

Abstract
Adenosine has been shown to decrease Ca2+ current (ICa) and attenuate the hypoxia-induced enhancement of intracellular free Ca2+ ([Ca2+]i) in oxygen-sensitive rat phaeochromocytoma (PC12) cells. These effects are mediated via the adenosine A2A receptor and protein kinase A (PKA). The current study was undertaken to determine the effects of adenosine on Ca2+ current and hypoxia-induced change in [Ca2+]i during chronic hypoxia.
Whole cell patch-clamp studies revealed that the effect of adenosine on ICa was significantly reduced when PC12 cells were exposed to hypoxia (10 % O2) for 24 and 48 h.
Ca2+ imaging studies using fura-2 revealed that the anoxia-induced increase in [Ca2+]i was significantly enhanced when PC12 cells were exposed to 10 % O2 for up to 48 h. In contrast, the inhibitory effects of adenosine on anoxia-induced elevation of [Ca2+]i was significantly blunted in PC12 cells exposed to hypoxia for 48 h.
Northern blot analysis revealed that mRNA for the A2A receptor, which is the only adenosine receptor subtype expressed in PC12 cells, was significantly upregulated by hypoxia. Radioligand binding analysis with [3H]CGS21680, a selective A2A receptor ligand, showed that the number of adenosine A2A receptor binding sites was similarly increased during exposure to 10 % O2 for 48 h.
PKA enzyme activity was significantly inhibited when PC12 cells were exposed to 10 % O2 for 24 and 48 h. However, we found that hypoxia failed to induce change in adenosine- and forskolin-stimulated adenylate cyclase enzyme activity. Chronic hypoxia also did not alter the immunoreactivity level of the G protein Gsα, an effector of the A2 signalling pathway.
Whole cell patch-clamp analysis showed that the effect of 8-bromo-cAMP, an activator of PKA, on ICa was significantly attenuated during 48 h exposure to 10 % O2.
We conclude therefore that the reduced effect of adenosine on ICa and [Ca2+]i in PC12 cells exposed to chronic hypoxia is due to hypoxia-induced downregulation of PKA. This mechanism may serve to reduce the negative feedback on ICa and [Ca2+]i by adenosine and therefore maintain enhanced membrane excitability of PC12 cells during long-term hypoxia.
The cellular mechanisms of hypoxic chemoreception have been intensively studied in O2 sensing cells (Gonzalez et al. 1994). In contrast, there have been relatively few studies on the molecular mechanisms that underlie the effect of chronic hypoxia on chemosensitivity in these cells. Type I cells in the carotid body are the major O2 sensing cells in mammals and are primarily responsible for the hyperventilation that occurs during exposure to hypoxia. It has been shown that chronic hypoxia induces both structural and functional changes in the type I cells, which may influence their response to hypoxia. For example, chronic hypoxia induces hypertrophy of the type I cells (Kusakabe et al. 1993) and enhances their catecholamine production (Hanbauer et al. 1981). In addition, chronic hypoxia modulates Na+, K+ and Ca2+ currents in type I cells and results in increased membrane excitability (Hempleman, 1995, 1996; Stea et al. 1995), although attenuated chemosensitivity due to reduced O2-sensitive K+ current was reported in rats which were born and reared in a hypoxic environment (Wyatt et al. 1995). In humans exposed to hypoxia for several days to weeks, the hyperventilatory response is increased (Weil, 1986). Therefore, the chemosensitivity of the O2 sensing cells is generally thought to be enhanced during chronic hypoxia. The molecular and cellular mechanisms for this are poorly understood, but could involve enhanced membrane excitability and/or increased release of neurotransmitter.
A number of endogenous mediators are involved in the process of carotid body chemoreception (Prabhakar, 1994). There is growing evidence that adenosine may regulate the carotid body response to hypoxia (McQueen & Ribeiro, 1986; Monteiro & Ribeiro, 1987; Watt et al. 1987; Runold et al. 1990). Adenosine is an intermediate metabolite of cellular ATP that is released from cells under conditions of inadequate tissue oxygenation (Winn et al. 1981; Zetterstrom et al. 1982; Phillis et al. 1987). To date, four adenosine receptor subtypes have been identified, including A1, A2A, A2B and A3 (Fredholm, 1995; Olah & Stiles, 1995). The A1 and A3 receptors are usually coupled to the inhibitory G protein, Gi, and mediate inhibition of adenylate cyclase (Fredholm, 1995; Olah & Stiles, 1995). The A2 receptor family, on the other hand, is primarily coupled to the stimulatory G protein, Gs, and thereby activates adenylate cyclase (Fredholm, 1995; Olah & Stiles, 1995). Our previous study revealed that adenosine modulates the cellular responses to acute hypoxia and stabilizes membrane excitability in rat phaeochromocytoma (PC12) cells, an O2-sensitive cell line (Kobayashi et al. 1998). We found that adenosine attenuates membrane depolarization and the increase in intracellular free Ca2+ ([Ca2+]i) induced by acute hypoxia by activating a K+ current and inhibiting a Ca2+ current (ICa). We further showed that these actions of adenosine are mediated by the A2A receptor and protein kinase A (PKA).
The current study was undertaken to investigate the effect of chronic hypoxia on the adenosine-induced cellular responses in PC12 cells. The PC12 cell line has been shown to be an ideal model to investigate the basic molecular and biochemical mechanisms for cellular adaptation to chronic hypoxia (Czyzyk-Krzeska et al. 1994; Zhu et al. 1996). It has been shown that PC12 cells express O2-sensitive K+ channels which are functionally very similar to those present in other chemosensitive cells (Zhu et al. 1996; Conforti & Millhorn, 1997). The present study was undertaken to determine if the effect of adenosine on ICa and [Ca2+]i is altered by pre-exposure of PC12 cells to chronic hypoxia. The results from this study revealed that this is indeed the case. We found that the inhibitory effect of A2A activation on [Ca2+]i was markedly reduced in cells exposed to chronic hypoxia and that reduced activity of PKA contributes to this downregulation. We propose that this is an important mechanism for maintenance of membrane excitability during prolonged hypoxia in the O2-sensitive PC12 cells.
METHODS
Cell culture
PC12 cells (Greene & Tischler, 1976) were grown in Dulbecco's modified Eagle's medium-Ham's F-12 (DMEM-F-12; Gibco) which contained 15 mm Hepes buffer, 2 mml-glutamine, 10 % v/v fetal bovine serum (Gibco) and penicillin-streptomycin (100 u ml−1 and 100 μg ml−1, respectively) in an incubator in which the environment (21 % O2 and 5 % CO2 (remainder N2); 37°C) was strictly maintained. Media were changed twice a week. Cells were grown to reach 70 % confluence before they were used for experiments.
Patch-clamp experiments
PC12 cells were plated on coverslips and kept in a normoxic (21 % O2) environment for at least 24 h. The cells were then exposed to either normoxia or hypoxia in an incubator (Forma Scientific, Marietta, OH, USA) in an environment of 21 % or 10 % O2, 5 % CO2 (balanced with N2) for 24 or 48 h.
The method for patch clamp was described previously (Kobayashi et al. 1998). Briefly, whole cell voltage-clamp recordings for ICa were performed in a conventional patch-clamp mode (Hamill et al. 1981) using an Axopatch 200A amplifier (Axon Instruments). The patch pipettes had resistances of 4–5 MΩ when filled with an internal solution. The cells were placed in a perfusion chamber (volume, 200–400 μl) which was mounted on the stage of an inverted interference microscope (ITM-2, Olympus). Cells were constantly perfused with the recording solution at a flow rate of 2–3 ml min−1. All experiments were performed at room temperature (23–25°C). In most experiments, series resistance before the electronic compensation was between 4 and 6 MΩ. Seventy to seventy five per cent of the series resistance was electronically compensated. Solutions containing adenosine were applied extracellularly after a stable recording of ICa had been obtained.
ICa was recorded using patch pipettes filled with (mm): 140 caesium gluconate, 1 CaCl2, 2 MgCl2, 10 EGTA, 10 Hepes, 3 Na-ATP and 0.2 GTP (pH adjusted to 7.2 with Tris base), while the external solution included (mm): 122 N-methylglucamine-glutamate, 20 BaCl2, 2 MgCl2, 10 Hepes and 10 glucose (pH adjusted to 7.4 with Tris base). Ba2+ was used as a charge carrier to augment the currents activated. A liquid junctional potential measured in this experimental condition was 14.0 ± 2.0 mV. The ICa was measured from a holding potential (Vh) of −80 mV with depolarizing steps to +20 mV (160 ms in duration). Leak and capacitance currents were subtracted by using currents elicited by small hyperpolarizing pulses (P/4 protocol) to ensure the ICa was not altered as a result of either run-down or a reduction of seal resistance. The current- voltage (I-V) relationship of ICa was measured with 100 ms long test pulses from a Vh of −80 mV to test potentials ranging from −50 to +60 mV (10 mV increments). This protocol induced an I-V relationship with 15–20 mV rightward shift in the position of the trough of the control and adenosine-stimulated ICa. This shift might be due to the presence of a junctional potential or to the charge screen effect of a high concentration of Ba2+. However, we did not correct for the junctional potential, because it does not affect the data interpretation.
Ca2+ imaging
Cytosolic free Ca2+ concentration ([Ca2+]i) was evaluated using the fluorescent Ca2+ indicator fura-2 (Molecular Probes) and a Ca2+ imaging system (Intracellular Imaging, Cincinnati, OH, USA). PC12 cells were exposed initially to either normoxia (21 % O2) or hypoxia (10 % O2) for 48 h. The cells were then incubated with serum-free DMEM-F-12 medium containing fura-2 AM (5 μm) and pluronic F-127 (0.01 %) for 40 min at 37°C. For the hypoxia experiments, the medium was equilibrated in advance with 10 % O2 and 5 % CO2. The cells were then rinsed twice with the same medium without fura-2 and left at 37°C for 30 min to allow for hydrolysis of the ester. Coverslips were placed in an environmental chamber mounted on the stage of a Nikon TMS microscope. Light from a 300 W xenon arc illuminator passed through a computer-controlled filter changer and shutter unit that contained 340 and 380 nm filters. Light from selected cells was collected by an integrating charge-coupled device video camera. The ratio of light intensity at the two wavelengths was calculated on-line and stored on the computer. The ratio of F340/F380 was used as an indicator of [Ca2+]i. This approach allows measurement of [Ca2+]i independent of intracellular fura-2 concentration. Experimental solutions (DMEM-F-12 medium without phenolphthalein) were perfused by a peristaltic pump at a flow rate of 5 ml min−1.
The response to anoxia was evaluated by perfusing cells with the medium saturated with 100 % N2 gas with 1 mm sodium dithionate (Na2S2O4), an oxygen chelator. To minimize the possible production of free radicals by dithionate (Archer et al. 1995), the medium was first bubbled with 100 % N2 to remove oxygen for more than 30 min and then dithionate was added into the solution. Our laboratory has shown that these conditions lead to a reduction in PO2 from 150 to approximately 0 mmHg (Zhu et al. 1996).
Northern blot analysis
Total cellular RNA was extracted from PC12 cells using TRI-REAGENT (Molecular Research Center, Cincinnati, OH, USA) according to the manufacturer's instructions. The RNA concentration and purity were determined by measurement of absorbance at 260 and 280 nm with a Genosys spectrophotometer (Fisher, Pittsburgh, PA, USA). A 30 μg aliquot of total RNA was fractionated by electrophoresis using a 1 % agarose gel in 1 × Mops buffer containing 0.4 m formamide. Gels were then stained with SYBR Green I dye (Molecular Probes) as a visual check that equal amounts of RNA had been loaded in each lane. RNAs were transferred by capillary blotting onto a nylon membrane (HybondTM-N+; Amersham, Chicago, IL, USA). The membrane was pre-hybridized for 2–4 h in buffer (0.05 m sodium phosphate, 10 × SCC (167 mm NaCl, 167 mm sodium citrate, pH 7.0), 10 × Denhardt's reagent (0.2 % w/v Ficoll, 0.2 % w/v polyvinylpyrrolidone, 0.2 % w/v bovine serum albumin) 0.1 μg ml−1 denatured salmon sperm DNA, 50 % formamide) and then hybridized overnight in the same buffer with 10 % dextran sulphate and 1.0 × 106 c.p.m. ml−1 of radiolabelled probe. Following hybridization, the membranes were washed three times at 55°C with 2 × SCC-0.1 % SDS and then exposed to a storage phosphor screen (Molecular Dynamics, CA, USA) for 4–5 h. The screen was scanned by an optical scanner (StormTM, Molecular Dynamics), and the signals were quantified using digital image analysing software (ImageQuaNTTM, Molecular Dynamics).
A full-length cDNA encoding adenosine A2A receptor was provided by Dr J. S. Fink (Fink et al. 1992). A fragment of the full-length A2A cDNA including the whole coding sequence of the A2A receptor was excised with Xho I and Xba I (1577 kb), and used as the probe for Northern blot analysis. The probe was labelled using a random-primed DNA labelling kit (Prime-A-GeneTM; Promega, Madison, WI, USA) and 2-[α-32P]desoxycytidine 5′-triphosphate (Dupont NEN, Boston, MA, USA) and then purified with a Sephadex G-50 column (Boehringer-Mannheim).
Radioligand binding assay
Binding of [3H]CGS21680, a specific adenosine A2A receptor agonist (Dupont NEN; Jarvis et al. 1989), to PC12 cell membranes was measured by a modification of the method described by Hide et al. (1992). To prepare membranes for radioligand binding assays, cells were initially washed with phosphate-buffered saline (PBS), pH 7.4, then harvested by scraping in a hypotonic lysis buffer containing 5 mm Tris-HCl (pH 7.4), 2 mm EDTA, 2 μg ml−1 leupeptin, 2 μg ml−1 aprotinin and 1 mm phenylmethylsulphonic fluoride (PMSF). Lysates were then homogenized in the same buffer, using a glass-glass homogenizer. The homogenate was centrifuged at 43 000 g for 20 min at 4°C. The pellet was resuspended in the same buffer and recentrifuged at 43 000 g for 20 min. The final pellet was resuspended in the same buffer and processed immediately for binding assay.
Assays for binding of [3H]CGS21680 (specific activity, 30.0 Ci mmol−1) were performed in duplicate in glass tubes containing 25 mm Tris-HCl (pH 7.5), 10 mm MgCl2, 100 mm NaCl, 2 mm EDTA and 0.2 units ml−1 adenosine deaminase. Non-specific binding of CGS21680 was defined in the presence of 100 μm 5′-N-ethylcarboxyamidoadenosine (NECA). For saturation studies, membrane fractions (protein amount, 0.08–0.1 mg) were incubated in a total volume of 0.25 ml at 25°C for 2 h with nine different concentrations of [3H]CGS21680, ranging from 0.25 to 64 nm. The binding assays were terminated by vacuum filtration on glass fibre filters (No. 34 glass; Schleicher & Schuell, Keene, NH, USA), followed by washing with 2 × 5 ml of an ice-cold buffer containing 10 mm Tris (pH 7.4). Filters were placed in scintillation vials, and bound radioactivity was determined using liquid scintillation counting. Protein concentrations were determined by the method of Bradford (Bradford, 1974).
Reverse transcriptase-polymerase chain reaction (RT-PCR)
RT-PCR was performed using a GeneAmpli kit (Perkin-Elmer, Norwalk, CT, USA) according to the instructions provided by the manufacturer. Briefly, for reverse transcriptase reactions, 1 μg of purified total RNA was incubated in the presence of 2.5 μm oligo-dT (16mer), 1 mm deoxynucleotide triphosphates, 1 unit RNase inhibitor and 2.5 units murine leukaemia virus (MuLV) reverse transcriptase, denatured at 85°C for 5 min, allowed to proceed for 15 min at 42°C, stopped by heating to 95°C for 5 min and then kept at 5°C for 5 min. Upstream and downstream specific primers for rat A1 type and A3 type adenosine receptors were synthesized. The primers for rat A1 and A3 adenosine receptors were as follows: A1 receptor, 5′-CGG CAG CAC CCA GAC GAA GA-3′ and 5′-CCC ACC ATG CCG CCC TAC AT-3′ (the predicted length of the amplified DNA fragment should be 579 bp); and A3 receptor, 5′-CAC ATC CTG CTG AAG AAG CAA CAG-3′ and 5′-GAG CTG GCT CTT TAT CTG TCA TGG-3′ (1045 bp). DNA amplification was done in the presence of 1.5 mm MgCl2, 1 × reaction buffer and 2.5 U AmpliTaq DNA polymerase. The PCR conditions were as follows: 2 min denaturation at 94°C, followed by 35 cycles consisting of 90 s at 94°C, 1 min at 50°C for A1 receptor or at 60°C for A3 receptor, and then 90 s at 72°C. Samples were then treated at 72°C for 7 min. The RT-PCR product was analysed by electrophoresis on 1 % agarose gels with ethidium bromide staining.
Adenylate cyclase activity assay
Cells were washed twice with ice-cold phosphate-buffered saline and harvested by scraping in 400 μl of a solution containing 0.25 m sucrose, 25 mm Tris (pH 7.2), 25 mm NaCl and 5 mm MgCl2. Cells were collected by centrifugation for 5 min at 3000 g at 4°C, and sonicated at 4°C for 2 s with a micro-ultrasonic cell disrupter (Kontes, Vineland, NJ, USA) in 200 μl of solution containing 0.25 m sucrose, 10 mm sodium phosphate (pH 7.0), 1 mm EDTA, freshly added leupeptin (2 μg ml−1), aprotinin (2 μg ml−1) and dithiothreitol (1 mm). The sample was centrifuged at 30 000 g for 10 min at 4°C and the pellet was resuspended in 250 μl of 10 mm sodium phosphate (pH 7.0) containing 1 mm EDTA, freshly added leupeptin (2 μg ml−1), aprotinin (2 μg ml−1) and dithiothreitol (1 mm) as the crude membrane fraction.
Adenylate cyclase assays were performed by the method of Salomon et al. (Salomon et al. 1974) as modified by Liggett et al. (1991). Crude membrane preparations were incubated in (mm): 40 Hepes (pH 8.0), 1.6 MgCl2, 0.9 EDTA, 100 NaCl, 0.12 ATP, 0.06 GTP, 2.8 phosphoenolpyruvate, 0.1 cAMP; 50 μg ml−1 myokinase and 2 mCi [α-32P]ATP in a final volume of 100 μl for 15 min at 37°C. The reaction was terminated by placing the tubes on ice and adding 1 ml of stop solution (containing 10 × 103 c.p.m. ml−1[3H]cAMP, 0.3 mm cAMP and 0.4 mm ATP) to each tube. [32P]cAMP was separated using sequential chromatography with Dowex and alumina columns (Salomon et al. 1974). The activity of adenylate cyclase was determined both under basal conditions and with the indicated concentrations of forskolin (2 μm) or adenosine (50 μm). Results were expressed as picomoles of cAMP formed per minute per milligram protein.
Immunoblotting of Gsα protein
The effect of chronic hypoxia on the amounts of the G protein, Gsα, was determined by immunoblotting. Crude membrane preparations were boiled for 3 min in sample buffer containing 50 mm Tris (pH 6.7), 2 % w/v SDS, 2 % v/v β-mercaptoethanol and Bromophenol Blue as a marker. Samples containing 20 μg of protein were separated by SDS-polyacrylamide gel and transferred to nitrocellulose membranes (Schleicher & Schuell) using standard electrophoresis and electroblotting procedures. Prestained molecular weight markers were obtained from Sigma. To reduce non-specific binding, blots were pre-incubated for 1 h in a blocking mixture (3 % non-fat dried milk, 10 mm sodium phosphate (pH 7.2), 140 mm NaCl and 0.1 % v/v Tween 20). Membranes were then incubated with an antibody directed against the COOH− terminal sequence (385–394) of Gsα (1 : 1000 dilution; Calbiochem) overnight at 4°C. The membranes were then washed three times in a buffer containing 10 mm sodium phosphate (pH 7.2), 140 mm NaCl and 0.1 % Tween 20 at room temperature and incubated with a donkey anti-rabbit horseradish peroxidase conjugated secondary antibody (1 : 2000 dilution; Amersham) for 1 h at room temperature. The membranes were washed three times over 1 h in the same buffer. Immunolabelling was detected by enhanced chemiluminescence (ECL; Amersham). Immunoreactivity was quantified using densitometric analysis with an ImagePro digital analysis system (Media Cybernetics, Silver Spring, MD, USA). Gsα immunoreactivity was linear over a 10-fold range of protein concentrations.
Protein kinase A activity assays
PKA enzyme activity was measured as previously described (Beitner-Johnson et al. 1998). Crude cellular extracts were prepared in a solution containing 0.25 m sucrose, 10 mm sodium phosphate (pH 7.0), 1 mm EDTA, freshly added leupeptin (2 μg ml−1), aprotinin (2 μg ml−1) and dithiothreitol (1 mm). For measurement of PKA activity, 2–10 μg of protein from crude cellular extracts was incubated for 6 min at 37°C in a final volume of 50 μl which included (mm): 25 Tris (pH 7.4), 1 MgCl2, 0.1 EDTA, 0.1 EGTA, 2.5 NaF, 5 dithiothreitol, 75 kemptide, 50 ATP (containing 0.1 μCi γ[32P]ATP per assay), 0.005 % v/v Nonidet P-40 (Boehringer-Mannheim) and either 10 μm 8-bromo-cAMP or 10 μm PKI6–22. Reactions were initiated by the addition of ATP and terminated by spotting 40 μl aliquots on 1.5 cm × 1.5 cm squares of Whatman p81 phosphocellulose papers followed by immersion in 75 mm H3PO4. Filter papers were washed twice for 5 min in 75 mm H3PO4, followed by one 5 min wash in water and one 5 min wash in 100 % ethanol. Filters were then allowed to air dry and were subjected to liquid scintillation counting. PKA-specific activity was calculated as the difference in 8-bromo-cAMP-stimulated phosphorylation of kemptide and phosphorylation found in the presence of PKI6–22, a specific inhibitor of PKA (Glass et al. 1989).
Data analysis
The results were expressed as the mean ±s.e.m. (n, number of observations). Analysis of variance was used for evaluating the significance of the obtained data. Statistical significance was accepted at the conventional P < 0.05 level by Student's two-tail t test evaluation.
Materials
Adenosine, 8-bromo-cAMP, PKI6–22 and NECA were purchased from Sigma.
RESULTS
Effect of chronic hypoxia on adenosine-induced inhibition of voltage-dependent Ca2+ current in PC12 cells
Whole cell patch-clamp analysis was undertaken to examine the effect of long-term hypoxia on adenosine-induced inhibition of ICa in PC12 cells. The upper panel in Fig. 1A shows representative traces illustrating the ICa response to adenosine (10 μm) in cells which were incubated either under normoxia (21 % O2, left panel) or hypoxia (10 % O2) for 24 and 48 h (middle and right panels, respectively). The inhibition of ICa by adenosine was reduced when the cells had been pre-exposed to chronic hypoxia. The lower panel in Fig. 1A shows the mean data from these experiments. The adenosine-induced inhibition of ICa was significantly smaller in the cells exposed to 10 % O2 for 48 h (P < 0.05). The mean membrane capacitance did not change significantly during chronic hypoxia (normoxia: 9.3 ± 0.5 pF (n = 6); 24 h hypoxia: 9.5 ± 1.0 pF (n = 6); 48 h hypoxia: 9.9 ± 0.7 pF (n = 6)). In addition, analysis of the current density of ICa revealed that there were no significant differences among the three groups (normoxia: 13.7 ± 3.0 pA pF−1 (n = 6); 24 h hypoxia: 12.5 ± 0.6 pA pF−1 (n = 6); 48 h hypoxia: 12.4 ± 1.3 pA pF−1 (n = 6)). Figure 1B shows the mean and s.e.m. of peak current- voltage relationships of ICa before and during the application of adenosine (10 μm) (n = 5). In normoxic cells, adenosine reduced peak ICa over a voltage range that leads to maximal activation of ICa (left panel). This inhibitory effect of adenosine was markedly smaller in the cells exposed to 48 h hypoxia. Note that adenosine did not induce a shift in the current-voltage relationship in either normoxic or hypoxic cells.
Figure 1. Attenuation of adenosine-induced inhibition of voltage-dependent Ca2+ current by long-term hypoxia in PC12 cells.
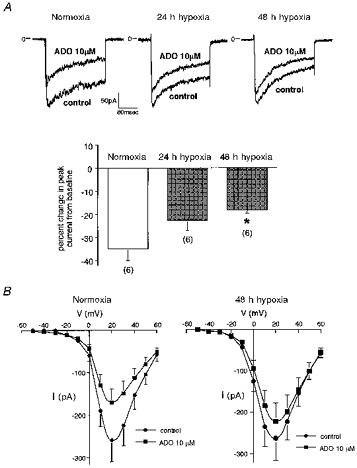
A (upper panel): traces showing the effect of adenosine (ADO) on ICa. ICa was measured every 30 s by 160 ms test pulses from a Vh of −80 to +20 mV. Peak current amplitude was measured for evaluation. The charge carrier was 20 mm Ba2+. In normoxic controls, adenosine (10 μm) elicited a decrease in the amplitude of ICa (left). This effect of adenosine was smaller when the cells had been pretreated with hypoxia (10 % O2) for 24 h (middle) or 48 h (right). A (lower panel): the effect of adenosine on ICa was significantly attenuated when the cells had been exposed to 10 % hypoxia for 48 h (*P < 0.05). The response to adenosine was evaluated as the percentage inhibition from baseline inward current. The numbers in parentheses indicate the number of cells examined. Means + s.e.m. are shown. B, effect of adenosine (ADO) on the peak current-voltage relationship of ICa. Means + s.e.m. are shown (n = 5 for each group). ICa was measured with 100 ms long test pulses from a Vh of −80 mV to test potentials ranging from −50 to +60 mV (10 mV increments). Peak currents were measured before and after steady-state inhibition by the application of adenosine (10 μm). Adenosine decreased ICa at the voltage range examined.
Effect of chronic hypoxia on the adenosine-mediated inhibition of intracellular free Ca2+ in PC12 cells
We performed Ca2+ imaging experiments to determine if long-term hypoxia modulates the effect of adenosine on elevation of [Ca2+]i induced by anoxia. A representative recording of [Ca2+]i in response to anoxia (100 % N2 and 1 mm sodium dithionate) in the absence and presence of adenosine (50 μm) in a cell that had been pre-exposed to normoxia is shown in Fig. 2A. Anoxia produced a rapid rise in [Ca2+]i which reached a peak within 3 min and returned to baseline following reoxygenation. The important finding was that the anoxia-induced enhancement of [Ca2+]i was reduced markedly by 50 μm adenosine. A separate set of experiments showed that adenosine (10 μm) attenuated the anoxia-induced elevation in [Ca2+]i, but the change was not significant (data not shown). Figure 2B shows the results from an identical experiment except that the cell had been pre-exposed to hypoxia (10 % O2) for 48 h prior to the experiment. Note that the preconditioning hypoxic stimulus blunted the inhibitory effect of adenosine on [Ca2+]i in response to anoxia. Figure 2C and D shows the overall results from the Ca2+ imaging studies. The increase in [Ca2+]i during anoxia was greater in cells that had been pre-exposed to 48 h of hypoxia (n = 14) than normoxic controls (n = 10) (Fig. 2C). On the other hand, the inhibitory effect of adenosine on the enhancement of [Ca2+]i during anoxia was significantly smaller in the cells exposed to hypoxia for 48 h (n = 14) than in normoxic cells (n = 10) (Fig. 2D). These results indicate that chronic hypoxia attenuates the effect of adenosine on anoxia-induced enhancement of intracellular free Ca2+ in PC12 cells.
Figure 2. Effect of chronic hypoxia on adenosine-mediated modulation of anoxia-induced increase of intracellular free Ca2+ in PC12 cells.
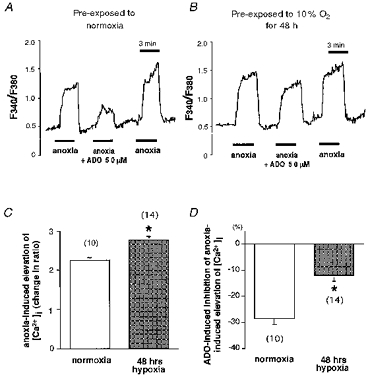
A, a representative measurement of cytosolic free Ca2+ concentration ([Ca2+]i) in response to anoxia in a cell which had been exposed to normoxia for 48 h. [Ca2+]i was estimated by using the fluorescent Ca2+ indicator fura-2. The ratio of F340/F380 was used to reflect [Ca2+]i. Anoxia was induced by saturating the medium with 100 % N2 with 1 mm sodium dithionate. Anoxia induced the elevation of [Ca2+]i which returned to the baseline level upon reoxygenation. The response to anoxia was reduced in the presence of 50 μm adenosine (ADO). B, a recording of [Ca2+]i in a cell that had been exposed to 10 % O2 for 48 h prior to the experiment. Anoxia could still induce an increase in [Ca2+]i. However, the inhibitory effect of 50 μm adenosine (ADO) on the anoxia-induced increase in [Ca2+]i was dramatically reduced. C and D, the overall data from Ca2+ imaging studies. The numbers in parentheses indicate the number of cells examined. The anoxia-induced increase in [Ca2+]i, evaluated as the relative change in the F340/F380 ratio, was significantly augmented when the cells had been pre-exposed to 10 % O2 for 48 h (C, *P < 0.05). On the other hand, the inhibitory effect of adenosine (ADO) on the anoxia-induced elevation of [Ca2+]i was significantly smaller in the cells exposed to hypoxia for 48 h (D, *P < 0.05). Means + s.e.m. are shown.
Effect of chronic hypoxia on adenosine A2A receptor gene expression and receptor binding in PC12 cells
Northern blot analysis was used to investigate the effect of hypoxia on adenosine A2A receptor mRNAs. PC12 cells were initially grown in normoxic conditions. When the cells reached 70 % confluency, they were placed in an incubator set to maintain an hypoxic environment (10 % O2) for 3, 6, 12, 24 or 48 h. Figure 3A shows the profile of adenosine A2A receptor mRNA expression during increasing duration of hypoxia in PC12 cells. It can be seen that A2A receptor mRNA gradually increased with hypoxia and peaked at 24 h. The mean results from these experiments are provided in Fig. 3B. A significant increase in A2A receptor mRNA was measured at 24 and 48 h (Fig. 3B). Thus, chronic hypoxia upregulates the expression of adenosine A2A receptor mRNA in PC12 cells.
Figure 3. Upregulation of adenosine A2A receptor mRNA expression by chronic hypoxia in PC12 cells.
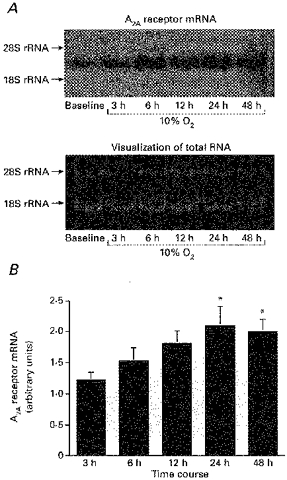
A, profiles of adenosine A2A receptor mRNA during exposure to hypoxia. Cells were harvested at the indicated time points after exposure to 10 % O2 and RNAs were analysed by Northern blots. The lower panel shows the SYBR Green I visualization to show that RNAs were equally loaded in each lane. B, the expression of A2A receptor gradually increased with hypoxia, and peaked at 24 h. The relative change from normoxic control in signal density was significant at 24 and 48 h (both *P < 0.05). Each bar shows the mean + s.e.m. of 6 samples.
In a previous study, we showed that PC12 cells express A2A and A2B receptors, but not A1 or A3 receptors (Kobayashi et al. 1998). To test the possibility that chronic hypoxia induces de novo expression of A1 and A3 receptors, which are generally coupled to Gi protein, RT-PCR analysis was performed to test for the presence of A1 and A3 receptor mRNAs after exposure to chronic hypoxia (Fig. 4). Neither the expression of A1 receptor nor that of A3 receptor was detected in the PC12 cells either before or after exposure to chronic hypoxia. The positive controls from rat brain (for A1 receptor) and rat lung (for A3 receptor) each showed a single band when the samples were treated with the same RT-PCR procedure as the PC12 cells. In each experiment, false amplification of the genomic DNA was ruled out by negative controls in which reverse transcriptase was not added to the reaction mixture. Therefore chronic mild hypoxia does not induce expression of either the A1 or A3 adenosine receptor in PC12 cells.
Figure 4. Effect of chronic hypoxia on expression of A1 and A3 receptors in PC12 cells.
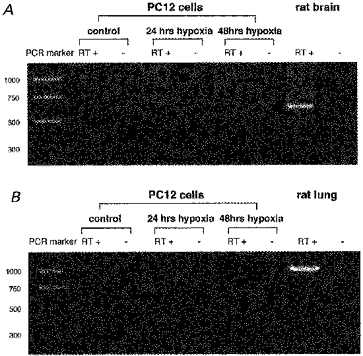
Analysis was performed to test for the presence of A1 and A3 receptor mRNAs after exposure to hypoxia. Ethidium bromide visualizations of PCR products obtained by RT-PCR of total RNA from PC12 cells for adenosine A1 receptor (A) and A3 receptor (B) are shown. The left lane corresponds to PCR (Promega) or pGEM marker (Promega) - the size of the DNA fragments (bp) for each marker is indicated on the left. The positive controls from rat brain (for A1 receptor) and rat lung (for A3 receptor) each showed a single band (the product predicted size 579 and 1045 bp, respectively). False amplification of the genomic DNA was ruled out by performing RT-PCR without reverse transcriptase as negative controls (shown as RT—).
To determine the effect of chronic hypoxia on A2A receptor function, we performed receptor binding studies. We measured ligand binding to the A2A receptor in control cells and in cells which had been exposed to prolonged hypoxia. Plasma membranes from PC12 cells incubated under normoxia or under 10 % O2 for 48 h were harvested, and receptor binding assays performed. We found that the binding of the A2A-selective adenosine receptor ligand [3H]CGS21680 was saturable and that the maximum number of binding sites (Bmax) for CGS21680 was increased during 48 h of 10 % O2 (P < 0.05). This corresponded to the increased A2A receptor mRNA level measured following chronic hypoxia. There was no significant change in the equilibrium dissociation constant (Kd). These results are summarized in Table 1. The values for Kd and Bmax were calculated from Scatchard analyses of binding data from three independent experiments.
Table 1.
Effect of chronic hypoxia on [3H]CGS21680 binding to membranes from rat PC12 cells
| Bmax (fmol (mg protein)−1) | Kd (nm) | |
|---|---|---|
| Normoxia | 110.5 ± 11.2 | 9.0 ± 4.0 |
| 48 h 10% hypoxia | 165.5 ± 10.7* | 9.8 ± 1.4 |
Bmax, maximum number of binding sites. Kd, equilibrium dissociation constant. Values shown are means ±s.e.m.
P < 0.05.
Effect of chronic hypoxia on Gsα protein levels in PC12 cells
We investigated the effect of chronic hypoxia on Gsα protein levels in PC12 cells exposed to 10 % O2 for 6, 12, 24 or 48 h. Figure 5 shows that chronic hypoxia had no effect on Gsα levels in PC12 cells, as measured by immunoblot analysis.
Figure 5. Effect of chronic hypoxia on Gsα protein levels in PC12 cells.

Profiles of Gsα-subunit protein during exposure to hypoxia. Cells were harvested at the indicated time points after exposure to 10 % O2 and proteins were analysed by immunoblot analysis. A representative immunoblot is shown from a total of n = 4 dishes in each group performed in a single experiment.
Effect of chronic hypoxia on adenylate cyclase activity in PC12 cells
We next examined the effect of hypoxia on the next step in the A2 adenosine receptor post-receptor signalling cascade, namely, adenylate cyclase enzyme activity. Basal, forskolin-stimulated (2 μm), and adenosine-stimulated (50 μm) adenylate cyclase activities were all unchanged in response to exposure to hypoxia (10 % O2) for either 24 or 48 h (Fig. 6).
Figure 6. Effect of chronic hypoxia on adenylate cyclase activity in PC12 cells.
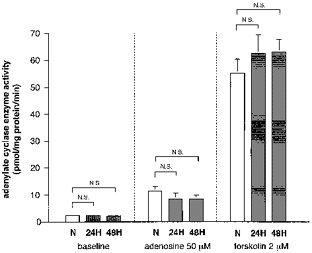
Cells were harvested at 24 and 48 h after exposure to 10 % O2 and the production of cAMP stimulated by forskolin (2 μm) or adenosine (50 μm) was measured. Basal production of cAMP was unchanged in response to 10 % O2 for either 24 or 48 h (left). Hypoxic exposure did not alter the activity of adenylate cyclase stimulated by forskolin (right). In addition, there was no significant change in the production of cAMP stimulated by adenosine in response to chronic hypoxia (middle). Each bar shows the mean + s.e.m. of 6 samples from 2 separate experiments.
Effect of chronic hypoxia on protein kinase A activity in PC12 cells
Regulation of PKA is the primary signalling mechanism by which adenosine receptors mediate their intracellular effects. For this reason, we next performed experiments to determine if the diminished effectiveness of adenosine in inhibition of [Ca2+]i in cells pre-exposed to hypoxia is due to decreased PKA activity. Basal phosphorylation of kemptide in the presence of PKI6–22, a specific inhibitor of PKA, was unchanged in response to 10 % O2 for either 24 or 48 h (Fig. 7). In contrast, maximal phosphorylation of kemptide stimulated by 8-bromo-cAMP (10 μm) was significantly reduced during exposure to 10 % O2 for 24 and 48 h. We found that PKA specific activity, calculated as the difference in 8-bromo-cAMP-stimulated phosphorylation of kemptide and phosphorylation found in the presence of PKI6–22, was significantly reduced during chronic hypoxia (24 h hypoxia: −27.7 ± 6.8 % from normoxic controls, P < 0.05; 48 h hypoxia: −32.6 ± 2.8 % from controls, P < 0.01). This finding is consistent with the reduced effectiveness of A2A receptor activation in inhibiting both ICa and [Ca2+]i. We found that the reduced PKA activity was not due to hypoxia-induced reduction in cell viability as measured by Trypan Blue exclusion as described previously (Beitner-Johnson et al. 1998) (48 h normoxia, 97.7 ± 3.2 %; 48 h hypoxia, 97.4 ± 2.9 %; n = 3 in each group). We conclude that chronic hypoxia (10 % O2) downregulates PKA activity in PC12 cells.
Figure 7. Chronic hypoxia downregulates protein kinase A enzyme activity in PC12 cells.
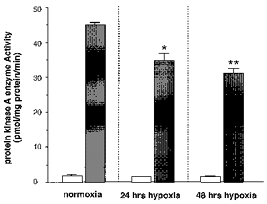
The cells were incubated under normoxia or under 10 % O2 for 24 and 48 h. For PKA assays, kemptide (75 mm) was used as substrate for PKA-activated phosphorylation. Basal non-specific phosphorylation of kemptide in the presence of PKI6–22, a specific inhibitor of PKA, was unchanged in response to 10 % O2 for either 24 or 48 h (□). In contrast, maximal phosphorylation of kemptide stimulated by 8-bromo-cAMP (8-Br-cAMP; 10 μm) was significantly reduced during pre-exposure to 10 % O2 for 24 and 48 h (▪; *P < 0.05, **P < 0.01). We found that PKA specific activity, calculated as the difference in 8-Br-cAMP-stimulated phosphorylation of kemptide and phosphorylation found in the presence of PKI6–22, was significantly reduced when the cells had been exposed to hypoxia for 24 and 48 h. Data are expressed as the mean + s.e.m. (n = 4 in each group).
Effect of chronic hypoxia on 8-bromo-cAMP-induced modulation of voltage-dependent Ca2+ current in PC12 cells
We next conducted whole cell patch-clamp experiments to determine if reduced PKA activity is the primary mechanism for the attenuated effect of adenosine on ICa during chronic hypoxia. We examined the effect of 8-bromo-cAMP (8-Br-cAMP) on ICa in PC12 cells which were incubated either under normoxia or 10 % O2 for 48 h. 8-Br-cAMP is a membrane-permeable derivative of cAMP and directly activates PKA. Figure 8A shows representative traces illustrating the ICa response to 8-Br-cAMP in PC12 cells. Extracellular application of 8-Br-cAMP (1 mm) induced a decrease in the peak amplitude of ICa in normoxic cells (left panel). Lower doses of 8-Br-cAMP did not induce a significant decrease in peak ICa (data not shown). The inhibition of ICa induced by 1 mm 8-Br-cAMP was blunted when the cells had been pre-exposed to chronic hypoxia (right panel). Figure 8B shows the mean data from these experiments. The 8-Br-cAMP-induced inhibition of ICa was significantly smaller in the cells exposed to 10 % O2 for 48 h (P < 0.05).
Figure 8. Effect of chronic hypoxia on 8-bromo-cAMP-induced modulation of Ca2+ current in PC12 cells.
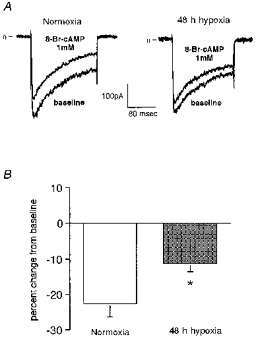
A, typical tracings showing the effect of 8-bromo-cAMP (8-Br-cAMP) on ICa. Voltage protocols were the same as those described in the legend to Fig. 1. ICa was recorded before and during extracellular application of 8-Br-cAMP (1 mm) in cells which were incubated either under normoxia or 10 % O2 for 48 h. The response to 8-Br-cAMP was evaluated after steady-state inhibition was achieved as the percentage inhibition from baseline inward current. In normoxic controls, the application of 8-Br-cAMP (1 mm) elicited a decrease in the amplitude of ICa (left). This effect of 8-Br-cAMP was smaller when the cells had been pretreated with mild hypoxia for 48 h (right). B, the effect of 8-Br-cAMP on ICa was significantly attenuated when the cells had been exposed to 10 % hypoxia for 48 h (*P < 0.05). The means + s.e.m. are shown (n = 4 in each group).
discussion
We reported previously that extracellularly applied adenosine inhibits ICa in PC12 cells (Kobayashi et al. 1998). Also we have reported that the elevation of [Ca2+]i induced by acute hypoxia is attenuated by adenosine through the inhibition of ICa. This is potentially an important mechanism because an increase in cytosolic Ca2+ during hypoxia is required for the regulation of certain genes (c-fos, Jun-B and tyrosine hydroxylase) and the release of neurotransmitter in PC12 cells during hypoxia (Czyzyk-Krzeska et al. 1994; Zhu et al. 1996; Raymond & Millhorn, 1997). In the current study, we have shown that the hypoxia-induced increase in [Ca2+]i in PC12 cells was significantly augmented when the cells were exposed to prolonged hypoxia. This result supports the idea that the chemosensitivity of the O2 sensing cells is enhanced during chronic hypoxia. The most important finding in the present study is that the exposure to chronic hypoxia attenuates the adenosine-induced modulation of ICa and [Ca2+]i in PC12 cells. Experiments were performed to attempt to identify the mechanism for downregulation of the effect of adenosine by chronic hypoxia.
It has been shown that the A2A and A2B type adenosine receptors are the only subtypes expressed in PC12 cells (Williams et al. 1987; Hide et al. 1992). PC12 cells do not express either the A1 or A3 subtype of adenosine receptors (Kobayashi et al. 1998). Signal transduction at both adenosine A2A and A2B subtypes is mediated via Gs, which stimulates adenylate cyclase to generate cAMP and subsequent activation of PKA (Fredholm, 1995; Olah & Stiles, 1995). In addition, our previous report showed that the action of adenosine on ICa is mediated via the A2A receptor and the activation of PKA (Kobayashi et al. 1998). In order to characterize the cellular mechanisms for the reduced actions of adenosine on ICa and [Ca2+]i, we examined the effect of chronic hypoxia on the adenosine A2A receptor-induced activation of signal transduction pathways.
We found that chronic hypoxia increased mRNA expression and ligand binding of A2A type adenosine receptor. This finding excluded the possibility that downregulation of A2A receptors was the mechanism for this effect. We also examined the effect of chronic mild hypoxia on the post-receptor elements involved in adenosine A2A receptor signalling. Prolonged hypoxia had no effect on Gsα immunoreactivity level, adenylate cyclase activity or the expression of A1 and A3 receptors. The reduced actions of adenosine on ICa and [Ca2+]i were not ascribed to the decreased numbers of functional Ca2+channels, since there were no significant changes in current densities for Ca2+ during 48 h hypoxia.
We next performed experiments to test the effect of chronic hypoxia on PKA activity, the intracellular mediator of A2A receptor function. Our laboratory reported recently that exposure of PC12 cells to 5 % O2 for at least 6 h downregulates both PKA enzyme activity and the level of both the regulatory and catalytic subunits of PKA (Beitner-Johnson et al. 1998). The current study revealed that PKA activity was similarly downregulated after chronic exposure to a milder level of hypoxia (10 %) and that downregulation of PKA occurred concomitantly with the inability of adenosine to inhibit ICa and [Ca2+]i. Since there was no difference in the cAMP production (e.g. adenylate cyclase activity) in response to adenosine before and after chronic hypoxic exposure, it is likely that downregulation of PKA activity is the primary mechanism by which ICa is reduced following chronic hypoxia. Evidence that this may be the case comes from our previous study (Kobayashi et al. 1998). We have found that the effect of activation of the A2A receptor on ICa was reduced by application of the PKA inhibitor peptide (PKI6–22) and in PKA-deficient PC12 cells. Furthermore, in the current study, we revealed that the inhibition of ICa induced by 8-Br-cAMP was attenuated when cells had been pre-exposed to chronic hypoxia. 8-Br-cAMP is a derivative of cAMP and directly activates PKA. Therefore, the action of 8-Br-cAMP is downstream of adenosine receptors, Gs protein or adenylate cyclase. 8-Br-cAMP is widely used for patch-clamp studies to investigate the role of PKA in modulating channel activities (Wickman & Clapman, 1995). It has been shown that 8-Br-cAMP decreases the activities of Ca2+ channels in vascular smooth muscle cells (Ishikawa et al. 1993; Xiong et al. 1994). Therefore, our findings strongly indicate that the reduced activity of PKA is the primary mechanism by which the actions of adenosine on ICa and [Ca2+]i are attenuated during chronic hypoxia in PC12 cells.
In cells exposed to long-term hypoxia, a series of defence and rescue mechanisms ensure the promotion of cell survival. The downregulation of the PKA signal transduction pathway has been hypothesized to be one of the defence mechanisms for hypoxia (Hochachka et al. 1996). The hypoxia-induced reduction in PKA activity suggests that a number of the substrate proteins for PKA are phosphorylated to a lesser degree during hypoxia. To our knowledge, the present study represents the first evidence that the downregulation of PKA activity is coupled to altered cellular function during hypoxia. In a previous study we showed that adenosine has an inhibitory effect on membrane excitability during hypoxia in PC12 cells (Kobayashi et al. 1998). The reduced actions of adenosine on Ca2+ current and [Ca2+]i following chronic hypoxia might function to enhance membrane excitability in PC12 cells during chronic hypoxia.
In contrast to PKA, the activity of adenylate cyclase was not altered during chronic hypoxia in PC12 cells. We found that exposure to 10 % O2 for 48 h did not alter the production of cAMP stimulated by forskolin, which directly activates adenylate cyclase in a receptor-independent manner. We also found that prolonged hypoxia did not alter adenylate cyclase activity stimulated by exogenously applied adenosine at the A2 receptor. We further found that the level of Gsα, the G protein that is coupled to the A2A receptor, did not change during long-term hypoxia. These findings indicate that unlike PKA, which was downregulated by chronic hypoxia, two more proximal effectors of adenosine A2 receptor signalling, Gsα and adenylate cyclase, were not altered in response to hypoxia.
We also found that adenosine A2A receptor mRNA expression was actually increased by hypoxia. We showed that mRNA message of A2A receptor increases during chronic hypoxia. This is the first known report that the expression of the A2A receptor gene is regulated by hypoxia. We also found that chronic hypoxia enhances A2A receptor protein numbers in PC12 cells, since ligand-binding studies revealed that the number of binding sites, but not the Kd, were increased during 48 h exposure to hypoxia. This finding suggests that the reduced effectiveness in adenosine-mediated inhibition of [Ca2+]i in cells exposed to chronic hypoxia was not due to downregulation of A2A receptor. We speculate that upregulation of A2A receptor gene and protein expression may be involved in a compensatory response to the reduced signalling function of adenosine on the effector proteins, including PKA and Ca2+ channel.
It should be noted that, although the number of A2A receptors was increased in PC12 cells during chronic hypoxia, adenosine-stimulated adenylate cyclase activity was not enhanced during this period. Our RT-PCR experiments revealed that chronic hypoxic exposure does not lead to expression of either the A1 or A3 receptors, which are normally not present in PC12 cells (Kobayashi et al. 1998). This is important because both A1 and A3 receptors are generally found to be coupled to Gi protein (Fredholm, 1995; Olah & Stiles, 1995). Thus, activation of these receptors could have resulted in the reduction in cAMP production by adenylate cyclase. The fact that adenosine-stimulated adenylate cyclase activity was not increased by hypoxia may suggest that coupling between the A2A receptor and adenylate cyclase is impaired during hypoxia. We showed that hypoxia had no effect on the amount of Gs protein α-subunit in PC12 cells. However, functional downregulation of Gsα protein activity during chronic hypoxia has been reported in rat hearts (Kacimi et al. 1995). This observation raises the possibility that chronic hypoxia may regulate A2A receptor coupling to Gs, potentially via receptor desensitization (Ramkumar et al. 1991; Chern et al. 1993).
Our previous and current papers clearly demonstrate that adenosine has an inhibitory effect on the excitability of PC12 cells (Kobayashi et al. 1998). These studies were performed for the express purpose of identifying basic mechanisms by which the A2A receptor modulates cellular excitability during hypoxia. Therefore, extreme care should be exercised in extrapolating the results from PC12 to other oxygen-sensitive tissues such as carotid body type I cells. There have been several reports which suggest that adenosine might actually stimulate carotid body activity in animals (McQueen & Ribeiro, 1986; Runold et al. 1990) and in humans (Watt et al. 1987). Several factors must be considered in explaining this potential discrepancy between the carotid bodies and PC12 cells. First, the action of adenosine in carotid bodies may vary among species; its action is still controversial in rats (Monteiro et al. 1987; McQueen, 1993; e Silva & Lewis, 1995). Most of the carotid body studies were performed in an in vivo model or in isolated whole carotid body preparations. These preparations include cells other than type I cells (vascular structures, afferent nerve terminals of carotid sinus nerves and type II cells, etc.) which may indirectly affect the adenosine-induced changes in carotid sinus nerve activities. Adenosine may increase the carotid sinus nerve activities through its action on vascular smooth muscles (improved perfusion to carotid bodies) or adenosine may have stimulatory effects primarily through activation of post-synaptic receptors on afferent nerve terminals. In addition, in the in vivo preparations it is difficult to control the concentration of adenosine which reaches type I cells and the action of adenosine may be concentration dependent. Therefore, it is likely that the direct action of adenosine on the excitability of type I cells remains to be fully uncovered. Further studies are required to characterize the cellular and molecular mechanisms for adenosine-induced modulation of hypoxic chemosensitivities in carotid body type I cells.
In summary, we found that the effect of A2A receptor activation on inhibition of ICa and [Ca2+]i is significantly attenuated in PC12 cells exposed to mild hypoxia for 48 h. We showed that the attenuated response was due to the downregulation of PKA activity and not due to reductions in Gsα, adenylate cyclase or A2A receptor level. We propose that the functional uncoupling of the A2A receptor with the Ca2+ channel during chronic hypoxia provides a mechanism to enhance membrane excitability and therefore the cellular response to hypoxia. These are novel findings which provide important insights concerning the molecular and cellular basis of O2 chemosensitivity in PC12 cells.
Acknowledgments
This study was supported by National Institutes of Health grants R37HL 33831 (D. E. M.) and HL 559945 (D. E. M.). D. B.- J. is the recipient of a Parker B. Francis Foundation fellowship. We thank Drs Stephen Liggett and Stuart Greene for providing technical assistance and reagents to perform adenylate cyclase assays and adenosine receptor binding studies.
References
- Archer SL, Hampl V, Nelson DP, Sidney E, Peterson DA, Weir EK. Dithionate increases radical formation and decreases vasoconstriction in the lung: evidence that dithionate does not mimic alveolar hypoxia. Circulation Research. 1995;77:174–181. doi: 10.1161/01.res.77.1.174. [DOI] [PubMed] [Google Scholar]
- Beitner-Johnson D, Leibold J, Millhorn DE. Hypoxia regulates the cAMP- and Ca2+/calmodulin signaling systems in PC12 cells. Biochemical and Biophysical Research Communications. 1998;241:61–66. doi: 10.1006/bbrc.1997.7907. [DOI] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantification of microgram quantities of protein utilizing the principle of protein-dye binding. Analytical Biochemistry. 1974;72:248–254. doi: 10.1006/abio.1976.9999. [DOI] [PubMed] [Google Scholar]
- Chern Y, Lai H-L, Fong JC, Liang Y. Multiple mechanisms for desensitization of A2a adenosine receptor-mediated cAMP elevation in rat pheochromocytoma PC12 cells. Molecular Pharmacology. 1993;44:950–958. [PubMed] [Google Scholar]
- Conforti L, Millhorn DE. Selective inhibition of a slow-inactivating voltage-dependent K+ channel in rat PC12 cells by hypoxia. The Journal of Physiology. 1997;502:293–305. doi: 10.1111/j.1469-7793.1997.293bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czyzyk-Krzeska MF, Furnari BF, Lawson EE, Millhorn DE. Hypoxia increases rate of transcription and stability of tyrosine hydroxylase mRNA in pheochromocytoma (PC12) cells. Journal of Biological Chemistry. 1994;269:760–764. [PubMed] [Google Scholar]
- e Silva MJM, Lewis DL. L- and N-type Ca2+ channels in the rat carotid body chemoreceptor type I cells. The Journal of Physiology. 1995;489:689–699. doi: 10.1113/jphysiol.1995.sp021083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, Reppert SM. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Molecular Brain Research. 1992;14:186–195. doi: 10.1016/0169-328x(92)90173-9. 10.1016/0169-328X(92)90173-9. [DOI] [PubMed] [Google Scholar]
- Fredholm BB. Purinoreceptors in the nervous system. Pharmacology and Toxicology. 1995;76:228–239. doi: 10.1111/j.1600-0773.1995.tb00135.x. [DOI] [PubMed] [Google Scholar]
- Glass DB, Lundquist LJ, Katz BM, Walsh DA. Protein kinase inhibitor-(6–22)-amide peptide analogs with standard and nonstandard amino acid substitutions for phenylalanine 10. Inhibition of cAMP-dependent protein kinase. Journal of Biological Chemistry. 1989;264:14579–14584. [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiological Reviews. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proceedings of the National Academy of Sciences of the USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hanbauer IF, Karoum F, Hellstrom S, Lahiri S. Effects of hypoxia lasting up to one month on the catecholamine content in rat carotid body. Neuroscience. 1981;6:81–86. doi: 10.1016/0306-4522(81)90245-1. [DOI] [PubMed] [Google Scholar]
- Hempleman SC. Sodium and potassium current in neonatal rat carotid body cells following chronic in vivo hypoxia. Brain Research. 1995;699:42–50. doi: 10.1016/0006-8993(95)00850-p. [DOI] [PubMed] [Google Scholar]
- Hempleman SC. Increased calcium current in carotid body glomus cells following in vivo acclimatization to chronic hypoxia. Journal of Neurophysiology. 1996;76:1880–1886. doi: 10.1152/jn.1996.76.3.1880. [DOI] [PubMed] [Google Scholar]
- Hide I, Padgett WL, Jacobson KA, Daly JW. A2a adenosine receptors from rat striatum and rat pheochromocytoma PC12 cells: characterization with radioligand binding and by activation of adenylate cyclase. Molecular Pharmacology. 1992;41:352–359. [PMC free article] [PubMed] [Google Scholar]
- Hochachka PW, Buck LT, Doll CJ, Land SC. Unifying theory of hypoxia tolerance: molecular/metabolic defense and rescue mechanisms for surviving oxygen lack. Proceedings of the National Academy of Sciences of the USA. 1996;93:9493–9498. doi: 10.1073/pnas.93.18.9493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishikawa T, Hume JR, Keef KD. Regulation of Ca2+ channels by cAMP and cGMP in vascular smooth muscle cells. Circulation Research. 1993;73:1128–1137. doi: 10.1161/01.res.73.6.1128. [DOI] [PubMed] [Google Scholar]
- Jarvis MF, Schultz R, Hutchinson AJ, Do E, Sills MA, Williams M. [3H] CGS21680, an A2 adenosine receptor agonist, directly labels A2 receptors in rat brain tissue. Journal of Pharmacology and Experimental Therapeutics. 1989;251:47–55. [PubMed] [Google Scholar]
- Kacimi R, Moalic JM, Aldashev A, Vatner DE, Richalet JP, Crozatier B. Differential regulation of G protein expression in rat hearts exposed to chronic hypoxia. American Journal of Physiology. 1995;269:H1865–1873. doi: 10.1152/ajpheart.1995.269.6.H1865. [DOI] [PubMed] [Google Scholar]
- Kobayashi S, Conforti L, Pun YK, Millhorn DE. Adenosine modulates hypoxia-induced responses in rat PC12 cells via the A2A receptor. The Journal of Physiology. 1998;508:95–107. doi: 10.1111/j.1469-7793.1998.095br.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kusakabe T, Powell FL, Ellisman MH. Ultrastructure of the glomus cells in the carotid body of chronically hypoxic rats: with special reference to the similarity of amphibian glomus cells. Anatomical Records. 1993;237:220–227. doi: 10.1002/ar.1092370209. [DOI] [PubMed] [Google Scholar]
- Liggett SB, Caron MG, Lefkowitz RJ, Hnatowich M. Coupling of a mutated form of the human β2-adrenergic receptor to Gi and Gs— requirement for multiple cytoplasmic domain in the coupling process. Journal of Biological Chemistry. 1991;266:4816–4821. [PubMed] [Google Scholar]
- McQueen DS. Does adenosine stimulate rat carotid body chemoreceptors? In: Data PG, Acker H, Lahiri S, editors. Neurobiology and Cell Physiology of Chemoreception, Advances in Experimental Medicine and Biology. New York: Plenum Press; 1993. pp. 289–293. [DOI] [PubMed] [Google Scholar]
- McQueen DS, Ribeiro JA. Pharmacological characterization of the receptor involved in chemoexcitation induced by adenosine. British Journal of Pharmacology. 1986;88:615–620. doi: 10.1111/j.1476-5381.1986.tb10242.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro EC, Ribeiro JA. Ventilatory effects of adenosine mediated by carotid body chemoreceptors in the rat. Naunyn-Schmiedeberg's Archives of Pharmacology. 1987;335:143–148. doi: 10.1007/BF00177715. [DOI] [PubMed] [Google Scholar]
- Olah ME, Stiles GL. Adenosine receptor subtypes: characterization and therapeutic regulation. Annual Review of Pharmacology and Toxicology. 1995;35:581–606. doi: 10.1146/annurev.pa.35.040195.003053. [DOI] [PubMed] [Google Scholar]
- Phillis JW, Walter GA, O'Regan MH, Stair RE. Increases in cerebral cortical perfusate adenosine and inosine concentrations during hypoxia and ischemia. Journal of Cerebral Blood Flow and Metabolism. 1987;7:679–686. doi: 10.1038/jcbfm.1987.122. [DOI] [PubMed] [Google Scholar]
- Prabhakar NR. Neurotransmitters in the carotid body. In: O'Regan RG, Nolan P, McQueen DS, Paterson DJ, editors. Arterial Chemoreceptors: Cell to System, Advances in Experimental Medicine and Biology. New York: Plenum Press; 1994. pp. 679–686. [Google Scholar]
- Ramkumar V, Olah M, Jacobson KA, Stiles GL. Distinct pathways of desensitization of A1- and A2-adenosine receptors in DDT1 MF-2 cells. Molecular Pharmacology. 1991;40:639–647. [PMC free article] [PubMed] [Google Scholar]
- Raymond R, Millhorn DE. Regulation of tyrosine hydroxylase gene expression during hypoxia: role of Ca2+ and PKC. Kidney International. 1997;51:536–541. doi: 10.1038/ki.1997.74. [DOI] [PubMed] [Google Scholar]
- Runold M, Cheniack NS, Prabhakar NR. Effect of adenosine on isolated and superfused cat carotid body activity. Neuroscience Letters. 1990;113:111–114. doi: 10.1016/0304-3940(90)90504-3. [DOI] [PubMed] [Google Scholar]
- Salomon Y, Londos C, Rodbell M. A highly sensitive adenylate cyclase assay. Analytical Biochemistry. 1974;58:541–548. doi: 10.1016/0003-2697(74)90222-x. [DOI] [PubMed] [Google Scholar]
- Stea A, Jackson A, Macintyre L, Nurse CA. Long-term modulation of inward currents in O2 chemoreceptors by chronic hypoxia and cAMP in vitro. Journal of Neuroscience. 1995;15:2192–2202. doi: 10.1523/JNEUROSCI.15-03-02192.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt AH, Reid PG, Stephens MR, Routledge PA. Adenosine-induced respiratory stimulation in man depends on site of infusion. Evidence for an action on the carotid body? British Journal of Clinical Pharmacology. 1987;23:486–490. doi: 10.1111/j.1365-2125.1987.tb03081.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weil JV. Ventilatory control at high altitude. In: Cherniack NS, Widdicombe JG, editors. Handbook of Physiology, section 3, The Respiratory System, Control of Breathing. II. Bethesda, MD, USA: American Physiological Society; 1986. pp. 701–728. part 2. [Google Scholar]
- Wickman K, Clapman DE. Ion channel regulation by G proteins. Physiological Reviews. 1995;75:865–885. doi: 10.1152/physrev.1995.75.4.865. [DOI] [PubMed] [Google Scholar]
- Williams M, Abreu M, Jarvis MF, Noronha-Blob L. Characterization of adenosine receptors in the PC12 cell pheochromocytoma cell line using radioligand binding: evidence for A-2 selectivity. Journal of Neurochemistry. 1987;48:498–502. doi: 10.1111/j.1471-4159.1987.tb04120.x. [DOI] [PubMed] [Google Scholar]
- Winn HR, Rubio R, Berne RM. Brain adenosine concentration during hypoxia in rats. American Journal of Physiology. 1981;241:H235–242. doi: 10.1152/ajpheart.1981.241.2.H235. [DOI] [PubMed] [Google Scholar]
- Wyatt CN, Wright C, Bee D, Peers C. O2-sensitive K+ currents in carotid body chemoreceptor cells from normoxic and chronically hypoxic rats and their roles in hypoxic chemotransduction. Proceedings of the National Academy of Sciences of the USA. 1995;92:295–299. doi: 10.1073/pnas.92.1.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Z, Sperelakis N, Fenoglo-Preiser C. Regulation of L-type calcium channels by cyclic nucleotides and phosphorylation in smooth muscle cells from rabbit portal vein. Journal of Vascular Research. 1994;31:271–279. doi: 10.1159/000159053. [DOI] [PubMed] [Google Scholar]
- Zetterstrom T, Vernet L, Ungerstedt U, Tossman U, Jonzon B, Fredholm BB. Purine levels in the intact rat brain. Studies with an implanted perfused hollow fibre. Neuroscience Letters. 1982;29:111–115. doi: 10.1016/0304-3940(82)90338-x. [DOI] [PubMed] [Google Scholar]
- Zhu WH, Conforti L, Czyzyk-Krzeska MF, Millhorn DE. Membrane depolarization in PC12 cells during hypoxia is regulated by an O2 sensitive K+ current. American Journal of Physiology. 1996;271:C658–665. doi: 10.1152/ajpcell.1996.271.2.C658. [DOI] [PubMed] [Google Scholar]