

Abstract
Modulation of the human ether-à-go-go-related gene (HERG) K+ channel was studied in two-electrode voltage-clamped Xenopus oocytes co-expressing the channel protein and the thyrotropin-releasing hormone (TRH) receptor.
Addition of TRH caused clear modifications of HERG channel gating kinetics. These variations consisted of an acceleration of deactivation, as shown by a faster decay of hyperpolarization-induced tail currents, and a slower time course of activation, measured using an envelope of tails protocol. The voltage dependence for activation was also shifted by nearly 20 mV in the depolarizing direction. Neither the inactivation nor the inactivation recovery rates were altered by TRH.
The alterations in activation gating parameters induced by TRH were demonstrated in a direct way by looking at the increased outward K+ currents elicited in extracellular solutions in which K+ was replaced by Cs+.
The effects of TRH were mimicked by direct pharmacological activation of protein kinase C (PKC) with β-phorbol 12-myristate, 13-acetate (PMA). The TRH-induced effects were antagonized by GF109203X, a highly specific inhibitor of PKC that also abolished the PMA-dependent regulation of the channels.
It is concluded that a PKC-dependent pathway links G protein-coupled receptors that activate phospholipase C to modulation of HERG channel gating. This provides a mechanism for the physiological regulation of cardiac function by phospholipase C-activating receptors, and for modulation of adenohypophysial neurosecretion in response to TRH.
The human ether-à-go-go (eag)-related gene (HERG)encodes a K+ channel (the HERG channel) that constitutes the molecular basis of the cardiac repolarizing K+ current, IKr (Sanguinetti et al. 1995; Trudeau et al. 1995). Malfunction of HERG channels is the cause of both inherited and acquired long-QT syndromes, characterized by an unusually slow repolarization of cardiac action potentials leading to cardiac arrhythmia and eventually ventricular fibrillation and sudden cardiac death (Curran et al. 1995; Sanguinetti et al. 1995; Spector et al. 1996a). Furthermore, HERG channels are molecular targets for widely used pharmacological agents such as class III anti-arrhythmics (Spector et al. 1996a) and some histamine receptor antagonists (Suessbrich et al. 1996). HERG channels were initially isolated from hippocampus, but their role in neuronal function is not completely understood. However, they have been implicated in the changes of the resting membrane potential associated with the cell cycle and in the control of neuritogenesis and differentiation in neuronal cells (Arcangeli et al. 1993, 1995; Faravelli et al. 1996). Finally, a recent report by Chiesa et al. (1997) indicated an important role for HERG channels in neuronal spike-frequency adaptation.
In spite of the physiological importance of HERG channels, little is known about their regulation by different neurotransmitters and/or hormone receptors. In GH3 rat anterior pituitary cells, regulation of an inwardly rectifying K+ current constitutes an important point for control of pacemaker activity in response to thyrotropin-releasing hormone (TRH; Barros et al. 1994, 1997). Such a regulation is exerted by means of a phosphorylation/dephosphorylation cycle triggered by a still unknown protein kinase, which is specifically reverted by protein phosphatase 2A (Barros et al. 1992, 1993; Delgado et al. 1992). Recent kinetic and pharmacological evidence indicates that a HERG-like K+ channel is the cause of the TRH-regulated inwardly rectifying K+ currents (Barros et al. 1997). The availability of cloned TRH receptors (TRH-Rs) and HERG channels allowed us to develop an in vitro assay to study the mechanism (s) of HERG regulation by co-expression of receptor and channel proteins.
Expression of HERG product in Xenopus oocytes yields depolarization-activated K+ currents which, as for GH3 cell currents, show strong inward rectification (Sanguinetti et al. 1995; Trudeau et al. 1995; Schönherr & Heinemann, 1996; Spector et al. 1996b; Wang et al. 1996, 1997). Recently it has been shown that this rectification arises from a C-type rapid inactivation mechanism (Schönherr & Heinemann, 1996; Smith et al. 1996; but see Wang et al. 1996, 1997) that reduces conductance at positive voltages and strongly limits the level of outward current after depolarizing the membrane. This precludes an accurate estimation of activation and inactivation parameters from direct measurements of outward currents, in which activation and inactivation properties overlap. In this report, we performed a characterization of the HERG gating properties by using an envelope of tail currents protocol. Both in oocytes and adenohypophysial cells, activation of phospholipase C (PLC) and generation of the two second messengers, inositol 1, 4, 5-trisphosphate (IP3) and diacylglycerol (DAG) are the prototypical consequences of TRH-R activation (de la Peña et al. 1992; Corette et al. 1995; Gershengorn & Osman, 1996). Our results with oocytes co-expressing HERG and TRH-R demonstrate clear alterations of HERG channel gating by TRH. Such alterations are manifested as an acceleration of deactivation and a slower time course of channel activation without any significant change in inactivation or inactivation recovery rates. The parallel between the effects of TRH and the protein kinase C (PKC)-specific activator β-phorbol 12-myristate, 13-acetate (PMA) indicates that a PKC-dependent pathway links the TRH-R to modulation of HERG. Our data also indicate that a phosphorylation triggered by activation of PKC is able to regulate channel gating properties by G protein-coupled receptors that generate PLC-dependent signals.
METHODS
Microinjection and electrophysiology of oocytes
Mature female Xenopus laevis (Nasco, Fort Atkinson, WI, USA) were anaesthetized by immersion in benzocaine solutions and subsequently maintained on ice in order to obtain oocytes. Ovarian lobes were removed through a small incision in the abdominal wall. After removal of the ovarian lobe, the frogs were sutured in the abdominal wall and in the external skin, and allowed to recover in a small water-filled container, with their heads elevated above water level. Once the animal had recovered from anaesthesia, it was placed in a separate aquarium by itself and periodically monitored until healed. Typically, lobes were obtained two or three times from a single frog, with several months in between. When individual frogs no longer yielded acceptable oocytes, anaesthetized frogs were killed by an overdose of benzocaine.
Procedures for microinjection and two-electrode voltage-clamp of oocytes have been described elsewhere (de la Peña et al. 1992, 1995; del Camino et al. 1997). Oocytes were maintained in OR-2 medium (mM: 82.5 NaCl, 2 KCl, 2 CaCl2, 2 MgCl2, 1 Na2HPO4 and 10 Hepes, at pH 7.5). Cytoplasmic microinjections were performed with 30–50 nl of in vitro synthesized cRNA per oocyte. HERG currents and TRH-induced responses were studied in manually defolliculated oocytes (de la Peña et al. 1992) using extracellular high-K+ OR-2 medium in which 50 mM KCl was substituted for an equivalent amount of NaCl. When indicated, a Cs+-containing extracellular solution was used constituted from OR-2 medium without K+, and in which NaCl was substituted with an equivalent amount of CsCl.
Functional expression was typically assessed 2–3 days after microinjection. Current measurements were performed at room temperature (20–25°C) using the two-electrode voltage-clamp method with a Turbo TEC 01C (NPI, Tamm, Germany) amplifier and 2 M KCl-filled glass capillaries showing DC resistances between 0.5 and 2.0 MΩ. Current recordings were obtained in an experimental chamber of 0.2 ml volume continuously perfused at 4–5 ml min−1. Membrane potential was clamped at −80 mV.
Channel deactivation and inactivation recovery kinetics were obtained from tail currents upon membrane repolarization, following depolarizing pulses that displaced the channels to open and inactive states. Repolarizations following depolarizing pulses lasted for 1–5 s to ensure total relaxation of tail currents and proper fits, but only the initial portion of the tails is shown in some graphs for clarity. By fitting an exponential to the initial rising phase and a second one to the decaying portion of the tails, both the rate of recovery from inactivation and the deactivation rate were measured, respectively.
The simultaneous occurrence of closed to open and open to inactive transitions at positive voltages due to the fast rate of HERG channel inactivation precludes an accurate estimation of activation and inactivation kinetic parameters by looking at the time-dependent development of outward currents. As an alternative, the time course of transitions from closed to open plus inactive states upon depolarization was monitored indirectly with an envelope of tail currents protocol, varying the duration of the depolarization pulse, fitting the relaxation of the tail currents recorded once the membrane is stepped back to a negative voltage, and extrapolating the current magnitude to the moment the depolarizing pulse was ended (Trudeau et al. 1995).
The time dependence of HERG fast inactivation from the open state can be determined by a dual-pulse protocol (Smith et al. 1996; Spector et al. 1996b; Schönherr & Heinemann, 1996) in which after a depolarization pulse to activate (and inactivate) the channels, the membrane is hyperpolarized briefly to allow them to recover from inactivation, entering the open state. Once the maximum inward current is obtained at negative voltages, the membrane is again depolarized to re-inactivate the channels. This re-inactivation is seen as a single exponential decline in current along the second depolarization pulse, from which the rate of the open to inactive transition can be obtained.
The data acquisition and analysis were performed using Apple Macintosh computers with Pulse/PulseFit (HEKA, Lambrecht, Germany) and IGOR (Wavemetrics, Lake Oswego, OR, USA) software. The output of the amplifier was digitized at 1 kHz. Records are shown without leak and capacitive correction unless otherwise indicated. In some cases, a P/4 protocol was performed for leak and capacitive current subtraction with a leak holding potential of −80 mV and a scaling factor of −0.2. Because the efficiency of the channel and receptor protein expression varied among oocyte batches, the data collected for the source batch of oocytes were compared for each set of experiment in the graphs. Nevertheless, the experiments were repeated at least three times with different batches from different donors. Data are presented as means ±s.e.m.
Plasmids and preparation of cRNA
Isolation of the TRH-R cDNA has been described previously (de la Peña et al. 1992). The plasmid containing the cDNA for the HERG channel was generously provided by Dr E. Wanke (University of Milano, Italy). Plasmids were linearized and capped cRNA was synthesized in vitro from the linear cDNA templates by standard methods using T7 or SP6 RNA polymerases as described (de la Peña et al. 1992).
Chemicals
TRH, PMA, KN-62, benzocaine and genistein were purchased from Sigma. GF109203X was from Calbiochem (San Diego, CA, USA). All other reagents were purchased from Sigma and were the highest quality available.
RESULTS
Effect of TRH-R activation on HERG channel gating transitions studied in K+-containing solutions
To determine whether the HERG channels may be modulated by receptor pathways that regulate PLC-dependent signals, we co-expressed the HERG channel protein with the TRH-R from rat adenohypophysial cells (de la Peña et al. 1992). Treatment of oocytes with 1 μM TRH resulted in fast activation of transient inward Cl− currents when the oocyte membrane was maintained at −80 mV (Fig. 1A, left). These currents, due to IP3-triggered release of Ca2+ from intracellular stores, were indistinguishable from those measured in oocytes without HERG channel transcripts (Fig. 1B, left; see de la Peña et al. 1992, 1995; del Camino et al. 1997). To test for receptor-mediated modulation of HERG currents, the co-injected oocytes clamped at −80 mV were submitted to a 400 ms depolarization at regular intervals followed by a repolarization to −100 mV. Without TRH addition, HERG-expressing oocytes bathed in solutions containing 50 mM K+ displayed only small outward currents (Fig. 1A, right). On repolarization to a negative membrane potential, a slow inward tail current is obtained with an initial rising phase as a result of fast inactivation removal, followed by a slow decay due to channel closing.
Figure 1. Effect of TRH-R activation on HERG tail current kinetics.
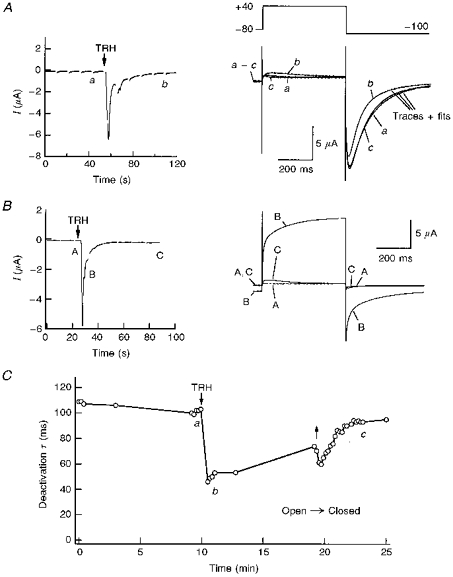
A, left, inward Ca2+-dependent Cl− currents in response to TRH. Representative inward Cl− currents in a voltage-clamped oocyte 2 days after co-injection of TRH-R and HERG channel cRNA are shown. The start of perfusion with high-K+ OR-2 plus 1 μM TRH is indicated. The current trace represents continuous segments obtained at −80 mV. The gaps on the trace indicate times at which a depolarization pulse was applied according to the voltage pulse diagram shown on the right. Right, membrane currents elicited by membrane depolarization at different times before (trace a) and after challenging the cell with TRH (traces b and c). Depolarization steps of 400 ms from −80 to +40 mV were delivered to the oocyte every 10 s, followed by a 400 ms repolarization to −100 mV. Membrane currents shown correspond to pulses delivered either 10 s before TRH addition (trace a), or 50 s after starting perfusion with 1 μM TRH (trace b), respectively. A current trace elicited with an identical protocol after 6 min of TRH washout (trace c) is also shown to illustrate the reversibility of the TRH effect. The TRH application lasted for about 9 min. Bi-exponential fits superimposed to the current traces are shown for −100 mV tails. Note the correspondence of current kinetics along the tail rising phase for control and TRH traces. B, left, inward Ca2+-dependent Cl− currents obtained in an oocyte injected with TRH-R cRNA but not with HERG channel messages. Right, membrane currents elicited by membrane depolarizations at the times marked A to C on the left. Identical conditions to those indicated in A were used. Note the fast monoexponential decay of the Cl− tail currents and their small magnitude 1 min after start of the TRH addition. C, time course of deactivation time constant variations in response to TRH. The time constant was quantified by fitting a double exponential to the tail as shown in A (right). Subsequently, the plotted deactivation time constant and the time constant for inactivation recovery were obtained from the decaying portion and the initial rising phase of the tail, respectively. The periods without data points correspond to times at which I-V curves were generated. Time constant values for the two pulses following introduction of TRH in the chamber, corresponding to periods of huge increases in inward Cl− currents, have not been included in the graph. Start of perfusion with 1 μM TRH and hormone washout is signalled by arrows. a-c correspond to the experimental times at which current traces marked with the same lettering in A were obtained.
Application of TRH caused a transient enhancement of outward currents recorded upon depolarization and of hyperpolarization-evoked inward tail currents. The transiently enhanced currents correspond to the Ca2+-activated Cl− currents triggered by the sudden release of Ca2+ from intracellular stores (see above). However, 1 or 2 min after the introduction of TRH, the hyperpolarization-evoked tail currents were obviously reduced (Fig. 1A, right). Such a reduction was mainly shown by a marked acceleration of decay. It is important to note that the reduction in tail currents was detected even before the basal current at −80 mV completely returned to its initial magnitude. However, an increased Cl− permeability in response to TRH would yield a bigger current at the negative voltages at which the tail currents are recorded. Further evidence that TRH-induced increases of oocyte Cl− conductance do not contribute significantly to the observed effects on the tail currents are as follows. (i) The modifications of the inward tail current kinetics lasted long after the initial Ca2+-dependent Cl− current increase was almost completely reverted (trace b in Fig. 1A). (ii) Delayed development of IP3-induced Cl− currents is associated with appearance of slowly rising non-inactivating hyperpolarization-evoked inward currents (Hartzell, 1996). This would cause an increase in the size of the tail currents and a slower rate of decay. However, the opposite effect is seen, and (iii) in oocytes expressing only TRH-Rs, 2 or 3 min after TRH addition the membrane currents recorded at −100 mV consisted exclusively of an instantaneous small inward current followed by a fast mono-exponential decay (Fig. 1B). This yields almost undetectable currents over the time range at which deactivation of HERG tail currents takes place. Altogether, this indicates that although the modifications induced by TRH treatment can be even larger than observed, the alterations in tail current kinetics are due to variations in the deactivation gating of the expressed HERG channels.
The results shown in Fig. 1A and C indicate that in spite of a certain variability in the kinetic parameters obtained from different oocytes, addition of TRH reduced the deactivation time constant by nearly a half (see also Table 1). The acceleration of the current deactivation was reversed several minutes after washing out TRH from the recording chamber. Interestingly, the reversion of the TRH effect was usually less than 50% during an exposure to the neuropeptide lasting up to 10 min. Furthermore, although a short (20–30 s) exposure to the hormone was systematically followed by a quite rapid recovery of the initial rates of tail current decay, only a slow and partial recovery was sometimes achieved following addition of TRH for several minutes (not shown).
Table 1.
Effect of thyrotropin-releasing hormone on HERG kinetic parameters
| Treatment | External solution | Activation t½ (ms) | n | N | Deactivation τ (ms) | Inactivation recovery τ (ms) | n | N |
|---|---|---|---|---|---|---|---|---|
| Control | 50 mm K+ | 30.6 ± 3.5 | 21 | 3 | 589 ± 64 | 9.4 ± 0.4 | 48 | 7 |
| TRH | 50 mm K+ | 66.0 ± 10.0* | 21 | 3 | 348 ± 30† | 10.4 ± 0.5 | 48 | 7 |
| Control | Cs+ | 13.3 ± 0.8 | 3 | — | 100 ± 4 | 14.2 ± 2.6 | 3 | — |
| TRH | Cs+ | 19.5 ± 1.8* | 3 | — | 64 ± 3* | 12.6 ± 2.7 | 3 | — |
P < 0.05
P < 0.001 vs. control
Student's paired t test. Times for half-maximal current activation (t½) were obtained from envelope of tail currents following the voltage protocol shown at the top of Fig. 2. Deactivation and inactivation recovery time constants (τ) were measured from bi-exponential fits to tail currents after repolarizing the membrane to −100 mV. n represents the number of oocytes used for the number of donors indicated by N.
HERG channel deactivation is a voltage-dependent process that occurs at a faster rate when the membrane potential is set at more negative voltages (Sanguinetti et al. 1995; Schönherr & Heinemann, 1996; Spector et al. 1996b). We measured a deactivation time constant of nearly 1 s at −60 mV, which was reduced to 20 ms at −140 mV. This time constant was 50% smaller in the presence of TRH at all voltages between −60 and −140 mV (data not shown).
As shown in Table 1, the rate of HERG recovery from inactivation was not significantly changed by TRH. Thus only minor variations in the time constant of the tail current rising phase were obtained in the presence of the hormone. A similar lack of effect was obtained at all voltages between −60 and −140 mV, at which the recovery process was studied (data not shown).
Due to superposition of relatively slow activation transitions and particularly fast inactivation rates upon depolarization, rigorous estimations of HERG activation rates can only be indirectly achieved by using an envelope of tail currents protocol (see Methods). As shown in Fig. 2, we obtained clear sigmoidal activation kinetics using this procedure. Furthermore, the time necessary to attain a half-maximal current magnitude was increased about twofold after treatment of the oocytes with TRH (Fig. 2 and Table 1).
Figure 2. Effect of TRH on HERG channel activation kinetics.
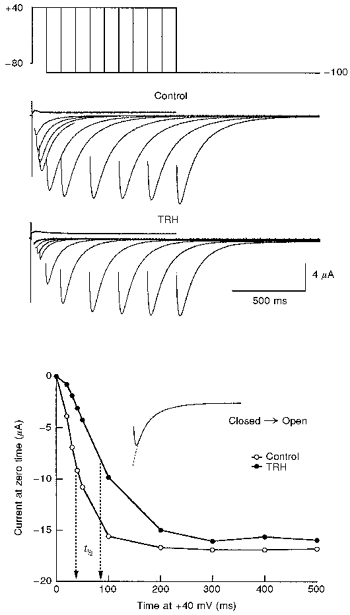
The time course of voltage-dependent activation was studied in the absence (control) or the presence of 1 μM TRH by varying the duration of a depolarizing prepulse to +40 mV according to the voltage protocol shown at the top. Test pulses were applied once every 20 s. For TRH, data collection started 2 min after challenging the cell with the neuropeptide. The magnitude of the instantaneous tail current at −100 mV was determined by fitting an exponential to the decaying portion of the tail as shown in the inset of the lower panel, and extrapolating the current to the moment the depolarizing pulse was ended. Capacitive transients have been blanked for clarity. Note the sigmoidal activation kinetics and the shift in the time necessary to attain a half-maximal current magnitude. For further explanation, see text.
The voltage dependence of the gating transitions during the depolarization pulses can be studied following a similar protocol in which the membrane potential is stepped to different voltages for a fixed time, and the extrapolated instantaneous tail current is measured after repolarizing the membrane to −100 mV (Fig. 3). The plot of the current magnitude vs. voltage was described by a Boltzmann equation with a V½ of near −20 mV. Treatment of the oocyte with TRH shifted the V½ value by 18 ± 2 mV (n = 3) in the depolarizing direction.
Figure 3. Effect of TRH on HERG channel voltage dependence of activation.
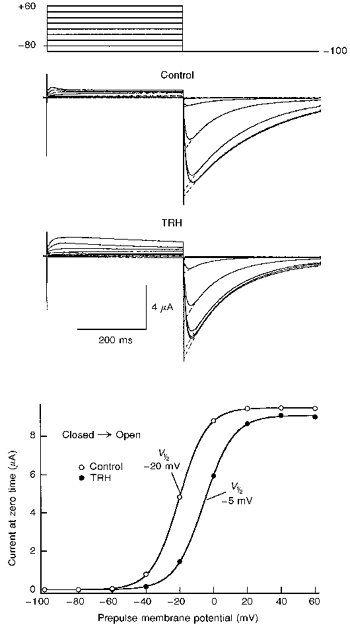
Voltage dependence of activation was studied in the absence (control) or the presence of 1 μM TRH by varying the magnitude of a 400 ms prepulse according to the voltage protocol shown at the top. Test pulses were applied once every 20 s. When the effect of TRH was tested, data collection started 2 min after challenging the cell with the neuropeptide. The magnitude of the instantaneous tail current at −100 mV was determined by fitting an exponential to the decaying portion of the tail as shown superimposed on the tail currents, and extrapolating the current to the moment the depolarizing pulse was ended. The continuous lines in the lower panel correspond to Boltzmann curves: h (V) =Imax (1/(1 + exp (V - V½)/k)), which best fitted to the data with V½ of −20 and −5 mV, Imax of 9.4 and 9.1 μA, and k values of −8.1 and −8.7 for control and TRH, respectively.
The time dependence of HERG fast inactivation from the open state was determined by a dual-pulse protocol, looking at the single exponential decline in current along the second depolarization pulse (see Methods). As shown in Fig. 4, the rate of inactivation was voltage dependent (see also Schönherr & Heinemann, 1996; Smith et al. 1996; Spector et al. 1996b; Wang et al. 1996). However, it remained unaltered at all voltages tested after challenging the cell with TRH. Failure to detect any change in the time course of inactivation was not due to a lack of TRH effects in this particular cell. As shown in the inset of Fig. 4, a clear acceleration of deactivation was induced in the same oocyte by the TRH treatment.
Figure 4. Lack of TRH effects on HERG channel inactivation kinetics.
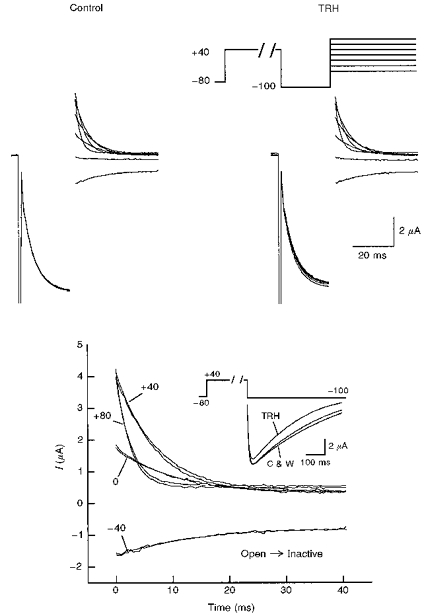
Onset of fast inactivation was studied using the voltage protocol shown at the top. HERG was activated and inactivated with a 400 ms prepulse to +40 mV. A 20 ms pulse to −100 mV was used to recover the channels from inactivation, followed by a test pulse to different voltages to re-inactivate the channels. Test pulses were applied once every 20 s. The decaying portion of the current during the test pulse is shown starting 2.5 ms after the hyperpolarizing pulse to −100 mV was ended. Membrane currents recorded at the end of the depolarization prepulse and along the 20 ms hyperpolarization pulse are also shown. The two families of current traces shown at the top were obtained in the absence (control) or starting 2 min after challenging the cell with 1 μM TRH. Inactivating decaying currents along the test pulses to −40, 0, +40 and +80 mV both in the absence or in the presence of TRH are shown superimposed in the lower panel. Note the almost exact correspondence of both currents at all tested voltages. TRH effects on inactivation kinetics were not observed even though the hormone induced a clear acceleration of the deactivation rate in the same oocyte, as shown in the inset. In this case, inward tail currents during long hyperpolarizations to −100 mV are shown. Currents were recorded either before (C), 90 s after start the TRH treatment (TRH), or after 10 min of hormone washout (W).
Altogether, these results indicate that activation of the TRH receptor modifies the activation and deactivation gating properties of HERG channels. Such a modification causes an acceleration of channel deactivation, a slower activation, and a shift in the voltage dependence of activation towards more positive voltages. However, neither the inactivation transitions from the open state nor the inactivation recovery process are influenced by treating the cells with TRH.
TRH-mediated modification of outward current kinetic parameters in Cs+-containing solutions
As indicated above, the superposition of quite unusual fast inactivation and slow activation kinetics makes it necessary to perform an indirect estimation of activation gating parameters through measurements of the recovery from open (O) plus inactive (I) states along the tail currents. However, as discussed below, this estimation can be biased by occurrence of inactivation directly from a closed state, that deviates from a sequential model for channel behaviour. To obtain a more direct demonstration of alterations in the activation rate, we took advantage from the fact that replacement of extracellular K+ by Cs+ causes a marked increase in outward currents (Schönherr & Heinemann, 1996). Although a direct effect of Cs+ replacement on closed (C) to O transitions cannot be completely excluded, we found that this outward current increase can be induced, at least in part, by a twofold slower rate of inactivation in the presence of external Cs+ (data not shown). In any case, we compared the speed of channel activation before and after challenging the oocytes with TRH, by measuring the time necessary to reach the peak of the outward current in extracellular Cs+. Figure 5A and B shows that, as predicted from the results obtained in K+-containing extracellular solutions, the activation time course was slowed by TRH at all voltages between −20 and 60 mV (see also Table 1). Furthermore, the neuropeptide caused a shift of near 20 mV in the voltage dependence for activation also in Cs+-containing solutions (Fig. 5C). Again, the kinetic alterations were not related to a modification of inactivation rates, since the time course of the O to I transitions remained unaltered after challenging the oocytes with TRH in Cs+-containing solutions (not shown). These results demonstrate in a more direct way that activation of the TRH-R is indeed able to slow down the activation kinetics of the HERG channels.
Figure 5. Effect of TRH on activation gating of HERG channels in Cs+-containing extracellular solutions.
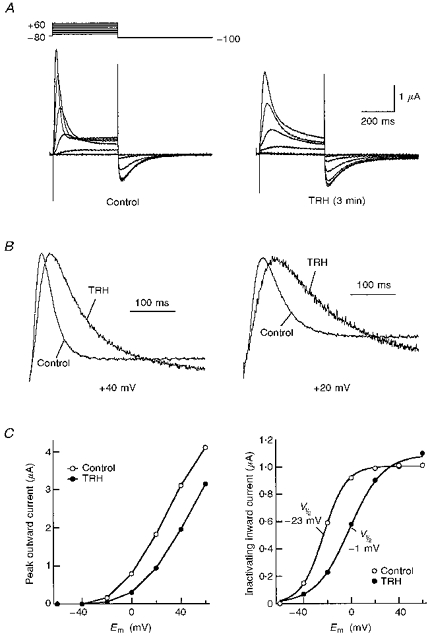
A, comparison of membrane currents elicited by membrane depolarization before (control) or 3 min after starting the treatment with 1 μM TRH (TRH). Pulses (400 ms) were applied to test potentials of −60 to +60 mV in 20 mV steps from a holding potential of −80 mV. Test pulses were applied once every 20 s. Current traces shown have been subtracted for leak. Extracellular solutions containing Cs+ instead of K+ were used. B, slower activation rates are induced by TRH on outward K+currents studied in Cs+-containing extracellular solutions. Outward currents obtained in the absence (control) or the presence of TRH are shown for depolarizing pulses to +40 (left) or +20 mV (right). For comparison, currents were normalized to peak. C, left, effect of TRH on the magnitude of outward currents as a function of membrane potential. Outward current magnitudes at the peak are plotted vs. test pulse potential (Em) for the currents shown in A. The data points are connected by straight lines. Right, effect of TRH on the magnitude of Cs+ inward tail currents as a function of membrane potential. The magnitude of the tail currents is plotted vs. test pulse potential for the currents shown in A. The continuous lines correspond to Boltzmann curves that best fitted the data with V½ of −23 and −1 mV, Imax of 1.0 and 1.1 μA, and k values of −9.5 and −14.2 for control and TRH, respectively.
The effects of TRH on activation and deactivation kinetics are mimicked by direct activation of PKC with β-phorbol 12-myristate, 13-acetate (PMA)
TRH-R activation of PLC leads to formation of diacylglycerol and activation of PKC, a Ca2+- and lipid-dependent kinase. To examine whether PKC modulates HERG channels we exposed oocytes expressing these channels to PMA, a pharmacologically specific activator of PKC. As with TRH, addition of PMA (100 nM) caused an acceleration of deactivation in a time-dependent manner. Although the PMA effect developed more slowly than that of TRH, 5–10 min after starting the treatment the time constant of tail current decay at −100 mV was reduced by 20% and 50–60% with 10 and 100 nM PMA, respectively (Fig. 6A). The effect of PMA was due to its action activating PKC. Thus pre-incubation of the oocytes with 3 μM GF109203X (a highly specific, non-toxic inhibitor of PKC; Crespo et al. 1994) for 1–3 h abolished the effect of 10 nM PMA and significantly reduced that of 100 nM PMA. Almost complete inhibition of the effect elicited by 100 nM PMA was achieved by raising the GF109203X concentration to 10 μM and the incubation time to 2–6 h (Fig. 6A). It is interesting that the effects of PMA were not accompanied by any modification of oocyte Ca2+-dependent Cl− currents. This further supports our conclusion that the alterations in current kinetics induced by TRH are not related to any effect of the hormone on Ca2+-dependent Cl− conductance.
Figure 6. Effect of PMA on HERG channel kinetics.
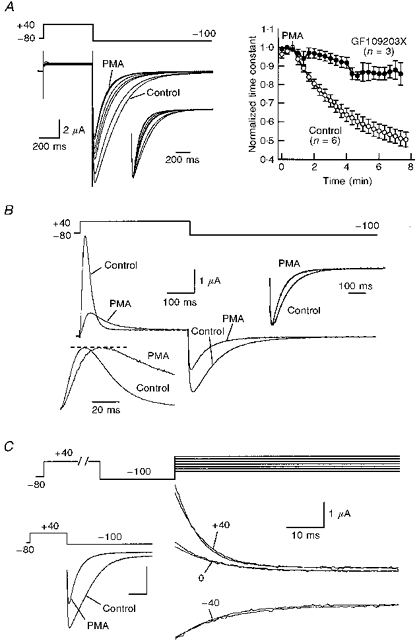
A, acceleration of deactivation rates in response to PMA. Superimposed current traces in response to the indicated potential protocol are shown on the left. Test pulses were delivered once every 20 s to an oocyte bathed in extracellular high-K+ medium. Only traces obtained before (control) and every minute after the start of 100 nM PMA addition are shown. Tail currents normalized to peak are shown in the inset for a better comparison of current kinetics. Note the correspondence of the initial rising phase of the tails. Averaged values of deactivation time constants for 6 control oocytes and 3 oocytes preincubated with 10 μM GF109203X for 2–3 h from the same donor are shown on the right. Similar results were obtained in 4 additional experiments with control oocytes (19 oocytes from 5 frogs) and 2 additional experiments with oocytes treated with GF109203X (11 oocytes from 3 frogs). Time constants were normalized to that measured at the time of PMA addition. B, slow-down of outward HERG current kinetics by PMA in Cs+-containing extracellular solutions. Membrane currents obtained with the voltage protocol shown at the top in the absence (control) or 5 min after addition of 100 nM PMA are shown superimposed. Outward currents normalized to peak are shown in the inset on the lower left for comparison. Normalized tail currents are also shown (inset, upper right). C, lack of PMA effect on HERG channel inactivation kinetics. Onset of fast inactivation was studied using an oocyte bathed in extracellular high-K+ medium, using the double-pulse protocol shown at the top (see legend to Fig. 5). Inactivating decaying currents along the test pulses to −40, 0 and +40 mV are shown both in the absence or presence of 100 nM PMA for 10 min. Note that currents in both conditions are practically indistinguishable. However, a clear effect of PMA on deactivation rates was observed in the same oocyte, as shown by a marked acceleration of hyperpolarization-induced tail current decay (inset, lower left corner).
To extend further the parallel between the actions of PMA and TRH, we also studied the effects of the PKC activator on activation and inactivation kinetic parameters. The rate of recovery from inactivation remained largely unaltered in the presence of PMA (not shown). This indicates that, as for TRH, the effects on tail current kinetics caused by direct activation of PKC with PMA are predominantly manifested in an acceleration of deactivation.
The results shown in Fig. 6B indicate that the activation gating transitions are slowed by challenging the cell with PMA. The time course of transitions from C to O plus I states was initially studied by varying the duration of the depolarization pulses and looking at the magnitude of the tail currents along a subsequent hyperpolarization (see above). In this case, a progressive increase in the activation time constant was obtained in the presence of PMA, leading to a twofold enhancement after 10 min of PMA treatment. This increase was significantly reduced in oocytes incubated with GF109203X (not shown). For a more direct demonstration that the C to O transition is slowed by PMA, we compared the speed of the outward current onset in Cs+-containing extracellular solutions. As shown in Fig. 6B, the activation time course of the current was clearly slowed by treatment with 100 nM PMA for 10 min. Furthermore, as with TRH, a nearly 20 mV shift in the depolarizing direction was also induced by PMA on the voltage dependence of activation in Cs+-containing solutions. Interestingly, the increase in the time to reach the peak of the outward current was paralleled by a 30% reduction in the time constant of the tail current decay (inset, Fig. 6B). This indicates that the acceleration of the deactivation process by PKC activation is exerted regardless of the nature of the permeant ion carrying the inward current: Cs+ instead of K+ in the experiment shown in Fig. 6B.
The clear parallel between the effects of TRH and PMA on kinetic properties of HERG channels is strengthened by the lack of PMA effects on fast inactivation. As shown in Fig. 6C, the time course of inactivation from the open state remained unaltered in the presence of PMA, although the tail inward currents were ostensibly modified by the drug in the same cell (Fig. 6C, inset). Altogether, this suggests that modulation of HERG channel gating properties by the PLC-activating TRH-R is probably achieved through a pathway at least partially dependent on PKC activation.
Effects of protein kinase inhibitors on regulation of HERG deactivation by TRH
As a final demonstration that a PKC-dependent mechanism participates in regulation of HERG gating by TRH, the effect of the neuropeptide was studied in the presence of GF109203X, under conditions known to eliminate the PKC-dependent PMA-mediated modification of HERG kinetics (see above). As shown in Fig. 7A, the acceleration of HERG tail current decay in response to TRH was abolished in oocytes treated with GF109203X. In this case, a limited but persistent increase of deactivation rates was usually induced by the hormone, which paralleled the reversion of the TRH effect in control untreated oocytes upon hormone washout. The reason for this behaviour is not known. However, it seems to be a consequence of the TRH treatment, since it was not observed upon addition of PMA to oocytes incubated with GF109203X.
Figure 7. Effect of different protein kinase inhibitors on TRH-induced modification of HERG deactivation kinetics.
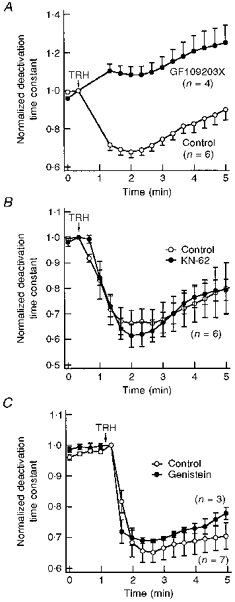
A, blockade of the TRH effect by the PKC inhibitor GF109203X. Deactivation time constants normalized to that measured at the moment of TRH addition are shown for untreated oocytes (control) or oocytes incubated for 4–6 h with 10 μM GF109203X. Test pulses were delivered to the cells every 20 s. Time constant values for the two pulses following introduction of TRH into the chamber have been deleted for clarity. The start of a 30 s perfusion with medium containing 1 μM TRH is indicated. B, TRH-induced effects in the presence of the Ca2+-calmodulin protein kinase II inhibitor KN-62. Variations in the deactivation time constant for untreated oocytes (control) and oocytes incubated for 1–3 h with 20 μM KN-62 are shown. The start of a 30 s perfusion with medium containing 1 μM TRH is indicated at the top of the graph. C, TRH-induced effects in the presence of the tyrosine kinase inhibitor genistein. Normalized deactivation time constant values are shown for oocytes injected with a micropipette filled with 100 μM genistein (final intracellular concentration approximately 5 μM genistein) or vehicle (control) 30 min before start of HERG current recordings. The start of perfusion with 1 μM TRH is indicated. In this case, the hormone remained present up to the end of the experiments.
Besides an activating effect of TRH on PKC-dependent protein phosphorylation, it could also be expected that elevations of cytoplasmic Ca2+ concentration after liberation of the cation from intracellular stores would lead to activation of Ca2+-dependent enzymatic activities such as Ca2+-calmodulin protein kinase II. This prompted us to check whether this Ca2+-dependent phosphorylating activity is involved in the effects of the neuropeptide. As shown in Fig. 7B, the TRH-dependent acceleration of deactivation kinetics remained unaltered in oocytes treated with the highly specific Ca2+-calmodulin protein kinase II inhibitor KN-62. Finally, a similar acceleration of tail current decay was also induced by TRH in the presence of the tyrosine kinase inhibitor genistein (Fig. 7C). This indicates that, unlike previous results obtained with Kv1.2 K+ channels (Huang et al. 1993), the PKC-dependent effects on HERG channel gating are not exerted through a transduction cascade, which implicates a tyrosine kinase that directly phosphorylates the channel protein.
DISCUSSION
In this study we show that the gating of HERG channels expressed in oocytes is modulated by the G protein-coupled TRH-R through a pathway involving PLC stimulation and activation of PKC. By co-expressing the cloned TRH-R and HERG channel in the oocyte model system, our results demonstrate that activation of TRH-R accelerates channel deactivation and slows down the activation rate. These effects are specifically exerted on voltage-dependent conformational changes leading to channel activation and closure, since neither the inactivation transitions from the open state nor the inactivation recovery rates are modified after challenging the oocytes with TRH.
PKC is activated physiologically by the co-ordinate action of DAG and Ca2+ in the presence of phosphatidylserine. Our data indicate that the PKC branch of the PLC signalling cascade participates in modulation of HERG channel gating. Thus the effect of TRH was mimicked by direct pharmacological activation of PKC with PMA. Interestingly, as with TRH, only channel activation and deactivation parameters, but not the onset or recovery rates of inactivation were modified by PMA, emphasizing the parallel between the actions of both regulators. Furthermore, the TRH effect was antagonized by GF109203X, a highly specific inhibitor of PKC that also abolished the PMA-dependent regulation of the channels, but not by the inhibitors of tyrosine kinases and Ca2+-calmodulin serine/threonine kinase, genistein and KN-62. However, it is important to note that our data do not allow us to distinguish between a direct phosphorylating action of PKC on the channel (or an auxiliary subunit associated to it), or an indirect effect of PKC on an early step of the transduction cascade.
A remarkable characteristic of HERG channels is the presence of a rapid and voltage-dependent inactivation process that reduces conductance at positive voltages and strongly limits the level of outward current after depolarizing the membrane. This precludes the accurate estimation of activation kinetic parameters by looking at the time-dependent development of outward currents. Assuming a sequential model for channel behaviour (C1 … Cn ⇌ O ⇌ I), the activation transitions can be characterized by using an envelope of tail currents protocol (see Methods). However, this would yield an estimation only of the rate at which the channels leave the closed state either through the open state or directly to the inactive state, if inactivation occurs directly from a closed state with a similar time dependence. Such a direct inactivation pathway has been previously observed for other structurally related voltage-dependent K+ channels such as Kv1.3 (Marom & Levitan, 1994) and Kv1.4 (F. Barros and P. de la Peña, unpublished data). It could also constitute a relevant pathway for HERG, in which inactivation seems to develop more quickly than activation (Spector et al. 1996b). Furthermore, this would complicate the interpretation of the regulatory effects of TRH and PMA on channel kinetics. Nevertheless, our results strongly suggest that both agents certainly induce a slowing of the activation time course besides an acceleration of deactivation. Thus the delaying actions of TRH and PMA were also demonstrated in a more direct way by looking at the markedly increased outward currents elicited in extracellular solutions in which K+ had been replaced by Cs+. It would be interesting to know whether similar effects to those reported here can be obtained with channel variants from which inactivation has been removed by site-directed mutagenesis (Schönherr & Heinemann, 1996; Smith et al. 1996).
The more generalized model for K+ channel behaviour (Zagotta & Aldrich, 1990; Hoshi et al. 1994) involves a voltage-dependent channel gating. Subsequently, many channels of the Shaker superfamily undergo inactivation by N-type or C-type mechanisms. The inactivation is generally regarded as a voltage-insensitive process in which any apparent voltage dependence arises from direct coupling to activation. In such a theoretical background it could be possible that, as shown here for HERG channels, phosphorylation may act by altering the voltage-dependent gating without significant modifications of the intrinsically voltage-independent transitions leading to inactivation. However, the fast time-dependent inactivation of HERG is strongly voltage dependent, both in the hyperpolarized and depolarized ranges of membrane potentials. Such a voltage dependence does not seem to arise from activation (Wang et al. 1996, 1997). Thus if phosphorylation of HERG exclusively disrupts the electrostatic interactions that control gating (Perozo & Bezanilla, 1990), it would be necessary that the activation and the inactivation apparatus lie in a different protein domain. Further knowledge of the structure of this channel would be necessary to prove such a hypothesis.
Previous work with Kv3.4 channels showed a reduction of channel inactivation through a direct PKC-dependent phosphorylation of sites located at the N-terminus (Covarrubias et al. 1994). The phosphorylation of the Kv3.4 inactivation gate modulates N-type inactivation in this channel. However, a C-type and not an N-type mechanism governs the inactivation of HERG (Schönherr & Heinemann, 1996; Smith et al. 1996; but see also Wang et al. 1996, 1997). Furthermore, the N-terminal domain has been implicated in the activation and deactivation of HERG, but not in the inactivation mechanism (Schönherr & Heinemann, 1996; Spector et al. 1996b). Thus the specificity of the TRH and PMA effects on activation and deactivation parameters would be coherent with the idea that a direct phosphorylation of the N-terminal domain by PKC regulates the gating properties of HERG channels. This is also supported by the marked acceleration of channel deactivation caused by deletion of the HERG cytoplasmic amino terminus without a severe change in inactivation kinetics (Schönherr & Heinemann, 1996; Spector et al. 1996b). As for other related channels, a net positive charge is present within the first thirty amino acids of HERG. It remains to be established whether phosphorylation of this or a nearby domain may disrupt some electrostatic interactions affecting channel gating.
It is important to note that although it is possible that PKC-dependent phosphorylation of the HERG N-terminal domain participates in channel regulation, other possibilities also exist. Thus phosphorylation of a C-terminal site by protein kinase A modifies interaction of the Shaker N-terminal domain with its receptor site(s) in the core of the protein (Drain et al. 1994). Similarly, phosphorylation of the ‘chain’ connecting the N-terminal inactivation ball of Kv1.4 with the core of the channel protein seems to regulate the interactions between the inactivation particle and its receptor site (Roeper et al. 1997). Clearly, further experiments are necessary to define the specific site(s) phosphorylated by PKC and its relevance in channel function.
The importance of HERG currents in regulating cardiac beat and action potential duration has been largely recognized. More recently, HERG (Curran et al. 1995; Sanguinetti et al. 1995; Spector et al. 1996a) and minK/KvLQT1 (Chouabe et al. 1997) channel malfunction has been implicated in some types of cardiac arrhythmia. The physiological relevance of the regulatory mechanism described here is twofold. Firstly, it is unlikely that TRH receptors are expressed in cardiac tissues, but the participation of a PKC-dependent pathway in channel regulation makes it generally applicable to other PLC-coupled receptors. Thus analogous effects to those caused by TRH-R activation are observed in response to serotonin when oocytes co-expressing HERG and serotonin-1c receptors are used (F. Barros and P. de la Peña, unpublished data). Since different transducers are used to couple TRH and serotonin-1c receptors to oocyte PLC activation (Quick et al. 1994; de la Peña et al. 1995), these results further emphasize the general significance of the mechanism described here for regulation of HERG channels, and subsequently of cardiac function. Interestingly, PMA-induced phosphorylation of minK by PKC also inhibits the cardiac slow delayed rectifier current IKs and shifts its activation voltage dependence to more depolarized voltages (Busch et al. 1992). Thus our results implicate for the first time the activation of physiologically active hormone and neurotransmitter receptors coupled to PLC in the concerted regulation of the two K+ current components that repolarize the cardiac action potential: IKr, contributed by HERG (Sanguinetti et al. 1995; Trudeau et al. 1995), and IKs, contributed by minK/KvLQT1 (Barhanin et al. 1996; Sanguinetti et al. 1996). Secondly, although obtained in the oocyte model system, our results can provide an answer to a long-standing question about how TRH modulates neurosecretion along the named second phase of hormone action. Inwardly rectifying K+ currents constitute an important TRH target for control of electrical activity in adenohypophysial cells (Barros et al. 1994, 1997). Recent kinetic and pharmacological evidence indicates that a HERG-like channel is the cause of such TRH-regulated currents (Barros et al. 1997). Furthermore, a phosphorylation mechanism is involved in both inwardly rectifying current inhibition and action potential frequency increases in response to TRH (Barros et al. 1992, 1993; Delgado et al. 1992). It should be noted, however, that the participation of PKC in electrical responses to TRH has remained controversial (Barros et al. 1992, 1993, 1994), and an unknown protein kinase distinct from PKC and protein kinase A has been implicated in regulation of GH3 cell HERG-like currents by TRH (Barros et al. 1993). Whether this is due to differences in some molecular characteristics between HERG channels and their adenohypophysial counterparts remains to be established. It would be interesting to know if a mechanism similar to that reported here modulates the function of the rat erg channel recently isolated from GH3/B6 cells (contributed by B. Engeland and J. Ludwig, under accession number Z96106).
Acknowledgments
We thank Dr Enzo Wanke for kindly providing the HERG channel-containing plasmid and Dr José López-Barneo and Dr Gary Yellen for critical comments on the manuscript. We also gratefully acknowledge the continuous encouragement and support from Dr Luis A. Pardo and Dr Walter Stühmer. C. G. V. holds a pre-doctoral fellowship from DGCYT of Spain. T. P. and T. G. are predoctoral fellows from FICYT of Asturias and the University of Oviedo, respectively. This work was supported by grants PB93-1076 and PB96-0316 from DGCYT of Spain.
References
- Arcangeli A, Becchetti A, Mannini A, Mugnai G, De Filippi P, Tarone G, Del Bene MR, Barletta E, Wanke E, Olivotto M. Integrin-mediated neurite outgrowth in neuroblastoma cells depends on the activation of potassium channels. Journal of Cell Biology. 1993;122:1131–1143. doi: 10.1083/jcb.122.5.1131. 10.1083/jcb.122.5.1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arcangeli A, Bianchi L, Becchetti A, Faravelli L, Coronnello M, Mini E, Olivotto M, Wanke E. A novel inward-rectifying K+ current with a cell-cycle dependence governs the resting potential of mammalian neuroblastoma cells. The Journal of Physiology. 1995;489:455–471. doi: 10.1113/jphysiol.1995.sp021065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barhanin J, Lesage F, Guillemare E, Fink M, Lazdunski M, Romey G. KvLQT1 and IsK (minK) proteins associate to form the IKs cardiac potassium current. Nature. 1996;384:78–80. doi: 10.1038/384078a0. [DOI] [PubMed] [Google Scholar]
- Barros F, del Camino D, Pardo LA, Palomero T, Giráldez T, de la Peña P. Demonstration of an inwardly rectifying K+ current component modulated by thyrotropin-releasing hormone and caffeine in GH3 rat anterior pituitary cells. Pflügers Archiv. 1997;435:119–129. doi: 10.1007/s004240050491. [DOI] [PubMed] [Google Scholar]
- Barros F, Delgado LM, Del Camino D, de la Peña P. Characteristics and modulation by thyrotropin-releasing hormone of an inwardly rectifying K+ current in patch-perforated GH3 anterior pituitary cells. Pflügers Archiv. 1992;422:31–39. doi: 10.1007/BF00381510. [DOI] [PubMed] [Google Scholar]
- Barros F, Mieskes G, de l Camino D, de la Peña P. Phosphatase 2A reverses inhibition of inward rectifying K+ currents by thyrotropin-releasing hormone in GH3 pituitary cells. FEBS Letters. 1993;336:433–439. doi: 10.1016/0014-5793(93)80851-k. [DOI] [PubMed] [Google Scholar]
- Barros F, Villalobos C, García-Sancho J, Del Camino D, de la Peña P. The role of the inwardly rectifying K+ current in resting potential and thyrotropin-releasing hormone-induced changes in cell excitability of GH3 rat anterior pituitary cells. Pflügers Archiv. 1994;426:221–230. doi: 10.1007/BF00374775. [DOI] [PubMed] [Google Scholar]
- Busch AE, Varnum MD, North RA, Adelman JP. An amino acid mutation in a potassium channel that prevents inhibition by protein kinase C. Science. 1992;255:1705–1707. doi: 10.1126/science.1553557. [DOI] [PubMed] [Google Scholar]
- Chiesa N, Rosati B, Arcangeli A, Olivotto M, Wanke E. A novel role for HERG K+ channels: spike-frequency adaptation. The Journal of Physiology. 1997;501:313–318. doi: 10.1111/j.1469-7793.1997.313bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chouabe C, Neyroud N, Guicheney P, Lazdunski M, Romey G, Barhanin J. Properties of KvLQT1 K+ channel mutations in Romano-Ward and Jervell and Lange-Nielsen inherited cardiac arrhythmias. EMBO Journal. 1997;16:5472–5479. doi: 10.1093/emboj/16.17.5472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corette B, Bauer J, Schwarz JR. Electrophysiology of anterior pituitary cells. In: Scherübl H, Hescheler J, editors. The Electrophysiology of Neuroendocrine Cells. Boca Raton, FL, USA: CRC Press Inc; 1995. pp. 101–143. [Google Scholar]
- Covarrubias M, Wei A, Salkoff L, Vyas TB. Elimination of rapid potassium channel inactivation by phosphorylation of the inactivation gate. Neuron. 1994;13:1403–1412. doi: 10.1016/0896-6273(94)90425-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo P, Xu N, Daniotti JL, Troppmair J, Rapp UR, Gutkind JS. Signaling through transforming G protein-coupled receptors in NIH 3T3 cells involves c-Raf activation. Evidence for a protein kinase C-independent pathway. Journal of Biological Chemistry. 1994;269:21103–21109. [PubMed] [Google Scholar]
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–804. doi: 10.1016/0092-8674(95)90358-5. [DOI] [PubMed] [Google Scholar]
- de la Peña P, Dela Camino D, Pardo LA, Domínguez P, Barros F. Gs couples thyrotropin-releasing hormone receptors expressed in Xenopus oocytes to phospholipase C. Journal of Biological Chemistry. 1995;270:3554–3559. doi: 10.1074/jbc.270.8.3554. [DOI] [PubMed] [Google Scholar]
- de la Peña P, Delgado LM, Dela Camino D, Barros F. Cloning and expression of the thyrotropin-releasing hormone receptor from GH3 rat anterior pituitary cells. Biochemical Journal. 1992;284:891–899. doi: 10.1042/bj2840891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Camino D, Barros F, Pardo LA, de la Peña P. Altered ligand dissociation rates in thyrotropin-releasing hormone receptors mutated in glutamine 105 of transmembrane helix III. Biochemistry. 1997;36:3308–3318. doi: 10.1021/bi9622534. [DOI] [PubMed] [Google Scholar]
- Delgado LM, de la Peña P, Del Camino D, Barros F. Okadaic acid and calyculin A enhance the effect of thyrotropin-releasing hormone on GH3 rat anterior pituitary cells excitability. FEBS Letters. 1992;311:41–45. doi: 10.1016/0014-5793(92)81362-p. [DOI] [PubMed] [Google Scholar]
- Drain P, Dubin AE, Aldrich RW. Regulation of Shaker K+ channel inactivation gating by the cAMP-dependent protein kinase. Neuron. 1994;12:1097–1109. doi: 10.1016/0896-6273(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Faravelli L, Arcangeli A, Olivotto M, Wanke E. A HERG-like K+ channel in rat F-11 DRG cell line: pharmacological identification and biophysical characterization. The Journal of Physiology. 1996;496:13–23. doi: 10.1113/jphysiol.1996.sp021661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gershengorn MC, Osman R. Molecular and cell biology of thyrotropin-releasing hormone receptors. Physiological Reviews. 1996;76:175–191. doi: 10.1152/physrev.1996.76.1.175. [DOI] [PubMed] [Google Scholar]
- Hartzell HC. Activation of different Cl currents in Xenopus oocytes by Ca liberated from stores and by capacitative Ca influx. Journal of General Physiology. 1996;108:157–175. doi: 10.1085/jgp.108.3.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi T, Zagotta WN, Aldrich R. Shaker potassium channel gating I: transitions near the open state. Journal of General Physiology. 1994;103:249–278. doi: 10.1085/jgp.103.2.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X-Y, Morielli AD, Peralta EG. Tyrosine kinase-dependent suppression of a potassium channel by the G protein-coupled m1 muscarinic acetylcholine receptor. Cell. 1993;75:1145–1158. doi: 10.1016/0092-8674(93)90324-j. [DOI] [PubMed] [Google Scholar]
- Marom S, Levitan IB. State-dependent inactivation of the Kv3 potassium channel. Biophysical Journal. 1994;67:579–589. doi: 10.1016/S0006-3495(94)80517-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo E, Bezanilla F. Phosphorylation affects voltage gating of the delayed rectifier K+ channel by electrostatic interactions. Neuron. 1990;5:685–690. doi: 10.1016/0896-6273(90)90222-2. [DOI] [PubMed] [Google Scholar]
- Quick MW, Simon MI, Davidson N, Lester HA, Aragay AM. Differential coupling of G protein α subunits to seven-helix receptors expressed in Xenopus oocytes. Journal of Biological Chemistry. 1994;269:30164–30172. [PubMed] [Google Scholar]
- Roeper J, Lorra C, Pongs O. Frequency-dependent inactivation of mammalian A-type K+ channel Kv1.4 regulated by Ca2+/calmodulin-dependent protein kinase. Journal of Neuroscience. 1997;17:3379–3391. doi: 10.1523/JNEUROSCI.17-10-03379.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanguinetti MC, Curran ME, Zou A, Shen J, Spector PS, Atkinson DL, Keating MT. Coassembly of KvLQT1 and minK (IsK) proteins to form cardiac IKs potassium channel. Nature. 1996;384:80–83. doi: 10.1038/384080a0. [DOI] [PubMed] [Google Scholar]
- Sanguinetti MC, Jiang C, Curran ME, Keating MT. A mechanistic link between an inherited and an acquired cardiac arrhythmia: HERG channel encodes the IKr potassium channel. Cell. 1995;81:299–307. doi: 10.1016/0092-8674(95)90340-2. [DOI] [PubMed] [Google Scholar]
- Schönherr R, Heinemann SH. Molecular determinants for activation and inactivation of HERG, a human inward rectifier potassium channel. The Journal of Physiology. 1996;493:635–642. doi: 10.1113/jphysiol.1996.sp021410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith PL, Baukrowitz T, Yellen G. The inward rectification mechanism of the HERG cardiac potassium channel. Nature. 1996;379:833–836. doi: 10.1038/379833a0. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Keating MT, Sanguinetti MC. Class III antiarrhythmic drugs block HERG, a human cardiac delayed rectifier K+ channel. Circulation Research. 1996a;78:499–503. doi: 10.1161/01.res.78.3.499. [DOI] [PubMed] [Google Scholar]
- Spector PS, Curran ME, Zou A, Keating MT, Sanguinetti MC. Fast inactivation causes rectification of the IKr channel. Journal of General Physiology. 1996b;107:611–619. doi: 10.1085/jgp.107.5.611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suessbrich H, Waldegger S, Lang F, Busch AE. Blockade of HERG channels expressed in Xenopus oocytes by the histamine receptor antagonists terfenadine and astemizole. FEBS Letters. 1996;385:77–80. doi: 10.1016/0014-5793(96)00355-9. [DOI] [PubMed] [Google Scholar]
- Trudeau MC, Warmke JW, Ganetzky B, Robertson GA. HERG, a human inward rectifier in the voltage-gated potassium channel family. Science. 1995;269:92–95. doi: 10.1126/science.7604285. [DOI] [PubMed] [Google Scholar]
- Wang S, Liu S, Morales MJ, Strauss HC, Rasmusson RL. A quantitative analysis of the activation and inactivation kinetics of HERG expressed in Xenopus oocytes. The Journal of Physiology. 1997;502:45–60. doi: 10.1111/j.1469-7793.1997.045bl.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Morales MJ, Liu S, Strauss HC, Rasmusson RL. Time, voltage and ionic concentration dependence of rectification of h-erg expressed in Xenopus oocytes. FEBS Letters. 1996;389:167–173. doi: 10.1016/0014-5793(96)00570-4. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Aldrich RW. Voltage-dependent gating of Shaker A-type potassium channels in Drosophila muscle. Journal of General Physiology. 1990;95:29–60. doi: 10.1085/jgp.95.1.29. [DOI] [PMC free article] [PubMed] [Google Scholar]