

Abstract
Inhibition of inositol 1,4,5-trisphosphate (IP3) receptor-mediated Ca2+ release by cGMP was examined in intact rat megakaryocytes, by using a combination of single cell fluorescence microscopy to monitor intracellular free calcium ([Ca2+]i) and flash photolysis of caged second messengers.
Sodium nitroprusside (SNP), a nitric oxide (NO) donor, and the hydrolysis-resistant cGMP analogue 8-(4-chlorophenylthio)guanosine 3′,5′-cyclic monophosphate (pCPT-cGMP) inhibited Ca2+ release induced by photolysis of caged IP3. Neither of them affected the rate of Ca2+ removal from the cytoplasm following photolysis of caged Ca2+.
Photolysis of the caged NO donor 3-morpholinosydnonimine (SIN-1) during agonist-induced [Ca2+]i oscillations inhibited Ca2+ release without affecting the rate of Ca2+ uptake and/or extrusion.
We conclude that the inhibition of IP3-induced Ca2+ release is the principal mechanism of NO-cGMP-dependent inhibition of [Ca2+]i mobilization.
IPG, a specific peptide inhibitor of cGMP-dependent protein kinase (cGMP-PK), blocked the inhibitory effect of pCPT-cGMP, indicating that the inhibition of IP3-induced Ca2+ release by pCPT-cGMP is mediated by cGMP-PK. However, the simultaneous application of both IPG and IP20, a specific peptide inhibitor of cAMP-dependent protein kinase (cAMP-PK), was required to block the inhibitory effect of SNP. These data strongly suggest that NO-cGMP-dependent inhibition of [Ca2+]i mobilization is mediated via the activation of both cGMP-PK and cAMP-PK.
Elevation of intracellular cGMP by endothelium-derived relaxing factor (EDRF) or nitric oxide (NO)-generating agents, as well as introduction of hydrolysis-resistant cGMP analogues, causes inhibition of platelet activation and relaxation of vascular smooth muscle. Although the precise mechanisms of cGMP-mediated inhibition are not well understood, it is clear that cGMP-dependent reduction of intracellular free calcium ([Ca2+]i) mobilization is a critical event involved in this inhibitory pathway (for review see Moncada et al. 1991; Walter et al. 1993; Lincoln et al. 1996; Clementi & Meldolesi, 1997).
Several mechanisms have been proposed to account for cGMP-dependent inhibition of [Ca2+]i mobilization, including inhibition of IP3 formation via inhibition of phospholipase C (PLC) or PLC-G-protein-receptor coupling (Deana et al. 1989; Hirata et al. 1990; Nguyen et al. 1991), stimulation of Ca2+ uptake and/or extrusion (Cornwell et al. 1991; Johansson & Haynes, 1992; Karczewski et al. 1992) and inhibition of Ca2+ entry (Geiger et al. 1992; Alioua et al. 1995). Recent studies point to the direct inhibition of Ca2+ release from the endoplasmic reticulum as a possible molecular mechanism that mediates the inhibitory effect of cyclic nucleotide-elevating agents on calcium mobilization (Cavallini et al. 1996; Komalavilas & Lincoln, 1996; Quinton et al. 1996). In this context the IP3 receptor has been found to be phosphorylated by cyclic nucleotide-dependent protein kinases in platelets and smooth muscle cells and its phosphorylation was associated with the cAMP- and cGMP-mediated inhibition of [Ca2+]i accumulation (Cavallini et al. 1996; El-Daher et al. 1996; Komalavilas & Lincoln, 1996). However, whether phosphorylation of the IP3 receptor actually regulates its ability to release calcium in the living cell has not yet been established.
The present experiments were designed to investigate whether elevation of intracellular cGMP actually inhibits IP3-induced Ca2+ release in an intact cell, and if so whether this inhibition requires activation of cyclic nucleotide-dependent protein kinases. In order to test this possibility we used flash photolysis of caged second messengers and selective inhibitors of cGMP- and cAMP-dependent protein kinases (cGMP-PK and cAMP-PK). This approach has proved valuable in studying the process of intracellular signalling in other cells types (Adams & Tsien, 1993). Previously we have reported that in megakaryocytes, the progenitor of platelets, agonist-induced [Ca2+]i oscillations are reversibly inhibited by agents that elevate intracellular cGMP and cAMP (Tertyshnikova & Fein, 1997). The present study provides direct evidence that elevation of cGMP inhibits IP3-induced Ca2+ release in intact cells, but does not affect the rate of Ca2+ removal from the cytoplasm. Furthermore our results indicate that cGMP-dependent inhibition of IP3-induced Ca2+ release is mediated by both cGMP-PK and cAMP-PK.
METHODS
Cell preparation and loading
Megakaryocytes were obtained from adult Wistar rats as previously described (Tertyshnikova & Fein, 1997). Briefly, bone marrow was obtained from rats anaesthetized by inhalation of an overdose of isoflurane and then killed by decapitation. This procedure is in accordance with University of Connecticut Health Centre guidelines. After filtration through a 75 μm nylon mesh to eliminate large masses of cells, the bone marrow suspension was spun and washed twice before incubation in standard external solution of composition (mm): 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 10 glucose, 10 Hepes, pH 7.4, supplemented by 0.1 % bovine serum albumin (BSA). Megakaryocytes were clearly distinguished from other bone marrow cells on the basis of their large size (25–50 μm) and multilobular nucleus. All experiments were done within 2–6 h after preparation at room temperature (23–25°C).
‘Cell permeant’ AM esters of o-nitrophenyl EGTA (caged calcium) and Oregon Green 488 BAPTA-1 (OGB488) were dissolved in DMSO, stored at −20°C and applied at 10–30 and 2.5–5 μm, respectively. For intracellular loading, megakaryocytes were resuspended in standard external solution containing caged calcium (the final concentration of DMSO was less then 0.1 %) and incubated for at least 2 h. Then cells were transferred onto glass coverslips and were additionally incubated with OGB488 AM for 30 min. The coverslips with adherent cells were then washed several times with the standard external solution and kept in the dark until use to avoid unwanted photolysis of the caged calcium.
Agonist application
Activators and inhibitors (ADP, pCPT-cGMP, carbacyclin, SNP, or the mixture of ADP with one of the others) were dissolved in the standard external solution and applied directly to single megakaryocytes using a DAD-6 computer controlled local superfusion system (ALA Scientific Instruments Inc., Westbury, NY, USA). The output tube of the micromanifold (100 μm inside diameter) was placed approximately within 200 μm of the cell, and the puff pressure was adjusted to achieve rapid agonist application while avoiding any mechanical disturbance of the cell. The time delay for arrival of agonists at the cell was measured and accounted for in the figures. For the experiments with caged SIN-1, the caged NO donor was added to the bath solution at a final concentration of 0.5 mm.
Caged IP3, IPG, IP20 and OGB488 (hexapotassium salt, ‘cell impermeant’), were included in the intrapipette solution at 100, 100, 100 and 200 μm, respectively, (solution composition, mm: 20 KCl, 120 potassium glutamate, 1 MgCl2, 2 Na-GTP, 10 Hepes, pH 7.3). Standard whole-cell patch-clamp recording techniques were used to voltage clamp and internally dialyse single megakaryocytes. Membrane current was monitored using an Axopatch-1D patch clamp amplifier (Axon Instruments). Recording pipettes were pulled from 1.5 mm borosilicate glass (no. 7052; Garner Glass Inc., Claremont, CA, USA) using a two-stage Narishige PB-7 vertical puller and then fire-polished on a Narishige MF-9 microforge. Pipette resistance was between 1 and 5 MΩ and the holding potential was −45 mV. For most cells 5–8 min was required for the OGB488 fluorescence signal to equilibrate in the patch clamped cell. Because of variability in cell responses to IP3, for every cell after equilibration of the intracellular and the pipette solution, the duration of the UV light pulse was adjusted to produce an IP3-induced rise in [Ca2+]i similar in magnitude and time course to that resulting from the agonist application. In control experiments we were able to use up to ten to fifteen IP3-releasing UV flashes without any significant degradation of the [Ca2+]i response.
Fluorescence measurement and flash photolysis
Megakaryocytes were viewed through a coverslip forming the bottom of the recording chamber using a Nikon Diaphot microscope equipped with a Nikon Fluor × 100, 1.3 NA oil-immersion lens. Single cell fluorometry was accomplished using an IonOptix photon counting fluorescence subsystem (designed by D. Tillotson, IonOptix, Milton, MA, USA) as previously described (Tertyshnikova & Fein, 1997), except that in these experiments we used OGB488 as the Ca2+ indicator instead of fura-2. For OGB488 excitation light was delivered from one channel of the chopper based dual excitation 75 W xenon arc light source via the light guide. Excitation and emission were centred on 485 nm and 535 nm, respectively. For caged SIN-1 and caged IP3 photolysis, pulses of ultraviolet light (290–370 nm) were applied to the cell through the second channel of the dual excitation light source. Caged calcium photolysis was produced by a 1 ms flash from an XF-10 high intensity xenon flash lamp (Hi-Tech Scientific, Salisbury, UK), focused through a 280–360 nm wide band filter from about 2.5 cm above the cell to produce a 2–3 mm spot. The flash energy was regulated by controlling the charging voltage of the capacitor bank used to fire the flash lamp. Fluorescence intensity was measured on-line using the PIA program (IonOptix). The time resolution was set at 0.1 s by averaging three points to obtain a better signal-to-noise ratio.
Chemicals
Oregon Green 488 BAPTA-1 AM (hexapotassium salt), NVOC-caged SIN-1 (N-(((4,5-dimethoxy-2-nitrobenzyl)oxy)carbonyl)-3-morpholinosydnoneimine) and caged NO (potassium nitrosylpentachlororuthenate) were obtained from Molecular Probes Inc. Caged IP3, o-nitrophenyl EGTA AM and caged cGMP (guanosine 3′,5′-cyclic monophosphate, P-1-(2-nitrophenyl)ethyl ester) were from Calbiochem. pCPT-cGMP (8-(4-chlorophenylthio)guanosine 3′,5′-cyclic monophosphate) was purchased from BioLog Life Science Institute (La Jolla, CA, USA). ODQ (1H-[1,2,4]oxadiazolo[4,3,-a]-quinoxalin-1-one) was obtained from Tocris Cookson Inc. (Ballwin, MO, USA). All other reagents were purchased from Sigma Chemical Co. Peptide inhibitor of cGMP-dependent protein kinase (IPG) (Leu-Arg-Arg-Arg-Arg-Phe-D-Ala-Phe-NH2) (Yan et al. 1996) was synthesized in D. S. Lawrence's laboratory (The Albert Einstein College of Medicine, Bronx, NY, USA).
RESULTS
Inhibition of IP3-induced Ca2+ release by pCTP-cGMP and SNP
To determine whether an elevation of intracellular cGMP inhibits IP3-induced Ca2+ release we first performed the experiment shown in Fig. 1. Caged IP3 and OGB488 were included in the patch pipette (see Methods). [Ca2+]i spikes produced by IP3-releasing flashes in cells bathed at least 1 h in Ca2+-free solution, containing 1 mm BAPTA, were similar to those obtained from megakaryocytes bathed in standard external solution, indicating that these IP3-induced [Ca2+]i spikes were the result of IP3-induced Ca2+ release and not Ca2+ influx (n = 4cells, data not shown).
Figure 1. pCPT-cGMP and SNP inhibit the IP3-induced rise in [Ca2+]i in megakaryocytes.

Caged IP3 (100 μm) and OGB488 (200 μm) were included in the patch pipette solution. In this and all other figures changes in [Ca2+]i were monitored by measuring OGB488 fluorescence intensity and expressed as ΔF (counts ms−1). IP3 was released by a pulse of UV light from the chopper based dual excitation light source, at the times indicated. SNP (0.25 mm) and pCPT-cGMP (1 mm) were applied to the cell via the local superfusion system as described in Methods.
To elevate intracellular cGMP we used either pCPT-cGMP, a membrane-permeant hydrolysis-resistant analogue of cGMP (Butt et al. 1992), or sodium nitroprusside (SNP), a NO donor that increases the intracellular level of cGMP through the activation of soluble guanylyl cyclase (for review see Buechler et al. 1994). Previous experiments have demonstrated that pCPT-cGMP and SNP reversibly inhibit agonist-induced [Ca2+]i oscillations in megakaryocytes (Tertyshnikova & Fein, 1997). As shown in Fig. 1, application of either pCPT-cGMP or SNP reversibly inhibited the rise in [Ca2+]i induced by photolysis of caged IP3 (n = 15cells).
To investigate whether the inhibition of the IP3-induced rise in [Ca2+]i by pCPT-cGMP and SNP involves the stimulation of Ca2+ uptake and/or extrusion we used caged Ca2+ for the experiment in Fig. 2. The time course of the fall in [Ca2+]i, following the flash-induced rise in [Ca2+]i, reflects the activity of Ca2+ sequestration and/or extrusion mechanisms. Ca2+ was photoreleased following a short application of adenosine 5′-diphosphate (ADP) that served as a positive control for the inhibitory effect of SNP or pCPT-cGMP. ADP has been shown to mobilize Ca2+ from intracellular stores in platelets (MacKenzie et al. 1996) and megakaryocytes (Uneyama et al. 1993) presumably through formation of IP3 as occurs in many other cell types. As can be seen in Fig. 2, both pCPT-cGMP (n = 6 cells) and SNP (n = 7 cells) reversibly inhibited the ADP-induced rise in [Ca2+]i while photoreleased [Ca2+]i declined with the same rate before, during and after application of SNP and pCPT-cGMP.
Figure 2. SNP and pCPT-cGMP reversibly inhibit Ca2+ mobilization by ADP without affecting the rate of Ca2+ removal from the cytoplasm.
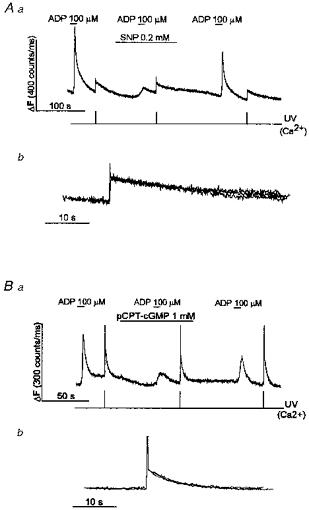
SNP (A) and pCPT-cGMP (B) reversibly inhibited ADP-induced Ca2+ mobilization, but had no effect on the time course of the fall in [Ca2+]i, following photolysis of caged Ca2+. SNP (0.2 mm), pCPT-cGMP (1 mm) and ADP (100 μm) were applied as shown. In Ab and Bb, the [Ca2+]i spikes resulting from photorelease of caged-Ca2+ in Aa and Ba, respectively, are shown superimposed on an expanded time scale. Ca2+ was released by a 1 ms flash of UV light from the xenon flash lamp at the times indicated (see Methods).
The experiment shown in Fig. 3 provides an additional test of whether or not elevation of cGMP stimulates Ca2+ removal from the cytoplasm. In these experiments we used SIN-1, a donor of NO, and a potent vasorelaxant and antithrombotic agent (for review see Feelisch & Stamler, 1996). Caged SIN-1 uncages in milliseconds after UV illumination to release SIN-1 which then spontaneously decomposes to release NO. This allows precise temporal control of the Ca2+ signal by SIN-1. We have previously reported that the falling phase of each [Ca2+]i spike that makes up the agonist-induced [Ca2+]i oscillation in megakaryocytes results from Ca2+ uptake and/or extrusion (Tertyshnikova & Fein, 1997). When SIN-1 was uncaged during the rising phase of a [Ca2+]i spike, further Ca2+ release was inhibited and the [Ca2+]i spike aborted (spike 2 in Fig. 3A) (n = 4cells). However, photolysis of SIN-1 did not cause any change in the time course of the falling phase of the spike (Fig. 3A). In Fig. 3B the [Ca2+]i spikes labelled 1 and 2 from Fig. 3A are shown superimposed on an expanded time scale. It can be seen that SIN-1 inhibits the development of the [Ca2+]i spike without affecting its falling phase. Similar results were obtained using caged NO (potassium nitrosylpentachlororuthenate) (n = 3cells) and caged cGMP (n = 8cells) (data not shown).
Figure 3. Photolysis of caged SIN-1 causes inhibition of ADP-induced [Ca2+]i oscillations without affecting the kinetics of the falling phase of the individual [Ca2+]i spikes.
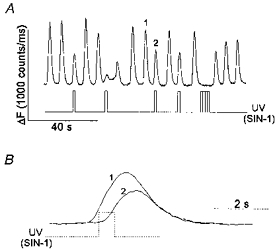
Megakaryocytes were loaded with OGB488 AM as described in Methods. Caged SIN-1 was added to the bath solution at a final concentration of 0.5 mm. A, [Ca2+]i oscillations were induced by 100 μm ADP as shown. SIN-1 was released by a 0.5 s pulse of UV light at the times indicated. B, the [Ca2+]i spikes labelled 1 and 2 in A are shown superimposed on an expanded time scale. The stepped line indicates the time of occurrence of the UV flash (SIN-1 release) during spike 2 (dotted line).
Taken together, the results in Figs 1–3 indicate that the principal mechanism of NO-cGMP-dependent inhibition of Ca2+ mobilization in megakaryocytes is by inhibition of IP3-induced Ca2+ release and not by stimulation of Ca2+ removal from the cytoplasm.
As mentioned above NO donors are thought to increase the intracellular level of cGMP via the activation of soluble guanylate cyclase (GC). However, in our experiments ODQ (1H-[1,2,4]oxadiazolo[4,3,-a]quinoxalin-1-one), a selective inhibitor of soluble guanylate cyclase (Moro et al. 1996), even at very high concentrations (10–100 μm), only partially blocked inhibition of agonist-induced [Ca2+]i oscillations by SNP in 2 out of 7 cells (data not shown). This finding may be due to a insufficient inhibition of guanylate cyclase by ODQ.
The inhibition of IP3-induced Ca2+ release by pCPT-cGMP is mediated by cGMP-dependent protein kinase
If the inhibition of IP3-induced Ca2+ release by cGMP is mediated by cGMP-PK, it should be possible to block the inhibitory effect of cGMP by inhibiting cGMP-PK. When 100 μm IPG, a specific peptide inhibitor of cGMP-PK (Yan et al. 1996), was added to the pipette solution together with caged IP3 and OGB488, pCPT-cGMP failed to inhibit IP3-induced Ca2+ release (n = 10cells) (Fig. 4). As a control for the specificity of IPG action we used carbacyclin, a chemically stable analogue of prostacyclin (PGI2) (Whittle et al. 1980), which is known to elevate intracellular cAMP through a Gs protein-dependent activation of adenylate cyclase (for review see Wise & Jones, 1996). Previous experiments have demonstrated that PGI2 inhibits both agonist-induced [Ca2+]i oscillations and IP3-induced Ca2+ release in megakaryocytes (Tertyshnikova & Fein, 1997, 1998). Assuming, that the effect of cAMP is mediated by cAMP-dependent protein kinase, it would be expected that IPG would not prevent the inhibitory effect of carbacyclin. Indeed, as shown in Fig. 4, carbacyclin reversibly inhibited IP3-induced Ca2+ release (n = 4cells) (similar results with carbacyclin were seen in 11 other cells without IPG in the pipette). In summary, we conclude that inhibition of IP3-induced Ca2+ release by pCPT-cGMP is mediated by cGMP-dependent protein kinase.
Figure 4. IPG prevents inhibition of IP3-induced Ca2+ release by pCPT-cGMP, but not by carbacyclin.

IPG (100 μm), caged IP3 (100 μm) and OGB488 (200 μm) were included in the intrapipette solution. IP3was released by UV flashes as indicated. Carbacyclin (100 nm) and pCPT-cGMP (1 mm) were applied to the cell as indicated.
The inhibition of IP3-induced Ca2+ release by SNP is mediated by both cGMP- and cAMP-dependent protein kinases
If the suppression of IP3-induced Ca2+ release by SNP involves only cGMP-dependent stimulation of cGMP-PK, then inhibition of cGMP-PK should block the inhibitory effect of both pCPT-cGMP and SNP. However, when SNP was applied to a cell in which cGMP-PK had been inhibited by IPG and pCPT-cGMP no longer suppressed IP3-induced Ca2+ release, SNP still reversibly inhibited IP3-induced Ca2+ release (Fig. 5, n = 7cells) (separate experiments showed that 0.2 mm SNP and 1 mm pCPT-cGMP were near the lowest concentrations which inhibited IP3-induced Ca2+ release in megakaryocytes).
Figure 5. IPG prevents inhibition of IP3-induced Ca2+ release by pCPT-cGMP, but not by SNP.
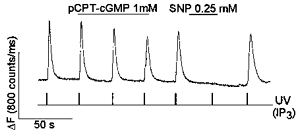
IPG (100 μm), caged IP3 (100 μm) and OGB488 (200 μm) were included in the intrapipette solution. IP3was released by UV flashes as indicated. SNP (0.25 mm) and pCPT-cGMP (1 mm) were applied to the cell as indicated.
It has been reported that SNP can cause an increase in platelet cAMP even in the absence of adenylate cyclase activation due to the inhibition of cAMP degradation by cGMP-inhibited cAMP phosphodiesterase (PDE III) (Maurice & Haslam, 1990). On the other hand, we have previously shown that inhibition of cAMP-PK with IP20, a specific peptide inhibitor of the catalytic subunit of cAMP-PK (Cheng et al. 1986), does not block the inhibitory effect of SNP (Tertyshnikova & Fein, 1998). In view of the above findings we tested the possibility that the inhibition of the IP3-induced Ca2+ release by SNP involves the activation of both cGMP-PK and cAMP-PK. When both 100 μm IPG and 100 μm IP20 were added to the pipette solution together with caged IP3 and OGB488, SNP failed to inhibit IP3-induced Ca2+ release at the concentration range of 0.25–0.5 mm, and caused only partial inhibition at 1–5 mm (n = 11cells) (Fig. 6). These data strongly suggest that SNP exerts its inhibitory effect via the activation of both cGMP-PK and cAMP-PK.
Figure 6. Simultaneous application of IP20 and IPG prevent inhibition of IP3-induced Ca2+ release by SNP.
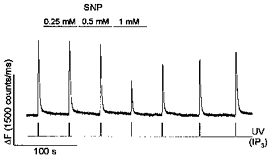
IPG (100 μm), IP20 (100 μm), caged IP3 (100 μm) and OGB488 (200 μm) were included in the intrapipette solution. IP3was released by UV flashes as indicated. SNP (0.25, 0.5 and 1 mm) were applied to the cell as indicated.
DISCUSSION
The data reported in this article provide the first direct demonstration that in the intact cell the NO-cGMP-dependent inhibition of Ca2+ release from IP3-sensitive stores requires the activation of both cAMP-PK and cGMP-PK.
Although it is usually assumed that cGMP-mediated inhibition of [Ca2+]i mobilization occurs predominantly via the activation of cGMP-PK (Butt et al. 1992; Geiger et al. 1992; for review see Walter et al. 1993; Lincoln et al. 1996), it has also been reported that at least in part cGMP exerts its inhibitory effect via inhibition of cAMP breakdown due to inhibition of PDE III (Maurice & Haslam, 1990; for review see Beavo, 1995). Studies have shown that cGMP-elevating agents potentiate the elevation of cAMP by prostaglandins when both cAMP and cGMP elevating agents are present at low concentrations (Radomski et al. 1987; De Caterina et al. 1988; Maurice & Haslam, 1990; Bowen & Haslam, 1991). Furthermore, the nitrovasodilators (SNP and SIN-1) cause increases in platelet cAMP level even in the absence of adenylate cyclase activators and this effect is due to the inhibition of PDE III (Maurice & Haslam, 1990), the major phosphodiesterase isozyme present in platelets and vascular smooth muscles (for review see Beavo, 1995). We have previously reported that in rat megakaryocytes inhibition of cAMP breakdown by the non-specific phosphodiesterase inhibitor 3-isobutyl-1-methyl-xanthine (IBMX) leads to inhibition of ATP-induced [Ca2+]i oscillations (Tertyshnikova & Fein, 1997). Therefore, we interpret our present results as evidence that NO donors exert their inhibitory effect via a direct elevation of cGMP and a secondary elevation of cAMP, mediated by the inhibition of PDE III.
Although pCPT-cGMP is a potent activator of cGMP-PK (Butt et al. 1992; Geiger et al. 1992), unlike cGMP or other hydrolysis-resistant cGMP analogues, such as 8-Br-cGMP, pCPT-cGMP has little or no effect on PDE III (Butt et al. 1992). This can explain our apparently contradictory findings that pCPT-cGMP inhibited Ca2+ release via the activation of cGMP-PK, whereas inhibition by SNP required the activation of both cGMP-PK and cAMP-PK (see Fig. 7).
Figure 7. Simplified diagram illustrating inhibition of IP3-mediated Ca2+ release by PGI2 and NO.
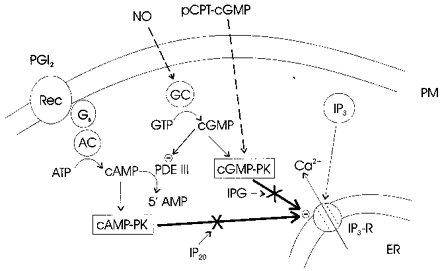
Prostacyclin (PGI2) is shown as activating adenylate cyclase (AC) via the GTP-binding protein Gs and NO is shown as directly activating soluble guanylate cyclase (GC). Elevation of cAMP levels by PGI2 inhibits IP3-mediated Ca2+ release via cAMP-PK. The membrane permeable hydrolysis-resistant analogue of cGMP, pCPT-cGMP, inhibits Ca2+ mobilization via the direct activation of cGMP-PK. Elevation of cGMP levels by NO inhibits IP3-induced Ca2+ release in two ways: by activating cGMP-PK and by inhibiting PDE III and thereby increasing cAMP, which inhibits IP3-induced Ca2+ release by activating cAMP-PK. Not shown are other modes of action by which PGI2 and NO might inhibit Ca2+ mobilization, including inhibition of IP3 production and protein nitrosylation.
Recently we have reported that in rat megakaryocytes elevation of cAMP by carbacyclin leads to the activation of cAMP-PK and thereby to the inhibition of IP3-induced Ca2+ release (Tertyshnikova & Fein, 1998). Our present findings imply that the activation of both cGMP-PK and cAMP-PK is required for the inhibition of IP3-induced Ca2+ release by NO-generating agents. These findings are summarized by the simplified diagram in Fig. 7. For simplicity we show cAMP-PK and cGMP-PK as directly inhibiting the IP3 receptor (IP3-R), although we have no evidence that the inhibition is not mediated via another protein that is phosphorylated by cyclic nucleotide-dependent protein kinases and that regulates IP3-R function.
The role of cyclic nucleotide-dependent protein kinases in regulating Ca2+ release by the IP3-R has been studied using the purified receptor. Both cGMP-PK and cAMP-PK catalyse the phosphorylation of serine 1755 on the IP3-R, and in addition, cAMP-PK phosphorylates serine 1589 (Ferris et al. 1991; Komalavilas & Lincoln, 1994). However, whether phosphorylation of the purified IP3-R in reconstituted systems potentiates or inhibits its ability to release Ca2+ remains controversial (Supattapone et al. 1988; Nakade et al. 1994).
The IP3-R from platelets has also been found to be a substrate for both cGMP and cAMP-dependent protein kinases (El-Daher et al. 1996). It was reported that PGI2 and SNP promoted phosphorylation of the IP3-R in platelets by activating cAMP- and cGMP-dependent protein kinases respectively, and that this phosphorylation was associated with the inhibition of IP3-evoked Ca2+ release (Cavallini et al. 1996). It was also reported that the platelets' IP3-R can be phosphorylated by cAMP-PK and endogenous membrane-bound kinases and that the additional phosphorylation by cAMP-PK inhibits the rate of the IP3-mediated Ca2+ release (Quinton et al. 1996).
We did not find any evidence that elevated cGMP stimulates Ca2+ uptake and/or extrusion in intact megakaryocytes (see Fig. 2). Cyclic nucleotides have been reported to contribute to the reduction of [Ca2+]i in aortic smooth muscle cells by phosphorylation of the regulatory protein phospholamban, thereby stimulating the activity of the sarcoplasmic reticulum Ca2+-ATPase (Cornwell et al. 1991; Karczewski et al. 1992). However, at present there is no evidence that cyclic nucleotide-dependent protein kinases regulate the Ca2+-ATPase by a similar mechanism in platelets. Although it has been suggested that cGMP and cAMP increase the rate of the calcium extrusion pump, and cAMP stimulates the dense tubular Ca2+ accumulation system (Johansson & Haynes, 1992), a more recent publication by Cavallini et al. (1996) failed to find any evidence for an effect of cyclic nucleotides either on platelet endomembrane or plasma membrane Ca2+-ATPases (Cavallini et al. 1996). This discrepancy, as to whether the activity of platelet Ca2+ uptake and/or extrusion mechanisms is regulated by cyclic nucleotide-dependent protein kinases, probably results from differences in the experimental methods used in these studies.
Our data do not rule out NO-dependent inhibition of [Ca2+]i mobilization at the sites upstream from the IP3-R, such as the inhibition of IP3 formation. The inhibition of IP3 and 1,2-diacylglycerol (DAG) formation via the inhibition of phospholipase C (PLC) or PLC-G-protein-receptor coupling has been proposed to account for the cGMP-PK-dependent reduction of [Ca2+]i in platelets (Deana et al. 1989; Nguyen et al. 1991) and smooth muscle tissues (Hirata et al. 1990).
Finally, some cGMP-independent but NO-dependent inhibition might contribute to the NO-mediated inhibition of [Ca2+]i mobilization, especially at the high concentration of the NO donor used (see Fig. 6) (for review see Lincoln et al. 1996). It has been reported that production of NO leads to a number of downstream reactions in which NO, in one of its redox states, reacts with free thiols in proteins of diverse functions with broad regulatory consequences (for review see Stamler et al. 1992). For example, the inhibitory action of peroxynitrite (derived from the interaction of NO and superoxide) on platelet aggregation has been reported to be independent of cGMP, and to be due to protein nitrosylation (Yin et al. 1995). However, whether the IP3-R undergoes S-nitrosylation by NO derivatives, and if so, how nitrosylation of the IP3-R would regulate its physiological functions is not known.
References
- Adams SR, Tsien RY. Controlling cell chemistry with caged compounds. Annual Review of Physiology. 1993;55:755–784. doi: 10.1146/annurev.ph.55.030193.003543. 10.1146/annurev.ph.55.030193.003543. [DOI] [PubMed] [Google Scholar]
- Alioua A, Huggins JP, Rousseau E. PKG-1a phosphoryalates the α-subunit and upregulates reconstituted GKCa channels from tracheal smooth muscle. American Journal of Physiology. 1995;268:L1057–1063. doi: 10.1152/ajplung.1995.268.6.L1057. [DOI] [PubMed] [Google Scholar]
- Beavo JA. Cyclic nucleotide phosphodiesterases: functional implications of multiple isoforms. Physiological Reviews. 1995;75:725–748. doi: 10.1152/physrev.1995.75.4.725. [DOI] [PubMed] [Google Scholar]
- Bowen R, Haslam RJ. Effects of nitrovasodilators on platelet cyclic nucleotide levels in rabbit blood; role of cyclic AMP in synergistic inhibition of platelet function by SIN-1 and prostaglandin E1. Journal of Cardiovascular Pharmacology. 1991;17:424–433. doi: 10.1097/00005344-199103000-00011. [DOI] [PubMed] [Google Scholar]
- Buechler WA, Ivanova K, Wolfram G, Drummer C, Heim J-M, Gerzer R. Soluble guanylyl cyclase and platelet function. Annals of the New York Academy of Sciences. 1994;714:151–157. doi: 10.1111/j.1749-6632.1994.tb12039.x. [DOI] [PubMed] [Google Scholar]
- Butt E, Nolte C, Schulz S, Beltman J, Beavo JA, Jastorff B, Walter U. Analysis of the functional role of cGMP-dependent protein kinase in intact human platelets using a specific activator 8-para-chlorophenylthio-cGMP. Biochemical Pharmacology. 1992;43:2591–2600. doi: 10.1016/0006-2952(92)90148-c. [DOI] [PubMed] [Google Scholar]
- Cavallini L, Coassin M, Borean A, Alexandre A. Prostacyclin and sodium nitroprusside inhibit the activity of the platelet inositol 1,4,5-trisphosphate receptor and promote its phosphorylation. Journal of Biological Chemistry. 1996;271:5545–5551. doi: 10.1074/jbc.271.10.5545. [DOI] [PubMed] [Google Scholar]
- Cheng HC, Kemp BE, Pearson RB, Smith AJ, Misconi L, Van Patten SM, Walsh DA. A potent synthetic peptide inhibitor of the cAMP-dependent protein kinase. Journal of Biological Chemistry. 1986;261:989–992. [PubMed] [Google Scholar]
- Clementi E, Meldolesi J. The cross-talk between nitric oxide and Ca2+: a story with a complex past and a promising future. Trends in Pharmacological Sciences. 1997;18:266–269. doi: 10.1016/s0165-6147(97)01087-0. [DOI] [PubMed] [Google Scholar]
- Cornwell TL, Pryzwansky KB, Wyatt TA, Lincoln TM. Regulation of sarcoplasmic reticulum protein phosphorylation by localized cyclic GMP-dependent protein kinase in vascular smooth muscle cells. Molecular Pharmacology. 1991;40:923–931. [PubMed] [Google Scholar]
- Deana R, Ruzzene M, Doni MG, Zoccarato F, Alexandre A. Cyclic GMP and nitroprusside inhibit the activation of human platelets by fluoroaluminate. Biochimica et Biophysica Acta. 1989;1014:203–206. doi: 10.1016/0167-4889(89)90035-9. 10.1016/0167-4889(89)90035-9. [DOI] [PubMed] [Google Scholar]
- De Caterina R, Giannessi D, Bernini W, Mazzone A. Organic nitrates: direct antiplatelet effects and synergism with prostacyclin. Thrombosis and Haemostasis. 1988;59:207–211. [PubMed] [Google Scholar]
- El-Daher SS, Eigenthaler M, Walter U, Furuichi T, Miyawaki A, Mikoshiba K, Kakkar VV, Authi KS. Distribution and activation of cAMP- and cGMP-dependent protein kinases in highly purified human platelet plasma and intracellular membranes. Thrombosis and Haemostatis. 1996;76:1063–1071. [PubMed] [Google Scholar]
- Feelisch M, Stamler JS. Donors of nitrogen oxides. In: Feelisch M, Stamler JS, editors. Methods in Nitric Oxide Research. New York: John Wiley & Sons Ltd; 1996. pp. 71–115. [Google Scholar]
- Ferris CD, Cameron AM, Bredt DS, Huganir RL, Snyder SH. Inositol 1,4,5-trisphosphate receptor is phosphorylated by cAMP-dependent protein kinase at serines 1755 and 1589. Biochemical and Biophysical Research Communications. 1991;175:192–198. doi: 10.1016/s0006-291x(05)81219-7. [DOI] [PubMed] [Google Scholar]
- Geiger J, Nolte C, Butt E, Sage SO, Walter U. Role of cGMP and cGMP-dependent protein kinase in nitrovasodilator inhibition of agonist-evoked calcium elevation in human platelets. Proceedings of the National Academy of Sciences of the USA. 1992;89:1031–1035. doi: 10.1073/pnas.89.3.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirata M, Kohse KP, Chang C-H, Ikebe T, Murad F. Mechanism of cyclic GMP inhibition of inositol phosphate formation in rat aorta segments and cultured bovine aortic smooth muscle cells. Journal of Biological Chemistry. 1990;265:1268–1273. [PubMed] [Google Scholar]
- Johansson JS, Haynes DH. Cyclic cGMP increases the rate of the calcium extrusion pump in intact human platelets but has no direct effect on the dense tubular calcium accumulation system. Biochimica et Biophysica Acta. 1992;1105:40–50. doi: 10.1016/0005-2736(92)90160-n. [DOI] [PubMed] [Google Scholar]
- Karczewski P, Kelm M, Hartmann M, Schrader J. Role of phospholamban in NO/EDRF-induced relaxation in rat aorta. Life Sciences. 1992;51:1205–1210. doi: 10.1016/0024-3205(92)90357-u. 10.1016/0024-3205(92)90357-U. [DOI] [PubMed] [Google Scholar]
- Komalavilas P, Lincoln TM. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic GMP-dependent protein kinase. Journal of Biological Chemistry. 1994;269:8701–8707. [PubMed] [Google Scholar]
- Komalavilas P, Lincoln TN. Phosphorylation of the inositol 1,4,5-trisphosphate receptor by cyclic GMP-dependent protein kinase. Journal of Biological Chemistry. 1996;271:21933–21938. doi: 10.1074/jbc.271.36.21933. 10.1074/jbc.271.36.21933. [DOI] [PubMed] [Google Scholar]
- Lincoln TM, Cornwell TL, Komalavilas P, MacMillan-Crow LA, Boerth N. The nitric oxide-cyclic GMP signaling system. In: Barany M, editor. Biochemistry of Smooth Muscle Contraction. San-Diego: Academic Press; 1996. pp. 257–268. [Google Scholar]
- MacKenzie AB, Mahaut-Smith MP, Sage SO. Activation of receptor-operated cation channel via P2X1 not P2T purinoceptors in human platelets. Journal of Biological Chemistry. 1996;271:2879–2881. doi: 10.1074/jbc.271.6.2879. 10.1074/jbc.271.6.2879. [DOI] [PubMed] [Google Scholar]
- Maurice DH, Haslam RJ. Molecular basis of the synergistic inhibition of platelet function by nitrovasodilators and activators of adenylate cyclase: inhibition of cyclic AMP breakdown by cyclic GMP. Molecular Pharmacology. 1990;37:671–681. [PubMed] [Google Scholar]
- Moncada S, Palmer RMJ, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacological Reviews. 1991;43:109–142. [PubMed] [Google Scholar]
- Moro MA, Russel RJ, Cellek S, Lizasoain I, Su Y, Darley-Usmar VM, Radomski MW, Moncada S. cGMP mediates the vascular and platelet actions of nitric oxide: Confirmation using an inhibitor of the soluble guanylyl cyclase. Proceedings of the National Academy of Sciences of the USA. 1996;93:1480–1485. doi: 10.1073/pnas.93.4.1480. 10.1073/pnas.93.4.1480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakade S, Rhee SK, Hamanaka H, Mikoshiba K. Cyclic AMP-dependent phosphorylation of an immunoaffinity-purified homotetrameric inositol 1,4,5-trisphosphate receptor (type 1) increases Ca2+ flux in reconstituted lipid vesicles. Journal of Biological Chemistry. 1994;269:6735–6742. [PubMed] [Google Scholar]
- Nguyen BL, Saiton M, Ware JA. Interaction of nitric oxide and cGMP with signal transduction in activated platelets. American Journal of Physiology. 1991;261:H1043–1051. doi: 10.1152/ajpheart.1991.261.4.H1043. [DOI] [PubMed] [Google Scholar]
- Quinton TM, Brown KD, Dean WL. Inositol 1,4,5-trisphosphate-mediated Ca2+ release from platelet internal membranes is regulated by differential phosphorylation. Biochemistry. 1996;35:6865–6871. doi: 10.1021/bi960128m. [DOI] [PubMed] [Google Scholar]
- Radomski MW, Palmer RMJ, Moncada S. The anti-aggregating properties of vascular endothelium: interactions between prostacyclin and nitric oxide. British Journal of Pharmacology. 1987;92:639–646. doi: 10.1111/j.1476-5381.1987.tb11367.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamler JS, Singel D, Loscalzo J. Biochemistry of nitric oxide and its redox-activated forms. Nature. 1992;258:1898–1902. doi: 10.1126/science.1281928. [DOI] [PubMed] [Google Scholar]
- Supattapone S, Danoff S, Theibert A, Joseph SK, Steiner J, Snyder SH. Cyclic AMP-dependent phosphorylation of a brain inositol trisphosphate receptor decreases its release of calcium. Proceedings of the National Academy of Sciences of the USA. 1988;85:8747–8750. doi: 10.1073/pnas.85.22.8747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tertyshnikova S, Fein A. [Ca2+]i oscillations and [Ca2+]i waves in rat megakaryocytes. Cell Calcium. 1997;21:331–344. doi: 10.1016/s0143-4160(97)90026-9. 10.1016/S0143-4160(97)90026-9. [DOI] [PubMed] [Google Scholar]
- Tertyshnikova S, Fein A. Inhibition of inositol 1,4,5-trisphosphate-induced Ca2+ release by cAMP-dependent protein kinase in a living cell. Proceedings of the National Academy of Sciences of the USA. 1998;95:1613–1617. doi: 10.1073/pnas.95.4.1613. 10.1073/pnas.95.4.1613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uneyama C, Uneyama H, Akaike N. Cytoplasmic calcium oscillation in rat megakaryocytes evoked by a novel type of purinoceptor. The Journal of Physiology. 1993;470:731–749. doi: 10.1113/jphysiol.1993.sp019885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter U, Eigenthaler M, Geiger J, Reinhard M. Role of cyclic nucleotide-dependent protein kinases and their common substrate VASP in the regulation of human platelets. In: Authi KS, Watson SP, Kakkar VV, editors. Mechanisms of Platelet Activation and Control. New York: Plenum Press; 1993. pp. 237–249. [DOI] [PubMed] [Google Scholar]
- Whittle BJ, Moncada S, Whiting F, Vane JR. Carbacyclin — a potent stable prostacyclin analogue for the inhibition of platelet aggregation. Prostaglandins. 1980;19:605–627. doi: 10.1016/s0090-6980(80)80010-4. [DOI] [PubMed] [Google Scholar]
- Wise H, Jones RL. Focus on prostacyclin and its novel mimetics. Trends in Pharmacological Sciences. 1996;17:17–21. doi: 10.1016/0165-6147(96)81565-3. 10.1016/0165-6147(96)81565-3. [DOI] [PubMed] [Google Scholar]
- Yan X, Lawrence DS, Corbin JD, Francis SH. Distinguishing between closely related protein kinases: a variation on the bisubstrate inhibitor theme. Journal of the American Chemical Society. 1996;118:6321–6322. 10.1021/ja9609213. [Google Scholar]
- Yin K, Lai P-S, Rodriguez A, Spur BW, Wong P Y.-K. Antithrombotic effect of peroxynitrite: inhibition and reversal of aggregation in human platelets. Prostaglandins. 1995;50:169–178. doi: 10.1016/0090-6980(95)00119-0. [DOI] [PubMed] [Google Scholar]