

Abstract
Using whole-cell patch-clamp techniques, the mechanisms of NMDA channel blockade by amino-adamantane derivatives (AADs) memantine (3,5-dimethyl-aminoadamantane, MEM) and amantadine (1-aminoadamantane, AM) have been studied in rat hippocampal neurons acutely isolated by the vibrodissociation method. A rapid concentration-jump technique was used to replace superfusing solutions.
The aspartate (Asp)-induced channel opening greatly accelerated but was not a prerequisite for the recovery from the block by MEM: it was able to leave the channel without agonist assistance. The co-agonist (glycine) as well as the competitive NMDA antagonist dl-2-amino-7-phosphonoheptanoic acid (APV), did not affect this recovery. Membrane depolarization accelerated it, strongly suggesting that this process proceeded via the hydrophilic pathway of the channel.
A comparison of the kinetics of the recovery from the block by AADs in the presence and absence of the agonist prompted a hypothesis that the blocker trapped in the channel increased the probability of its transition to the open state.
Both MEM and AM were able to block NMDA channels not only in the presence but also in the absence of Asp, although in the latter case the effective blocking concentrations were much higher and the rate of the block development was much smaller than in the former case. The extent of the block increased with the duration of the blocker application. Glycine enhanced this block, while APV attenuated it. The MEM-induced blockade of agonist-unbound channels was enhanced by membrane hyperpolarization and weakened by external Mg2+. These findings strongly suggested that the blocker reached its binding sites via the same hydrophilic pathway both in the presence and absence of the agonist.
A comparative analysis of the channel unblocking kinetics in the presence of Asp after their blockade with or without the agonist assistance led us to conclude that in the two cases AADs were bound to the same blocking sites in the channel.
Recently it has been established that the amino-adamantane derivatives (AADs), memantine (MEM) and amantadine (AM) belong to the class of blockers manifesting the so-called ‘trapping block’ of NMDA channels (Johnson et al. 1995). Other representatives of this group are the well-known non-competitive NMDA-receptor antagonists MK-801, phencyclidine and ketamine (Huettner & Bean, 1987; Kemp et al. 1987; MacDonald et al. 1991). When applied externally, these drugs can enter into an open NMDA channel and bind to its ‘blocking site’ located deep in the pore. This binding, however, does not prevent the subsequent channel closure after the fast removal of the agonist from the medium. Therefore the blocking molecules can remain in the pore for a relatively long time being trapped ‘behind the closed activation gate’. Agonist reapplication opens the gate and thus allows the blocker to leave the channel. The simplified kinetic model of this block appears as follows:
where C, D and O represent the channel in closed, desensitized and open states, respectively; the subscripts A and B indicate the binding of all agonists and blockers to every possible site, respectively; and the asterisk indicates the conducting state. Proceeding from this assumption, one could expect that in the absence of the agonist both MEM and AM will be unable to block NMDA channels. However, this is not the case.
The data presented in this work show that in acutely isolated rat hippocampal neurons the aspartate (Asp)-induced opening of NMDA channels is not a prerequisite for their blockade by MEM and AM. These cationic compounds have proved to be able, although much more slowly, to enter and leave the NMDA channel via the ‘hydrophilic route’ without the assistance of the agonist. Moreover, the externally applied blocker reaches the same blocking site to which it binds in open channels. Some preliminary results from this study have been published in abstract form (Sobolevsky et al. 1996).
METHODS
Two- to four-week-old Wistar rats were killed by cervical dislocation. Hippocampal slices were prepared according to the procedure described by Vorobiev (1991). Pyramidal neurons were mechanically isolated from the CA-1 region of the slice by vibrodissociation (Vorobiev, 1991). The experiments were started no earlier than 3 h after incubation of the hippocampal slices in a medium containing (mm): NaCl, 124; KCl, 3; CaCl2, 1.4; MgCl2, 2; glucose, 10; NaHCO3, 26. The solution was bubbled with carbogen and maintained at 32°C. During the whole period of isolation and current recording, nerve cells were washed with a Mg2+-free solution (mm): NaCl, 140; KCl, 5; CaCl2, 2; glucose, 15; Hepes, 10; pH 7.3. All the drugs were dissolved in water. Concentrated drug stock solutions were prepared and kept frozen until use. Fast replacement of the superfusing solutions (τ < 30 ms) was achieved by using the concentration-jump technique (Benveniste et al. 1990; Vorobiev, 1991). The currents were recorded at 18°C in the whole-cell configuration by using micropipettes made from Pyrex tubes and filled with an ‘intracellular’ solution (mm): CsF, 140; NaCl, 4; Hepes, 10; pH 7.2. Electrical resistance of the filled micropipettes was 3–7 MΩ. The analog current signals were digitized at 1 kHz frequency.
Statistical analysis was performed with the aid of Origin 3.5 (Microcal Software Inc., MA, USA) software. All the data are presented as means ±s.e.m. and comparisons were made using Student's paired t test except as noted. To distinguish between one- and two-exponential fits, Fischer's test was used.
Amino-adamantane derivatives were synthesized at MERZ (Eckenheimer Landstr. 100–104, 60318 Frankfurt-am-Main, Germany). The pKa value for MEM is 10.27. Under our experimental conditions (pH 7.3) MEM and the more hydrophilic AM were almost completely dissociated and carried the charge of +1.
RESULTS
Blockade of open NMDA channels by MEM and AM
In agreement with previous reports (Chen et al. 1992; Parsons et al. 1993, 1995; Bresink et al. 1996; Blanpied et al. 1997; Chen & Lipton, 1997), MEM and AM produced a concentration-, time- and voltage-dependent blockade of open NMDA channels.
Ionic currents through NMDA channels were elicited by fast application of 100 μm aspartate (Asp) in a Mg2+-free, 3 μm glycine-containing solution. In all experiments except for those studying the voltage dependencies, the membrane potential was held at −100 mV. Asp induced an inward current which, after an initial fast rise (τ < 30 ms) up to the value IC, indicating the opening of NMDA channels, decreased gradually (τD= 374 ± 26 ms) down to a certain plateau level I0 (Fig. 1A, first trace). Such a current decay under continued action of the agonist is a result of desensitization of the receptor-channel complex. The rate of recovery from desensitization was fast (Fig. 1A). The time constant of this process measured in six cells with a fraction of desensitized channels, d = 1 - I0/IC= 0.50 ± 0.03, was 1.17 ± 0.05 s (Fig. 1B).
Figure 1. Desensitization of NMDA channels.
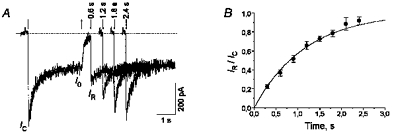
A, inward current through NMDA channels was elicited by a 3 s application of 100 μm Asp at the membrane potential of −100 mV. The dotted line indicates zero current level. The desensitization-induced current decay during the Asp pulse was fitted with the single exponential function with the time constant τD= 374 ± 26 ms (continuous line). The second to the fifth traces are the currents elicited by the test Asp application 0.6–2.4 s after the termination of the first (conditioning) Asp application. The initial current (IR) recovered with an increase in the time interval between the Asp applications. The downward and upward arrows indicate the beginning and termination of the Asp applications, respectively. B, time course of the recovery from desensitization. The continuous line is the single exponential fitting of IR/IC with the time constant τ= 1.17 ± 0.05 s.
MEM was applied at different concentrations in the continuous presence of Asp (100 μm). The two-exponential fitting of the current traces (Fig. 2A) made it possible to reveal the existence of two (fast and slow) kinetic components in both blocking and recovery processes. The average values of the fast and slow time constants (τfast and τslow, respectively) and the amplitude of the fast component (Afast) for the recovery from the MEM block are presented in Table 1. Afast decreased with a rise in the blocker concentration (the values of Afast at any two different concentrations were significantly different, P < 0.0002). This decrease is the evidence for two distinct blocking sites of MEM in open NMDA channels which can be simultaneously occupied by two blocker molecules (Sobolevsky & Koshelev, 1998). For the sake of simplicity, this point is not reflected in Model 1.
Figure 2. Kinetics, concentration and voltage dependencies of the amino-adamantane derivative-induced blockade of open NMDA channels.
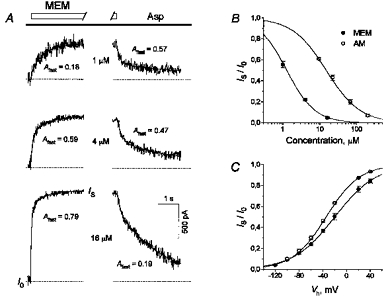
A, kinetics of the open NMDA channel interaction with MEM. Asp (100 μm) was applied continuously. MEM was co-administered at different concentrations for 5 s with Asp. Original NMDA responses were recorded at MEM concentrations of 1–16 μm. The current traces presented in the left and right panels show the onset and offset kinetics of MEM, respectively. The continuous lines show the fitting of these traces with two-exponential functions. Afast, the fraction of the fast component, rose with the MEM concentration at the onset and diminished at the offset. B, concentration dependence of the stationary blockade by MEM and AM. AADs were applied at different concentrations in the continuous presence of Asp, as shown in A. Plateau current responses (IS) divided by the control plateau value (I0) were plotted against the concentration of the blockers. The continuous lines show the fittings of the data with the logistic equation (eqn (1)). The fit parameters are: IC50= 1.28 μm, nH= 1.15 for MEM and IC50= 14.9 μm, nH= 0.94 for AM. C, voltage dependence of the open-channel blockade by AADs. MEM (10 μm) or AM (200 μm) at different membrane potentials were applied in the continuous presence of Asp, as shown in Fig. 2A. The IS/I0 values were plotted against the membrane potential. The continous lines show the fitting of the experimental data with eqn (2). The fit parameters are: Kd(0)= 17.9 μm, δ= 0.73 for MEM and Kd(0)= 694 μm, δ= 0.90 for AM.
Table 1.
Asp-induced dissociation kinetics of MEM under different conditions
| [MEM]μm | Afast | τfast (S) | τslow (S) |
|---|---|---|---|
| In the continuous presence of agonist | |||
| 1 | 0.55±0.05 | 1.5±0.3 | 20.4±4.3 |
| 4 | 0.42±0.03 | 1.3±0.5 | 18.8±1.1 |
| 16 | 0.19±0.01 | 1.3±0.3 | 17.0±1.9 |
| After blockade, without agonist assistance | |||
| 50 | 0.24±0.02 | 1.7±0.5 | 21.3±4.4 |
Model 1.
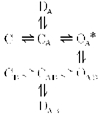
The kinetics of AM were much faster than those of MEM and were well fitted with monoexponential functions. The value of the time constant for the recovery from the AM block did not depend on the AM concentration, being on average 0.25 ± 0.04 s (n = 8).
Figure 2B shows the concentration dependencies of the stationary block of open NMDA channels by MEM and AM measured according to the experimental protocol shown in Fig. 2A. The fitting was performed in accordance with a two-parameter logistic equation:
| (1) |
where Io is the stationary current amplitude in the absence of the blocker, [C] is the blocker concentration, IC50 is the concentration resulting in 50 % block and nH is the Hill coefficient. The respective IC50 values and the Hill coefficient were: 1.28 ± 0.10 μm and 1.15 ± 0.07 (n = 6) for MEM and 14.9 ± 0.3 μm and 0.94 ± 0.04 (n = 9) for AM.
The extent of the block increased with membrane hyperpolarization. Figure 2C demonstrates the voltage dependence of the stationary blockade by MEM (10 μm) and AM (200 μm) examined using the experimental protocol shown in Fig. 2A. The fitting was performed according to the equation:
| (2) |
where I0 is the stationary current amplitude in the absence of the blocker, [C] is the blocker concentration, Vh is the holding membrane potential, Kd(0) is the equilibrium dissociation constant at Vh= 0 mV, and δ is the fraction of the membrane electric field seen by the blocker bound to a single blocking site. F, R and T have their usual physical meanings. The voltage dependence had the following parameters: Kd(0)= 17.9 ± 1.0 μm and δ= 0.73 ± 0.02 (n = 5) for MEM; Kd(0)= 694 ± 17 μm and δ= 0.90 ± 0.01 (n = 4) for AM.
Recovery of NMDA channels from the block without agonist assistance
Model 1 predicts that the blocker can leave the closed blocked state of the channel, CB, only after the binding of the agonist to the receptor. However, the data presented below show that the channel unblocking may also proceed without agonist assistance, the agonist greatly accelerating this process. The time course of this unblocking was monitored using the experimental protocol shown in Fig. 3A. The specific blocker of the NMDA receptor, APV (100 μm), was added to the washout solution in order to avoid possible activation of the channels by putative traces of the agonist in the medium. At the beginning of the experiment, Asp (100 μm) and MEM (25 μm) were co-applied once or several times up to a practically complete inhibition of the stationary current (IF). At this point, the value of IF/IC was 0.026 ± 0.002 (n = 19). At the end of Asp and MEM co-application, the majority of channels were in states OAB, CAB and DAB and only a small number of them were in states OA*, CA and DA (see Model 1). After washout during the time interval t (from 10 to 300 s), the cell was stimulated with an Asp (100 μm) test pulse. The latter elicited a fast current increase (IB) followed by its gradual elevation. The slow current increase up to its control stationary level (I0) evidently reflected the process of Asp-induced recovery from the block (the transitions from states CB, CAB, DAB and OAB to OA* in Model 1). The value of the IB time constant, τini, was equal to 118 ± 6 ms (n = 18), which exceeded that measured in the absence of APV in the washout solution (τ= 70 ± 10 ms, n = 11). This slow-down of the initial fast component of the current recovery was evidently determined from the dissociation kinetics of APV. As τini was much smaller than τfast of the current recovery kinetics in the presence of Asp (Fig. 2A, Table 1), IB may be considered as a current through those NMDA channels which had already reached the closed state C by the beginning of the Asp test pulse (see Model 1) and were ready to open right after APV dissociation. To estimate the value of IB, the following procedure was used. To exclude the influence of desensitization of non-blocked channels (transitions to state D) on the Asp-induced current recovery from the blocked states (CB, CAB, DAB and OAB), the test current trace (2) was divided by the control current trace (1). The IB/IC value was estimated via linear approximation of the initial phase of this quotient to the beginning of the Asp application after complete APV dissociation (Fig. 3A, inset). The fast component, IB, increased with the lengthening of the washout time interval t (Fig. 3B). The values of IB/IC at t = 0 s and 5 min were significantly different (P < 0.00025, n = 10); their difference was on average 0.32 (s.d.= 0.17). Figure 3C shows the time dependence of the IB/IC values. The time dependence of the mean IB/IC values included two components: (1) the fast component with a time constant smaller than 10 s and (2) the slow component with a time constant, τ*slow, greater than 100 s. To estimate the amplitude of the fast component, A*fast, and the time constant of the slow component, the data were fitted with the following equation:
| (3) |
where A is 1, A*fast is 0.13 ± 0.03 and τ*slow is 592 ± 129 s (n = 18). The parameter A was taken as unity because the fitting of the mean data and the individual fittings in the majority of cells (n = 11/15) with free A gave the value greater than or equal to 1 within the error. So in the majority of cells, the recovery from the MEM block in the absence of the agonist was complete. The individual fittings of MEM kinetics performed separately for each cell yielded the values of A*fast in the range 0 to 0.29 and τ*slow from 2.3 to 40 min. Thus, the kinetics of the recovery varied greatly in different cells. This variability can be explained by possible heterogeneity of NMDA receptors and their developmental changes. This seems to be quite probable taking into account the large number of females from which the young animals were taken (> 100) and the range of their age (14–28 days) during which time the properties of NMDA receptors vary greatly (McBain & Mayer, 1994).
Figure 3. NMDA channel recovery from AAD blockade without agonist assistance.
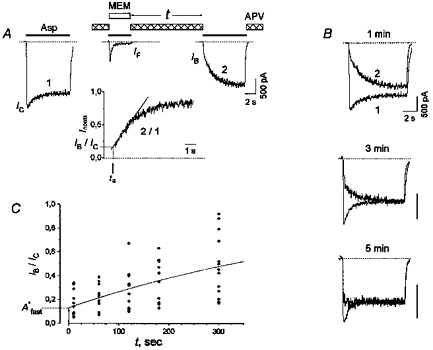
A, the experimental protocol was used to study the unblocking kinetics of the channels in the absence of the agonist. After MEM (25 μm) and Asp (100 μm) co-application, the cell was washed with an agonist-free control solution for 1 min. To ensure the absence of the agonist contamination, APV (100 μm) was added to the washout solution. The inset shows the ratio of test (2) and control (1) current traces. The linear part of the slow gradual increase of this ratio from the moment when APV dissociated from the channel was fitted with the linear equation Inorm= a + b (t - ta) (continous line). The value ‘a’ on this line corresponding to the beginning of the test Asp application (t = ta) is IB/IC. B, the superposition of control and test current traces obtained by using the experimental protocol shown in A at different (1, 3 and 5 min) washout time intervals. The dissociation of APV was fast; therefore the distortion of the initial current increase is not seen on this time scale. Note that the slow component of the current recovery decreases, while the fast component rises with an increase in t. C, the time course of the IB/IC recovery in the absence of the agonist. The continous line shows the fitting of the time dependence of mean IB/IC values with eqn (3). The fit parameters are: A = 1, A*fast= 0.13 and τ*slow= 592 s.
When both the agonist and the blocker were removed from the medium after their co-application, all the channels started to transit from the agonist-bound states OA*, CA, DA, OAB, CAB and DAB to states C and CB (see Model 1). In response to the test Asp application after the washout interval, the activation of the channels in state C yielded the fast component of the current (IB), whereas the transition of the channels from state CB to state OA* yielded the slow component of the current (IC - IB). Thus, the fast component of the IB/IC recovery with the amplitude of A*fast included NMDA channels which returned quickly to state C. What is the origin of the slow component? One possibility is the exit from desensitization (state DA). However, the recovery from the desensitization state was shown to be fast (Fig. 1). The other possibility which seems to reflect the real situation is that the slow component of the IB/IC recovery reflects the slow channel unblocking from state CB to state C without the agonist assistance via a pathway that is not shown in Model 1 (see Discussion).
The following experiments were carried out without addition of APV to the washout solution. This did not affect the rate of the NMDA channel recovery from the MEM-induced blockade in the absence of the agonist. Thus, the values of IB/IC at t = 1 min were 0.20 ± 0.03 and 0.20 ± 0.02 in the presence and absence of APV (100 μm), respectively (these values were not significantly different, P > 0.92, n = 10). This observation provided strong evidence that our solutions were not contaminated with traces of NMDA receptor agonists.
The recovery from the MEM block without agonist assistance was found to be voltage dependent (Fig. 4). Membrane depolarization accelerated this process. Thus, after a 1 min washout at −100 mV, IB/IC was 0.19 ± 0.03, whereas in the case when the membrane potential was switched to 0 mV during the washout time interval, IB/IC was 0.30 ± 0.05 (these values were significantly different, P < 0.01, n = 9). The switch-over of Vh to 0 mV was performed 10 s after the Asp plus MEM co-application and the reversal switch-over to −100 mV was performed 2 s before the test Asp pulse.
Figure 4. Voltage dependence of the recovery of the NMDA channels from the MEM block in the absence of the agonist.
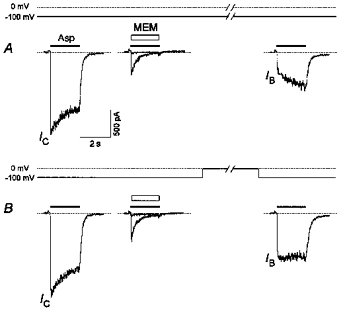
A, washout of the cell for 1 min at −100 mV after a practically complete blockade by MEM (25 μm) led to the clearing of a relatively small fraction of the channels (IB/IC= 0.19 ± 0.03, n = 9). B, many more channels (IB/IC= 0.30 ± 0.05, n = 9) were recovered when the membrane potential was switched to 0 mV during the same washout interval.
Non-addition of glycine to the washout solution did not affect the rate of the unblocking of the agonist-independent channels. Thus, after 3 min washout of the cell with a glycine (3 μm)-containing solution and in the case when during the washout time interval this solution was switched to a nominally glycine-free solution, the values of IB/IC were 0.32 ± 0.04 and 0.31 ± 0.03, respectively (these values were not significantly different, P > 0.5, n = 9). The switching of the solutions was performed 10 s after the Asp plus MEM co-application and 20 s before the test Asp pulse.
The recovery of the Asp-induced current after a practically complete inhibition by Asp and AM (250 μm) co-application (IF/IC= 0.029 ± 0.006, n = 8) proceeded much faster than after the Asp plus MEM co-application. Thus, already after 10 s washout of the cell, the value of IB/IC was 0.63 ± 0.02 (n = 8).
AAD-induced blockade of NMDA channels in the absence of the agonist
As MEM is able to leave the NMDA channel without agonist assistance, it may be supposed that it can enter the channel in the absence of the agonist. Indeed, prolonged MEM application in the absence of Asp in the superfusing solution caused a pronounced blockade of NMDA channels. The experimental protocol used in this study is shown in Fig. 5B. At first, the control current was elicited by an 8 s 100 μm Asp pulse. Not earlier than 20 s after this pulse, the cell was exposed to MEM (50 μm) for 1 min, then washed for 10 s with the control solution to remove all possible traces of MEM in the medium and, finally, once again stimulated with the Asp test pulse. Glycine (3 μm) was present in all these solutions. In control experiments, glycine failed to induce even a weak current through NMDA channels (Fig. 5A). Moreover, addition of APV (100 μm) to the glycine-containing solution did not cause any change in the zero-level current (the mean and s.d. values were not significantly different at P > 0.26, n = 15). These observations provide strong evidence that the glycine-containing control solution was not contaminated with traces of NMDA receptor agonists. The current responses to the test Asp application following MEM washout were similar to those observed in studies of recovery of unliganded channels. When the inhibition of the initial current was considerable and desensitization was not very strong, the initial fast current increase was followed by the slow current elevation resulting from Asp-induced channels unblocking. However, when the inhibition of the Asp-induced current by MEM in the absence of the agonist was modest, desensitization caused a slow current decrease masking the slow current recovery (see Figs 6B, 9 and 10B). The latter became evident with more prolonged test Asp applications (not shown). As the initial fast current increase (IB) reflects the opening of unblocked channels, the value IC- IB may be considered as a measure for the fraction of NMDA channels which trapped the blocker without agonist assistance (the number of channels in state CB of Model 1). A more thorough analysis of the Asp-induced slow current recovery revealed two kinetic components: one fast and one slow. As in the case of recovery of the NMDA channels in the absence of the agonist we fitted not the original current trace but the result of its division by the control trace, Inorm (Fig. 5B, inset). The parameters of the two-exponential fitting equation:
| (4) |
proved to be: Afast= 0.24 ± 0.02; τfast= 1.7 ± 0.5 s; and τslow= 21.3 ± 4.4 s (n = 7). A comparison of these fitting parameters with the corresponding kinetic parameters for the open-channel blockade is given in Table 1. As in the case of the recovery of the channels in the absence of the agonist (see above), the value of IB/IC and, correspondingly, the rate of blockade of the NMDA channels by MEM in the absence of the agonist varied greatly in different cells. Thus, the mean value of IB/IC for all our experiments, in which MEM was applied for 1 min in the absence of the agonist, was 0.35 (s.d.= 0.19, n = 33).
Figure 5. MEM-induced blockade of NMDA channels in the absence of Asp.
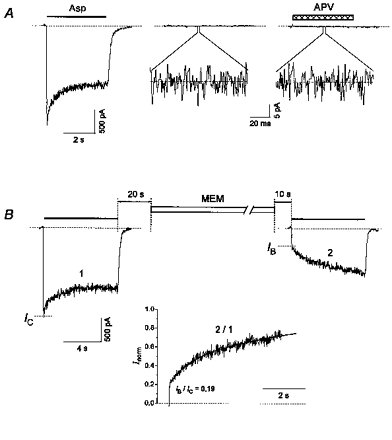
A, control experiment, which shows that the external solution was not contaminated with traces of NMDA receptor agonists. In the continuous presence of glycine (3 μm), Asp (100 μm) elicited a current through NMDA channels with a peak amplitude of 2 nA. Glycine alone failed to induce even a weak current (Iglycine= 0). Addition of APV (100 μm) to a glycine-containing solution did not cause any change in the zero-level current (IAPV= 0). Standard deviations of Iglycine (3.6 ± 0.2 pA) and IAPV (3.4 ± 0.1 pA) were not significantly different at P > 0.26, n = 15. B, a 1 min application of 50 μm MEM in the absence of the agonist caused significant inhibition of the initial current in response to the test Asp application (IB/IC= 0.19). The inset shows the ratio of the test (2) and control (1) current responses. The slow gradual increase of this ratio was well fitted by eqn (4) (continuous line) with Afast= 0.24, τfast= 1.18 s and τslow= 10.9 s.
Figure 6. Effect of glycine on the MEM-induced blockade of NMDA channels in the absence of Asp.
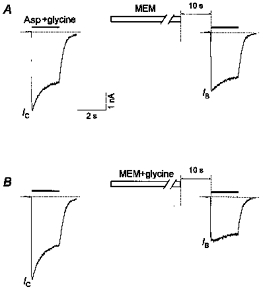
A, a 1 min application of MEM (50 μm) in the absence of glycine produced only a modest decrease in the initial current response caused by the test application of Asp (100 μm) plus glycine (3 μm) (IB/IC= 0.78 ± 0.14, n = 6). B, a much more profound block was produced by the same MEM application in the continuous presence of glycine (IB/IC= 0.51 ± 0.12, n = 6).
Figure 9. Kinetics of the MEM-induced blockade of NMDA channels in the absence of Asp.
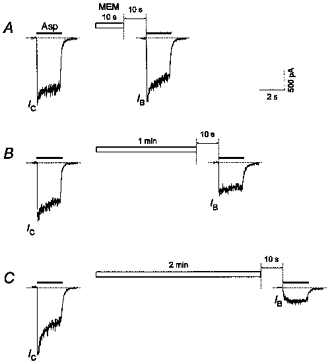
Applications of MEM (50 μm) for 10 s (A), 1 min (B) and 2 min (C) produced an increasing inhibition of the initial current (IB/IC= 0.95, 0.58 and 0.14, respectively) in response to the test Asp (100 μm) application. All recordings were made from the same cell.
Figure 10. Voltage dependence of the MEM-induced blockade of NMDA channels in the absence of Asp.
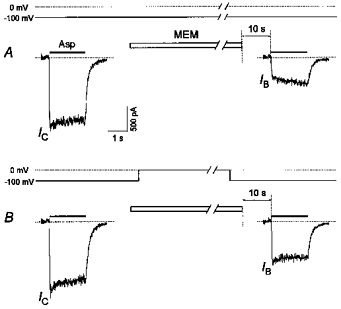
A, a 1 min application of MEM (50 μm) at −100 mV caused significant inhibition of the initial current in response to the test application of Asp (100 μm) (IB/IC= 0.21 ± 0.03, n = 4). B, a less profound MEM block (IB/IC= 0.39 ± 0.07, n = 4) was produced when the membrane potential was switched to 0 mV during MEM application.
Addition of 3 μm glycine to the MEM-containing solution enhanced the blocking effect of MEM (Fig. 6). Thus a 1 min application of MEM (50 μm) in the presence of glycine induced a stronger blockade of the channels without agonist assistance (IB/IC= 0.51 ± 0.12, n = 6) than in the nominally glycine-free solution (IB/IC= 0.78 ± 0.14, n = 6). These values were significantly different (P < 0.003, n = 6).
Addition of APV (100 μm) to the MEM-containing solution considerably diminished the inhibition of the initial current elicited by test application of Asp (Fig. 7). The IB/IC values after a 1 min application of MEM in the presence (0.79 ± 0.05) and absence of APV (0.32 ± 0.04) were significantly different (P < 10 −6, n = 16).
Figure 7. Effect of APV on the MEM-induced blockade of NMDA channels in the absence of Asp.
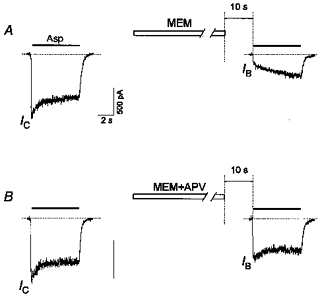
A, a 1 min application of MEM (50 μm) caused significant inhibition of the initial current in response to the test Asp (100 μm) application (IB/IC= 0.32 ± 0.04, n = 16). B, addition of APV (100 μm) to a MEM-containing solution greatly decreased the inhibition of the initial current response (IB/IC= 0.79 ± 0.05, n = 16).
AM also proved to be able to block the channels in the absence of the agonist (Fig. 8). In this case, however, most of the channels became cleared during a 10 s washout interval (see above). Therefore, the AM-induced blockade of the agonist-unbound channels was manifested only as a reduction of the initial current value (IB/IC < 1) in response to the test application of Asp. Because of the fast recovery from the block by AM, the Asp application 4 s after the test one induced a current response identical to that of the control (Fig. 8, last trace).
Figure 8. AM-induced blockade of NMDA channels in the absence of Asp.
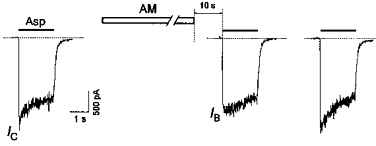
A 1 min application of AM (1 mm) in the absence of the agonist produced only a modest reduction of the initial current (IB/IC= 0.8) in response to the test application of Asp (100 μm). The next test application of Asp 4 s after the first one induced a current response identical to the control.
The blockade of NMDA channels by MEM in an Asp-free medium increased with the duration of the blocker application (Fig. 9). In Fig. 9 the desensitization of the channels during the control Asp pulse increased slowly over the time course of the experiment (cf. control responses in A and C). In this case we increased the duration of MEM application from A to C. However, we also conducted another experiment in which the longest MEM application was used at first and then its duration was decreased. The result was the same: the fraction of blocked channels increased with the lengthening of the MEM application. Thus, the fact that the agonist-induced desensitization increased with time did not affect the results of our experiments. The single-exponential fit of the IB/IC time dependence gave the value of the time constant of 84 ± 6 s (n = 5). Neglecting the small fast component of recovery of the channels from the MEM-induced block in the absence of the agonist (13 %, see Fig. 3C), the unblocking of the channels can be also considered as a single-exponential process. Thus, within the frame of a bimolecular reaction process, the association and dissociation rate constants for MEM binding and unbinding in the absence of the agonist can be estimated as 2.04 (± 0.44) × 102 M−1 s−1 and 1.68 (± 0.129) × 10−3 s−1, respectively. The apparent Kd value (8.3 μm) proved to be six times higher than the IC50 for the MEM-induced blockade of open channels (1.3 μm). Therefore, the affinity of NMDA channels to MEM in the absence of the agonist is about six times smaller than in its presence.
Membrane depolarization attenuated this block (Fig. 10). Thus, at −100 mV a 1 min MEM (50 μm) treatment induced a profound blockade (IB/IC= 0.21 ± 0.03). Membrane depolarization to 0 mV during the period of MEM application diminished this block (IB/IC= 0.39 ± 0.07; these values were significantly different, P < 0.03, n = 4). The switching of the membrane potential was performed 2 s after the beginning and 2 s before the termination of the MEM application.
Magnesium antagonized the MEM-induced blockade of NMDA channels in both the presence and the absence of Asp
In a Mg2+-free Asp (100 μm)-containing solution, a 5 s application of MEM (15 μm) caused a practically complete blockade of open NMDA channels. In this case, only a small fast component (17 ± 3 %, n = 5) was observed in the offset kinetics of the MEM block (Fig. 11A, first trace). Addition of Mg2+ (2.5 mm) to the MEM-containing solution (Fig. 11A, third trace) greatly increased this fast component (48 ± 1 %, n = 5). The offset kinetics of Mg2+ (Fig. 11A, second trace) is known to be very fast (Ascher & Nowak, 1988). Therefore, the difference between the above-mentioned fast component values (they were significantly different, P < 0.001) reflects the minimal hindrance of Mg2+ to the blockade of open channels by MEM (31 ± 3 %, n = 5).
Figure 11. Effect of Mg2+ on the MEM-induced blockade of NMDA channels.
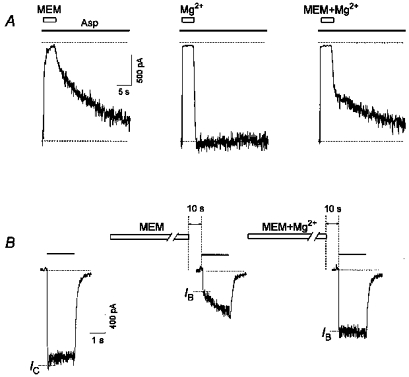
A, effect of Mg2+ on the MEM-induced open-channel blockade. Asp (100 μm) was applied continuously. MEM (15 μm) applied for 5 s manifested mainly slow, while Mg2+ (2.5 mm) manifested very fast open-channel kinetics. After MEM and Mg2+ co-application, the double-exponential unblocking kinetics (intermediate between those mentioned above) reflects the minimal hindrance of Mg2+ to the MEM-induced open-channel blockade. B, effect of Mg2+ on the MEM-induced blockade in the absence of Asp. A 1 min application of MEM (50 μm) caused significant inhibition of the initial current in response to the test application of Asp (100 μm) (IB/IC= 0.33 ± 0.05, n = 6). Addition of Mg2+ (2.5 mm) to a MEM-containing solution caused a less profound inhibition of the initial current response (IB/IC= 0.67 ± 0.04, n = 6).
In the second series of our experiments, we examined the effect of Mg2+ on the MEM-induced blockade of NMDA channels in the absence of Asp (Fig. 11B). MEM (50 μm) and Mg2+ (2.5 mm) co-application induced a less profound blockade (IB/IC= 0.67 ± 0.04) of NMDA channels than MEM itself (IB/IC= 0.33 ± 0.05). These values were significantly different (P < 0.03, n = 6). Thus Mg2+ prevented the MEM-induced blockade of NMDA channels not only in the presence but also in the absence of the agonist.
DISCUSSION
Blockade of NMDA channels by AAD does not prevent the subsequent closure of the channel after removal of the agonist from the medium (Johnson et al. 1995; Chen & Lipton, 1997). The finding that Asp reapplication readily cleared the channels was considered as an indication that the blocking molecule had been trapped in the channel by the closed activation gate. Our experiments showed, however, that agonist-induced channel openings greatly accelerated but were not a prerequisite for the recovery from the AAD blockade (Fig. 3). What is the pathway whereby the blocker leaves the channel without agonist assistance? The finding that membrane depolarization accelerates recovery of the channels from AADs not only in the presence (Fig. 2C) but also in the absence of Asp (Fig. 4) allows us to conclude that in the two cases the blocker exits the channel via the same route (the ‘hydrophilic’ pathway in Hille's (1977) terminology). If so, provided that the blocker cannot leave the closed channel, we have to assume that in addition to the agonist-induced openings of NMDA channels there exist some infrequent agonist-independent transitions between the closed and open states of the channel. Therefore, in the absence of the agonist the unblocking of the channels is thought to be as follows: CB→ OB→ O → C, where all the transitions are agonist independent and the transition from OB→ O is voltage dependent. The CB→ OB transition provides a much slower recovery from the AAD-induced block in the absence of the agonist than the transition CAB→ OAB (see Model 1) in its presence. Thus, for MEM the slow recovery time constant in the absence of the agonist (Fig. 3C) was approximately thirty times greater than that for the unblocking of open channels (Table 1). If the unblocking of the channels in the absence of Asp resulted from spontaneous transitions of channels from the closed to the open states, these openings would generate a permanent inward current of only 30 times smaller magnitude than that elicited by 100 μm Asp. However, this is not the case (Fig. 5A). To our knowledge, up to now there is still no evidence for the existence of spontaneous NMDA channel openings (i.e. C → O transitions), although such openings are well known for acetylcholine-activated ligand-gated channels (Jackson, 1986). Glycine alone was shown to activate recombinant heteromeric NMDA channels (Meguro et al. 1992; Monyer et al. 1992). In our experiments, glycine alone did not induce a current through the NMDA channels (Fig. 5A). Moreover, the glycine independence of recovery of the NMDA channels from the MEM block in the absence of the agonist strongly indicates that glycine alone does not induce channel opening. Therefore, we have to assume that the blocking molecule trapped in the channel promotes in some way random channel openings. This supposition is in good agreement with the recent finding that MEM binding inside the channel pore shifts the open-closed equilibrium towards the channel opening by ∼ 4.81 kJ m−1 (Chen & Lipton, 1997).
The ability of AADs to leave the unliganded NMDA channel led us to examine their capability of blocking the channels without agonist assistance. It has been found that MEM and AM are able to block the agonist-unbound channels, although this process is much slower (Fig. 9) and requires much higher concentrations than the open channel blockade.
Membrane hyperpolarization enhanced the MEM-induced blockade of agonist-unbound NMDA channels (Fig. 10) as well as the open-channel blockade (Fig. 2C). External Mg2+ effectively antagonized the MEM block development in both the presence (Fig. 11A) and the absence (Fig. 11B) of Asp. These findings strongly support the hypothesis that the blocker reaches its binding site in the agonist-unbound channel via the same ‘hydrophilic pathway’ as in the presence of the agonist.
By drawing an analogy with recovery from the AAD block for unliganded channels, we may assume that the blocker alone is able to promote agonist-independent channel transitions to the open state which provides a subsequent channel blockade. A possible mechanism of blockade of the channels and, correspondingly, the unblocking in the absence of the agonist is shown in Fig. 12. AAD binding to the modulatory site (M-site) promotes the agonist-independent channel openings. During these openings, AAD ‘jumps’ to the blocking site (B-site) and becomes trapped behind the closed activation gate. The OM state can be the non-conducting or short-living (comparing with state OA* in Model 1) conducting state of the NMDA channel which cannot be detected at our time resolution. It is therefore not surprising that no openings were observed in our experiments with the AAD-induced blockade and recovery from it in the absence of the agonist. Thus, the new states (CM, OM and OB) should be added to Model 1, resulting in the simplified Model 2.
Figure 12. Schematic presentation of a possible mechanism of AAD interaction with the NMDA channel in the absence of the agonist.
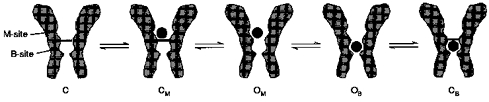
AAD binding to the modulatory site (M-site) or the blocking site (B-site) promotes the agonist-independent channel openings. During these openings, AAD ‘jumps’ from the M-site to the B-site or back, and either becomes trapped by the closed activation gate or leaves the channel. OM can be the non-conducting or short-lived conducting state of the NMDA channel.
Model 2.
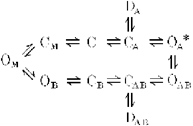
The fact that APV effects in the absence of the agonist were not symmetrical (APV hindered the MEM-induced blockade but did not influence unblocking of the channels) can be easily explained in terms of Model 2. APV can prevent the MEM binding to the M-site but does not affect its binding to the blocking site.
There are at least two other possible answers to the question ‘How can externally applied AAD reach its binding site located deep in the channel pore via the hydrophilic pathway without agonist assistance?’. (1) MEM and AM block NMDA channels during their infrequent spontaneous openings. (2) The NMDA receptor co-agonist glycine induces random channel openings. As discussed above, the involvement of spontaneous and glycine-induced openings in the recovery of unliganded channels from the AAD-induced blockade seems to be doubtful. As far as the blockade of the agonist-unbound channels is concerned, our data do not allow us to make a choice between spontaneous, glycine-induced or blocker-induced openings.
The kinetics of recovery of the channels from the MEM block in the presence of the agonist deserve special attention. In our experiments, NMDA channels were blocked by MEM in the presence (see Fig. 2A) or absence of Asp (see Fig. 5B). In the two cases, the agonist-induced current recovery appeared to be identical (Table 1). A good coincidence of the corresponding time constants for single- or two-exponential fittings allowed us to conclude that the blocking sites for MEM in the channel did not depend on whether the channels were blocked in the presence or absence of the agonist. The fact that the 10 s washout interval did not change either of the components of the Asp-induced unblocking kinetics (Table 1) suggests that these binding sites are located ‘behind the activation gate’ of the channel and that MEM being bound to these sites did not prevent the channel closure after removal of the agonist from the medium.
Our interpretation of the interaction of MEM with the majority of NMDA channels differs from that of Blanpied et al. (1997) who proposed two different mechanisms for MEM action. According to their opinion, MEM is able to produce (1) a trapping block where the blocker cannot leave without the agonist assistance and (2) non-competitive inhibition of NMDA channels where the blocker reaches its site via the hydrophobic pathway. In our opinion, MEM blocks and leaves the agonist-unbound channel via the same hydrophilic pathway as it does in the open channel. Even the binding sites remain the same and only the rate of the MEM interaction with the channels in the absence of the agonist is slowed down. However, in our experiments the recovery from the MEM block in the absence of the agonist was not complete in some cells (4/15). In these cases, the mechanism of MEM action can probably be described as in Blanpied et al. (1997). The existence of different mechanisms of MEM action can be explained by the heterogeneity of NMDA channels in the neuronal membrane.
It is also necessary to discuss the major methodological differences between our study and that of Blanpied et al. (1997). We used a saturating agonist concentration (100 μm Asp), while Blanpied et al. (1997) used a very low agonist concentration (5 μm NMDA). Therefore, in the latter study the blockade reached its steady-state level very slowly: after 1 min agonist and blocker co-application even at a high MEM concentration (50 μm). In contrast, in our experiments 2–5 s co-application appeared to be quite sufficient to induce an almost complete block.
As MEM affinity for the NMDA channel in the absence of the agonist is only about six times smaller than in its presence, the wide therapeutic application of memantine (Danysz et al. 1995) can be partly due to its slow interaction with NMDA channels during the intervals between glutamate releases.
A more profound analysis is required to explain the difference in the mechanisms of MEM interaction with the gating machinery of closed NMDA channels in two cases: when the blocker trapped in the channel ‘attempts’ to leave it and when the blocker is present in the external medium and ‘knocks’ at the closed channel gate. Different effects of APV point to different mechanisms of MEM action in these two cases.
Acknowledgments
The authors thank Dr P. Behe (Department of Pharmacology, University College London, UK) for critical discussion and helpful comments on an earlier version of this manuscript. We are very grateful to our colleagues at MERZ & Co. who have kindly provided us with amino-adamantanes. This work was supported by the Russian Fund of Fundamental Research (Nos 960449227 and 960449228) and an ISSEP grant to A. I. S. (No. a98–2018). The financial help of Dr Vadim Bogomolov is also highly appreciated.
References
- Ascher P, Nowak L. The role of divalent cations in the N-methyl-D-aspartate responses of mouse central neurones in culture. The Journal of Physiology. 1988;399:247–266. doi: 10.1113/jphysiol.1988.sp017078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benveniste M, Clements J, Vyklicky J, Mayer ML. A kinetic analysis of the modulation of NMDA receptors by glycine in mouse cultured hippocampal neurones. The Journal of Physiology. 1990;428:333–357. doi: 10.1113/jphysiol.1990.sp018215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanpied TA, Boeckman F, Aizenman E, Johnson JW. Trapping channel block of NMDA-activated responses by amantadine and memantine. Journal of Neurophysiology. 1997;77:309–323. doi: 10.1152/jn.1997.77.1.309. [DOI] [PubMed] [Google Scholar]
- Bresink I, Benke TA, Collett VJ, Seal AJ, Parsons CG, Henley JM, Collingridge GL. Effects of memantine on recombinant rat NMDA receptors expressed in HEK 293 cells. British Journal of Pharmacology. 1996;119:195–204. doi: 10.1111/j.1476-5381.1996.tb15971.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-SV, Lipton SA. Mechanism of memantine block of NMDA-activated channels in rat retinal ganglion cells: uncompetitive antagonism. The Journal of Physiology. 1997;499:27–46. doi: 10.1113/jphysiol.1997.sp021909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H-SV, Pellegrini JW, Aggarwal SK, Lei SZ, Warach S, Jensen FE, Lipton SA. Open-channel block of NMDA responses by memantine: therapeutic advantage against NMDA receptor-mediated neurotoxicity. Journal of Neuroscience. 1992;12:4427–4436. doi: 10.1523/JNEUROSCI.12-11-04427.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danysz W, Parsons CG, Bresink I, Quack G. Glutamate in CNS disorders. Drug News and Perspectives. 1995;8:261–277. doi: 10.1358/dnp.1998.11.9.863689. [DOI] [PubMed] [Google Scholar]
- Hille B. Local anesthetics: hydrophilic and hydrophobic pathways for the drug-receptor reaction. Journal of General Physiology. 1977;69:497–515. doi: 10.1085/jgp.69.4.497. 10.1085/jgp.69.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: selective binding to open channels. Proceedings of the National Academy of Sciences of the USA. 1987;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson MB. Kinetics of unliganded acetylcholine receptor channel gating. Biophysical Journal. 1986;49:663–672. doi: 10.1016/S0006-3495(86)83693-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Antonov SM, Blanpied TS, Li-Smerin Y. Channel block of NMDA receptor. In: Wheal HV, editor. Excitatory Amino Acids and Synaptic Transmission. London: Academic Press, Inc.; 1995. pp. 99–113. [Google Scholar]
- Kemp JA, Foster AC, Wong EHF. Non-competitive antagonists of excitatory amino acid receptors. Trends in Neurosciences. 1987;10:294–298. 10.1016/0166-2236(87)90176-7. [Google Scholar]
- McBain CJ, Mayer ML. N-methyl-D-aspartic acid receptor structure and function. Physiological Reviews. 1994;74:723–760. doi: 10.1152/physrev.1994.74.3.723. [DOI] [PubMed] [Google Scholar]
- MacDonald JF, Bartlett MC, Mody I, Pahapill P, Reynolds JN, Salter MW, Schneiderman JH, Pennefather PS. Actions of ketamine, phencyclidine and MK-801 on NMDA receptor currents in cultured mouse hippocampal neurones. The Journal of Physiology. 1991;432:483–508. doi: 10.1113/jphysiol.1991.sp018396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meguro H, Mori H, Araki K, Kushina E, Kutsuwada T, Yamazaki M, Kumanishi T, Arakawa M, Sakimura K, Mishina M. Functional characterization of a heteromeric NMDA receptor channel expressed from cloned cDNAs. Nature. 1992;357:70–74. doi: 10.1038/357070a0. 10.1038/357070a0. [DOI] [PubMed] [Google Scholar]
- Monyer H, Sprengel R, Schoepfer R, Herb A, Higuchi M, Lomeli H, Burnashev N, Sakmann B, Seeburg PH. Heteromeric NMDA receptors: molecular and functional distinction of subtypes. Science. 1992;256:1217–1221. doi: 10.1126/science.256.5060.1217. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Gruner R, Rozental J, Millar J, Lodge D. Patch clamp studies on the kinetics and selectivity of NMDA receptor antagonism by memantine. Neuropharmacology. 1993;32:1337–1350. doi: 10.1016/0028-3908(93)90029-3. 10.1016/0028-3908(93)90029-3. [DOI] [PubMed] [Google Scholar]
- Parsons CG, Quack G, Bresink I, Baran L, Przegalinski E, Kostowski W, Krzascik P, Hartmann S, Danysz W. Comparison of the potency, kinetics and voltage-dependency of a series of uncompetitive NMDA receptor antagonists in vitro with anticonvulsive and motor impairment activity in vivo. Neuropharmacology. 1995;34:1239–1258. doi: 10.1016/0028-3908(95)00092-k. 10.1016/0028-3908(95)00092-K. [DOI] [PubMed] [Google Scholar]
- Sobolevsky A, Koshelev SG. Two blocking sites of amino-adamantane derivatives in open N-methyl-D-aspartate channels. Biophysical Journal. 1998;74:1305–1319. doi: 10.1016/S0006-3495(98)77844-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobolevsky A, Koshelev SG, Khodorov BI. Memantine-induced blockade of NMDA channels without agonist assistance. The Journal of Physiology. 1996;495.P:49P. [Google Scholar]
- Vorobiev V. Vibrodissociation of sliced mammalian nervous tissue. Journal of Neuroscience Methods. 1991;38:145–150. doi: 10.1016/0165-0270(91)90164-u. 10.1016/0165-0270(91)90164-U. [DOI] [PubMed] [Google Scholar]