

Abstract
The effects of NO donors on Ca2+-dependent Cl− currents (ICl(Ca)) were investigated in freshly isolated cat tracheal myocytes using the whole-cell patch clamp technique.
With nystatin-perforated whole-cell recording, carbachol (CCh, ≥ 1 μm) induced a transient inward current (ICCh) with a reversal potential of about -20 mV. Activation of ICCh probably occurred through the M3 muscarinic receptor, since nanomolar concentrations of 4-diphenylacetoxy-N-methylpiperidine methobromide (4-DAMP) greatly inhibited this current, while 11-(2-(diethylamino)methyl)-1-piperidinylacetyl)-5,11-dihydro-6H-pyrido (2,3β) (1,4)benzodiazepine-6-one (AF-DX 116) or pirenzepine at concentrations of up to 1 μm were almost ineffective.
Chloride channel/transporter blockers such as DIDS (100 μm), anthracene-9-carboxylic acid (9-AC, 100 μm) and niflumic acid (100 μm) greatly inhibited ICCh, but cation channel blockers, such as nifedipine (10 μm), Zn2+ (500 μm) or Gd3+ (500 μm), were without effect.
Activation of ICCh was strongly attenuated by pretreatment with ryanodine (4 μm) plus caffeine (10 mM). Addition of neomycin (1 mM) into the bath or inclusion of heparin (3 mg ml−1) in the pipette abolished a substantial part of ICCh. These results suggest that ICCh is ICl(Ca), which is activated by inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release.
The nitric oxide donor S-nitroso-N-acetyl penicillamine (SNAP) reduced the amplitude of ICCh dose dependently (IC50, ≈10 μm). Similar inhibition was also exerted by other types of NO donor such as glyceryl trinitrate (GTN) and (±)-E-methyl-2-(E-hydroxyimitol)-5-nitro-6-methoxy-3-hexeneamide (NO-R).
SNAP-induced ICCh inhibition was effectively antagonized by Methylene Blue (1-100 nM), and mimicked by dibutyryl cGMP (db-cGMP) (0.5-1 mM), whereas two structurally distinct types of cGMP-dependent (G)-kinase inhibitor, N-(2-aminoethyl)-5-isoquinilinesulphonamide (H-8, 2.5 μm) and KT5823 (1 μm), failed to counteract the inhibitory effects of SNAP or db-cGMP. Another G-kinase-specific inhibitor Rp-8-(para-chlorophenylthio)guanosine-3′,5′-cyclic monophosphorothioate (Rp-8-pCPT-cGMPS; 1 μm) itself caused a marked reduction in ICCh.
SNAP (100 μm) or db-cGMP (100 μm) exhibited no inhibitory actions, when caffeine (10 mM) or photolytically released IP3 were used instead of CCh to activate the inward current.
These results suggest that inhibition of ICCh by NO donors involves a cGMP-dependent but G-kinase-independent mechanism, which may operate at a site(s) between the muscarinic (M3) and IP3 receptors.
Muscarinic receptors play a central role in the excitatory regulation of airway tone. In airway tissue, activation of muscarinic receptors leads to a number of important cellular consequences, including inositol 1,4,5-trisphosphate (IP3)-mediated Ca2+ release from internal stores (Hashimoto et al. 1985) and Ca2+ entry through both voltage-dependent and -independent pathways (Murray & Kotlikoff, 1991; Wang & Kotlikoff, 1997a). It has recently been demonstrated by patch clamp experiments that, in tracheal smooth muscle cells (SMCs) from various species, Ca2+ released from the sarcoplasmic reticulum or entering from the extracellular space can induce several Ca2+-dependent conductances such as Cl−, K+ and other cationic currents (Janssen & Sims, 1992, 1993, 1995; Henmi et al. 1996; Fleischmann et al. 1997; Wang & Kotlikoff, 1997a, b). Amongst these, the Ca2+-dependent Cl− current (ICl(Ca)), which may be large enough to depolarize the membrane to the Cl− equilibrium potential (-30 to -20 mV; Aickin, 1990), may contribute to membrane depolarization and subsequent muscle tension development during muscarinic receptor activation (see review by Large & Wang, 1996), although the physiological significance of this mechanism in the tracheal smooth muscle remains to be determined.
Nitric oxide (NO) and compounds capable of liberating NO relax various types of smooth muscle (Lincoln, 1989; Kuriyama et al. 1995); this probably also applies to airway smooth muscle, where not only the pivotal role of NO in the non-adrenergic, non-cholinergic (NANC) transmission, but also strong tracheo-relaxing actions of NO-related compounds have been demonstrated (Hamaguchi et al. 1992; Bialecki & Stinton-Fisher, 1995; Jing et al. 1995; Takahashi et al. 1995). These relaxing actions, in part, appear to be associated with membrane hyperpolarization (Hamaguchi et al. 1992; Bialecki & Stinton-Fisher, 1995), which would in turn decrease Ca2+ entry through voltage-dependent Ca2+ channels (which exhibit a steep voltage dependence close to the resting membrane potential; Nelson et al. 1990), although in cat tracheal smooth muscle endogenous and exogenous NO potently relax the muscle with no discernible changes in membrane potential (Jing et al. 1995; Takahashi et al. 1995). The mechanism underlying NO-induced or NO-related compound-induced hyperpolarization is likely to involve the increased activity of large-conductance, Ca2+-dependent K+ (BK) channels (Hamaguchi et al. 1992; Bialecki & Stinton-Fisher, 1995), through increased phosphorylation of BK subunits by an altered balance between phosphatase (Zhou et al. 1996) and cGMP-dependent kinase (G-kinase) activities (Robertson et al. 1993; Yamakage et al. 1996). This is analogous to the actions of another cyclic nucleotide, cAMP (Kume et al. 1989). However, to our knowledge, little information is yet available about the mechanisms by which cyclic nucleotides affect the Cl− conductance in airway SMCs, especially ICl(Ca), which may critically control membrane excitability of smooth muscle (Large & Wang, 1996). Furthermore, it has recently been shown that in cat airways, the excitatory (ACh) and inhibitory (NO and vasoactive intestinal polypeptide) neurotransmitters may co-exist in the same nerve terminal, and the thresholds for activating the cholinergic and nitrergic responses are indistinguishable (Takahashi et al. 1995). These facts provide the important idea that there may be considerable cross-interaction between the cholinergic and nitrergic nervous systems.
The main objectives of this study were therefore twofold: firstly to characterize the biophysical and pharmacological properties of ICl(Ca) activated through muscarinic receptor stimulation in cat tracheal SMCs, and secondly to examine the effects of NO-releasing compounds on ICl(Ca), and explore the mechanism underlying them. Part of this study has been communicated to the 70th annual meeting of the Japanese Pharmacological Society in abstract form (Waniishi et al. 1997b).
METHODS
Materials and cell dispersion
Adult mongrel cats of either sex weighing 2–3 kg were anaesthetized with intraperitoneal sodium pentobarbitone (30-40 mg kg−1) and killed by exsanguination. A segment of cervical trachea (3-4 cm) was excised and quickly transferred into modified Krebs solution (see below for composition) aerated with 95 % O2-5 % CO2. After removing attached connective tissue from its outer surface (the cartilage was left attached to keep tension on the muscle), the segment was allowed to fully relax in modified Krebs solution under oxygenated conditions at room temperature (22-25°C); this normally took more than 2 h. The relaxed segment was then incubated in modified Krebs solution (1.5 mM Ca2+) containing 0.5-1 mg ml−1 collagenase (Sigma Type I) and 2.5-10 μg ml−1 actinase (Tokyo Kasei), for 35 min at 34°C, and then stored in modified Krebs solution (1.5 mM Ca2+) supplemented with 1 mg ml−1 bovine serum albumin and 1 mg ml−1 soybean trypsin inhibitor (Sigma) in a refrigerator at 4-10°C. Just before an experiment, a small patch of tracheal muscle was mechanically detached from the cartilage in nominally Ca2+-free Krebs solution, and then cut into pieces (∼2 mm3). These pieces were then gently triturated with a blunt tipped Pasteur pipette, until a sufficient number of single cells were released. Single tracheal cells were used within 6 h from the time of cell dispersion. All experiments were performed at room temperature (22-25°C).
Electrophysiology
The patch clamp system employed was very similar to that described elsewhere (Waniishi et al. 1997a). In brief, a high impedance, low noise patch clamp amplifier (EPC7, List Electronic) was used to apply voltages and sample voltage or current signals from the clamped cell, through an A/D converter (TL-1, Axon Instruments; low-pass filtered at 500 Hz and digitized at 1 kHz) which was driven by a 32 bit computer (Aptiva, IBM) using pCLAMP version 5.0 (Axon Instruments). In some experiments, in which a long period of recording was required, the signals from the amplifier were directly stored on a computer hard disk after digitization (100 Hz; MacLab/4, ADInstruments, New South Wales, Australia; low-pass filtered at 50 Hz). Data analysis was performed and illustrations made using Clampfit version 6.03 (Axon Instruments) or Chart version 2.0 (ADInstruments).
Flash photolysis of caged IP3
Single tracheal myocytes were loaded with 100–200 μm caged IP3 via the patch pipette until equilibrated (10-15 min). A high-pressure mercury lamp (intensity, 320 mW 10 mm−2 at 360 nm, the wavelength to cause a selective photolysis of caged IP3) was used as the source of UV light, and flashes of varying durations were generated by an electronic shutter (HB-10103AF, Nikon, Tokyo). The flash durations required for threshold and maximum current activation were estimated in each loaded cell (0.2-0.5 ms and 10–20 ms, respectively). All experiments were performed by choosing a flash of sufficiently short duration to generate a nearly constant amplitude of inward current (in our experimental conditions, 1–2 ms turned out to be optimal), otherwise progressive run-down occurred on repeated photorelease of IP3. Under these conditions, the amplitude of the photolysis-induced inward current averaged -560 ± 150 pA at -70 mV (n = 10), which was a value intermediate between the amplitudes of 10 and 100 μm CCh-induced inward currents (see Fig. 1B).
Figure 1. CCh activates ICCh in cat tracheal myocytes.
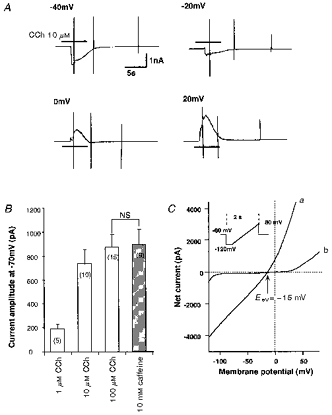
The bath contained modified Krebs solution. A, nystatin-perforated recording with Cs+ internal solution. Holding potentials are indicated above each trace. Horizontal bars indicate the approximate timing of application of 10 μm CCh. Vertical deflections are currents in response to short ramp and step voltages (100 ms). Nifedipine (10 μm) was added in the bath to block the voltage-dependent Ca2+ current. B, histograms showing the amplitude of CCh- and caffeine-induced inward currents at -70 mV. Nystatin-perforated whole-cell recording with K+ internal solution and elevated K+ external solution (for composition see Methods). Values in parentheses indicate the number of experiments. NS, no significant difference between the paired data sets with Student's t test (P > 0.05). C, current-voltage relationship of inward current induced by 10 μm CCh (same experimental conditions as in A). Curves a and b indicate the net membrane currents responding to a slow rising ramp voltage (inset) in the presence and absence of CCh, respectively.
Solutions
The inward current activated by CCh (ICCh) showed a rapid and progressive decline upon repeated application of CCh, when the conventional whole-cell recording was employed. This made it impossible to evaluate the effects of various compounds on ICCh using sequential applications. We therefore employed the nystatin-perforated whole-cell recording technique (Horn & Marty, 1988), in which ICCh is more stable, throughout the present study, except for experiments using flash photolysis. Furthermore, the amplitude of ICCh stayed much more constant when K+ rather than Cs+ was used as the major cation in the pipette solution. Thus, we used K+ internal solution in most parts of this study (except for Fig. 1A and C), and raised the external K+ concentration to 9 mM to set the K+ equilibrium potential close to the holding potential of -70 mV (assuming an intracellular K+ concentration of 120–130 mM).
The composition of solutions used in the present study was as follows. Modified Krebs solution (mM): 137 Na+, 5.9 K+, 1.2 Mg2+, 1.5 Ca2+, 15.5 HCO3−, 2.5 H2PO4−, 130.3 Cl−, 12 glucose, continuously aerated with 95 % O2-5 % CO2. Elevated K+ external solution (mM): 134 Na+, 9 K+, 1.2 Mg2+, 1.5 Ca2+, 148.3 Cl−, 5 glucose, 10 Hepes (adjusted to pH 7.4 with Tris base). K+ (or Cs+) internal solution for nystatin-perforated whole-cell recording (mM): 140 K+ (or Cs+), 1.2 Mg2+, 142.4 Cl−, 10 glucose, 10 Hepes (adjusted to pH 7.2 with Tris base). K+ internal solution for conventional whole-cell recording (mM): 140 K+, 1.2 Mg2+, 142.4 Cl−, 2 Na2ATP, 5 phosphocreatine, 0.05 EGTA, 10 Hepes (adjusted to pH 7.2 with Tris base).
Chemicals
S-nitroso-N-acetyl penicillamine (SNAP), (±)-E-methyl-2-(E-hydroxyimitol)-5-nitro-6-methoxy-3-hexeneamide (NO-R), glyceryl trinitrate (GTN), Methylene Blue, ethyleneglycol-bis-(β-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) and ZnCl2 were purchased from Dojin; carbachol (CCh), ryanodine, niflumic acid, anthracene-9-carboxylic acid (9-AC), 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS), pirenzepine, 11-(2-(diethylamino)methyl)-1-piperidinylacetyl)-5,11-dihydro-6H-pyrido (2,3β) (1,4)benzodiazepine-6-one (AF-DX 116) and 4-diphenylacetoxy-N-methylpiperidine methobromide (4-DAMP) were purchased from Sigma; N-(2-aminoethyl)-5-isoquinolinesulphonamide (H-8, dihydrochloride), KT5823 (Nocardiopsis sp.), N2,2′-O-dibutyryl-guanosine 3′,5′-cyclic monophosphate (db-cGMP, sodium salt), Rp-8-(para-chlorophenylthio)guanosine-3′,5′-cyclic monophosphorothioate (Rp-8-pCPT-cGMPS) and D-myo-inositol 1,4,5-trisphosphate, P4(5)-1(2-nitrophenyl)ethyl ester (caged IP3; trisodium salt) were purchased from Calbiochem.
Statistics
All data are expressed as means ±s.e.m. Statistical significance was evaluated using Student's paired and unpaired t tests as indicated in each figure.
RESULTS
CCh activates Ca2+-dependent Cl− currents through the M3 muscarinic receptor
A single myocyte dissociated from the cat trachea had a smooth and elongated appearance and rapidly contracted in response to CCh (≥ 1 μm) added to the bath. This contraction was reversible and could be repeated several times, if sufficiently long intervals were chosen (usually 5–10 min) between applications. When the myocytes were current clamped using the nystatin-perforated whole-cell recording technique (K+ internal solution), the resting membrane potential ranged between -70 and -45 mV (-62.2 ± 4.7 mV, n = 6), and spontaneous oscillations of 20–30 mV in magnitude were occasionally observed. Bath-applied CCh produced a rapid and transient (∼10 s) depolarization of between -20 and -10 mV (-12.4 ± 1.3 mV, n = 4).
Switching from current-clamp to voltage-clamp mode under the same experimental conditions revealed that rapidly developing smooth inward currents (ICCh, -873 ± 107 pA at -70 mV, range: -425 to -3780 pA; see Figs 1–5 for actual traces) were activated in response to CCh with a threshold concentration of about 1 μm. The amplitude of ICCh was increased abruptly to the maximum, within a narrow range of CCh concentrations (1-10 μm), with an apparent Kd of several micromolar (Fig. 1B). This type of activation is reminiscent of the all-or-none-like characteristics of agonist-induced Ca2+ release described elsewhere (Bootman et al. 1992; Iino et al. 1993). When the conventional voltage-clamp technique, instead of nystatin-perforated recording, was used, the amplitude of ICCh declined in a manner dependent on the time elapsed after establishing the whole-cell configuration. This is probably due to washout of intracellular constituents (Horn & Marty, 1988). Thus in the rest of this study, we adopted the nystatin-perforated technique.
Figure 5. Inhibitory effects of SNAP on ICCh and antagonism by Methylene Blue.
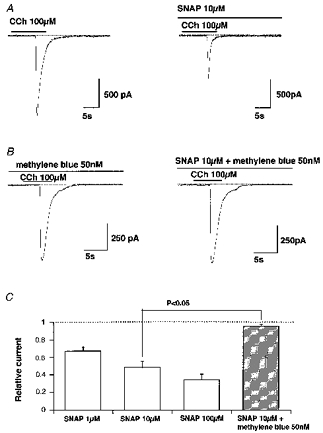
A and B, examples of actual traces. SNAP and/or Methylene Blue were added 10 min before application of CCh. C, summary of experiments shown in A and B. Vertical axis shows the amplitude of ICCh in the presence of the drugs normalized to their controls. The P value indicates the results of Student's unpaired t test. All columns represent the results of 6 experiments.
The time course of ICCh was, in most cases, characterized by rapidly developing and decaying phases (the current terminating within 10 s in the continuing presence of CCh; Figs 1–5). An oscillatory activating pattern was occasionally observed (Fig. 2B), usually at a near-threshold concentration of CCh (1 μm). Activation of ICCh was abolished, when millimolar EGTA (4-10 mM) was included in the pipette under the conventional voltage-clamp conditions (10 observations), thus suggesting that the conductance underlying ICCh is Ca2+ dependent.
Figure 2. Effects of muscarinic subtype-specific antagonists on ICCh.
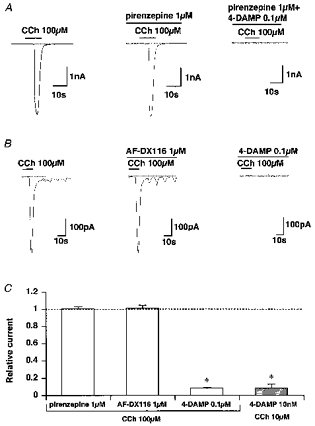
Here and in subsequent figures, unless otherwise stated, recording conditions are as for Fig. 1B. In A and B, the indicated antagonists were added at least 10 min before applying CCh. Here and in subsequent figures the dotted line indicates the zero current level. C, summary of inhibition of ICCh by pirenzepine, AF-DX 116 and 4-DAMP. The vertical axis shows the amplitude of ICCh in the presence of antagonists normalized relative to their controls (i.e. before application of drugs). Asterisks indicate significant differences using Student's paired t test (P < 0.005).
The amplitude of ICCh was not affected by removing external Ca2+ or Na+ from the bath (for summary, see Fig. 4B), or by setting the K+ equilibrium potential close to the holding potential of -70 mV (i.e. when [K+]o was raised to about 9 mM; see Methods), thus suggesting involvement of a Cl− conductance. We therefore tried to evaluate the reversal potential of ICCh less equivocally with Cs+ internal solution, thus largely eliminating the contribution of K+. By varying the holding potential (Fig. 1A) or applying a slow rising ramp voltage (Fig. 1C), the reversal potential of ICCh was found to be -20 mV (-18 ± 2 mV, n = 6), which is close to the reported equilibrium potential of Cl− in various airway smooth muscles (Aickin, 1990). This value did not change significantly upon substituting all external cations with an impermeant cation, N-methyl-D-glucamine (-16 ± 2 mV, n = 4; P > 0.05, Student's unpaired t test). It is thus highly likely that a substantial part of ICCh is carried through Cl−-selective channels. In addition, ICCh appears to exhibit little voltage dependence, especially at membrane potentials more negative to the reversal potential (see the inward portion of curve a in Fig. 1C).
Figure 4. Pharmacological profile of ICCh.
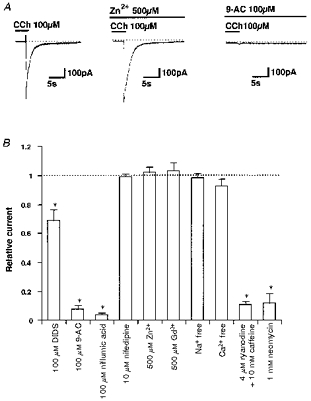
A, examples of actual traces. Zn2+ (500 μm) was added shortly before application of CCh, as it blocks cation channels almost immediately. In contrast, the inhibitory effects of 100 μm 9-AC on ICCh take much longer to develop and so 9-AC was applied > 20 min before CCh. B, summary of inhibition by various blockers. Vertical axis shows the amplitude of ICCh in the presence of the blockers normalized to their controls. Note that the values for DIDS, 9-AC and niflumic acid were obtained 20–30 min after the start of application to confirm their maximal effects. The data in Ca2+-free solution (0.1 mM EGTA) were obtained 5 min after its introduction. Repeated activation of ICCh in Ca2+-free solution resulted in a gradual decline in amplitude (not shown). Asterisks indicate significant differences with Student's paired t test (P < 0.005).
To determine the subtype of muscarinic receptors involved in ICCh activation, the effects of three relatively subtype-specific muscarinic antagonists, pirenzepine, AF-DX 116 and 4-DAMP (Eglen et al. 1996) were tested. As demonstrated in Fig. 2A and B, 10 min pretreatment with 1 μm pirenzepine or AF-DX 116 (concentrations 10–100 times higher than the Kd values for M1 and M2 muscarinic receptors, respectively; Eglen et al. 1996), failed to inhibit ICCh induced by 100 μm CCh (Fig. 2C). In contrast, 100 nM 4-DAMP, the M3 receptor-specific antagonist, greatly reduced the amplitude of inward current activated by the same concentration of CCh (Fig. 2C). Furthermore, when CCh concentration was reduced to one-tenth (i.e. 10 μm), the concentration of 4-DAMP required to substantially abolish ICCh activation was also reduced to one-tenth (10 nM, Fig. 2C). Although thorough inspection of the antagonism between CCh and the three muscarinic antagonists over the full concentration range was not feasible due to technical difficulties, these results are consistent with the muscarinic receptor involved in ICCh activation being of M3 subtype.
Inward currents having similar properties to ICCh, e.g. magnitude, time course of activation, reversal potential and dependence on intracellular [Ca2+] (data not shown), were also induced by application of 10 mM caffeine (-894 ± 107 pA at -70 mV, n = 9, hatched column in Fig. 1B; for actual records, see Figs 3A and 7). In the continued presence of 10 mM caffeine, or after combined application of 10 mM caffeine plus 4 μm ryanodine, CCh (100 μm) was almost ineffective (Figs 3 and 4B). Furthermore, 10 min pretreatment of cells with neomycin, which non-specifically inhibits phospholipases including phospholipase C, resulted in a marked diminution of ICCh (Fig. 4B). In four other experiments, we applied 3 mg ml−1 heparin intracellularly via a patch pipette. After 10 min, no detectable inward currents were evoked by 10–100 μm CCh. These results collectively suggest that increased production of IP3 upon M3 muscarinic receptor stimulation and subsequent release of stored Ca2+ is responsible for ICCh activation.
Figure 3. Store depletion abolishes ICCh.
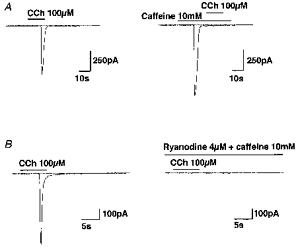
In both A and B, drugs were added as indicated by the bars except in the right panel of B, where 4 μm ryanodine and 10 mM caffeine were introduced into the bath about 5 min before application of CCh.
Figure 7. SNAP or db-cGMP are unable to inhibit the caffeine-induced Cl− current.
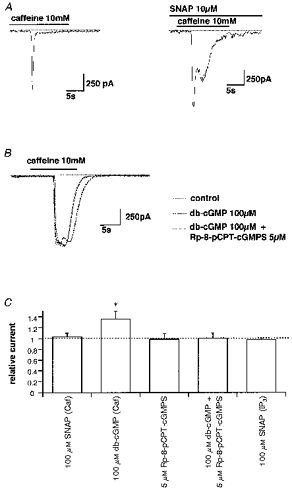
A, actual traces of caffeine-induced currents. SNAP was added in the bath 10 min before application of CCh. B, actual traces of caffeine-induced currents in the absence (continuous curve) and presence of db-cGMP (bold dotted curve) or db-cGMP plus Rp-8-pCPT-cGMPS (dotted curve). C, summary of experiments as shown in A and B. Vertical axis shows the amplitude of ICCh in the presence of the drugs normalized to their controls. Asterisk indicates a significant difference evaluated by Student's paired t test (P < 0.05). Caf and IP3 indicate inward currents activated by 10 mM caffeine or photolytically released IP3, respectively.
The pharmacological profile of ICCh seems to accord with that of Ca2+-dependent Cl− currents so far identified in smooth muscle (for review see Large & Wang, 1996). As illustrated and summarized in Fig. 4, ICCh was not significantly affected by the cation channel blockers such as 500 μm Zn2+ or Gd3+ (for review see Kuriyama et al. 1995), and 10 μm nifedipine. However, the amplitude of ICCh was significantly decreased with 100 μm DIDS, and greatly suppressed by 100 μm 9-AC or 100 μm niflumic acid, the concentrations of which were similar to those reported to block Ca2+-dependent Cl− currents in other types of smooth muscle (Large & Wang, 1996). It thus looks reasonable to designate ICCh as the Ca2+-dependent Cl− current (ICl(Ca)) in the rest of this paper.
NO donors greatly suppress ICl(Ca) through the cGMP-dependent mechanism
Nitric oxide (NO) or its related compounds are thought to affect the activity of a wide variety of ion channels (Kuriyama et al. 1995; McDonald & Murad, 1996). We therefore tested how these agents influenced the activity of ICl(Ca) in cat tracheal smooth muscle, about which very little is yet known.
Figure 5A demonstrates a typical example of the effects of SNAP (10 μm) on CCh (100 μm)-induced ICl(Ca). In the case shown, SNAP inhibited ICl(Ca) amplitude (by 48 % of control) and the total charge carried and appeared to accelerate the deactivation time course of ICl(Ca). Inhibition of ICl(Ca) amplitude was concentration dependent with 50 % inhibition of ICl(Ca) by SNAP occurring at about 10 μm (Fig. 5C). Furthermore, the fact that SNAP inhibits ICl(Ca) maximally activated by 100 μm CCh suggests that a reduced sensitivity of muscarinic receptors to CCh may not be the main cause of this inhibition. A similar dose-dependent inhibition of ICl(Ca) was also observed with different types of NO donors, GTN (39 ± 13 % of control with 10 μm, n = 5) and NO-R (56 ± 7 % of control with 10 μm, n = 3).
Inhibition of CCh-induced ICl(Ca) by NO donors was effectively antagonized by simultaneous addition of the soluble guanylate cyclase inhibitor Methylene Blue. At a concentration of 50 nM, Methylene Blue abolished the inhibitory effects of 10 μm SNAP (Fig. 5B and C). A further increase in Methylene Blue concentration caused a significant reduction in ICl(Ca) (by 35 ± 5 % of control at 1 μm, n = 6). In contrast, a membrane permeable analogue of cGMP, dibutyryl cGMP (db-cGMP), mimicked the inhibitory effects of NO donors on CCh-induced ICl(Ca) (Fig. 6A). Five minutes pretreatment with 100 μm db-cGMP reduced the amplitude of ICl(Ca) by about 80 % (Fig. 6C). These results suggest that NO donors exerted the inhibitory actions on ICl(Ca) via an increased production of cGMP.
Figure 6. Inhibitory effects of cGMP analogues on ICCh and inability of G-kinase inhibitors to antagonize them.
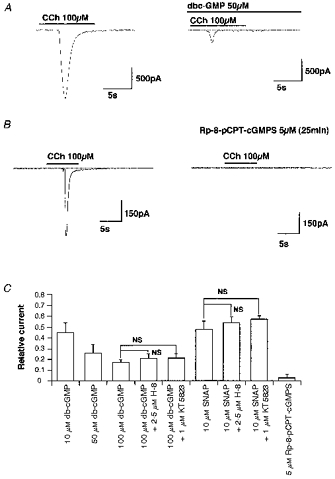
A and B, actual traces. In A, db-cGMP was added 10 min before application of CCh. C, summary of the experiments. Vertical axis shows the amplitude of ICCh in the presence of the drugs normalized to their controls. NS, no significant difference with Student's unpaired t test (P > 0.05). All the columns shown were significantly different from their controls in the absence of drug (Student's paired t test, P < 0.05). The number of experiments was 4–10 for each column.
Cyclic GMP-dependent inhibition of ICl(Ca) does not require protein phosphorylation
It has generally been thought that the cellular effects of cGMP are mediated by a G-kinase which phosphorylates a variety of functional proteins including ion channels and thereby alters their function (McDonald & Murad, 1996). We therefore tested this possibility for the case of NO donor-induced inhibition of ICl(Ca).
As summarized in Fig. 6C, potent inhibitors of cGMP-dependent kinase, H-8 and KT5823, did not significantly affect the inhibitory effects of SNAP or db-cGMP, at concentrations known to block the G-kinase almost maximally and relatively specifically (2.5 and 1 μm, respectively). In addition, a recently synthesized membrane-permeable Rp-diastereomer of cGMP Rp-8-pCPT-cGMPS which is a highly specific antagonist of the activation of cGMP-dependent kinase by cGMP that does not affect cAMP-dependent kinase or cGMP-regulated phosphodiesterases (Butt et al. 1994), itself produced markedly decreased ICl(Ca) (Fig. 6B and C). These results strongly suggest that inhibition of CCh-induced ICl(Ca) may involve a mechanism depending on cGMP but not requiring the G-kinase (thus phosphorylation independent) (see Discussion).
cGMP-dependent ICl(Ca) inhibition may involve a reduced IP3 production
To gain more insight into the mechanism underlying these observations, we tested the effects of NO donors and db-cGMP on ICl(Ca) induced by 10 mM caffeine, a compound known to release stored Ca2+ without stimulating IP3 production. In contrast to CCh, neither 10 μm SNAP nor 100 μm db-cGMP were able to inhibit the caffeine-induced ICl(Ca), on the contrary, they seemed to slightly increase its amplitude and duration (Fig. 7A and B). This observation could reflect, in part, an increased Ca2+ content in the store as a result of a cGMP-dependent increase in Ca2+ re-uptake (Raeymaekers et al. 1990). We also tested Rp-8-pCPT-cGMPs, which had potently inhibited CCh-induced ICl(Ca), but no inhibitory effects were observed on caffeine-induced ICl(Ca) (Fig. 7C). This compound reversed the augmentative effects of SNAP (not shown) or db-cGMP (Fig. 7B) on caffeine-induced ICl(Ca).
Finally, we tested whether or not the sensitivity of the IP3 receptor (channel) could be affected by SNAP using flash photolysis of caged IP3 under conventional whole-cell voltage-clamp conditions. For this, a sufficiently short duration of flash that activates the inward current only partially (1-2 ms given at an interval of 3–5 min; see also Methods) was chosen to minimize run-down. As illustrated in Fig. 8, the amplitude of the IP3-induced inward current was not significantly affected 10 min after application of 10 μm SNAP (98 ± 3 % of control, n = 6).
Figure 8. SNAP does not affect the Cl− current activated by photolytically released IP3.
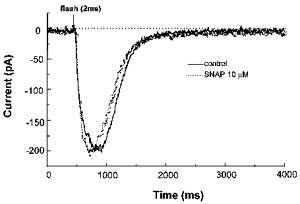
Caged IP3 (200 μm) was introduced into the cell via the patch pipette and allowed to equilibrate for 20 min. The holding potential was -70 mV. In this particular cell, repeated flashes (2 ms) at an interval of 3 min produced almost constant inward currents over 30 min. SNAP (10 μm) was added into the bath between the 4th and 5th flashes.
Taken together, the results obtained above suggest that the inhibitory effects of NO donors or db-cGMP on CCh-induced Ca2+ release and Cl− current are not mediated by alterations in the IP3 receptor, the amount of stored Ca2+ or Ca2+-dependent Cl− channels or their accessory proteins.
DISCUSSION
The main conclusions of this study are that: (1) in cat tracheal myocytes the predominant effect of CCh is to activate a Ca2+-dependent Cl− current (ICl(Ca)) through the M3 muscarinic receptor, via an IP3-mediated Ca2+ release, and (2) NO donors such as SNAP, GTN and NO-R potently inhibit ICl(Ca) via an increased intracellular cGMP level, which seems to act independently of G-kinase on site(s) located downstream of the muscarinic receptors and upstream of the IP3 receptor. The second conclusion is novel, but sheds light on the modulation by the NO-cGMP cascade of agonist-induced Ca2+ mobilization, not only in smooth muscle but perhaps in other types of tissue (see below).
In tracheal smooth muscle cells from several species, one of the major actions of CCh is to activate a Cl− conductance produced as a result of Ca2+ release from internal stores by stimulating IP3 production (dog: Janssen & Sims, 1992; guinea-pig: Henmi et al. 1996; human: Janssen, 1996; horse; Wang & Kotlikoff, 1997b). Consistent with these observations, we found that, in cat tracheal myocytes, pretreatment with ryanodine (plus caffeine) or neomycin or intracellular application of heparin, abolished the activation of the Cl− current produced by CCh (ICCh or ICl(Ca)), whereas caffeine and photo-released IP3 mimicked its effect. The fact that among the three subtype-specific antagonists, the M3 muscarinic receptor antagonist 4-DAMP alone potently inhibited ICCh, supports this conclusion, since the M3 receptor is thought to link via a PTX-insensitive G protein (Gq/G11) with the phospholipase Cβ which catalyses the conversion of phosphatidylinositides to IP3 and diacylgylerol in various types of smooth muscle including the trachea (Eglen et al. 1996; Wang & Kotlikoff, 1997b). On the other hand, the large averaged conductance of ICCh (more than 10 nS; -870 pA at -70 mV) suggests that only partial activation of ICCh (10 % or less) would depolarize the membrane sufficiently to increase Ca2+ entry through the voltage-dependent, dihydropyridine-sensitive Ca2+ channels and thereby evoke contraction (on the assumption that the resting conductance is more than 10 times lower (about 1 nS cell−1; unpublished data) than the conductance underlying ICCh and the Cl− equilibrium potential about -20 mV). Indeed, voltage-dependent Ca2+ currents with a threshold of about -40 mV have been identified in tracheal smooth muscle of various species (e.g. Hisada et al. 1990). Recently, growing attention has been directed to the importance of Cl− conductance (not restricted to Ca2+-dependent Cl− channels) in the control of membrane excitability and muscle tone in various vascular smooth muscles (Large & Wang, 1996; Nelson et al. 1997). In these studies, it has been shown that selective blockers of Cl− channels/transporters such as DIDS, 9-AC, niflumic acid and indanyloxyacetic acid (IAA-94), effectively attenuate the agonist- or pressure- induced contractions. We explored such a role for ICl(Ca) in cat trachea, by employing a similar approach. Prolonged treatment (> 30 min) with 10–100 μm niflumic acid caused a variable inhibition (by 10–80 % of control) of neurally evoked atropine-sensitive contractions of cat tracheal muscle, but the voltage-dependent Ca2+ channel blocker nifedipine (10-100 μm) exerted a much less inhibitory effect (by 5–10 % of control). This discrepancy between the actions of niflumic acid and nifedipine could be accounted for by an additional (inhibitory) action of niflumic acid on the muscarinic receptor-mediated Ca2+ mobilization. Thus, our tentative conclusion is that the role played by ICl(Ca) activation in increasing Ca2+ entry is much less important than that of IP3-mediated Ca2+ release in muscarinic receptor-mediated [Ca2+]i elevation in cat tracheal muscle.
Inhibition of CCh-induced ICl(Ca) (or Ca2+ mobilization) by SNAP or db-cGMP is so potent that even when maximally activated (by 100 μm CCh) ICl(Ca) was inhibited by more than half. This suggests that desensitization of the muscarinic receptor, albeit present, cannot account for the major part of SNAP- or db-cGMP-induced ICl(Ca) inhibition. Similarly, involvement, in this inhibition, of decreased stored Ca2+ content or reduced Ca2+-dependent Cl− channel activity can clearly be precluded by the experiments shown in Figs 7 and 8, where ICl(Ca) induced by 10 mM caffeine was not inhibited by SNAP or db-cGMP. Thus, the most probable mechanisms involved are an impairment of process(es) tightly associated with IP3 production (Murthy et al. 1993; Hirata & Murad, 1994) or alteration of essential properties of the IP3 receptor such as reduced IP3 affinity and decreased Ca2+-mobilizing efficacy (Murthy et al. 1993). The experiment shown in Fig. 8 greatly reduces the second possibility, since no significant inhibition occurred of the submaximal current activated by an intermediate flash duration, although in this experiment lack of a precise relationship between the photo-released IP3 dose and the resultant ICl(Ca) amplitude obscures the validity of this conclusion. The first possibility, on the other hand, seems more likely. It has been shown that in bovine aortic smooth muscle, cGMP inhibits the vasopressin- or GTPγS-induced activation of phospholipase C. This effect is likely to occur at the level of the G protein or the interaction of activated G protein with phospholipase C, since inhibition was not observed when phospholipase C was directly activated by Ca2+ (Hirata & Murad, 1994). However, in this case, the hydrolysis of ATP was a prerequisite (i.e. G-kinase mediated), which is inconsistent with our observations (also see below). We are now trying to measure IP3 using a radioisotope technique, to delineate more exactly the effects of NO on IP3 synthesis during muscarinic receptor stimulation in cat tracheal muscle.
In general, inhibitory or relaxing actions of NO donors or cGMP-increasing agents on smooth muscle are thought to be mediated by altered properties of various cellular proteins which participate in Ca2+ homeostasis as well as those directly associated with contractile events (Lincoln, 1989; Kuriyama et al. 1995). Well-established targets of cGMP include BK (augmentation: Robertson et al. 1993; Yamakage et al. 1996), voltage-dependent Ca2+ channels (inhibition: Sperelakis & Ohya, 1991), sarcoplasmic and sarcolemmal Ca2+-ATPases (stimulation: Raeymaekers et al. 1990; Wuytak & Raeymaekers, 1992), myosin light chain kinase (inhibition: Kuriyama et al. 1995), and possibly the IP3 receptor (Komalavilas & Lincoln, 1994). These cGMP-mediated changes have been attributed mostly to protein phosphorylation by G-kinase (Lincoln, 1989; Kuriyama et al. 1995), although cGMP also regulates phosphodiesterases (type II and III) and cyclic nucleotide-gated cation channels (McDonald & Murad, 1996). For example, the open probability of BK in porcine tracheal myocytes has been shown to increase about 13-fold by 10 μm sodium nitroprusside, and this is prevented by the G-kinase inhibitor Rp-8-pCPT-cGMPS (20 μm) (Yamakage et al. 1996). A more complicated but also G-kinase-dependent model of BK regulation by cGMP has also been proposed in bovine tracheal myocytes, in the light of inhibitors for G-kinase (Rp-8-pCPT-cGMPs) and phosphatase (okadaic acid; Zhou et al. 1996). Against this line of interpretation, however, we found using essentially the same approach, that two structurally distinct G-kinase inhibitors KT5823 and H-8 each failed to antagonize the inhibitory effects of SNAP or db-cGMP. This could not be due to inadequate application of the inhibitors, since small increases in caffeine-induced ICl(Ca) by db-cGMP (Fig. 7) were effectively blocked by KT5823 (authors' unpublished observation) or Rp-8-pCPT-cGMPs (Fig. 7B). It has been reported that cAMP can cross-activate G-kinase at its higher concentrations (Lincoln et al. 1990). Thus, the reverse, i.e. cGMP cross-activation of A-kinase, might also occur. However, this possibility is also unlikely, since a non-specific inhibitor for both A- and G-kinases, H-8 (2.5 μm; Ki= 1.2 and 0.48 μm, respectively), did not significantly affect the inhibitory effects of NO donors (Fig. 6). Taken together, the most likely interpretation at present is that inhibition of IP3 synthesis by cGMP involves a more direct process(es) than the phosphorylation-dephosphorylation cycle, e.g. direct interaction of cGMP with phospholipase C or its associated G protein (Hirata & Murad, 1994). This type of regulation represents a novel modulation by cGMP of agonist-induced Ca2+ mobilization, and is reminiscent of the actions of cyclic nucleotides on retinal and olfactory cyclic nucleotide-gated cation channels, hyperpolarization-activated cation channels and inward-rectifying K+ channels (Finn et al. 1996; Ito et al. 1997). Interestingly, in cardiac myocytes, Rp-cAMP, one of the ‘antagonistic’ diastereometric phosphorothioate derivatives of cAMP, has very recently been found to increase the activity of hyperpolarization-activated cation currents (Bois et al. 1997). This is strikingly similar to our finding that Rp-8-pCPT-cGMPs, like db-cGMP, caused a marked reduction in ICl(Ca). Obviously, further investigation will be required before an exact understanding of these observations is possible.
In conclusion, we have demonstrated the importance of ICl(Ca) in the cat tracheal smooth muscle membrane, which can be activated by the Ca2+ release triggered by IP3 and caffeine. Interestingly, NO donors were able to suppress ICl(Ca) only when this was activated through the muscarinic receptor, and thus the inhibition seems to reflect mainly a reduced synthesis of IP3 caused by an increased cGMP concentration, in a G-kinase-independent fashion. These results provide a novel mechanism for the inhibitory actions of NO in the airway, and may suggest a vital role for NO as a brake against bronchoconstriction caused by cholinergic overdrive (Jing et al. 1995).
Acknowledgments
The authors are grateful to Professor A. F. Brading (University Department of Pharmacology, Oxford, UK), Miss Y. Taniguchi for critically reading the manuscript and typing the text and Miss M. Yoshikawa for technical assistance. Thanks are also due to Drs H. Tanaka and M. Fujisawa, who helped us obtain cat tracheal segments. This work was supported, in part, by grants-in-aid from the Ministry of Science, Education and Culture, Japan, and from the Mitsui Life Social Welfare Foundation, awarded to Y. I.
References
- Aickin C. Chloride transport across the sarcolemma of vertebrate smooth and skeletal muscle. In: Alvarez-Leefmans FJ, Russell JM, editors. Chloride Channels and Carriers in Nerve, Muscle and Glial Cells. New York: Plenum Press; 1990. pp. 209–249. [Google Scholar]
- Bialecki RA, Stinton-Fisher C. KCa channel antagonists reduce NO donor-mediated relaxation of vascular and tracheal smooth muscle. American Journal of Physiology. 1995;268:L152–159. doi: 10.1152/ajplung.1995.268.1.L152. [DOI] [PubMed] [Google Scholar]
- Bois P, Renaudon B, Baruscotti M, Lenfant J, Difrancesco D. Activation of f-channels by cAMP analogues in macropatches from rabbit sino-atrial node myocytes. The Journal of Physiology. 1997;501:565–571. doi: 10.1111/j.1469-7793.1997.565bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bootman M, Berridge MJ, Taylor CW. All-or-nothing Ca2+ mobilization from the intracellular stores of single histamine-stimulated Hela cells. The Journal of Physiology. 1992;450:163–178. doi: 10.1113/jphysiol.1992.sp019121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butt E, Eigenthaler M, Genieser H-G. (Rp)-8-pCPT-cGMPS, a novel cGMP-dependent protein kinase inhibitor. European Journal of Pharmacology. 1994;269:265–268. doi: 10.1016/0922-4106(94)90095-7. [DOI] [PubMed] [Google Scholar]
- Eglen RM, Hedge SS, Watson N. Muscarinic receptor subtypes and smooth muscle function. Pharmacological Reviews. 1996;48:531–565. [PubMed] [Google Scholar]
- Finn JT, Grunwald ME, Yau K-W. Cyclic nucleotide-gated ion channels: an extended family with diverse functions. Annual Review of Physiology. 1996;58:395–426. doi: 10.1146/annurev.ph.58.030196.002143. 10.1146/annurev.ph.58.030196.002143. [DOI] [PubMed] [Google Scholar]
- Fleischmann BK, Wang Y-X, Kotlikoff MI. Muscarinic activation and calcium permeation of nonselective cation currents in airway myocytes. American Journal of Physiology. 1997;272:C341–349. doi: 10.1152/ajpcell.1997.272.1.C341. [DOI] [PubMed] [Google Scholar]
- Hamaguchi M, Ishibashi T, Imai S. Involvement of charybdotoxin-sensitive K+ channel in the relaxation of bovine tracheal smooth muscle by glyceryl trinitrate and sodium nitroprusside. Journal of Pharmacology and Experimental Therapeutics. 1992;262:263–270. [PubMed] [Google Scholar]
- Hashimoto T, Hirata M, Ito Y. A role for inositol 1,4,5-trisphosphate in the initiation of agonist-induced contractions of dog tracheal smooth muscle. British Journal of Pharmacology. 1985;86:191–199. doi: 10.1111/j.1476-5381.1985.tb09449.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henmi S, Imaizumi Y, Muraki K, Watanabe M. Time course of Ca2+-dependent K+ and Cl− currents in single smooth muscle cells of guinea-pig trachea. European Journal of Pharmacology. 1996;306:227–236. doi: 10.1016/0014-2999(96)00193-8. 10.1016/0014-2999(96)00193-8. [DOI] [PubMed] [Google Scholar]
- Hirata M, Murad F. Interrelationships of cyclic GMP, inositol phosphates, and calcium. Advances in Pharmacology. 1994;26:195–216. doi: 10.1016/s1054-3589(08)60055-1. [DOI] [PubMed] [Google Scholar]
- Hisada T, Kurachi Y, Sugimoto T. Properties of membrane currents in isolated smooth muscle cells from guinea-pig trachea. Pflügers Archiv. 1990;416:151–161. doi: 10.1007/BF00370237. [DOI] [PubMed] [Google Scholar]
- Horn R, Marty A. Muscarinic activation of ionic currents measured by a new whole-cell recording method. Journal of General Physiology. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino M, Yamazawa T, Miyashita Y, Endo M, Kasai H. Critical intracellular Ca2+ concentration for all-or-none Ca2+ spiking in single smooth muscle cells. EMBO Journal. 1993;12:5287–5291. doi: 10.1002/j.1460-2075.1993.tb06224.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito H, Tsuchimochi H, Tada Y, Kurachi Y. Phosphorylation-independent inhibition by intracellular cyclic nucleotides of brain inwardly rectifying K+ current expressed in Xenopus oocytes. FEBS Letters. 1997;402:12–16. doi: 10.1016/s0014-5793(96)01458-5. [DOI] [PubMed] [Google Scholar]
- Janssen LJ. Acetylcholine and caffeine activate Cl− and suppress K+ conductances in human bronchial smooth muscle. American Journal of Physiology. 1996;270:L772–781. doi: 10.1152/ajplung.1996.270.5.L772. [DOI] [PubMed] [Google Scholar]
- Janssen LJ, Sims SM. Acetylcholine activates non-selective cation and chloride conductances in canine and guinea-pig tracheal smooth muscle cells. The Journal of Physiology. 1992;453:197–218. doi: 10.1113/jphysiol.1992.sp019224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LJ, Sims SM. Histamine activates Cl− and K+ currents in guinea-pig tracheal myocytes: convergence with muscarinic signalling pathway. The Journal of Physiology. 1993;465:661–677. doi: 10.1113/jphysiol.1993.sp019699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen LJ, Sims SM. Ca2+-dependent Cl− current in canine tracheal smooth muscle cells. American Journal of Physiology. 1995;269:C163–169. doi: 10.1152/ajpcell.1995.269.1.C163. [DOI] [PubMed] [Google Scholar]
- Jing R, Inoue R, Tashiro K, Takahashi S, Ito Y. Role of nitric oxide in non-adrenergic, non-cholinergic relaxation and modulation of excitatory neuroeffector transmission in cat airway. The Journal of Physiology. 1995;483:225–237. doi: 10.1113/jphysiol.1995.sp020580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisophosphate receptor by cyclic GMP-dependent protein kinase. Journal of Biological Chemistry. 1994;269:8701–8707. [PubMed] [Google Scholar]
- Kume H, Takai A, Tokuno H, Tomita T. Regulation of Ca2+-dependent K+ channel activity in tracheal myocytes by phosphorylation. Nature. 1989;341:152–154. doi: 10.1038/341152a0. [DOI] [PubMed] [Google Scholar]
- Kuriyama H, Kitamura K, Nabata H. Pharmacological and physiological significance of ion channels and factors that modulate them in vascular tissues. Pharmacological Reviews. 1995;47:387–573. [PubMed] [Google Scholar]
- Large WA, Wang Q. Characteristics and physiological role of the Ca2+-activated Cl− conductance in smooth muscle. American Journal of Physiology. 1996;271:C453–454. doi: 10.1152/ajpcell.1996.271.2.C435. [DOI] [PubMed] [Google Scholar]
- Lincoln TM. Cyclic GMP and mechanisms of vasodilation. Pharmacology and Therapeutics. 1989;41:479–502. doi: 10.1016/0163-7258(89)90127-7. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Cornwell TL, Taylor AE. cGMP-dependent protein kinase mediates the reduction of Ca2+ by cAMP in vascular smooth muscle cells. American Journal of Physiology. 1990;258:C399–407. doi: 10.1152/ajpcell.1990.258.3.C399. [DOI] [PubMed] [Google Scholar]
- McDonald LJ, Murad F. Nitric oxide and cyclic GMP signaling. Proceedings of the Society for Experimental Biology and Medicine. 1996;211:1–6. doi: 10.3181/00379727-211-43950a. [DOI] [PubMed] [Google Scholar]
- Murray RK, Kotlikoff MI. Receptor-activated calcium influx in human airway smooth muscle cells. The Journal of Physiology. 1991;435:123–144. doi: 10.1113/jphysiol.1991.sp018501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy KS, Severi C, Grider JR, Makhlouf GM. Inhibition of IP3 and IP3-dependent Ca2+ mobilization by cyclic nucleotides in isolated gastric muscle cells. American Journal of Physiology. 1993;264:G967–974. doi: 10.1152/ajpgi.1993.264.5.G967. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Conway MA, Knot HJ, Brayden JE. Chloride channel blockers inhibit myogenic tone in rat cerebral arteries. The Journal of Physiology. 1997;502:259–264. doi: 10.1111/j.1469-7793.1997.259bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson MT, Patlak JB, Worley JF, Standen NB. Calcium channels, potassium channels, and voltage-dependence of arterial smooth muscle cell tone. American Journal of Physiology. 1990;259:C3–18. doi: 10.1152/ajpcell.1990.259.1.C3. [DOI] [PubMed] [Google Scholar]
- Raeymaekers L, Eggermont JA, Wuytack F, Casteels R. Effects of cyclic nucleotide dependent protein kinases on the endoplasmic reticulum Ca2+ pump of bovine pulmonary artery. Cell Calcium. 1990;11:261–268. doi: 10.1016/0143-4160(90)90002-c. [DOI] [PubMed] [Google Scholar]
- Robertson BE, Schubert R, Hescheler J, Nelson MT. cGMP-dependent protein kinase activates Ca-activated K channels in cerebral artery smooth muscle cells. American Journal of Physiology. 1993;265:C299–303. doi: 10.1152/ajpcell.1993.265.1.C299. [DOI] [PubMed] [Google Scholar]
- Sperelakis N, Ohya Y. Regulation of calcium slow channels in vascular smooth muscle cells. In: Sperelakis N, Kuriyama H, editors. Ion Channels of Vascular Smooth Muscle Cells and Endothelial Cells. Elsevier; 1991. pp. 27–38. [Google Scholar]
- Takahashi N, Tanaka H, Abdullah NA, Jing L, Inoue R, Ito Y. Regional difference in the distribution of L-NAME-sensitive and -insensitive NANC relaxation in cat airway. The Journal of Physiology. 1995;488:709–720. doi: 10.1113/jphysiol.1995.sp021002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y-X, Kotlikoff MI. M2 receptor activation of nonselective cation channels in smooth muscle cells: calcium and Gi/Go requirements. American Journal of Physiology. 1997a;272:C341–349. doi: 10.1152/ajpcell.1997.273.2.C500. [DOI] [PubMed] [Google Scholar]
- Wang Y-X, Kotlikoff MI. Muscarinic signaling pathway for calcium release and calcium-activated chloride current in smooth muscle. American Journal of Physiology. 1997b;273:C509–519. doi: 10.1152/ajpcell.1997.273.2.C509. [DOI] [PubMed] [Google Scholar]
- Waniishi Y, Inoue R, Ito Y. Preferential, potentiation by hypotonic cell swelling of muscarinic cation current in guinea-pig ileum. American Journal of Physiology. 1997a;472:C240–253. doi: 10.1152/ajpcell.1997.272.1.C240. [DOI] [PubMed] [Google Scholar]
- Waniishi Y, Morita H, Inoue R, Ito Y. NO donors inhibit activation of CCh-induced Cl− current through a cGMP-dependent mechanism in cat tracheal myocytes. Japanese Journal of Pharmacology. 1997b;73(suppl. I):55. [Google Scholar]
- Wuytack F, Raeymaekers L. The Ca2+-transport ATPases from the plasma membrane. Journal of Bioenergetics and Biomembranes. 1992;24:285–300. doi: 10.1007/BF00768849. [DOI] [PubMed] [Google Scholar]
- Yamakage M, Hirshman CA, Croxton TL. Sodium nitroprusside stimulates Ca2+-activated K+ channels in porcine tracheal smooth muscle cells. American Journal of Physiology. 1996;270:L338–345. doi: 10.1152/ajplung.1996.270.3.L338. [DOI] [PubMed] [Google Scholar]
- Zhou X-B, Ruth P, Achlossmann J, Hofmann F, Korth M. Protein phosphatase 2A is essential for the activation of Ca2+-activated K+ currents by cGMP-dependent protein kinase in tracheal smooth muscle and chinese hamster ovary cells. Journal of Biological Chemistry. 1996;271:19760–19767. doi: 10.1074/jbc.271.33.19760. [DOI] [PubMed] [Google Scholar]