

Abstract
We examined whether transmitter release could be modified by the activation of protein kinase C (PKC) of retinal bipolar cells. A bipolar cell with a large axon terminal was isolated from the goldfish retina. The presynaptic Ca2+ current was measured under whole-cell voltage clamp, and the released transmitter (probably glutamate) was detected electrophysiologically by using the response of NMDA receptors of catfish horizontal cells as a reporter.
Transmitter release was potentiated by a PKC activator, phorbol 12-myristate 13-acetate (PMA), but not by an ineffective phorbol ester, 4α-phorbol 12,13-didecanoate. A PKC inhibitor, bisindolylmaleimide I, did not affect the transmitter release by itself but blocked the PMA-induced potentiation of transmitter release. These results suggest that the actions of PMA were mediated via the activation of PKC.
Introduction of 5 mm EGTA into the presynaptic terminals of bipolar cells revealed two separate components of transmitter release. A rapid component was triggered immediately after depolarization while a slow component appeared with a delay. Application of PMA selectively potentiated the slow component without affecting the Ca2+ dependence of exocytosis.
We suggest that the activation of PKC may modify the recruitment process of synaptic vesicles in retinal bipolar cells.
In some synapses transmitter release from presynaptic terminals is modulated by the activation of protein kinases, such as protein kinase A and protein kinase C (PKC) (see reviews by Nicoll & Malenka, 1995; Byrne & Kandel, 1996). These protein kinases seem to affect not only the amount of Ca2+ influx (Parfitt & Madison, 1993) or the excitability of presynaptic terminals (Sugita et al. 1997), but also the exocytotic machinery itself (Capogna et al. 1995; Kondo & Marty, 1997).
A subtype of goldfish retinal bipolar cells is suitable for investigating Ca2+-transmitter release coupling since the bipolar cell has a large presynaptic terminal (∼10 μm in diameter). The presynaptic Ca2+ current can be measured under voltage clamp, and at the same time, the release of an excitatory amino acid transmitter, probably glutamate, from a single presynaptic terminal can be detected with a bioassay (Tachibana & Okada, 1991). Since PKC is present in this subtype of bipolar cell (Negishi et al. 1988; Suzuki & Kaneko, 1990), we examined whether transmitter release could be modulated by the activation of PKC. We found that the activation of PKC potentiated the amount of releasable transmitter from bipolar cells.
METHODS
Horizontal cells were dissociated from catfish (Ictalurus punctatus) retina as described previously (Tachibana & Okada, 1991). In brief, a catfish (body length, 15–40 cm) was killed by decapitation followed by immediate pithing of the brain and spinal cord, following ‘Guiding Principles for the Care and Use of Animals in the Field of Physiological Sciences, The Physiological Society of Japan’. Retinas detached from the pigment epithelium of the enucleated eyes were treated with hyaluronidase (0.1 mg ml−1), and then with cysteine-activated papain (2.5–4 mg ml−1) at 28°C for ∼20 min. These agents were dissolved in a Ca2+-free solution, which consisted of (mm): NaCl, 110; KCl, 2.6; NaHCO3, 1; NaH2PO4, 0.5; sodium pyruvate, 1; Hepes, 4; and glucose, 16 (pH 7.2, 260 mosmol l−1). The retinas were then dissociated mechanically with a glass pipette. A few drops of cell suspension were plated on a culture dish (Falcon No. 3001; Becton Dickinson & Co., Franklin Lakes, NJ, USA) containing 2 ml Ames' medium (Sigma). The cells were maintained at 15°C for 1–10 days before use.
On-type bipolar cells with large bulbous axon terminals were similarly dissociated from the retinas of decapitated and pithed goldfish (Carassius auratus; body length, 15–20 cm). The cell suspension, including isolated bipolar cells, was put into the dish in which catfish horizontal cells were maintained. Experiments were carried out within 2 h after the dissociation of the goldfish retina.
Dissociated cells were continuously superfused with a control solution composed of (mm): NaCl, 125; KCl, 2.6; CaCl2, 2.5; MgCl2, 1; glucose, 10; Hepes, 10; and 0.1 mg ml−1 bovine serum albumin (pH 7.4, 270 mosmol l−1). In high-Ca2+ solution, the concentration of Ca2+ was raised from 2.5 to 3.0 mm without osmolarity compensation. Glutamate was simply dissolved in the control solution. Phorbol 12-myristate 13-acetate (PMA; Sigma), 4α-phorbol 12,13-didecanoate (4α-PDD; Sigma) and bisindolylmaleimide I (BIS; Calbiochem, San Diego, CA, USA) were dissolved in dimethyl sulphoxide (DMSO). Test solutions were made by diluting these concentrated stock solutions with the control solution before experiments. The concentration of DMSO in the test solutions was less than 0.02 %. Each test solution was applied locally to a pair of voltage-clamped cells by a Y-tube microflow system (Suzuki et al. 1990). The pipette solution for horizontal cells contained (mm): CsCl, 125; Hepes, 10; EGTA, 5; CaCl2, 0.5; MgCl2, 2; and Na2-cAMP, 0.5 (pH 7.2, 270 mosmol l−1). The pipette solution for bipolar cells usually contained (mm): CsCl, 30; caesium glutamate, 100; Hepes, 10; BAPTA, 0.2; MgCl2, 2; Na2-ATP, 5; and Na3-GTP, 0.2 (pH 7.2, 270 mosmol l−1). In some experiments, 0.2 mm BAPTA was replaced with 5 mm EGTA; the osmolarity was adjusted by reducing the concentration of caesium glutamate.
The bioassay technique has been described in detail by Tachibana & Okada (1991). Two separate patch pipettes were attached to the axon terminal of a single bipolar cell and the cell body of a horizontal cell, respectively, and each cell was whole-cell voltage clamped. The axon terminal of the bipolar cell was closely apposed to the horizontal cell by manipulating the patch pipette of the bipolar cell. Its series resistance ranged between 20 and 40 MΩ, and was compensated by 20–40 % with the electrical circuitry of the patch-clamp amplifier (EPC-7; List-Electronic, Darmstadt, Germany). Catfish horizontal cells have N-methyl-D-aspartate (NMDA) receptors, and are sensitive to glutamate in the submicromolar range (Johnson & Ascher, 1987; Tachibana & Okada, 1991). We have already shown that NMDA receptors in horizontal cells are not saturated or desensitized seriously by the transmitter released from single bipolar cells (Tachibana & Okada, 1991; Sakaba et al. 1997; see also Fig. 2). The horizontal cell was maintained at +30 mV with a second patch-clamp amplifier (CEZ-2200; Nihon Kohden Corp., Tokyo, Japan) to avoid blockage of NMDA receptors by extracellular Mg2+ (Nowak et al. 1984). Glycine (10 μm) was always added to solutions ejected from the Y-tube microflow system to augment the current through NMDA receptors (Tachibana & Okada, 1991). Depolarizing pulses were applied to the bipolar cell every 30–60 s, during which time releasable synaptic vesicle pools mostly recovered from their depletion (Sakaba et al. 1997; von Gersdorff & Matthews, 1997).
Figure 2. No significant effect of PMA on the glutamate-induced current in horizontal cells.
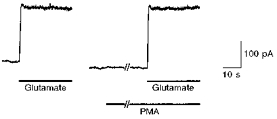
The glutamate-induced current was recorded from a horizontal cell voltage clamped at +30 mV. Glutamate (1 μm) and PMA (100 nm) were applied from the Y-tube microflow system. The second application of glutamate started 160 s after the application of PMA.
RESULTS
Potentiation of transmitter release by a PKC activator
A single bipolar cell was voltage clamped with a patch pipette, which was filled with a solution containing Cs+ to block K+ currents and a low concentration of Ca2+ buffer (0.2 mm BAPTA). A depolarizing pulse (from −60 to −10 mV for 100 ms) induced a sustained Ca2+ current (ICa) in the bipolar cell (Fig. 1). At the same time, an outward current was evoked in the horizontal cell, which was closely apposed to the axon terminal of the bipolar cell and maintained at +30 mV. It has been shown that the bipolar cell ICa is of the dihydropyridine-sensitive type, flows mostly into the axon terminal and mediates transmitter release (Heidelberger & Matthews, 1992; Tachibana et al. 1993). The outward current induced in the horizontal cell has been demonstrated to result from the activation of NMDA receptors by the transmitter released from the bipolar cell (Itr; Tachibana & Okada, 1991). When the same depolarizing pulse was repeatedly applied to the bipolar cell every ∼30 s in control solution, the amplitude of Itr reached a stable value within a few minutes, and then gradually decreased. Itr usually disappeared in approximately 20 min, though ICa was still evoked.
Figure 1. PMA-induced potentiation of transmitter release from bipolar cells.
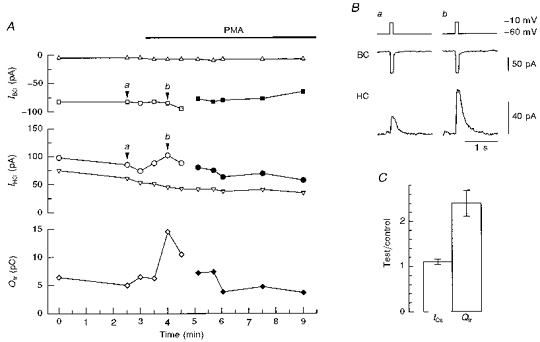
The axon terminal of a bipolar cell (BC) was closely apposed to a horizontal cell (HC), and the bipolar cell was stimulated with a 100 ms depolarizing pulse to evoke transmitter release. The membrane currents were simultaneously recorded from the bipolar cell (IBC) and horizontal cell (IHC), before and after the application of PMA (100 nm). The horizontal cell was held at +30 mV. The pipette solution for the bipolar cell contained 0.2 mm BAPTA. A, IBC, IHC and the charge of Itr (Qtr; diamonds) as a function of time. The amplitude of ICa is defined as the difference between a square and a triangle (the holding current of the bipolar cell; top) and that of Itr as the difference between a circle and a triangle (the holding current of the horizontal cell; middle). The bipolar cell was initially depolarized to −10 mV (open symbols) and then to −20 mV (filled symbols) to reduce the amplitude of PMA-potentiated ICa. B, ICa (middle) and Itr (bottom) before (a) and after (b) the application of PMA. Data were obtained at the time indicated by arrowheads in A. C, effects of PMA on the amplitude of ICa and the Qtr. The ratio of the values with PMA (test) to those of control is illustrated. Means ±s.e.m. were calculated from the data obtained from 9 cell pairs.
PMA (50 or 100 nm) was applied extracellularly to activate PKC in the bipolar cell after Itr was stably evoked (Fig. 1A). One to two minutes after PMA application, the amplitude of Itr was potentiated to more than twice that of control (Fig. 1A and B). The holding currents in both cells were not affected by PMA (Fig. 1A). The amplitude of ICa was slightly increased by PMA (Fig. 1A and C). However, the PMA-induced potentiation of Itr was not due to the increase in ICa because Itr was still potentiated after ICa had been reduced in amplitude to a level equivalent to that of control by decreasing the intensity of depolarization (Fig. 1A). To evaluate the effect of PMA on the amount of transmitter release, the time integral of Itr (i.e. the charge of Itr; Qtr) was calculated (Fig. 1A and C). PMA increased Qtr to 2.40 ± 0.30 (mean ±s.e.m., n = 9) of control (Fig. 1C). After PMA treatment, the potentiated Itr showed run-down in a few minutes though ICa was still evoked by a depolarizing pulse (Fig. 1A). Since the effects of PMA were mostly irreversible, the culture dish was discarded once PMA was introduced into the dish.
It is possible that the potentiation of Itr was due to the modification of NMDA receptors of horizontal cells by PMA. However, PMA changed neither the amplitude (1.04 ± 0.03 of control, n = 12) nor desensitization of glutamate (1 or 10 μm)-induced responses in horizontal cells (Fig. 2). This observation indicates that the potentiation of Itr was caused by the PMA-induced modification of presynaptic mechanisms.
Since it has been reported that phorbol esters may act in a PKC-independent manner (Doerner et al. 1990), the actions of PMA may not be ascribed exclusively to the activation of PKC. To examine this possibility, 4α-PDD, a phorbol ester ineffective in activating PKC (Gillis et al. 1996), was applied to cell pairs. Neither ICa nor Itr was affected by 100 nm 4α-PDD (Fig. 3A; 4α-PDD). The amplitude of ICa and the Qtr in the presence of 4α-PDD were 0.98 ± 0.01 and 1.09 ± 0.07 (n = 4) of control, respectively. After washout of 4α-PDD, subsequent application of PMA was able to augment the Qtr to 1.58 ± 0.10 (n = 4) of control (Fig. 3A; PMA), indicating that the ineffectiveness of 4α-PDD was not due to run-down of transmitter release. Therefore, it is likely that transmitter release from bipolar cells is potentiated by the PMA-induced activation of PKC.
Figure 3. Effects of phorbol esters and a PKC inhibitor on transmitter release.
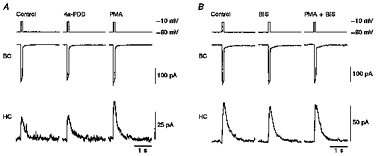
A bipolar cell (BC) was depolarized for 100 ms, and ICa and Itr were simultaneously recorded from the bipolar cell and an apposed horizontal cell (HC), respectively. The horizontal cell was maintained at +30 mV. The pipette solution for the bipolar cell contained 0.2 mm BAPTA. A, a cell pair was superfused with control solution, then with control solution containing 100 nm 4α-PDD, and finally with control solution containing 100 nm PMA after washout of 4α-PDD. B, another cell pair was superfused with control solution, then with control solution containing a PKC inhibitor (500 nm BIS), and finally with a mixture of 500 nm BIS and 100 nm PMA.
Effect of a PKC inhibitor on the PMA-induced potentiation of Itr
To confirm the action of PKC on transmitter release, BIS, an inhibitor specific to PKC (Toullec et al. 1991), was applied to cell pairs. Neither ICa nor Itr amplitude was altered by BIS (500 nm; Fig. 3B; BIS). The amplitude of ICa and the Qtr were 0.94 ± 0.02 and 0.94 ± 0.09 (n = 5) of control, respectively. These slight decreases were probably due to run-down of ICa and transmitter release with time. Since transmitter release was still evoked after the application of BIS, PKC may not play a major role in evoked transmitter release without PMA.
Subsequent addition of 100 nm PMA failed to potentiate Itr or to augment ICa (Fig. 3B; PMA + BIS). The amplitude of ICa and the Qtr were decreased to 0.86 ± 0.10 and 0.83 ± 0.03 (n = 5) of control, respectively, probably due to gradual run-down. This result further indicates that transmitter release was potentiated by the PMA-induced activation of PKC in bipolar cells.
Increase in releasable synaptic vesicles by the activation of PKC
It has been demonstrated that transmitter release from goldfish retinal bipolar cells consists of at least two components and that only a limited number of synaptic vesicles (approximately 6000 synaptic vesicles; von Gersdorff et al. 1996) can be released when the duration of the depolarizing pulse is increased to more than a few hundred milliseconds (Mennerick & Matthews, 1996; Sakaba et al. 1997). Introduction of a high concentration of BAPTA or EGTA into the presynaptic terminals of bipolar cells is an effective way of separating the two components of transmitter release (Sakaba et al. 1997). A rapid component is triggered immediately after depolarization and quickly depleted; its pool size is estimated to be ∼1200 synaptic vesicles. This component corresponds to the ultrafast component revealed by capacitance measurements (Mennerick & Matthews, 1996), which was not detected in their previous study (von Gersdorff & Matthews, 1994). A slow component, which corresponds to the slower component detected by Mennerick & Matthews (1996), appears with a delay after the onset of depolarization; its pool size is assumed to be ∼4800 synaptic vesicles. The slow component appears with a shorter delay or fused to the rapid component when the amplitude of ICa becomes large and/or the intracellular free Ca2+ concentration is weakly buffered by exogenously applied Ca2+ buffers (Fig. 1B; see Fig. 3 of Sakaba et al. 1997).
We examined how these two components of transmitter release could be affected by the activation of PKC. The two components were separated by introducing 5 mm EGTA into the presynaptic terminal of a bipolar cell through a recording pipette. When a long depolarizing pulse (3 s in duration) was applied to the bipolar cell, the evoked Itr displayed two consecutive peaks, one which appeared with short latency and a second which appeared more than 1 s after the pulse onset (Fig. 4A, HC; continuous trace). Once the slow component reached a peak, the amplitude of Itr declined to the baseline, though ICa was still activated (Fig. 4A, BC; continuous trace). Thus, the 3 s pulse was strong and long enough to totally deplete the releasable synaptic vesicles even with 5 mm internal EGTA.
Figure 4. Effects of PMA on the rapid and slow components of transmitter release.
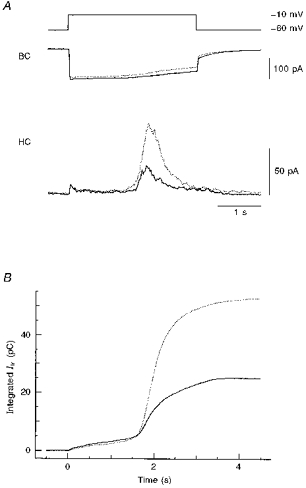
A, simultaneous recordings of ICa and Itr from a single bipolar cell (BC) and a horizontal cell (HC), respectively. The pipette solution for the bipolar cell contained 5 mm EGTA to separate the rapid and slow components of Itr. The bipolar cell was depolarized to −10 mV for 3 s. The horizontal cell was maintained at +30 mV. The continuous and dotted traces were obtained before and after the application of 100 nm PMA, respectively. B, relationship between the integration time and the time integral of Itr before (continuous trace) and after (dotted trace) the application of 100 nm PMA. The bipolar cell was depolarized between 0 and 3 s. The data shown in A were used for calculating the time integrals of Itr.
When 100 nm PMA was applied to the same cell pair, the slow component of Itr was strongly potentiated while the rapid component did not change appreciably (Fig. 4A, HC; dotted trace). It is clear that PMA increased the amount of releasable transmitter and that PMA potentiated mainly the slow component of transmitter release.
To further clarify the effect of PMA on each component of Itr, time integrals of Itr were calculated before and after the application of PMA, and plotted against the corresponding integration time (Fig. 4B). The relationship between the integration time and the time integral of Itr may reflect the relationship between the amount of Ca2+ influx and the amount of transmitter release, since both inactivation of ICa and desensitization of NMDA receptors are slow. The two curves closely overlapped up to approximately 1.5 s after the onset of the depolarizing pulse (Fig. 4B), by which time the rapid component was mostly depleted (Fig. 4A, HC). The value of the time integral of Itr by this time (< 1.5 s) was not augmented by PMA (0.90 ± 0.06 of control, n = 3). However, the two curves diverged when the second component started developing under each condition. The time integrals of Itr increased slightly even after the termination of the depolarizing pulse (> 3 s; Fig. 4B), probably due to the slow deactivation time constant of NMDA receptors (∼200 ms; Lester & Jahr, 1992; Sakaba et al. 1997). The final value of the time integral of Itr was increased to 1.81 ± 0.14 (n = 3) of control.
If PMA increased the Ca2+ concentration at the release site or changed the Ca2+ dependence of exocytosis, the relationship between the integration time and the time integral of Itr in the presence of PMA should have shifted to the left along the abscissa. However, such a shift was not observed; instead, the maximal value of the time integral of Itr was increased (Fig. 4B). Therefore, it is likely that PMA increased the pool size of the slow component of transmitter release.
Effects of ICa augmentation on the total amount of releasable transmitter
PMA showed a tendency to slightly augment the amplitude of ICa (Fig. 1C; 1.10 ± 0.06 of control, n = 9). To examine a possible contribution of the augmented ICa to the amount of releasable transmitter, the amplitude of ICa was increased by raising the extracellular Ca2+ concentration from 2.5 to 3.0 mm in the absence of PMA. Although the amplitude of ICa evoked by a 3 s depolarizing pulse increased to approximately 1.2 times that of control (1.18 ± 0.03 of control, n = 8) in 5 mm EGTA-filled bipolar cells, neither the amplitude of the rapid component nor that of the slow component of transmitter release was increased. The slow component appeared slightly earlier with 3.0 mm Ca2+ than with 2.5 mm Ca2+ since the second component is initiated when the amount of Ca2+ influx reaches a certain value (Sakaba et al. 1997). The Qtr was almost identical for both Ca2+ concentrations (1.03 ± 0.05 of control, n = 8), presumably due to saturation of transmitter release (Sakaba et al. 1997). This observation indicates that the slight augmentation of Ca2+ influx could not be a main factor for the PMA-induced potentiation of transmitter release.
DISCUSSION
In the present study it was demonstrated that application of PMA potentiated transmitter release from bipolar cells. The potentiation seemed to be due to an increase in the amount of releasable transmitter without significant alteration of the Ca2+ dependence of exocytosis, since application of PMA increased the maximal value of transmitter release but did not shift the relationship between the amount of Ca2+ influx and the amount of transmitter release along the abscissa (Fig. 4B). The accompanying slight augmentation of ICa cannot account for the increase in transmitter release by PMA. The major target of PMA seemed to be PKC, the presence of which has been demonstrated in bipolar cells (Negishi et al. 1988; Suzuki & Kaneko, 1990). However, PKC activators and inhibitors interact with proteins with C1- and C2-domains, such as Munc13, which may play an important role in transmitter release (Brose et al. 1995; Orita et al. 1997). It is possible that the PMA-induced potentiation of transmitter release was due to the activation of proteins such as Munc13 in bipolar cells.
It has been shown that phorbol esters increase evoked transmitter release from hippocampal neurons by affecting events downstream of Ca2+ influx (Capogna et al. 1995). Gillis et al. (1996) suggested that the potentiation of secretion from chromaffin cells by the activation of PKC was due to an increase in the size of releasable pools of secretory granules. The present results show that PMA mainly potentiated the slow component of transmitter release. Since the slow component has been assumed to reflect the recruitment of synaptic vesicles from the synaptic ribbons to the active zones (Mennerick & Matthews, 1996; von Gersdorff et al. 1996; Sakaba et al. 1997), the potentiation of transmitter release by application of PMA may be due to the modification of the recruitment process of vesicles towards synaptic ribbons or the increase in the number of vesicles attached to synaptic ribbons. Alternatively, application of PMA may facilitate the fusion of docked vesicles located in regions away from Ca2+ channels, since simulation studies have shown that the delayed secretion in neuroendocrine cells can be interpreted as the fusion of such docked vesicles in the remote regions (Klingauf & Neher, 1997).
The present results suggest that the activation of PKC in bipolar cells may increase the gain of synaptic transfer from bipolar cells to postsynaptic cells. In the intact retina, the axon terminals of bipolar cells receive inputs from amacrine cells via conventional chemical synapses (Yazulla & Studholme, 1991). It has been reported that PKC is activated by a neurotransmitter via second messenger cascades (Feigenspan & Bormann, 1994) or modulated by a membrane-permeable substance (Shearman et al. 1991; Coffey et al. 1994). How PKC of bipolar cells is endogenously activated in the intact retina should be the subject of future studies.
Acknowledgments
We thank Laurence H. Pinto, Henrique von Gersdorff and Erwin Neher for critical reading of the manuscript. This work was supported by Grant-in Aid for Scientific Research from The Ministry of Education, Science, Sports and Culture to M. T. (No. 07458218 and No. 09480238).
References
- Brose N, Hofmann K, Hata Y, Südhof TC. Mammalian homologues of Caenorhabditis elegans unc-13 gene define novel family of C2-domain proteins. Journal of Biological Chemistry. 1995;270:25273–25280. doi: 10.1074/jbc.270.42.25273. 10.1074/jbc.270.42.25273. [DOI] [PubMed] [Google Scholar]
- Byrne JH, Kandel ER. Presynaptic facilitation revisited: state and time dependence. Journal of Neuroscience. 1996;16:425–435. doi: 10.1523/JNEUROSCI.16-02-00425.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capogna M, Gähwiler BH, Thompson SM. Presynaptic enhancement of inhibitory synaptic transmission by protein kinases A and C in the rat hippocampus in vitro. Journal of Neuroscience. 1995;15:1249–1260. doi: 10.1523/JNEUROSCI.15-02-01249.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coffey TE, Herrero I, Sihra TS, Sánchez-Prieto J, Nicholls DG. Glutamate exocytosis and MARCKS phosphorylation are enhanced by a metabotropic glutamate receptor coupled to a protein kinase C synergistically activated by diacylglycerol and arachidonic acid. Journal of Neurochemistry. 1994;63:1303–1310. doi: 10.1046/j.1471-4159.1994.63041303.x. [DOI] [PubMed] [Google Scholar]
- Doerner D, Abdel-Latif M, Rogers TB, Alger BE. Protein kinase C-dependent and -independent effects of phorbol esters on hippocampal calcium channel current. Journal of Neuroscience. 1990;10:1699–1706. doi: 10.1523/JNEUROSCI.10-05-01699.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigenspan A, Bormann J. Modulation of GABAC receptors in rat retinal bipolar cells by protein kinase C. The Journal of Physiology. 1994;481:325–330. doi: 10.1113/jphysiol.1994.sp020442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis KD, Mößner R, Neher E. Protein kinase C enhances exocytosis from chromaffin cells by increasing the size of the readily releasable pool of secretory granules. Neuron. 1996;16:1209–1220. doi: 10.1016/s0896-6273(00)80147-6. 10.1016/S0896-6273(00)80147-6. [DOI] [PubMed] [Google Scholar]
- Heidelberger R, Matthews G. Calcium influx and calcium current in single synaptic terminals of goldfish retinal bipolar neurons. The Journal of Physiology. 1992;447:235–256. doi: 10.1113/jphysiol.1992.sp019000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JW, Ascher E. Glycine potentiates the NMDA response in cultured mouse brain neurons. Nature. 1987;325:529–531. doi: 10.1038/325529a0. 10.1038/325529a0. [DOI] [PubMed] [Google Scholar]
- Klingauf J, Neher E. Modeling buffered Ca2+ diffusion near the membrane: implications for secretion in neuroendocrine cells. Biophysical Journal. 1997;72:674–690. doi: 10.1016/s0006-3495(97)78704-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S, Marty A. Protein kinase A-mediated enhancement of miniature IPSC frequency by noradrenaline in rat cerebellar stellate cells. The Journal of Physiology. 1997;498:165–176. doi: 10.1113/jphysiol.1997.sp021849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lester RA, Jahr CE. NMDA channel behavior depends on agonist affinity. Journal of Neuroscience. 1992;12:635–643. doi: 10.1523/JNEUROSCI.12-02-00635.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mennerick S, Matthews G. Ultrafast exocytosis elicited by calcium current in synaptic terminals of retinal bipolar neurons. Neuron. 1996;17:1241–1249. doi: 10.1016/s0896-6273(00)80254-8. 10.1016/S0896-6273(00)80254-8. [DOI] [PubMed] [Google Scholar]
- Negishi K, Kato S, Teranishi T. Dopamine cells and rod bipolar cells contain protein kinase C-like immunoreactivity in some vertebrates. Neuroscience Letters. 1988;94:247–252. doi: 10.1016/0304-3940(88)90025-0. 10.1016/0304-3940(88)90025-0. [DOI] [PubMed] [Google Scholar]
- Nicoll RA, Malenka RC. Contrasting properties of two forms of long-term potentiation in the hippocampus. Nature. 1995;377:115–118. doi: 10.1038/377115a0. 10.1038/377115a0. [DOI] [PubMed] [Google Scholar]
- Nowak L, Bregestovski P, Ascher P, Herbet A, Prochiantz A. Magnesium gates glutamate-activated channels in mouse central neurons. Nature. 1984;307:462–465. doi: 10.1038/307462a0. [DOI] [PubMed] [Google Scholar]
- Orita S, Naito A, Sakaguchi G, Maeda M, Igarashi H, Sasaki T, Takai Y. Physical and functional interactions of Doc2 and Munc13 in Ca2+-dependent exocytotic machinery. Journal of Biological Chemistry. 1997;272:16081–16084. doi: 10.1074/jbc.272.26.16081. 10.1074/jbc.272.26.16081. [DOI] [PubMed] [Google Scholar]
- Parfitt KD, Madison DV. Phorbol esters enhance synaptic transmission by a presynaptic, calcium-dependent mechanism in rat hippocampus. The Journal of Physiology. 1993;471:245–268. doi: 10.1113/jphysiol.1993.sp019900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaba T, Tachibana M, Matsui K, Minami N. Two components of transmitter release in retinal bipolar cells: exocytosis and mobilization of synaptic vesicles. Neuroscience Research. 1997;27:357–370. doi: 10.1016/s0168-0102(97)01168-1. 10.1016/S0168-0102(97)01168-1. [DOI] [PubMed] [Google Scholar]
- Shearman MS, Shinomura T, Oda T, Nishizuka Y. Protein kinase C subspecies in adult rat hippocampal synaptosomes. Activation by diacylglycerol and arachidonic acid. FEBS Letters. 1991;279:261–264. doi: 10.1016/0014-5793(91)80163-w. 10.1016/0014-5793(91)80163-W. [DOI] [PubMed] [Google Scholar]
- Sugita S, Baxter DA, Byrne JH. Differential effects of 4-aminopyridine, serotonin, and phorbol esters on facilitation of sensorimotor connections in Aplysia. Journal of Neurophysiology. 1997;77:177–185. doi: 10.1152/jn.1997.77.1.177. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kaneko A. Identification of bipolar cell subtypes by protein kinase C-like immunoreactivity in the goldfish retina. Visual Neuroscience. 1990;5:223–230. doi: 10.1017/s0952523800000298. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Tachibana M, Kaneko A. Effects of glycine and GABA on isolated bipolar cells of the mouse retina. The Journal of Physiology. 1990;421:645–662. doi: 10.1113/jphysiol.1990.sp017967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Okada T. Release of endogenous excitatory amino acids from ON-type bipolar cells from the goldfish retina. Journal of Neuroscience. 1991;11:2199–2208. doi: 10.1523/JNEUROSCI.11-07-02199.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tachibana M, Okada T, Arimura T, Kobayashi K, Piccolino M. Dihydropyridine-sensitive calcium current mediates neurotransmitter release from bipolar cells of goldfish retina. Journal of Neuroscience. 1993;13:2898–2909. doi: 10.1523/JNEUROSCI.13-07-02898.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toullec D, Pianetti P, Coste H, Bellevergue P, Grand-Perret T, Ajakane M, Baudet V, Boissin P, Boursier E, Loriolle F, Duhamel L, Charon D, Kirilovsky J. The bisindolylmaleimide GF 109203X is a potent and selective inhibitor of protein kinase C. Journal of Biological Chemistry. 1991;266:15771–15781. [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Dynamics of synaptic vesicle fusion and membrane retrieval in synaptic terminals. Nature. 1994;367:735–739. doi: 10.1038/367735a0. 10.1038/367735a0. [DOI] [PubMed] [Google Scholar]
- von Gersdorff H, Matthews G. Depletion and replenishment of vesicle pools at a ribbon-type synaptic terminal. Journal of Neuroscience. 1997;17:1919–1927. doi: 10.1523/JNEUROSCI.17-06-01919.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Vardi E, Matthews G, Sterling P. Evidence that vesicles on the synaptic terminals of retinal bipolar neurons can be rapidly released. Neuron. 1996;16:1221–1227. doi: 10.1016/s0896-6273(00)80148-8. 10.1016/S0896-6273(00)80148-8. [DOI] [PubMed] [Google Scholar]
- Yazulla S, Studholme KM. Glycine-receptor immunoreactivity in retinal bipolar cells is postsynaptic to glycinergic and GABAergic amacrine cell synapses. Journal of Comparative Neurology. 1991;310:11–20. doi: 10.1002/cne.903100104. [DOI] [PubMed] [Google Scholar]