

Abstract
To examine the contributions of the putative Ca2+ releasers, inositol 1,4,5-trisphosphate (InsP3), cyclic ADP ribose (cADPR), and nicotinate adenine dinucleotide phosphate (NAADP), to carbachol (CCh)-induced contraction in airway smooth muscle, we measured force development of permeabilized rabbit tracheal smooth muscle, human bronchial smooth muscle and guinea-pig ileum longitudinal smooth muscle.
In the presence of 50 μm GTP, CCh and InsP3 contracted α-toxin-permeabilized tracheal smooth muscle dose dependently; the EC50 values for CCh and InsP3 were 1.84 μm and 363 μm, and the maximum responses (normalized to the 30 mM caffeine response) to 100 μm CCh and to 800 μm InsP3 were 206 ± 13.4 % (mean ± s.e.m.) and 84.4 ± 5.3 %, respectively.
However, cADPR (10-300 μm), β-NAD+ (2.5 mM), FK506 (30 μm) and NAADP (100 μm) neither contracted the strip by themselves nor affected the subsequent CCh (1 μm) response. α-Toxin-permeabilized bronchial smooth muscle and ileum smooth muscle also responded to caffeine, InsP3 and CCh but not to cADPR.
Both 100 μm 8-amino-cADPR, a selective cADPR antagonist, and 100 μm thionicotinamide-NADP, a selective NAADP antagonist, failed to inhibit the CCh response, although procaine abolished the caffeine, InsP3 and CCh responses in the permeabilized tracheal smooth muscle.
Although inhibition of the caffeine response by 30 μm ryanodine was nearly complete, approximately 30 % of the InsP3 (300 μm) plus GTP (50 μm) response was retained, and the resultant response disappeared after the caffeine response was evoked in the presence of ryanodine.
Heparin (300 μg ml−1) blocked InsP3 (300 μm) and CCh (3 μm) responses in β-escin-permeabilized tracheal smooth muscle, while Ruthenium Red (100 μm) partially inhibited the CCh response.
Collectively, InsP3 but not cADPR or NAADP plays a key role in CCh-initiated contraction, and InsP3 utilizes a single compartment of the caffeine/ryanodine-sensitive stored Ca2+ in airway smooth muscle.
It is widely accepted that inositol 1,4,5-trisphosphate (InsP3) plays an important role in the initiation of the agonist-induced contraction via Ca2+ release from the sarcoplasmic reticulum (SR) of vascular and other visceral smooth muscle (SM) cells (Abdel-Latif et al. 1977; Somlyo et al. 1985; Somlyo & Somlyo, 1994). Established evidence is based on phosphoinositide metabolism (InsP3 generation) (Seager et al. 1994), the capability of Ca2+ mobilization from SR, contraction by InsP3 (Kitazawa et al. 1989; Kobayashi et al. 1989; Iino, 1990; Seager et al. 1994) and its kinetic analysis by caged compound experiments in these types of SM cells (Somlyo et al. 1992).
In airway SM, there has been speculation about similar mechanisms by analogy with other types of SM. Indeed in animal (Baron et al. 1984; Hashimoto et al. 1985) and in human airway SM (Meurs et al. 1989; Van Amsterdam et al. 1990), phosphoinositide metabolism has been studied extensively; InsP3 production was accompanied by agonist-induced contraction (Baron et al. 1984), and InsP3 evoked 45Ca2+ release from SR of saponin-permeabilized canine (Hashimoto et al. 1985) and rabbit (Chopra et al. 1991) tracheal SM cells which were enzymatically dispersed and cultured. However, InsP3 levels produced by agonists varied in airway SM (Hashimoto et al. 1985; Meurs et al. 1989). Furthermore, in intact guinea-pig trachea 5-hydroxytryptamine mobilized Ca2+ in the absence of extracellular Ca2+ without changes in InsP3 generation (Watts et al. 1994). To our knowledge, direct evidence of InsP3-induced contraction is still lacking in airway SM. Thus, the role of InsP3 in the initiation of agonist-induced contraction of airway SM remains to be elucidated.
Recently, cyclic ADP ribose (cADPR) has been highlighted as another potent Ca2+ releaser from intracellular Ca2+ stores in sea urchin eggs (Lee et al. 1989), neuronal cells, heart, brain, pancreatic β-cells (Galione & Sethi, 1996), and alveolar macrophages (Ebihara et al. 1997). cADPR is thought to be a candidate intrinsic regulator for the ryanodine receptor (RyR) in the SR (Dousa et al. 1996; Galione & Sethi, 1996). In intestinal ileum longitudinal but not circular SM cells, agonist-induced cADPR generation and SM contraction have also been suggested (Kuemmerle & Makhlouf, 1995; Murthy et al. 1995). More recently, the third Ca2+ mobilizing agent, nicotinate adenine dinucleotide phosphate (NAADP) was discovered in sea urchin eggs (Chini et al. 1995; Lee & Aarhus, 1995). However, the effects of cADPR and NAADP on Ca2+ mobilization from the intracellular Ca2+ pool in airway SM has not been studied.
The aim of this study was to determine whether the classical (InsP3) and novel (cADPR and NAADP) Ca2+ releasers contribute to muscarinic acetylcholine agonist-induced contraction of airway SM via Ca2+ mobilization. We permeabilized rabbit tracheal SM with Staphylococcus aureusα-toxin or β-escin, and measured transient isometric force development in response to InsP3, caffeine, cADPR, NAADP, aluminum fluoride (AlF4−), and a muscarinic acetylcholine receptor agonist, carbachol (CCh). We also examined the responses to InsP3, caffeine, cADPR, and CCh in α-toxin-permeabilized human bronchial SM and guinea-pig ileum longitudinal SM. Furthermore, inhibitory actions of heparin, Ruthenium Red, procaine, and ryanodine were determined in either α-toxin- or β-escin-permeabilized tracheal SM. Human bronchial SM was used to estimate species difference and to compare responses to the reagents in airway SM obtained from a different portion of the bronchial tree. We also employed guinea-pig ileum longitudinal SM because cADPR was reported as a putative Ca2+ releaser in this tissue (Murthy et al. 1995).
METHODS
Preparation of airway SM strips
The trachea was removed from Japanese albino rabbits weighing 2.5-3.0 kg under halothane anaesthesia. The anaesthesia was administered by placing the animals in an anaesthetic chamber until the animals became anaesthetized and unresponsive to corneal stimulation. When the tracheal tissue had been removed, the animals were killed by rapid exsanguination through the carotid artery, in accordance with the recommendations of the Council of Animal Care in Gunma University, Japan. The airways were first cut longitudinally at the centre of the cartilage opposite the SM. Small strips of tracheal SM (width 400 μm; thickness 40–50 μm; length 3.0 mm) were carefully separated from connective tissue, epithelium and cartilage with a razor-blade under a binocular microscope. The strips were then tied with silk monofilaments to two tungsten needles, one of which was connected to an isometric force transducer (Akers, AE801, Horten, Norway), and mounted in wells on a bubble plate whose surface was coated with Teflon. The transducer was connected to an amplifier and the output was connected to a Macintosh computer through an analog-digital converter (MacLab, Analog-Digital Instruments, Castle Hill, Australia). Unless noted otherwise, all experiments were performed at 24°C in the presence of 2 μm ibuprofen to prevent the release and influence of cyclo-oxygenase products (Iizuka et al. 1997). Bronchial SM was prepared from a macroscopically normal part of lung tissue which was obtained at surgery for lung cancer. We obtained consent from all of the patients before surgery. The surgically resected tissue was put in ice-cold Dulbecco's modified Eagle's medium, and small bronchi with an outer diameter of 2–4 mm were carefully dissected as mentioned above. Ileum SM was taken from male guinea-pigs (350-400 g) which had been anaesthetized with halothane in a similar manner to the rabbits, and dissected as described previously (Iizuka et al. 1994).
Solutions and permeabilization with α-toxin
The composition of the solutions has been reported elsewhere (Iizuka et al. 1994). In brief, the normal relaxing solution (G1) contained (mM): 74.1 potassium methanesulphonate, 2 Mg2+, 4.5 ATP (Mg2+ salt), 1 [ethylenebis(oxyethylenenitrilo)]-tetraacetic acid (EGTA), 10 creatine phosphate and 30 Pipes-KOH (pH 7.1 at 24°C, ionic strength 0.2). The same solution containing 10 mM rather than 1 mM EGTA and various amounts of calcium methanesulphonate was used to achieve the desired concentration of free Ca2+.
The tracheal, bronchial and ileum SM strips were exposed to α-toxin (16.4 μg ml−1) in the pCa 6.5 solution for 30–45 min at 30°C, and then washed in G1 for 5 min. The temperature was kept at 24°C after permeabilization to obtain a reproducible contractile response. We first confirmed the feasibility of the α-toxin-permeabilized strip by the pCa 5.0-induced maximum contraction. The pCa 5.0 response was repeated three times; the first response was small but the second and third responses were comparable and larger than the first response. Next we tested the 30 mM caffeine response to estimate the semi-quantitative index of the amount of Ca2+ in the SR (Saida & van Breemen, 1987), and then applied the putative second messengers, AlF4−, or CCh. Unless noted otherwise, the peak amplitude evoked by the Ca2+ releasing agents was normalized to that of the 30 mM caffeine response in every α-toxin-permeabilized preparation.
Rabbit tracheal SM permeabilized with β-escin
Cells were permeabilized with β-escin rather than α-toxin to allow the passage of heparin and Ruthenium Red across the plasma membrane (Kobayashi et al. 1989; Iizuka et al. 1994, 1997). The formation of homogeneous β-escin-induced pores in the plasma membrane of SM cells was achieved with the use of the cold pre-incubation method described by Masuo et al. (1994) with minor modification (Iizuka et al. 1997). Briefly, tracheal SM strips were treated for 45 min with 30 μmβ-escin in G1 at 4°C, after which the temperature was increased rapidly to 30°C by exchange of the cold bubble plate for one that had been prewarmed, and permeabilization was continued for an additional 15 min. The temperature was then readjusted to 24°C. To avoid deterioration of contractile function, we added calmodulin (CaM, 0.1 μm) to the solutions used in the experiments with muscle permeabilized with β-escin (Kobayashi et al. 1989; Iizuka et al. 1994, 1997). Because the time-dependent decrease in contraction (run-down) and earliest loss of receptor-coupled signalling occur at the receptor and/or its interface with the G-protein in β-escin-treated smooth muscle (Iizuka et al. 1994), the peak of the transient contractions was normalized to the final contraction at pCa 5.0 with CaM (5 μm) rather than the 30 mM caffeine response.
Agonist-induced Ca2+ release from the SR in airway SM permeabilized with α-toxin or β-escin
The effect of CCh on Ca2+ mobilization in α-toxin- or β-escin-permeabilized airway SM was performed according to a previously described protocol (Kobayashi et al. 1989; Iizuka et al. 1994) with minor modification. The permeabilized tracheal SM strips were incubated in G1, and then for 7 min at a pCa of 6.3 to load Ca2+ to the SR. After switching solutions to G0.4 (G0.4 containing 0.4 rather than 1 mM EGTA) for 2–3 min, and then to G0.1 (G0.1 containing 0.1 mM EGTA) for 1–3 min, CCh at a range of 0.3-100 μm with 50 μm GTP as indicated in Fig. 2A was applied to the permeabilized strip.
Figure 2. CCh-induced contraction in α-toxin-permeabilized tracheal SM.
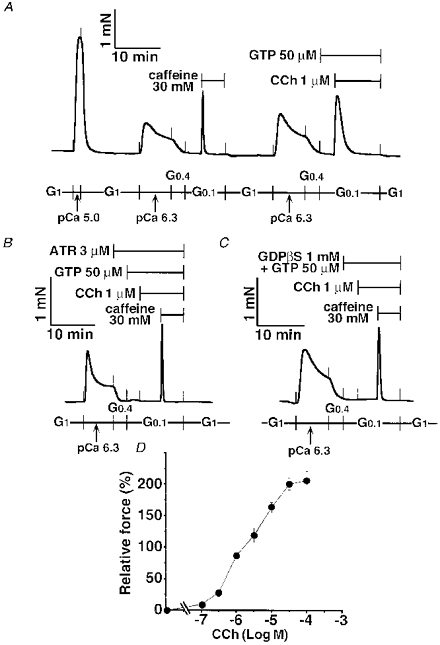
Following the maximum contraction at pCa 5.0, the caffeine response was observed as a semi-quantitative marker for Ca2+ release. GTP was present 3 min before and during the 1 μm carbachol (CCh) response (A). In the presence of 3 μm atropine (ATR) and 50 μm GTP as indicated, 1 μm CCh was added to the strip, followed by the 30 mM caffeine response (B). GTP (50 μm) and GDPβS (1 mM) were present 3 min before and during CCh and caffeine responses (C). The traces in A, B and C represent at least five separate experiments. The dose-response relationship for CCh was determined using 37 strips. Data are expressed as means ±s.e.m., and the number of experiments at each concentration was 4–8 (D).
Caffeine-, InsP3-, cADPR- and NAADP-induced Ca2+ release in airway SM permeabilized with α-toxin
We investigated the effect of different Ca2+ releasers in α-toxin-permeabilized tracheal SM. The tracheal SM strips were exposed to caffeine (0.3-100 mM), InsP3 (30-800 μm), cADPR (10-300 μm), and CCh (0.1-100 μm) with or without GTP (50 μm). Although desensitization to caffeine was not observed, CCh responses apparently declined by repeating the Ca2+ release protocol. InsP3 also showed a small degree of desensitization. However, non-cumulative experiments with InsP3 were too expensive. Thus, to determine the dose-response relationship for caffeine and InsP3, we allowed the same preparation to be exposed to different concentrations of the reagents repeatedly. In contrast, different preparations were definitely required to construct the dose- response curves for CCh. α-Toxin-permeabilized ileum SM was also exposed to caffeine (30 mM), InsP3 (300 μm) plus GTP (50 μm), cADPR (300 μm) plus GTP (50 μm) and CCh (1 μm) plus GTP (50 μm).
Effects of pretreatment with β-NAD+, α-NAD+ and FK506 on CCh-induced contraction in tracheal SM permeabilized with α-toxin
We examined the effects of cADPR-related substances, β-NAD+, α-NAD+ and FK506 on the CCh (1 μm) plus GTP (50 μm)-induced contraction. β-NAD+ is the precursor of cADPR, and β-NAD+ but not α-NAD+ was converted to cADPR by cADPR cyclase leading to a release of Ca2+ with a time lag (Dousa et al. 1996; Galione & Sethi, 1996). Thus, we added 2.5 mM β-NAD+ or α-NAD+ as indicated in Fig. 6, and subsequently the CCh response was observed. A potent immunosuppressant, FK506, released Ca2+ from islet microsomes mediated by the dissociation of 12.6 FK506-binding protein from RyR, and pretreatment with FK506 prevented the subsequent cADPR response (Noguchi et al. 1997). To examine whether FK506-binding protein might contribute to CCh-induced Ca2+ release, we applied FK506 (30 μm) 6 min before and during the CCh-induced contraction. FK506 at higher concentrations (> 30 μm) became insoluble. A similar protocol was followed to determine the effect of NAADP on Ca2+ release.
Figure 6. Effects of α-NAD+ and β-NAD+ on the CCh response in α-toxin-permeabilized tracheal SM.
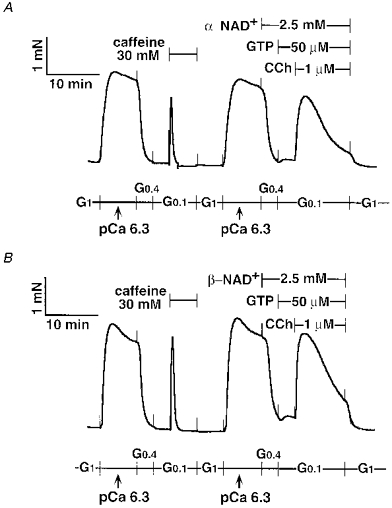
Either 2.5 mM α-NAD+ (A) or 2.5 mM β-NAD+ (B) was present 6 min before and during the CCh response. Note that more of a tonic CCh response was observed in both traces. The traces represent at least four separate experiments.
Effects of 8-NH2-cADPR, thionicotinamide-ADP, Ruthenium Red and procaine on the CCh-induced contraction in tracheal SM permeabilized with α-toxin
A selective cADPR antagonist, 8-amino-cADPR (8-NH2-cADPR, 100 μm), a selective NAADP antagonist, thionicotinamide-ADP (thio-NADP, 100 μm), a selective inhibitor of RyR, Ruthenium Red (100 μm) (Dousa et al. 1996) and a potent Ca2+-induced Ca2+ release channel inhibitor, procaine (20-60 mM), were added to the α-toxin-permeabilized tracheal SM 6 min before and during the caffeine, InsP3 or CCh application.
Effect of ryanodine on caffeine and InsP3-induced contractions in tracheal SM permeabilized with α-toxin
To clarify the mechanism further, we examined the effect of ryanodine on the caffeine and InsP3 responses in α-toxin-permeabilized tracheal SM. After the first caffeine response was obtained, ryanodine (30 μm) was present 30 min before and during the second caffeine response or the InsP3 response. In some experiments the strips were washed in the relaxing solution at 37°C for 60 min. Subsequently temperature was kept at 24°C again, after which the final caffeine response was evoked. Control experiments were run in parallel with 0.3 % dimethyl sulphoxide (DMSO), a vehicle of ryanodine.
Inhibitory effects of heparin and Ruthenium Red on CCh-induced contraction in tracheal SM permeabilized with β-escin
An InsP3 inhibitor, heparin (300 μg ml−1), was added to the β-escin-permeabilized tracheal SM 13 min before and during CCh and caffeine responses as indicated in Fig. 13. Ruthenium Red (100 μm) was present 6 min before and during the CCh response. To confirm the permeability of the β-escin-treated tracheal SM, the contractile effect of 5 μm CaM was tested in every tracheal SM permeabilized with β-escin.
Figure 13. Inhibition of InsP3- and CCh-induced contractions by heparin in β-escin-permeabilized tracheal SM.
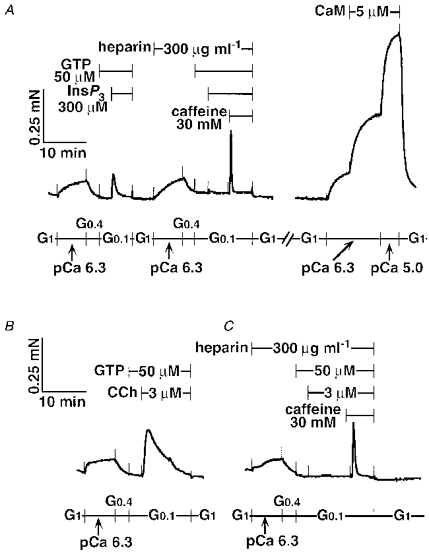
Heparin (300 μg ml−1) was present 13 min before and during the second exposure to 300 μm InsP3 with 50 μm GTP and to 30 mM caffeine. After the caffeine-induced contraction, the strip was washed in G1, then incubated with A23187 (10 μm) for 15 min (trace not shown). Calmodulin (CaM, 5 μm) was added to the submaximum contraction at pCa 6.3. Unless noted otherwise, CaM (0.1 μm) was present in all solutions (A). Control CCh (3 μm) plus the GTP (50 μm) response is shown in B. Complete inhibition of the CCh response by heparin (300 μg ml−1) is shown in C. The traces represent four separate experiments.
Reagents
Staphylococcus aureusα-toxin was obtained from RBI (Natick, MA, USA); guanosine 5′-O-(2′-thiodiphosphate)(GDPβS) was from Boehringer Mannheim. InsP3 and ryanodine were purchased from Calbiochem. cADPR, 8-NH2-cADPR, Ruthenium Red, NAADP, thio-NADP, low molecular weight heparin, procaine and GTP were purchased from Sigma. FK506 was a gift from Fujisawa Pharmaceutical Co. Ltd (Osaka, Japan). All other chemicals were of reagent grade.
Statistical analysis
All data are presented as means ±s.e.m., and were compared by the Mann-Whitney U test or Student's t test with the Bonferroni correction for multiple comparisons. A P value of < 0.05 was considered to be statistically significant in the Mann-Whitney U test, and a significance level of P < 0.05/m (where m is the number of comparisons) was used in the Bonferroni method. EC50 values and 95 % confidence limits (95 % CL) were determined using the probit method.
RESULTS
Characterization of caffeine-induced contraction in tracheal SM permeabilized with α-toxin
The maximum contraction at pCa 5.0 was 3.9 ± 0.5 mN (n = 10) in α-toxin-permeabilized tracheal SM. Caffeine (30 mM) evoked a sharp and monophasic response which was highly reproducible; the amplitude of contraction was identical during four repetitions of the protocol in the same strip (Figs 1A and 3B). The top force generated by 30 mM caffeine was independent of GTP (Fig. 1B); the responses to 30 mM caffeine with or without GTP (50 μm) were 59.5 ± 2.1 % and 55.4 ± 1.8 % (normalized to the pCa 5.0-induced contraction, n = 10). We constructed the dose- response curves for caffeine in which the α-toxin-permeabilized tracheal SM was repeatedly exposed to different concentrations of caffeine. The caffeine response achieved a plateau at 30 mM with contraction at 67.7 ± 3.4 % of the maximum contraction at pCa 5.0 (n = 7). The EC50 was 4.96 mM (95 % CL, 3.82-6.27 mM, Fig. 1C). Following the 30 mM caffeine-induced contraction, the subsequent response to CCh at a supra-maximum concentration (100 μm) disappeared (Figs 1D and 2D). To exclude the possibility that inhibitory effects of caffeine on SM contractility might be sustained after the 3 min washing with G1, we tested the pCa 5.0 response at this point. The amplitude of the pCa 5.0 response was 111 ± 6.2 % (n = 6) of the prior pCa 5.0 response in the same strips, indicating that the 3 min washing was enough to eliminate the caffeine effects (trace not shown). Thus, the 30 mM caffeine response was employed as a semi-quantitative marker to assess the Ca2+ release capacity of the α-toxin-permeabilized tracheal SM.
Figure 1. Caffeine-induced contraction in α-toxin-permeabilized tracheal SM.
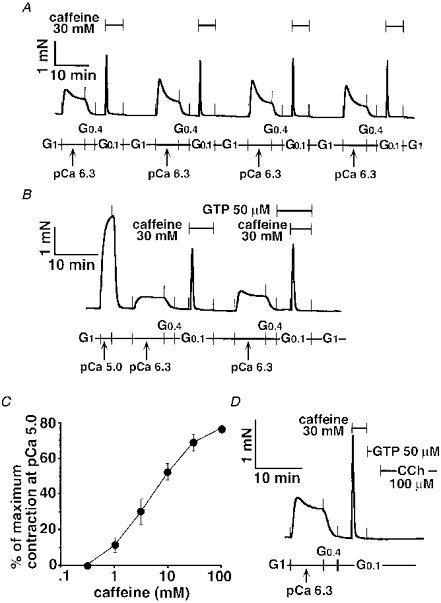
The 30 mM caffeine response without GTP was repeated four times in the same strip (A). The caffeine response was compared in the presence or absence of GTP in the same strip (B). The dose-response relationship for caffeine was determined in seven strips. The peak amplitude was normalized to the maximum contraction at pCa 5.0, and expressed as the mean ±s.e.m. (C). After the 30 mM caffeine response and its washing out for 3 min, 100 μm carbachol (CCh) was added to the strip in the presence of 50 μm GTP (D). The traces in A, B and D represent at least four separate experiments.
Figure 3. Decrease in the responses to CCh, InsP3 and AlF4− but not to caffeine in α-toxin-permeabilized tracheal SM.
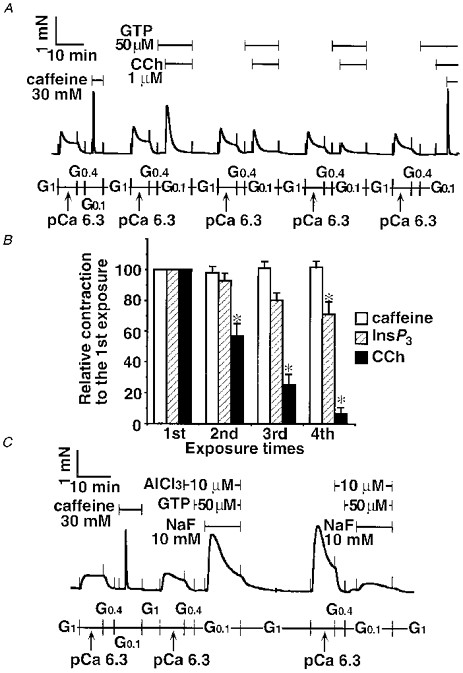
After the caffeine-induced contraction, Ca2+ loading with pCa 6.3 and Ca2+ release induced by 1 μm CCh plus 50 μm GTP was repeated four times, followed by the second caffeine-induced contraction in the same strip (A). Ca2+ release protocol with 30 mM caffeine alone (n = 4), 300 μm InsP3 with 50 μm GTP (n = 4), or 1 μm CCh with 50 μm GTP (n = 4) was repeated four times in the same strip (B). Ca2+ loading with pCa 6.3 and Ca2+ release induced by AlF4− (AlCl3 plus NaF) was repeated twice (C). Data are expressed as means ±s.e.m. and statistical significance was determined by Student's t test with the Bonferroni correction for multiple comparisons (B). * P < 0.05. The traces in A and C represent at least 4 separate experiments.
CCh-induced contraction mediated by Ca2+ release from the SR in tracheal SM permeabilized with α-toxin
As shown in Fig. 2A, CCh (1 μm) evoked a transient contraction in the presence of GTP (50 μm). Pretreatment with a muscarinic acetylcholine receptor antagonist, atropine (3 μm), abolished the CCh response, although the caffeine (30 mM) response was not affected by treatment with this receptor antagonist (Fig. 2B). A non-permeating G-protein competitor, GDPβS (1 mM), also inhibited the CCh-initiated contraction completely, whereas the subsequent caffeine response appeared again in the presence of GDPβS (Fig. 2C). Both CCh and caffeine responses disappeared after treatment with the calcium ionophore A23187 at 10 μm for 15 min (data not shown), which is known to irreversibly deplete the Ca2+ content of the SR (Kitazawa et al. 1989; Iizuka et al. 1994, 1997). The dose-response relationship for CCh was determined using thirty-seven different strips. As shown in Fig. 2D, the CCh response reached a peak at 30 μm with a 206 ± 13.4 % contraction of the 30 mM caffeine response (n = 4-8), and the EC50 of CCh was 1.84 μm (95 % CL, 1.50-2.28 μm).
Desensitization of CCh-induced contraction in tracheal SM
The response to 1 μm CCh with 50 μm GTP was reduced after the repetitive protocol of Ca2+ loading in SR and CCh-triggered Ca2+ mobilization (Fig. 3A). The fourth CCh response decreased to 6.5 ± 4.3 % of the first CCh response (P < 0.05, n = 4, Fig. 3B). Even in this situation the final caffeine response persisted at 102 ± 6.4 % of the initial caffeine response (n = 4, Fig. 3A). To clarify whether the decreased CCh response occurred at the muscarinic acetylcholine receptor site or downstream of the G-protein level in tracheal SM permeabilized with α-toxin, we examined the effect of AlF4− on Ca2+ release, since AlF4− is known to directly activate the trimeric G-protein(s) (Kahn, 1991), such as Gq/11, leading to phospholipase C activation. This in turn produces two second messengers, diacylglycerol and InsP3. The latter messenger, InsP3, evokes Ca2+ release from the SR (Somlyo & Somlyo, 1994; Bárány & Bárány, 1996). After the caffeine response was obtained, AlF4− (10 mM NaF plus 10 μm AlCl3) with GTP (50 μm) caused a large transient contraction (Fig. 3C, 172 ± 8.4 % of the caffeine response, n = 4). The strip was washed in G1 for 10 min, followed by Ca2+ loading with pCa 6.3 again. The second pCa 6.3-induced contraction was larger than the first, indicating that AlF4−-induced Ca2+ sensitization (Gong et al. 1996; Iizuka et al. 1997) was sustained at this point. The second exposure to AlF4− evoked a smaller contraction (37.5 ± 24.3 % of the first AlF4− response; P < 0.05, n = 4, Mann-Whitney U test).
InsP3-induced Ca2+ release from the SR in airway SM permeabilized with α-toxin
Figure 4A and B shows that InsP3 with GTP (50 μm) contracted the α-toxin-permeabilized tracheal SM dose dependently. The EC50 value of InsP3 with GTP (50 μm) was 363 μm (95 % CL, 328–402 μm), and the maximum response was 84.4 ± 5.3 % (normalized to the 30 mM caffeine response in the same strip, n = 6) at 800 μm InsP3. In the absence of GTP, 300 μm InsP3 caused a small contraction (data not shown). As shown in Fig. 3B, the repeated exposure to InsP3 (300 μm) with GTP (50 μm) revealed a small but significant degree of desensitization at the fourth application (70.9 ± 8.2 % of the initial InsP3 response, P < 0.05, n = 4). These results were not due to InsP3 hydrolysis because we used fresh solutions containing InsP3 in these experiments.
Figure 4. InsP3-induced contraction in α-toxin-permeabilized tracheal SM.
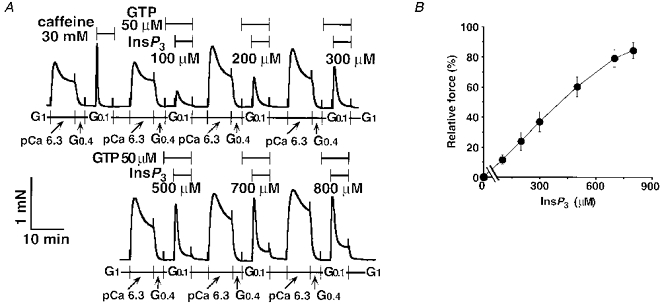
After the 30 mM caffeine response, InsP3 at a range of 100–800 μm with 50 μm GTP was added to the same strip repeatedly (A). The dose-response relationship for InsP3 was determined using six strips. Data are expressed as means ±s.e.m. (n = 6, B).
Comparison of the contractile responses to caffeine, cADPR, InsP3 and CCh in tracheal, bronchial and ileum SM permeabilized with α-toxin
Although 100 μm InsP3 required GTP to obtain detectable force development in tracheal SM (Figs 4A and 5A), 100 μm InsP3 alone evoked a small contraction in bronchial SM (Fig. 5B). cADPR (10 μm) failed to contract either tracheal SM or bronchial SM. We increased the concentration of cADPR up to 300 μm with GTP (50 μm) in tracheal SM. However, cADPR never changed the force of contraction by itself, and never changed contractions induced either by CCh plus GTP or by 30 mM caffeine (data not shown). Thus, we decided to apply cADPR to ileum SM permeabilized with α-toxin, because cADPR was proposed as an intrinsic ligand for RyR in guinea-pig ileum longitudinal SM (Murthy et al. 1995). cADPR (300 μm) with GTP (50 μm) was ineffective even in this tissue, although caffeine (30 mM), InsP3 (300 μm) plus GTP (50 μm) and CCh (1 μm) plus GTP (50 μm) evoked transient contractions as in tracheal and bronchial SM (Fig. 5C). Quantitative data are summarized in Table 1.
Figure 5. Comparison of the Ca2+ mobilization among α-toxin-permeabilized tracheal, bronchial and ileum longitudinal SM.
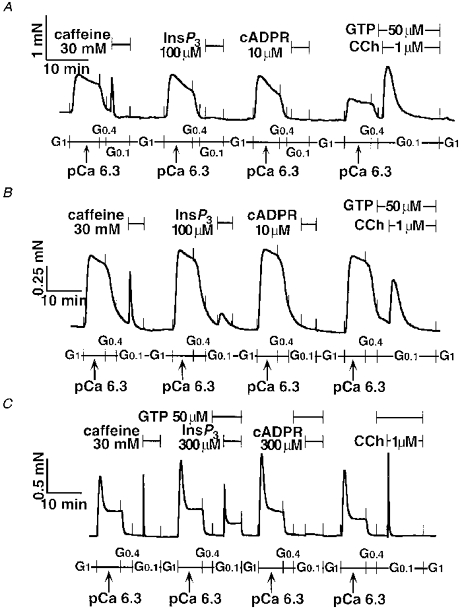
Caffeine (30 mM), InsP3 (100-300 μm), cADPR (10-300 μm), and CCh (1 μm) were added to the α-toxin-permeabilized rabbit tracheal smooth muscle (SM, A), human bronchial SM (B), and guinea-pig ileum longitudinal SM (C). Note that GTP was present with InsP3 and cADPR in ileum SM. The traces represent at least three separate experiments.
Table 1.
Transient contractions mediated by Ca2+ release in tracheal, bronchial and ileum SM permeabilized with α-toxin
| Material | Reagent | Dose (μm) | 50 μm GTP | Percentage of caffeine response | n |
|---|---|---|---|---|---|
| Tracheal SM | InsP3 | 100 | – | 0.1 ± 0.3 | 4 |
| cADPR | 10 | – | 0.2 ± 0.3 | 4 | |
| CCh | 1 | + | 96.3 ± 5.5 | 4 | |
| Bronchial SM | InsP3 | 100 | – | 32.5 ± 9.1 | 12 |
| cADPR | 10 | – | −3.7 ± 2.0 | 4 | |
| CCh | 1 | + | 85.6 ± 14 | 5 | |
| Ileum SM | InsP3 | 300 | + | 105 ± 12 | 3 |
| cADPR | 300 | + | 0.5 ± 0.7 | 3 | |
| CCh | 1 | + | 102 ± 6.5 | 3 |
Rabbit tracheal smooth muscle (SM), human bronchial SM and guinea-pig ileum longitudinal SM were studied. InsP3, inositol 1,4,5-trisphosphate; cADPR, cyclic ADP ribose; CCh, carbachol; GTP, guanosine triphosphate. The amplitudes of the caffeine (30 mm) responses of tracheal, bronchial and ileum SM strips were 0.8 ± 0.12 mN (n = 4), 0.5 ± 0.21 mN (n = 12) and 0.8 ± 0.12 mN (n = 3).
Inhibition of CCh-induced contraction by procaine, but not by cADPR-related compounds
β-NAD+ itself did not contract the strip, and there was no difference in the peak amplitude of the CCh response between α-NAD+ (2.5 mM)- and β-NAD+ (2.5 mM)-treated tracheal SM, although the transient contractions showed a more tonic pattern after the α- or β-NAD+ treatment (Fig. 6A and B). We observed both α-NAD+ (2.5 mM) and β-NAD+ (2.5 mM) causing additional contractions at pCa 6.5 in α-toxin-permeabilized and A23187-treated tracheal SM (trace not shown). These findings were similar to Ca2+ sensitization. However, we noticed that these reagents decreased pH from 7.1 to approximately 6.7, leading to an increase in the free Ca2+ concentration. 8-NH2-cADPR (100 μm) was not effective. Although 8-NH2-cADPR showed a tendency to attenuate the CCh response (Fig. 7), this was not statistically significant (Table 2), and was apparently different from the inhibitory actions of procaine and ryanodine in the α-toxin-permeabilized tracheal SM (see below). To exclude the possibility that 8-NH2-cADPR might not pass through the α-toxin-produced pores, we added 8-NH2-cADPR (100 μm) to the β-escin-treated tracheal SM. However, 8-NH2-cADPR had no effect on CCh-induced contraction (data not shown). FK506 (30 μm) also had no remarkable effect on CCh- (Fig. 8A and B) and caffeine-induced contractions (Fig. 8C). We prolonged incubation time with FK506 by 60 min. However, there was still no inhibitory effect of FK506.
Figure 7. No inhibitory effect of 8-NH2-cADPR on CCh responses in α-toxin-permeabilized tracheal SM.
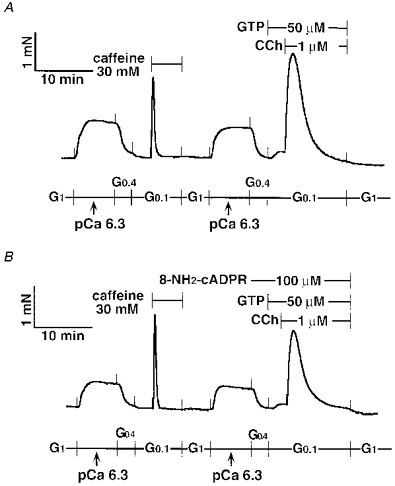
8-NH2-cADPR (100 μm) was not present in the control strip (A), but was present 6 min before and during CCh response in the treated strip (B).
Table 2.
Effects of cADPR- and NAADP-related compounds on CCh-induced contraction via Ca2+ release in tracheal SM permeabilized with α-toxin
| Reagent | Dose | 30 mm caffeine response (% of maximum contraction) | 1 μm CCh (% of caffeine response) | n |
|---|---|---|---|---|
| 8-NH2-cADPR | 100 μm | 54.1 ± 5.9 | 98.2 ± 6.3 | 5 |
| Control | 53.3 ± 6.6 | 111 ± 12 | 6 | |
| Ruthenium Red | 100 μm | 56.4 ± 5.4 | 125 ± 8.9 | 4 |
| Control | 60.0 ± 4.2 | 118 ± 14.5 | 4 | |
| FK506 | 30 μm | 59.3 ± 8.5 | 95.9 ± 9.1 | 4 |
| Control | 55.2 ± 9.0 | 92.3 ± 8.0 | 4 | |
| α-NAD+ | 2.5 mm | 61.2 ± 8.0 | 111 ± 18 | 4 |
| β-NAD+ | 2.5 mm | 59.3 ± 6.8 | 105 ± 14 | 6 |
| Control | 58.0 ± 6.5 | 109 ± 19 | 4 | |
| Procaine | 20 mm | 23.0 ± 3.2 * | 0.5 ± 0.7 * | 4 |
| 60 mm | 0.2 ± 0.3 * | n.d. | 4 | |
| Control | 59.3 ± 8.5 | 105 ± 10 | 4 | |
| NAADP | 100 μm | 60.2 ± 7.6 | 115 ± 30 | 4 |
| thio-NADP | 100 μm | 59.8 ± 7.6 | 138 ± 14 | 4 |
| Control | 63.2 ± 8.5 | 127 ± 5.5 | 4 |
Data are presented as means ±s.e.m. and were compared by Mann-Whitney U test or Student's t test with the Bonferroni correction as appropriate. A P value of < 0.05
was considered statistically significant. CCh, carbachol. n.d., not determined.
Figure 8. Lack of effect of FK506 on CCh responses in α-toxin-permeabilized tracheal SM.
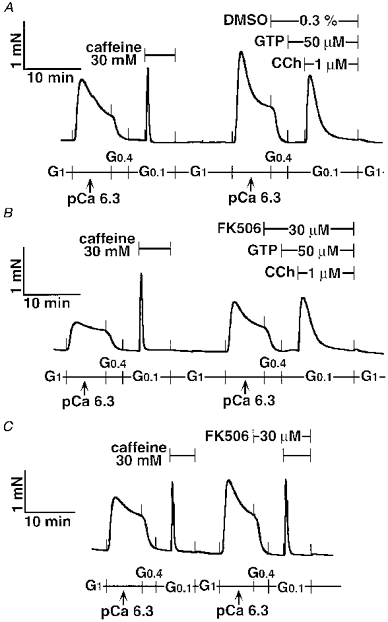
Dimethyl sulphoxide (DMSO) is the vehicle of FK506. DMSO (0.3 %) in the control strip (A) or FK506 (30 μm) in the control strip (B) was present 6 min before and during the CCh response. After the control caffeine response was obtained, FK506 (30 μm) was added to the tracheal SM 6 min before and during the second caffeine response (C). The traces represent four separate experiments.
As shown in Fig. 9B, procaine (20 mM) inhibited contractions induced by InsP3 (600 μm) plus GTP (50 μm) or CCh (100 μm) plus GTP (50 μm) completely. Although a higher concentration was required, the 30 mM caffeine response also was completely and reversibly inhibited by 60 mM procaine (Fig. 9A). Procaine at more than 20 mM suppressed and then potentiated the pCa 6.3-induced contraction of the α-toxin and A23187-treated tracheal SM where the Ca2+ contraction was clamped at a pCa of 6.3 (data not shown). Ruthenium Red (100 μm) did not affect CCh and caffeine responses in α-toxin-permeabilized tracheal SM. These quantitative data are summarized in Table 2.
Figure 9. Inhibitory effect of procaine on caffeine-, InsP3- and CCh-induced contractions in α-toxin-permeabilized tracheal SM.
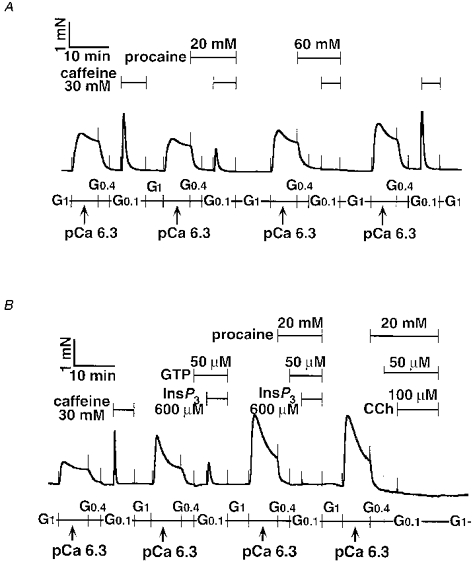
Procaine (20-60 mM) was present 6 min before and during the second and third caffeine responses (A). Procaine (20 mM) completely inhibited both InsP3- and carbachol (CCh)-induced contractions (B). The traces represent at least four separate experiments.
Difference in inhibition of caffeine and InsP3 responses by ryanodine
As shown in Fig. 10 pretreatment with ryanodine markedly reduced the caffeine responses (106.1 ± 7.0 % in control group vs. 4.9 ± 3.1 % in ryanodine group, P < 0.05, n = 4). In contrast, the InsP3 response was only partially (approximately 70 %) inhibited by ryanodine treatment (58.8 ± 5.0 % in the control group vs. 18.7 ± 2.2 % in the ryanodine group, P < 0.01, n = 6). Although the InsP3 response was evoked in the presence of GTP (Ca2+ sensitized), the caffeine response was determined in the absence of GTP (non-sensitized). It was possible that the caffeine response in Fig. 10B might be underestimated; tension development induced by caffeine might be below the detectable level (Fig. 10B), even if the amount of Ca2+ released by caffeine was similar to that by InsP3 (Fig. 10D). Therefore, next we tested the caffeine response in the presence of 50 μm GTP according to the protocol in Fig. 1B. The caffeine plus GTP response after ryanodine treatment was 36.4 ± 6.7 % (n = 3). These results indicate that some part of RyR still escaped the ryanodine treatment. To confirm whether InsP3-releasable Ca2+ came from the caffeine/ryanodine-sensitive pool in airway SM, the caffeine response was evoked with ryanodine, after which InsP3-induced contraction was carried out in the presence of ryanodine. In this protocol, the majority of RyRs would be expected to lock in an open state, resulting in functional removal of the caffeine/ryanodine-sensitive SR more completely. As shown in Fig. 11, the InsP3 response disappeared entirely (72.4 ± 4.5 % in the control group vs. 1.7 ± 0.5 % in prior treatment with caffeine plus ryanodine group, P < 0.05, n = 4). This inhibitory effect of ryanodine was not caused by its toxic effect, because after extensive washing at 37°C the final caffeine response was recovered (140 ± 11 % in control group vs. 115 ± 9.2 % in caffeine plus ryanodine group, n = 4).
Figure 10. Inhibitory action of ryanodine on caffeine and InsP3 responses.
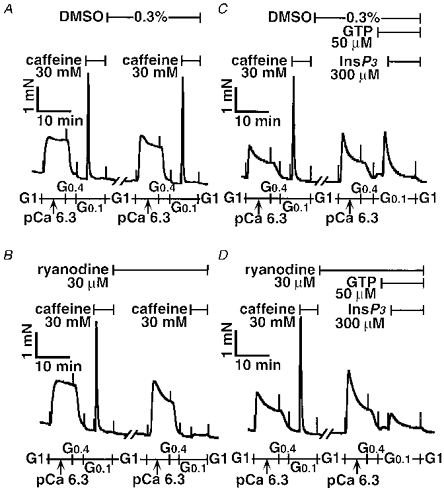
After the first caffeine response was obtained, the tracheal SM strips were incubated in the relaxing solution G1 containing either dimethyl sulphoxide (DMSO; A) or ryanodine (B) for 30 min at 24 °C. The second caffeine response was evoked in the absence (A) or presence (B) of ryanodine. A similar protocol was carried out for the InsP3 response in the absence (C) or presence (D) of ryanodine. Note that GTP was present 3 min before and during the InsP3 response as indicated. The traces represent at least four separate experiments.
Figure 11. Disappearance of InsP3 response after treatment with ryanodine plus caffeine.
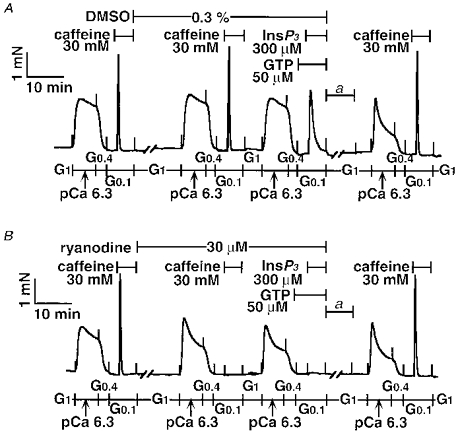
After the first caffeine response was obtained, the tracheal SM strips were incubated in the relaxing solution G1 containing either dimethyl sulphoxide (DMSO; A) or ryanodine (B) for 30 min at 24 °C. The second caffeine response was evoked in the absence (A) or presence (B) of ryanodine, followed by Ca2+ loading with the pCa 6.3 solution for 7 min, and then the GTP plus InsP3 response was evoked. Ryanodine or the vehicle (DMSO) was present as indicated. To eliminate the ryanodine effect, the strips were incubated in G1 for 60 min at 37 °C, and then the temperature was returned to 24 °C (a). The final caffeine response was determined in control (A) and post-ryanodine-treated strips (B). The traces represent four separate experiments.
The absence of effects of NAADP and thio-NADP on CCh-induced contraction in tracheal SM permeabilized with α-toxin
Neither NAADP (100 μm) nor thio-NADP (100 μm) affected the CCh response in tracheal SM permeabilized with α-toxin (Fig. 12 and Table 2).
Figure 12. No influence of NAADP on the subsequent CCh response in α-toxin-permeabilized tracheal SM.
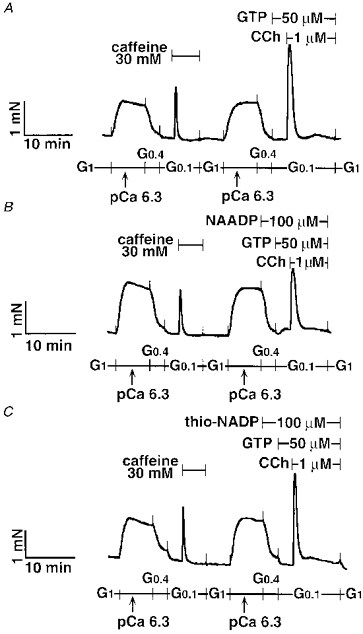
Typical traces are shown in the control (A), 100 μm NAADP-treated (B), and 100 μm thio-NADP-treated strip (C). NAADP or thio-NADP was present 6 min before and during the CCh response. The traces represent four separate experiments.
Inhibitory effects of heparin and Ruthenium Red on CCh-induced contraction in tracheal SM permeabilized with β-escin
The maximum contraction at pCa 5.0 with 5 μm CaM was 0.8 ± 0.2 mN (n = 9) in β-escin-permeabilized tracheal SM. β-Escin-permeabilized tracheal SM was incubated with heparin (300 μg ml−1) 13 min before and during InsP3 (300 μm) or CCh (3 μm) with GTP (50 μm)-induced Ca2+ release (Fig. 13A). The heparin treatment completely inhibited the InsP3 but not the caffeine response. The same strip was incubated with A23187 (10 μm) for 15 min to deplete the stored Ca2+ in the SR, and then was contracted with CaM (5 μm) in the presence of constant free Ca2+ at pCa 6.3 (Fig. 13A). The increment of force by CaM was 37.4 ± 6.1 % (n = 4) of the final maximum contraction at pCa 5.0 with CaM (5 μm). Treatment with heparin inhibited the CCh response completely, although the caffeine response was still retained (Fig. 13B and C). Ruthenium Red also attenuated the CCh (3 μm) plus GTP (50 μm) and caffeine responses in β-escin-permeabilized rabbit tracheal smooth muscle (Table 3). The CaM response was not different between heparin, Ruthenium Red and control groups.
Table 3.
Inhibitory effects of heparin and Ruthenium Red on CCh-induced contraction mediated by Ca2+ release in tracheal SM permeabilized with β-escin
| Percentage of final contraction | ||||
|---|---|---|---|---|
| Reagent | Dose | CCh response | CaM response | n |
| Heparin | 300 μg ml−1 | 0.1 ± 0.3 * | 37.4 ± 6.1 | 4 |
| Ruthenium Red | 100 μm | 6.1 ± 4.8 * | 36.5 ± 5.3 | 4 |
| Control | 25.3 ± 5.5 | 38.9 ± 4.5 | 4 | |
Rabbit tracheal smooth muscle (SM) was permeabilized with β-escin. After loading Ca2+ in pCa 6.3 for 7 min, 3 μm carbachol (CCh) plus 50 μm guanosine triphosphate (GTP) evoked a transient contraction, followed by washing in a relaxing solution, and treatment with A23187 (10 μm) to deplete the intracellular store of Ca2+. At the steady-state contraction at pCa 6.3, calmodulin (CaM, 5 μm) evoked a further contraction at a constant Ca2+ level of pCa 6.3. Final contraction was obtained at pCa 5.0 in the presence of 5 μm CaM. Data are presented as means ± s.e.m., and were compared by Student's t test with the Bonferroni correction for multiple comparisons. A P value of < 0.05
was considered statistically significant.
DISCUSSION
Feasibility of experimental conditions
Treatment with α-toxin is the most successful permeabilization to maintain the force with receptor coupling (Iizuka et al. 1994). Indeed, the maximum contraction at pCa 5.0 in tracheal SM treated with α-toxin was approximately five times greater than with β-escin. Thus, we primarily employed α-toxin permeabilization in this study. The CCh plus GTP-induced transient contractions were inhibited effectively by atropine or GDPβS treatment, while the caffeine response was insensitive to both atropine and GDPβS. All contractile responses disappeared after A23187 treatment. These results indicate that the CCh response contained components of muscarinic acetylcholine receptor stimulation, GDPβS-sensitive G-protein(s) activation, and utilization of Ca2+ from the A23187-sensitive SR in completely permeabilized tracheal SM (Iizuka et al. 1997); caffeine-induced contraction was dependent on Ca2+ content in the SR, but independent of the signalling from the receptor to the GDPβS-sensitive G-protein(s) activation.
The cold pre-incubation method is useful for obtaining homogeneous pore formation in the smooth muscle strip with β-escin (Masuo et al. 1994; Iizuka et al. 1997). Small tension development in the β-escin-treated preparations described above were due to the relatively heavy permeabilization protocol (exposure to 30 μmβ-escin for a total of 60 min), which was required for successful introduction of heparin (molecular mass < 5 kDa) into each cell of the strip. Permeability of the β-escin-treated strips was confirmed by the contractile effect of 5 μm CaM (molecular mass 17 kDa) on the force at pCa 6.3 with CaM (0.1 μm) and by the inhibitory effect of heparin on the InsP3-induced contraction.
Desensitization to CCh in tracheal SM permeabilized with α-toxin
Repetition of the CCh response caused desensitization. Similar findings were observed in rat anococcygeus (Crichton & Smith, 1991). The reduced response to CCh is not due to a fall in the Ca2+ content of the SR, because the caffeine response was maintained even after CCh-induced desensitization was observed. AlF4− apparently induced desensitization as with CCh, whereas InsP3 with GTP showed desensitization to a lesser extent. Thus, desensitization to CCh occurred primarily between post-muscarinic acetylcholine receptor sites and upstream of InsP3 receptors on the SR in tracheal SM permeabilized with α-toxin, although downstream of InsP3 receptors might be, at least in part, responsible for this phenomenon.
The role of InsP3 in CCh-induced transient contraction
Chopra et al. (1991) have shown that InsP3 evokes 45Ca2+ release from SR in saponin-permeabilized rabbit tracheal SM. In β-escin-permeabilized porcine tracheal SM, InsP3-induced Ca2+ mobilization was detected by a fluorescent Ca2+ indicator, fura-2 (Kannan et al. 1997). To our knowledge, however, this is the first report of InsP3-induced force generation of airway SM in a dose-dependent manner.
In hepatocytes GTP markedly mimicked the InsP3-induced Ca2+ release (Dawson et al. 1986), and in rabbit mesenteric artery permeabilized with saponin, InsP3-induced contraction was dependent on GTP (Saida & van Breemen, 1987; Seager et al. 1994). By contrast, GTP-independent, InsP3-induced contraction was reported (Iino, 1990; Crichton & Smith, 1991). In the present study 100 μm InsP3-induced contraction of tracheal SM required 50 μm GTP, although human bronchial SM showed a small response to 100 μm InsP3 in the absence of exogenously applied GTP. However, it is not likely that GTP is essential for InsP3-induced Ca2+ release in rabbit SM, because 300 μm InsP3 alone caused a small contraction in the SM. Presumably GTP increased the Ca2+ sensitivity, rather than enhancing the InsP3-induced Ca2+ release, resulting in detectable force generation. Indeed, bronchial SM showed more tonic responses than tracheal SM. The resultant content of endogenous GTP after permeabilization may account for this difference in GTP dependency between tracheal and bronchial SM strips (Seager et al. 1994).
To determine the dose-response relationship for InsP3, we added GTP to tracheal SM permeabilized with α-toxin, because it was difficult in practice to obtain a saturating concentration of InsP3 (800 μm) in the absence of GTP. Note that the concentration of InsP3 around the SR may be different from that in the solution, because it is expected that the second messenger, InsP3, would be degenerated rapidly before detectable force development in the multicellular strip. To test this possibility, further experiments using a smaller strip (ideally single cell), caged InsP3, or a more stable InsP3 analogue are required.
In β-escin-permeabilized tracheal SM, heparin abolished InsP3- and CCh-, but not caffeine-induced contraction. Under the same experimental conditions, the CCh response was only partially inhibited by Ruthenium Red. Kannan et al. (1997) have observed that acetylcholine-triggered Ca2+ oscillations were inhibited completely by Ruthenium Red and attenuated partially by heparin in β-escin-permeabilized porcine airway SM. These results therefore indicate that InsP3 may contribute to force generation, although Ruthenium Red-sensitive (presumably RyR-mediated) Ca2+ release may be responsible for muscarinic acetylcholine receptor-induced Ca2+ oscillations.
Lack of contribution of cADPR to CCh-induced contraction in airway SM
Ca2+-induced Ca2+ release is thought to be mediated by RyRs, and expression of the RyR mRNA isoform was verified recently in swine airway SM (Kannan et al. 1997). Caffeine-induced Ca2+ release is associated with RyRs rather than InsP3 receptor activation. Thus, dose-dependent and reproducible responses to caffeine strongly suggest the presence of caffeine-sensitive, RyR-linked mechanisms of Ca2+ release in tracheal SM. cADPR has been proposed as a primary candidate for the role of intrinsic regulator for the RyR in not only sea urchin eggs but also in several mammalian cells (Dousa et al. 1996; Galione & Sethi, 1996). However, it is not likely that cADPR plays an important role in contractions mediated by Ca2+ release from the SR of airway SM. This conclusion arises from the following observations: (1) cADPR up to a concentration of 300 μm had no contractile effect in tracheal, bronchial, or ileum SM; (2) no difference in the subsequent CCh response between α-NAD+- and β-NAD+-treated tracheal SM was observed; (3) 8-NH2-cADPR failed to block the CCh response in α-toxin-permeabilized tracheal SM, although procaine completely and reversibly inhibited caffeine, InsP3 and CCh responses; (4) FK506 neither contracted the preparation by itself nor affected the subsequent Ca2+-releasing events. We first considered the possibility that cADPR could not be introduced into the α-toxin-treated tracheal SM, because Ruthenium Red, whose molecular mass is less than 1 kDa, was effective in β-escin- but not in α-toxin-permeabilized tracheal SM, indicating that Ruthenium Red was not permeable through the pore generated by α-toxin. Not only molecular size but also its shape and/or charge may affect the permeability of a substance in α-toxin-treated cells. In a preparation appropriately treated with β-escin, up to 150 kDa proteins, such as IgG, are permeable with retention of receptor coupling (Iizuka et al. 1994). However, cADPR had no effect in tracheal SM (present study) and guinea-pig vas deferens (Nixon et al. 1994) permeabilized with β-escin, indicating that the lack of effect of cADPR is independent of its permeability in the α-toxin-treated preparation. The negative results with FK506 also support this aspect, because FK506 is cell permeable. Second, some unknown essential co-factor whose molecular size is less than 1 kDa may not remain within the α-toxin- and β-escin-permeabilized tracheal SM. If this is the case, GTP probably is not the co-factor, because 300 μm cADPR plus 50 μm GTP in combination failed to contract ileum SM permeabilized with α-toxin. Third, we could not exclude the possibility that exogenously applied cADPR and 8-NH2-cADPR are inactivated rapidly by cADPR hydrolase in airway SM. Indeed, 8-NH2-cADPR tended to reduce the CCh response, although this was not significant. More stable cADPR analogues, such as 7-deaza-cADPR and 7-deaza-8-bromo-cADPR would be helpful in testing this possibility (Sethi et al. 1997). Nevertheless, because CCh plus GTP and InsP3 plus GTP dose-dependently contracted the α-toxin-permeabilized tracheal SM under the same experimental conditions, the lack of difference in CCh response between the α- and β-NAD+-treated groups, inability of FK506 to have an effect, and the complete inhibition of the CCh response by heparin indicates that InsP3 rather than cADPR plays a major role in CCh-initiated force generation via Ca2+ release from the SR in airway SM.
It has been proposed that in the rabbit (Kuemmerle & Makhlouf, 1995) and the guinea-pig (Murthy et al. 1995) ileum longitudinal SM cADPR but not InsP3 is the important second messenger to release Ca2+ from the SR. However, we have reported that InsP3 effectively contracted guinea-pig ileum longitudinal SM permeabilized with β-escin (Iizuka et al. 1994). In this study we confirmed the apparent effect of InsP3 in the ileum SM permeabilized with α-toxin. The reason for this discrepancy is not known. It may be due to the methodological differences (e.g. strip preparation vs. enzymatically dispersed cells; force generation vs. Ca2+ detection with fura-2 and cell shortening; and differences in the agonists, CCh vs. cholecystokinin octapeptide). However, InsP3 generation in response to CCh and GTPγS was reported in ileum longitudinal SM permeabilized with α-toxin (Prestwich et al. 1995), suggesting the importance of InsP3 in the generation of force via Ca2+ release from the SR in both airway and ileum longitudinal SM. Another possibility is that cADPR binds only to the peripheral SR near the plasma membrane, because simultaneous measurements of force and intracellular Ca2+ ([Ca2+]i) with aequorin suggest contractile and non-contractile [Ca2+]i compartments in vascular smooth muscle (Abe et al. 1996). Thus, cADPR may contribute to the regulation of this non-contractile [Ca2+]i compartment in airway SM.
NAADP is another candidate as a Ca2+ releaser. However, NAADP like cADPR failed to contract in tracheal SM. Although the ready occurrence of homologous desensitization is a unique characteristic of NAADP in sea urchin eggs (Dousa et al. 1996), pre-incubation with NAADP did not attenuate the subsequent CCh response. Furthermore, the lack of inhibitory effect of thio-NADP suggests that NAADP is not involved in the CCh-induced contraction.
Inhibition of Ca2+ release-mediated contractions by procaine and ryanodine in tracheal SM permeabilized with α-toxin
Procaine completely and reversibly inhibited the responses to CCh and InsP3 at 20 mM, and to caffeine at 60 mM in tracheal SM. Ueno et al. (1987) have reported that procaine preferentially inhibited the InsP3 response to caffeine, and procaine also blocked acetylcholine-induced InsP3 production in the porcine coronary artery. We observed that procaine attenuated the Ca2+ sensitivity of tracheal SM treated with α-toxin and A23187. Thus, it should be noted that procaine has multiple inhibitory actions, such as inhibition of Ca2+-induced Ca2+ release coupled to RyR and/or InsP3 receptors and attenuation of Ca2+ sensitivity. In β-escin-permeabilized tracheal SM, heparin alone and Ruthenium Red alone prevented the CCh response, indicating that heparin-sensitive and Ruthenium Red-sensitive components are involved in CCh-induced contraction. Thus, although caffeine-, Ruthenium Red- and procaine-sensitive (presumably RyR-associated) mechanisms are involved in the CCh-induced contraction, cADPR and NAADP are not likely to play a significant role in the force generation of airway SM.
To study the mechanism of the Ca2+ release-mediated contractions in more detail, we tested the effect of ryanodine. We treated the strip with ryanodine at 30 μm (a relatively high concentration) for 30 min at 24°C, resulting in effective inhibition of the caffeine response but in only partial inhibition of the InsP3 response. To remove the caffeine/ryanodine-sensitive SR more effectively, the InsP3 response was preceded by ryanodine plus the caffeine response. As a result, the InsP3 response was no longer observed. It is known that ryanodine does not inhibit the InsP3 receptor (Ehrlich et al. 1994). Therefore, we supposed that after caffeine and ryanodine were concomitantly present, most RyRs were clamped in an open state, resulting in better functional removal of the SR, similar to A23187 treatment. While the ryanodine effect is irreversible at room temperature (Iino, 1990), the effect was eliminated by washing in the relaxing solution at 37°C for 60 min, leading to a complete recovery of the final caffeine response. Hence, in airway SM CCh stimulation produces InsP3, which in turn releases Ca2+ from the caffeine/ryanodine-sensitive pool, leading to contraction.
The present results were not in agreement with the two-compartment Ca2+ store model proposed by Iino (1990), since stored Ca2+ in the caffeine/ryanodine-sensitive pool was essential for the CCh- (Fig. 1D) and InsP3- (Fig. 11B) induced contractions. The reason for this discrepancy is unknown at present. It is possible that non-contractile [Ca2+]i compartments contribute to the difference, because we did not measure [Ca2+]i. Further studies are required to clarify this point.
Conclusion
In conclusion, we confirmed a major contribution of InsP3, but not of cADPR and NAADP, to the CCh-induced contraction via Ca2+ release from intracellular stores which are sensitive to caffeine/ryanodine in airway SM.
Acknowledgments
We thank I. Yoshida for preparing human lung tissue. This work was partly supported by the Ministry of Education, Science and Culture of Japan.
References
- Abdel-Latif AA, Akhtar RA, Hawthorne JN. Acetylcholine increases the breakdown of triphosphoinositide of rabbit iris muscle prelabelled with [32P] phosphate. Biochemical Journal. 1977;162:61–73. doi: 10.1042/bj1620061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe F, Karaki H, Endoh M. Effects of cyclopiazonic acid and ryanodine on cytosolic calcium and contraction in vascular smooth muscle. British Journal of Pharmacology. 1996;118:1711–1716. doi: 10.1111/j.1476-5381.1996.tb15596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bárány M, Bárány K. Inositol 1,4,5-trisphosphate production. In: Bárány M, editor. Biochemistry of Smooth Muscle Contraction. San Diego, CA, USA: Academic Press; 1996. pp. 269–282. [Google Scholar]
- Baron CB, Cunningham M, Strauss JF, III, Coburn RF. Pharmacomechanical coupling in smooth muscle may involve phosphatidylinositol metabolism. Proceedings of the National Academy of Sciences of the USA. 1984;81:6899–6903. doi: 10.1073/pnas.81.21.6899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chilvers ER, Challiss RA, Barnes PJ, Nahorski SR. Mass changes of inositol(1,4,5)trisphosphate in trachealis muscle following agonist stimulation. European Journal of Pharmacology. 1989;164:587–590. doi: 10.1016/0014-2999(89)90269-0. [DOI] [PubMed] [Google Scholar]
- Chini EN, Beers KW, Dousa TP. Nicotinate adenine dinucleotide phosphate (NAADP) triggers a specific calcium release system in sea urchin eggs. Journal of Biological Chemistry. 1995;270:3216–3223. doi: 10.1074/jbc.270.7.3216. [DOI] [PubMed] [Google Scholar]
- Chopra LC, Twort CH, Cameron IR, Ward JP. Inositol 1,4,5-trisphosphate- and guanosine 5′-O-(3-thiotriphosphate)-induced Ca2+ release in cultured airway smooth muscle. British Journal of Pharmacology. 1991;104:901–906. doi: 10.1111/j.1476-5381.1991.tb12524.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crichton CA, Smith GL. GTP and noradrenaline-induced force in isolated toxin-permeabilized rat anococcygeus and guinea-pig portal vein. The Journal of Physiology. 1991;185:543–561. doi: 10.1113/jphysiol.1991.sp018610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson AP, Comerford JG, Fulton DV. The effect of GTP on inositol 1,4,5-trisphosphate-stimulated Ca2+ efflux from a rat liver microsomal fraction. Is a GTP-dependent protein phosphorylation involved. Biochemical Journal. 1986;234:311–315. doi: 10.1042/bj2340311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dousa TP, Chini EN, Beers KW. Adenine nucleotide diphosphates: emerging second messengers acting via intracellular Ca2+ release. American Journal of Physiology. 1996;271:C1007–1024. doi: 10.1152/ajpcell.1996.271.4.C1007. [DOI] [PubMed] [Google Scholar]
- Ebihara S, Sasaki T, Hida W, Kikuchi Y, Oshiro T, Shimura S, Takasawa S, Okamoto H, Nishiyama A, Akaike N, Shirato K. Role of cyclic ADP-ribose in ATP-activated potassium currents in alveolar macrophages. Journal of Biological Chemistry. 1997;272:16023–16029. doi: 10.1074/jbc.272.25.16023. 10.1074/jbc.272.25.16023. [DOI] [PubMed] [Google Scholar]
- Ehrlich BE, Kaftan Ed, Bezprozvannaya S, Bezprozvanny I. The pharmacology of intracellular Ca2+-release channels. Trends in Pharmacological Sciences. 1994;15:145–149. doi: 10.1016/0165-6147(94)90074-4. 10.1016/0165-6147(94)90074-4. [DOI] [PubMed] [Google Scholar]
- Galione A, Sethi J. Cyclic ADP-ribose and calcium signaling. In: Bárány M, editor. Biochemistry of Smooth Muscle Contraction. San Diego, CA, USA: Academic Press; 1996. pp. 295–305. [Google Scholar]
- Gong MC, Iizuka K, Nixon G, Browne JP, Hall A, Eccleston JF, Sugai M, Kobayashi S, Somlyo AV, Somlyo AP. Role of guanine nucleotide-binding proteins - ras-family or trimeric proteins or both - in Ca2+ sensitization of smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1996;93:1340–1345. doi: 10.1073/pnas.93.3.1340. 10.1073/pnas.93.3.1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto T, Hirata M, Ito Y. A role for inositol 1,4,5-trisphosphate in the initiation of agonist-induced contractions of dog tracheal smooth muscle. British Journal of Pharmacology. 1985;86:191–199. doi: 10.1111/j.1476-5381.1985.tb09449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M. Calcium release mechanisms in smooth muscle. Japanese Journal of Pharmacology. 1990;54:345–354. doi: 10.1254/jjp.54.345. [DOI] [PubMed] [Google Scholar]
- Iizuka K, Dobashi K, Yoshii A, Horie T, Suzuki H, Nakazawa T, Mori M. Receptor-dependent G protein-mediated Ca2+ sensitization in canine airway smooth muscle. Cell Calcium. 1997;22:21–30. doi: 10.1016/s0143-4160(97)90086-5. 10.1016/S0143-4160(97)90086-5. [DOI] [PubMed] [Google Scholar]
- Iizuka K, Ikebe M, Somlyo AV, Somlyo AP. Introduction of high molecular weight (IgG) proteins into receptor coupled, permeabilized smooth muscle. Cell Calcium. 1994;16:431–445. doi: 10.1016/0143-4160(94)90073-6. 10.1016/0143-4160(94)90073-6. [DOI] [PubMed] [Google Scholar]
- Kahn RA. Fluoride is not an activator of the smaller (20-25 kDa) GTP-binding proteins. Journal of Biological Chemistry. 1991;266:15595–15597. [PubMed] [Google Scholar]
- Kannan MS, Prakash YS, Brenner T, Mickelson JR, Sieck GC. Role of ryanodine receptor channels in Ca2+ oscillations of porcine tracheal smooth muscle. American Journal of Physiology. 1997;272:L659–664. doi: 10.1152/ajplung.1997.272.4.L659. [DOI] [PubMed] [Google Scholar]
- Kitazawa T, Kobayashi S, Horiuti K, Somlyo AV, Somlyo AP. Receptor-coupled, permeabilized smooth muscle: role of the phosphatidylinositol cascade, G-proteins, and modulation of the contractile response to Ca2+ Journal of Biological Chemistry. 1989;264:5339–5342. [PubMed] [Google Scholar]
- Kobayashi S, Kitazawa T, Somlyo AV, Somlyo AP. Cytosolic heparin inhibits muscarinic and α-adrenergic Ca2+ release in smooth muscle. Journal of Biological Chemistry. 1989;264:17997–18004. [PubMed] [Google Scholar]
- Kuemmerle JF, Makhlouf GM. Agonist-stimulated cyclic ADP ribose. Endogenous modulator of Ca2+-induced Ca2+ release in intestinal longitudinal muscle. Journal of Biological Chemistry. 1995;270:25488–25494. doi: 10.1074/jbc.270.43.25488. 10.1074/jbc.270.43.25488. [DOI] [PubMed] [Google Scholar]
- Lee HC, Aarhus R. A derivative of NADP mobilizes calcium stores insensitive to inositol trisphosphate and cyclic ADP-ribose. Journal of Biological Chemistry. 1995;270:2152–2157. doi: 10.1074/jbc.270.5.2152. 10.1074/jbc.270.5.2152. [DOI] [PubMed] [Google Scholar]
- Lee HC, Walseth TF, Bratt GT, Hayes RN, Clapper DL. Structural determination of a cyclic metabolite of NAD+ with intracellular Ca2+-mobilizing activity. Journal of Biological Chemistry. 1989;264:1608–1615. [PubMed] [Google Scholar]
- Masuo M, Reardon S, Ikebe M, Kitazawa T. A novel mechanism for the Ca2+-sensitizing effect of protein kinase C on vascular smooth muscle: inhibition of myosin light chain phosphatase. Journal of General Physiology. 1994;104:265–286. doi: 10.1085/jgp.104.2.265. 10.1085/jgp.104.2.265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meurs H, Timmermans A, Van Amsterdam RG, Brouwer F, Kauffman HF, Zaagsma J. Muscarinic receptors in human airway smooth muscle are coupled to phosphoinositide metabolism. European Journal of Pharmacology. 1989;164:369–371. doi: 10.1016/0014-2999(89)90480-9. 10.1016/0014-2999(89)90480-9. [DOI] [PubMed] [Google Scholar]
- Murthy KS, Kuemmerle JF, Makhlouf GM. Agonist-mediated activation of PLA2 initiates Ca2+ mobilization in intestinal longitudinal smooth muscle. American Journal of Physiology. 1995;269:G93–102. doi: 10.1152/ajpgi.1995.269.1.G93. [DOI] [PubMed] [Google Scholar]
- Nixon GF, Mignery GA, Somlyo AV. Immunogold localization of inositol 1,4,5-trisphosphate receptors and characterization of ultrastructural features of the sarcoplasmic reticulum in phasic and tonic smooth muscle. Journal of Muscle Research and Cell Motility. 1994;15:682–700. doi: 10.1007/BF00121075. [DOI] [PubMed] [Google Scholar]
- Noguchi N, Takasawa S, Nata K, Tohgo A, Kato I, Ikehata F, Yonekura H, Okamoto H. Cyclic ADP-ribose binds to FK506-binding protein 12.6 to release Ca2+ from islet microsomes. Journal of Biological Chemistry. 1997;272:3133–3136. doi: 10.1074/jbc.272.6.3133. 10.1074/jbc.272.6.3133. [DOI] [PubMed] [Google Scholar]
- Prestwich SA, Miyazaki H, Bolton TB. Effects of GTPγS on muscarinic receptor-stimulated inositol phospholipid hydrolysis in permeabilized smooth muscle from the small intestine. British Journal of Pharmacology. 1995;115:147–157. doi: 10.1111/j.1476-5381.1995.tb16332.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saida K, van Breemen C. GTP requirement for inositol-1,4,5-trisphosphate-induced Ca2+ release from sarcoplasmic reticulum in smooth muscle. Biochemical and Biophysical Research Communications. 1987;144:1313–1316. doi: 10.1016/0006-291x(87)91453-7. [DOI] [PubMed] [Google Scholar]
- Seager JM, Murphy TV, Garland CJ. Importance of inositol (1,4,5)-trisphosphate, intracellular Ca2+ release and myofilament Ca2+ sensitization in 5-hydroxytryptamine-evoked contraction of rabbit mesenteric artery. British Journal of Pharmacology. 1994;111:525–532. doi: 10.1111/j.1476-5381.1994.tb14769.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sethi JK, Empson RM, Bailey VC, Potter BV, Galione A. 7-Deaza-8-bromo-cyclic ADP-ribose, the first membrane-permeant, hydrolysis-resistant cyclic ADP-ribose antagonist. Journal of Biological Chemistry. 1997;272:16358–16363. doi: 10.1074/jbc.272.26.16358. 10.1074/jbc.272.26.16358. [DOI] [PubMed] [Google Scholar]
- Somlyo AV, Bond M, Somlyo AP, Scarpa A. Inositol trisphosphate-induced calcium release and contraction in vascular smooth muscle. Proceedings of the National Academy of Sciences of the USA. 1985;82:5231–5235. doi: 10.1073/pnas.82.15.5231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somlyo AV, Horiuti K, Trentham DR, Kitazawa T, Somlyo AP. Kinetics of Ca2+ release and contraction induced by photolysis of caged D-myo-inositol 1,4,5-trisphosphate in smooth muscle. The effects of heparin, procaine, and adenine nucleotides. Journal of Biological Chemistry. 1992;267:22316–22322. [PubMed] [Google Scholar]
- Somlyo AP, Somlyo AV. Signal transduction and regulation in smooth muscle. Nature. 1994;372:231–236. doi: 10.1038/372231a0. 10.1038/372231a0. [DOI] [PubMed] [Google Scholar]
- Ueno H, Sumimoto K, Hashimoto T, Hirata M, Kuriyama H. Effects of procaine on pharmaco-mechanical coupling mechanisms activated by acetylcholine in smooth muscle cells of porcine coronary artery. Circulation Research. 1987;60:356–366. doi: 10.1161/01.res.60.3.356. [DOI] [PubMed] [Google Scholar]
- Van Amsterdam RG, Meurs H, Ten Berge RE, Veninga NC, Brouwer F, Zaagsma J. Role of phosphoinositide metabolism in human bronchial smooth muscle contraction and in functional antagonism by beta-adrenoceptor agonists. American Review of Respiratory Disease. 1990;142:1124–1128. doi: 10.1164/ajrccm/142.5.1124. [DOI] [PubMed] [Google Scholar]
- Watts SW, Cox DA, Johnson BG, Schoepp DD, Cohen ML. Contractile serotonin-2A receptor signal transduction in guinea pig trachea: importance of protein kinase C and extracellular and intracellular calcium but not phosphoinositide hydrolysis. Journal of Pharmacology and Experimental Therapeutics. 1994;271:832–844. [PubMed] [Google Scholar]