. doi: 10.1111/j.1469-7793.1998.245bi.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X, Sachs F. Block of stretch-activated ion channels in Xenopus oocytes by gadolinium and calcium. Science. 1989;243:1068–1071. doi: 10.1126/science.2466333. [DOI] [PubMed] [Google Scholar]