

Abstract
The effects of diadenosine polyphosphates (APnA, where n = 4–6) were studied on beating frequency of perfused guinea-pig hearts and on muscarinic K+ current (IK(ACh)) and ATP-regulated K+ current (IK(ATP)) in atrial myocytes from guinea-pig hearts using whole-cell voltage clamp.
Bradycardia induced by APnA in perfused hearts was completely inhibited by 8-cyclopentyl- 1,3-dipropylxanthine (CPX, 20 μm), a selective antagonist at A1 adenosine receptors, and was augmented by dipyridamole (Dipy), an inhibitor of cellular adenosine (Ado) uptake.
Whereas exposure of atrial myocytes to Ado (100 μm) within about 1 s induced a significant whole-cell IK(ACh), APnA up to 1 mm applied for some tens of seconds failed to activate IK(ACh). If present for periods > 2 min, APnA caused inhibition of agonist-evoked IK(ACh) and activation of a weakly inward rectifying K+ current, which was identified as IK(ATP) by its sensitivity to glibenclamide and its current-voltage curve.
The actions of extracellular APnA on IK(ACh) and IK(ATP) were mimicked by intracellular loading of compounds via the patch clamp pipette and by intracellular loading of AMP.
The results from isolated myocytes exclude APnA acting as A1 agonists. It is suggested that myocytes can take up APnA, which are degraded to AMP. In the presence of ATP, AMP is converted to ADP, a physiological activator of ATP-regulated K+ channels, by adenylate kinase. A similar mechanism resulting in a reduction of the [GTP]/[GDP] ratio might be responsible for inhibition of IK(ACh).
In the perfused heart and other multicellular cardiac preparations the actions of APnA are mediated by Ado via A1 receptors. It is suggested that APnA in multicellular cardiac tissue are hydrolysed by an ectohydrolase to yield AMP which is converted to Ado by ectonucleotidases.
Diadenosine polyphosphates (APnA) represent a family of compounds composed of two molecules of adenosine bridged by a variable number of phosphates. These compounds are produced and released from platelets and chromaffin cells and are considered to act as mediators in various tissues including the cardiovascular system and the CNS (for recent reviews see Ogilvie et al. 1996; Kisselev et al. 1998). AP5A and AP6A have been shown to elicit constriction of different vascular smooth muscle preparations (Tepel et al. 1996). AP3A and AP4A exert relaxing effects on isolated arteries and in the coronary vasculature, which seem to be mediated by the endothelium (Busse et al. 1988; Pohl et al. 1991). In different preparations it has been demonstrated that these compounds act on different subtypes of P2 purinoceptors (Ogilvie et al. 1996). A specific APnA receptor has been suggested to exist on heart cells (Hildermann et al. 1991). More recently, negative chronotropic and inotropic actions in isolated atria and multicellular ventricular preparations of AP5A and AP6A have been described which were similar to those of adenosine (Ado) and which were completely or partially sensitive to specific A1 receptor antagonists (Hoyle et al. 1996; Rubino & Burnstock, 1996; Vahlensick et al. 1996). From these data it was concluded that APnA act as A1 receptor agonists.
Effects of APnA (where n = 4–6) were investigated in perfused hearts and analysed in isolated atrial myocytes by measurement of two K+ currents expressed in these cells, namely IK(ACh) and IK(ATP). IK(ACh) represents the prototype of a G protein-gated K+ channel (see Kurachi 1995a for review; Dascal, 1997). In supraventricular cardiac cells opening activity of IK(ACh) channels is increased by interaction of the channel, or its GIRK1/GIRK4 subunits with the βγ-subunits of a pertussis toxin-sensitive G protein (Gi/Gk) (Huang et al. 1995). Whole-cell IK(ACh) in cardiac myocytes can be activated by the muscarinic (M2) receptor, by the A1 Ado receptor (Kurachi, 1995b) and a recently discovered sphingolipid receptor (Bünemann et al. 1996a). Due to the membrane-delimited nature of this signalling pathway, measurement of IK(ACh) provides a most sensitive and fast on-line assay for activation of Gi/Gk or receptors converging on these G protein(s), such as the A1 receptor. APnA in a range of concentrations (from 10 μm to 1 mm) which are effective in multicellular cardiac preparations causing sinus bradycardia or negative inotropism in isolated atria, failed to cause activation of atrial IK(ACh) in cells which normally responded to Ado. However, in the presence of APnA for periods of time > 2 min, there was a slow inhibition of IK(ACh) which was independent of the activating ligand/receptor (A1, M2, intracellular GTP-γ-S). Inhibition of IK(ACh) was paralleled by activation of a K+ current with the current-voltage characteristics and pharmacological properties of the ATP-regulated K+ current (IK(ATP)).
Our results suggest that APnA in multicellular cardiac tissues are hydrolysed to yield Ado, which acts via A1 receptors. In isolated myocytes APnA are taken up resulting in a rise in intracellular concentrations of nucleotide diphosphates, which cause activation of IK(ATP) and inhibition of IK(ACh).
METHODS
Isolation and culture of atrial myocytes
Experiments were performed with local ethics committee approval. Guinea-pigs of either sex weighing 200–250 g were killed by cervical dislocation. The method of enzymatic isolation of atrial myocytes has been described in detail previously (e.g. Bünemann et al. 1996b). The culture medium was bicarbonate-buffered M199 (Gibco) containing gentamicin and kanamycin (each at 25 μg ml−1) (Sigma); culture medium was not supplemented with fetal calf serum (FCS). Cells were plated at a low density (several hundred cells per dish) on 36 mm culture dishes. Medium was changed 24 h after plating and then every second day. Cells were used experimentally from about 4 h after isolation up to 6 days in culture. As for the phenomena investigated, no influence of time in vitro was found, except for an increase in sensitivity to ACh reflecting recovery of M2 receptors from in vivo downregulation (Bünemann et al. 1997).
Solutions and chemicals
For the patch clamp measurements an extracellular solution of the following composition was used (mm): NaCl, 120; KCl, 20; CaCl2, 2.0; MgCl2, 1.0; Hepes/NaOH, 10.0, pH 7.4. The solution for filling the patch clamp pipettes for whole-cell voltage clamp experiments contained (mm): potassium aspartate, 110; KCl, 20; NaCl, 10, MgATP, 2.0; EGTA, 2.0; GTP, 0.01; Hepes/KOH, 10.0, pH 7.4. K+ equilibrium potential (EK) under these conditions is −50 mV. For the experiments on perfused hearts a solution of the following composition was used (mm): NaCl, 118; KCl, 4.7; CaCl2, 2.5; MgSO4, 1.65; NaHCO3, 24.88; KH2PO4, 1.18; glucose, 5.0; sodium pyruvate, 2.0. The solution was continuously gassed with 95 % O2- 5 % CO2. pH was 7.37–7.42. Standard chemicals were from Merck. EGTA, Hepes, MgATP, adenosine GTP and acetylcholine chloride were from Sigma. CPX and glibenclamide (Gli) were from RBI. APnA (n = 4–6) were purchased from Sigma. To remove ATP and other impurities (up to 10 %) from the commercially available diadenosine polyphosphates, the substances were purified by means of reversed-phase HPLC displacement chromatography. Twenty milligrams of diadenosine polyphosphate were dissolved in 1 ml eluent A (40 mm triethylammonium acetate in water, pH 6.5; carrier). The sample was injected into a HPLC system equipped with a reversed-phase column (Superspher RP-18, Merck), equilibrated with eluent A. The flow rate was 0.1 ml min−1. After sample injection the eluent was changed to 100 % B (160 mm butanol in eluent A). The fraction size was 1 ml. The effluate was detected using a UV detector at 254 nm. The resulting fractions were analysed with a gradient reversed-phase system and with matrix-assisted laser desorption/ionization mass spectrometry (MALDI-MS) to confirm the purity. Fractions with impurities larger than 0.1 % were discarded.
Current measurement
Membrane currents were measured under voltage clamp by means of patch clamp pipettes (whole-cell mode, Hamill et al. 1981). Pipettes were fabricated from borosilicate glass with a filament (Clark, Pangbourne, UK) on a vertical puller (DMZ, Munich, Germany) and were filled with the solution listed above. The DC resistance of the filled pipettes ranged from 2 to 6 MΩ. Current measurements were performed by means of a patch clamp amplifier (List LM/EPC 7). Signals were analog filtered (corner frequency of 1–3 kHz), digitally sampled at 5 kHz and stored on the hard disk of an IBM compatible computer, equipped with a hardware/software package (ISO2 by MFK, Frankfurt/Main, Germany) for voltage control (pulse and ramp generation), data acquisition, and data evaluation. Experiments were performed at ambient temperature (22–24°C). Cells were voltage clamped at a holding potential of −90 mV, i.e. negative to EK. K+ channel currents under this condition are in the inward direction. Current-voltage relations were determined by means of a ramp protocol. From the holding potential membrane potential was stepped to −120 mV, followed by a voltage ramp at 360 mV s−1 to +60 mV and a step back to −90 mV. Rapid superfusion of the cells for application and withdrawal of different solutions was performed by means of a solenoid-operated flow system that permitted switching between up to six different solutions. Half-time of exchange of solution seen by the superfused cell was less than 100 ms and was not limiting activation kinetics of IK(ACh).
Experiments on perfused hearts
Guinea-pigs were killed as described above. Hearts were cannulated through the aorta and mounted on a Langendorff apparatus (Hugo Sachs, March Hugstetten, Germany). Perfusion at 37°C was performed at constant pressure (50 mmHg). Each heart was allowed to equilibrate for 30 min. Surface ECG was recorded between two silver wires, one placed at the apex of the ventricle, the other on the right atrium. Ventricular frequency was determined by measuring the intervals between consecutive ventricular (R) signals. Bolus injections of compounds were made through an injection port in the aortic cannula, about 4 cm above the aortic valve. Continuous perfusion of compounds was performed via a cannula inserted in the main perfusion pathway at 1/10 of the basal flow of the main perfusion. For HPLC analysis perfusates were collected, immediately frozen in liquid nitrogen and stored at −80°C until use for HPLC analysis.
HPLC analysis
To 100 μl of each sample triethylammonium acetate (TEAA, final concentration 40 mm) and di(1,N6-ethenoadenosine) hexaphosphate (ɛ-AP6A) as internal standard were added. In order to desalt the samples, the mixture was injected into a C18 reversed-phase column (LiChrospher, Merck; 100 × 2.1 mm i.d.). For desalting an isocratic HPLC pump (Bai, Bensheim-Auerbach, Germany) was used at a rate of 200 μl min−1. The analytes were eluted from the reversed-phase column with 30 % acetonitrile. Speed-Vac-dried samples were dissolved in 300 μl eluent A (2 mm tetrabutylammonium hydrogensulphate (TBA) in a phosphate buffer, 10 mm K2HPO4, pH 6.8). One hundred microlitres of each sample was injected by an autosampler (AS 2000, Merck). The mobile phase was pumped at a rate of 300 μl min−1 by a gradient pump system (L6200 A, Merck). The analytes were separated by gradient elution (0–0.8 min, 100 % eluent A; 0.8–4.8 min, 0–10 % B (water-acetonitrile, 20 : 80, v/v); 4.8–16.8 min, 10–22 % eluent B; 16.8–17.4 min, 22–50 % eluent B) on a reversed-phase column (Poros R2/H, 100 × 2.1 mm i.d., PerSeptive Biosystems, Wiesbaden, Germany). The column effluate was monitored by means of a UV detector at 254 nm (L 4250 UV-VIS, Merck, Darmstadt, Germany). Data were recorded and processed using an integrator (D 2520 GPC Integrator, Merck, Darmstadt, Germany). Peak areas were used for quantitation. The content of the individual analytes were calculated by comparing their peak heights with that of the internal standard of known concentration of ɛ-AP6A.
RESULTS
Evidence that APnA-induced bradycardia in perfused hearts is mediated by Ado
To address the question of whether APnA in perfused hearts exert effects which can be explained by an A1 agonistic action, bolus injections of Ado, AP6A, and - for comparison - ACh were applied and the resulting changes in frequency of ventricular action potentials were recorded. A representative experiment is illustrated in Fig. 1. Under control conditions all three compounds caused a marked transient increase in the R-R interval (Fig. 1A). For Ado and AP6A this was completely blocked by simultaneous perfusion of the A1 selective antagonist CPX (20 μm), which neither affected the ACh-induced bradycardia (Fig. 1B) nor baseline R-R intervals. The inhibitory action of CPX was almost completely reversible within about 2 min (Fig. 1C). This result, which is qualitatively representative of four hearts studied this way, confirms previously published data on other types of multicellular cardiac preparations and is in line with the notion that cardiac actions of APnA are mediated by A1 receptors.
Figure 1. Inhibition of Ado- and AP6A-induced bradycardia by CPX in a perfused heart.
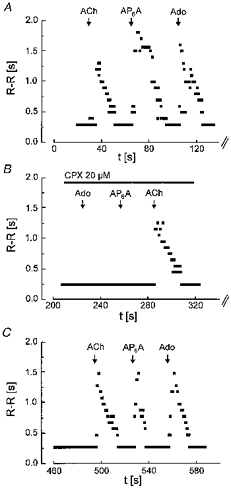
R-R intervals from surface ECG, averaged over three cycles, have been plotted against time; t = 0 corresponds to the end of the equilibration time (30 min after mounting the heart in the Langendorff apparatus). Boli of 300 μl of solutions containing ACh, AP6A and Ado (each at 100 μm) were injected at the times indicated. A, control; B, in the presence of CPX (20 μm) in the perfusing fluid; C, recovery after washout of CPX.
APnA in other tissues can be cleaved by endothelial ectoenzymes to yield AMP and adenosine-5′-(n - 1)-phosphate (Mateo et al. 1997) which can be further metabolized by the ectonucleotidase system to adenosine. It is conceivable, therefore, that the bradycardia described above is, at least partly, mediated by adenosine. If that is the case, Dipy, an inhibitor of cellular uptake of Ado, should augment the effect of a given dose of APnA.
In Fig. 2 frequency of ventricular action potentials has been plotted against time after starting perfusion with either 100 μm Ado or 100 μm AP5A. (In contrast to the experiment illustrated in Fig. 1, continuous perfusion of the compounds was used instead of bolus injection.) Under control conditions, i.e. without Dipy, both compounds exerted moderate negative chronotropic effects in the steady state, R-R intervals being increased from 0.31 to 0.39 s (AP5A) and 0.78 s (Ado). In the presence of Dipy the mean steady-state R-R interval was 2.84 s during Ado perfusion and 1.89 s during perfusion with AP5A. Mean ±s.d. values of increases in R-R intervals measured in four hearts (n = 4) were (%): 23.9 ± 7.3 (AP5A), 101 ± 29.5 (Ado), 460 ± 156 (AP5A plus Dipy), and 648 ± 186 (Ado plus Dipy). The strong enhancement of the effects of AP5A by Dipy supports the notion that the active compound in multicellular systems is Ado rather than APnA. However, some role for APnA as substrates of the Dipy-sensitive transporter cannot be completely ruled out. Further confirmation for substantial degradation of APnA during perfusion was obtained by HPLC analysis of the effluent collected from the hearts. Summarized data are illustrated in Fig. 3. The hearts were perfused with either 100 μm AP5A or 100 μm Ado. The concentration of AP5A in the effluent (Fig. 3A) was determined as ∼3 μm, i.e. 97 % of the compound was lost during passage of the heart. The concentration of Ado was determined as ∼0.5 μm. In the initial solution to which AP5A had been added no contamination by Ado was found (detection limit ∼0.05 μm). If AP5A was perfused in the presence of Dipy, there was a fourfold higher concentration of Ado (1.91 μm) in the effluent and an increase in the concentration of AP5A by 55 %. Corresponding concentrations of Ado from Ado-perfused hearts were 2.6 μm without Dipy and 30.5 μm in the presence of the uptake inhibitor (Fig. 3B). These data should be considered more qualitatively, since the compounds were injected at constant rate, whereas total flow was changing to various extents. Nevertheless they clearly show that (i) AP5A was almost completely degraded during cardiac passage, and (ii) was, at least partly, converted to Ado.
Figure 2. Enhancement of Ado- and AP5A-induced bradycardia by dipyridamole in a perfused heart.
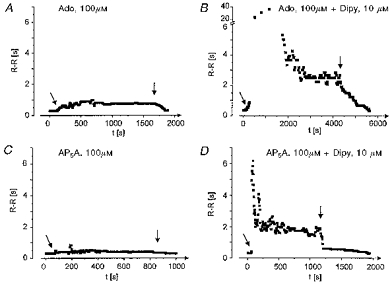
R-R intervals have been plotted against time; t = 0 only denotes the beginning of the individual plots. Responses to Ado in the absence (A) and presence (B) of 10 μm dipyridamole and to AP5A in the absence (C) and presence (D) of 10 μm dipyridamole are shown. Compounds were continuously perfused as described in Methods for periods of time indicated by the arrows. The heart was perfused with standard solution for ∼10 min between two tests.
Figure 3. HPLC analysis of fluids from perfused hearts.
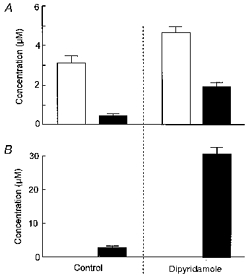
Hearts were perfused with Ado (100 μm, ▪) or AP5A (100 μm, □) as described in Methods. Samples of the effluent were collected 3 min after starting perfusion with either of the two compounds. A, concentrations of AP5A and Ado from AP5A-perfused heart with and without Dipy (20 μm). B, concentrations of Ado from Ado-perfused hearts with and without Dipy.
APnA do not activate IK(ACh) in atrial myocytes
As stated in the Introduction, in supraventricular myocytes IK(ACh) represents a major target of the signalling pathway linked to A1 receptors. Thus, if A1 receptors are involved in mediating the bradycardia induced by intact APnA, these compounds should cause activation of this current.
In line with previous findings, maximum current density available to Ado was significantly less than the steady-state current available to activation via M2 receptors (Bünemann & Pott, 1995; Takano & Noma, 1997). The mean ratio of the steady-state (non-desensitizing) current amplitudes available to Ado and ACh in those cells where both agonists have been tested at saturating concentrations, was 0.66 ± 0.19 (n = 16). Evidence has been provided that the difference between saturating responses to ACh and Ado reflects a difference in densities of these two receptors (Pott & Bünemann, 1994). Independent of time in culture, each cell tested in the context of the present and previous studies responded to Ado at concentrations ≥ 5 μm by activation of IK(ACh). As shown previously (Bünemann & Pott, 1995) this required the absence of serum supplements in the culture medium. In the presence of FCS (≥ 5 %), a complete loss of sensitivity to Ado, but not to ACh, was observed within 3 days. A representative recording of membrane current from an atrial myocyte showing the effects of a saturating concentration of Ado (100 μm) is illustrated in Fig. 4. As stated in Methods, K+ currents in the present experimental conditions are measured in the inward direction. The background I–V relation and the I–Vrelation of the Ado-activated IK(ACh) are characterized by their strongly inward rectifying properties (Fig. 4B) at membrane potentials positive to EK (−50 mV). Background current in atrial myocytes is assumed to reflect predominantly basal (agonist-independent) opening activity of ACh-regulated K+ (KACh) channels (e.g. Kaibara et al. 1991). Upon switching to the Ado-containing solution, IK(ACh) was fully activated within about 1 s. In the experiment illustrated in Fig. 4, between two challenges by Ado the cell was superfused for a period of 35 s with a solution containing 1 mm AP6A, a concentration which resulted in significant bradycardia in the perfused heart (compare Figs 1 and 2). No effect of the compound on the holding current or the I–Vrelation between −110 mV and + 60 mV (Fig. 4B) was observed, i.e. AP6A failed to cause any measurable activation of IK(ACh). This was found in all thirty cells from six different animals over a range of concentrations between 10−5 and 10−3 M, provided exposure to AP6A was limited to some tens of seconds only. The same negative result was obtained with AP5A in eight out of eight cells and with AP4A in eight out of eight cells. The concentration-response curve of Ado and the negative results obtained with APnA, using the same experimental protocols as depicted in Fig. 4A, are summarized in Fig. 4C.
Figure 4. AP6A fails to activate IK(ACh).
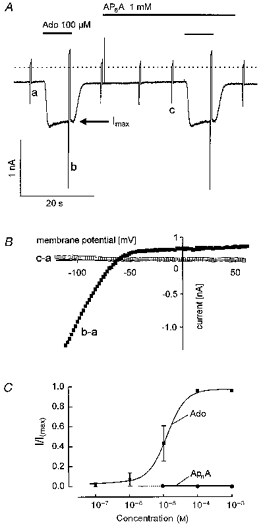
A continuous recording of membrane current at −90 mV holding potential. Ado (100 μm) and AP6A were applied as indicated. Rapid deflections represent changes in membrane current (I–V relations) caused by voltage ramps from −120 mV to +60 mV. The dotted line in this and subsequent figures denotes zero current level. B, difference I–V relations obtained by subtraction of the curves labelled in A. Curve labelled b - a represents the Ado-activated current, whereas c - a, yielding a straight line at 0 pA, indicates that AP6A had no effect on the background I–V relation. C, concentration- response curve for Ado (▪) and APnA (•). Mean values ± s.d. have been plotted against concentration. Currents have been normalized to Imax, the current evoked by either 100 μm or 1 mm Ado at the end of the fast desensitizing component, marked by the arrow in A (n = 4 to 9 for each concentration of the two compounds).
Slow activation of a weakly inward rectifying K+ current and inhibition of IK(ACh) by APnA
Exposure of atrial myocytes to AP6A (50–100 μm) for longer periods of time (as a rule > 2 min) in the majority of cells resulted in significant delayed changes in membrane currents. A representative result is illustrated in Fig. 5A. The cell was repeatedly challenged by 10 μm ACh, a saturating concentration of this agonist for activation of IK(ACh) (Bünemann et al. 1996b). Comparable results were obtained if Ado was used as stimulating agonist (see below). Superfusion with AP5A (50 μm) using the same protocol as for AP6A in Fig. 4A had no immediate effect on membrane current. However, in the continuous presence of the compound for about 4 min a slow shift of the holding current in the inward direction was recorded, concomitant with a reduction in the amplitude of the ACh-evoked current to about 10 % of its initial amplitude (Fig. 6A). Whereas IK(ACh) was characterized by strong inward rectification (Fig. 6B, cf. Fig. 4), the increased ‘background current’ had a linear dependence on voltage between −130 mV and around 0 mV, with some rectification at more positive membrane potentials (Fig. 6C). The reversal potential of this current also corresponded to EK (−50 mV), i.e. it represents a K+-selective pathway which is, however, clearly different from IK(ACh). Similar results were obtained in three out of seven cells with AP5A and two out of four cells with AP4A. In eight out of thirty-three cells no effects of AP6A at 50 or 100 μm on IK(ACh) and on background current were observed within 5 min. Due to limited stocks of the purified compounds, episodes longer than 5 min without measurable effects were not tested. For the same reason concentrations larger than 100 μm have not been used for exposures longer than 20 s. The changes in membrane currents in the presence of APnA were not, or very little, reversible after washout for up to 22 min (not shown).
Figure 5. Inhibition of IK(ACh) and activation of a weakly inward rectifying current by AP6A.
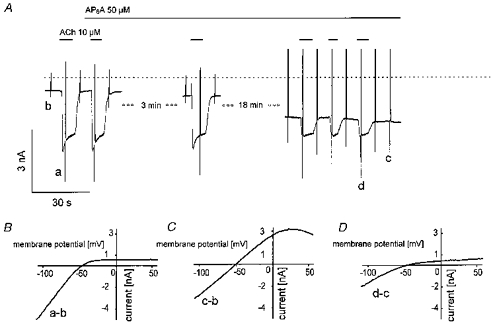
Sections of membrane current recordings at −90 mV holding potential with superimposed voltage ramps (A). Large ramp-induced peak currents at this time resolution were sometimes graphically truncated or distorted by reduction of data points in the graphics files and thus do not always represent true currents. ACh (10 μm) and AP6A (50 μm) were applied as indicated. B-D, difference I–V curves obtained by subtraction of voltage ramp-induced currents corresponding to the labelling in A (B, ACh-induced current; C, AP6A-induced current; D, fraction of ACh-induced current inhibited by AP6A).
Figure 6. Effects of AP5A are sensitive to glibenclamide.
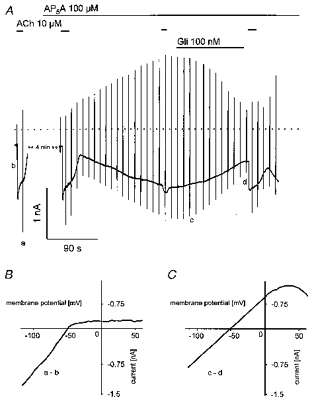
A, current recording at −90 mV holding potential with superimposed voltage ramps. ACh, AP5A and Gli were applied as indicated. B, difference I–V curve of ACh-activated current. C, difference I–V curve of the fraction of current inhibited by 100 nm Gli.
Identification of the APnA-activated K+ current as IK(ATP)
The voltage dependence of the current activated by APnA was reminiscent of the weakly inward rectifying ATP-regulated K+ current (IK(ATP)), first discovered in cardiac myocytes (Noma, 1983). To test for this hypothesis, the sulphonylurea compound Gli, a classical inhibitor of cardiac KATP channels, was used (e.g. Fosset et al. 1988). As shown in Fig. 6A, Gli at a concentration of 100 nm reversibly inhibited the AP5A-induced current by about 65 % (62.7 ± 8.4 %). Complete inhibition was observed at concentrations ≥ 1 μm (not shown). At concentrations < 100 nm development of the inhibitory action of Gli was too slow to obtain reliable values for the degree of inhibition. The order of magnitude of the estimated EC50 between 50 and 100 nm is in line with previous studies on inhibition of single identified cardiac KATP channels (e.g. 180 nm; Brady et al. 1998). The inhibited fraction of current obtained by subtraction (Fig. 6C) was characterized by the same I–Vcharacteristics as the total activated current shown in Fig. 5C. Surprisingly, inhibition of Ado-evoked IK(ACh) was also partially reversed by Gli. Identity of the Gli-sensitive background current with IK(ATP) is further supported by the result illustrated in Fig. 7. In this representative cell the metabolic uncoupler 2,4-dinitrophenol (DNP, 100 μm) was used to activate IK(ATP). The current activated by superfusion with DNP-containing solution reversed at EK (−50 mV) and was characterized by a weakly inward rectifying I–Vrelation, which was indistinguishable from that of the current activated by APnA. Moreover, the metabolic uncoupler not only caused activation of IK(ATP) but also a concomitant inhibition of IK(ACh) elicited by ACh (10 μm). In the final stage, reached after about 3 min in the presence of DNP, the cell was completely insensitive to ACh. Both effects were almost fully reversible, provided exposure to DNP was limited to less than ∼5 min. After longer exposures the cells appeared to become damaged. In summary, metabolic inhibition by means of DNP induced effects of striking similarity to those induced by APnA.
Figure 7. Activation of IK(ATP) and inhibition of IK(ACh) by DNP.
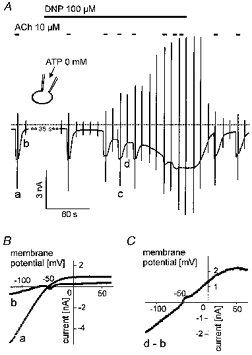
A, recording of membrane current at −90 mV holding potential with superimposed voltage ramps. ACh and DNP were applied as indicated. B, ACh-activated current (a) and background current (b) before exposure of the cell to DNP. C, current-voltage relation of DNP-activated current (difference of ramp currents labelled d and b in A). The deflections in the I–V curve result from activation of a small inward Na+ current by the positive voltage ramp, which sometimes occurred with some variability during the course of an experiment. Pipette solution not supplemented with ATP was used.
Intracellular APnA mimics the effects of extracellular APnA
Because of the similarity of the effects of APnA and DNP and because of the slow and variable time course of the response to the diadenosine compounds it was hypothesized that they might represent some intracellular site of action. Therefore in a series of experiments APnA were applied intracellularly by inclusion in the pipette solution. A representative result is illustrated in Fig. 8. AP5A used in this experiment at a concentration of 10 μm at standard [ATP]i of 2 mm resulted in an increasing inhibition of IK(ACh) activated by 100 μm Ado, followed by activation of IK(ATP). After about 4 min in the whole-cell mode the cell became completely insensitive to Ado. Before activation of IK(ATP) became discernible, the holding current transiently became less inward. This is likely to reflect inhibition of background (agonist-independent) IK(ACh), which represents the major background conductance pathway in these cells. There was a certain tendency that activation of IK(ATP) in this type of experiment occurred with a lag after the effect on IK(ACh). This was seen in none of the measurements using extracellular application of APnA or DNP, where inhibition of IK(ACh) and activation of IK(ATP) appeared to be more tightly coupled. Figure 8B-D confirms the difference in the I–Vcurves of IK(ACh) and the current activated by intracellular AP5A. As with extracellular application of the diadenosine polyphosphates, the increased ‘background’ current was characterized by a weakly inward rectifying I–V curve, whereas the voltage dependence of the reduced IK(ACh) remained unchanged. This result is qualitatively representative of eleven of fifteen cells (5/5 with 10 μm AP5A, 6/10 with 10 μm AP6A). In the remaining four cells IK(ACh) was inhibited during internal loading with APnA (50 or 100 μm) without detectable activation of the weakly inward rectifying K+ current (not shown). The results described so far clearly demonstrate that the effects of extracellular APnA are not mediated by a membrane receptor, but are likely to reflect intracellular site(s) of action.
Figure 8. Inhibition of IK(ACh) and activation of IK(ATP) by intracellular AP5A.
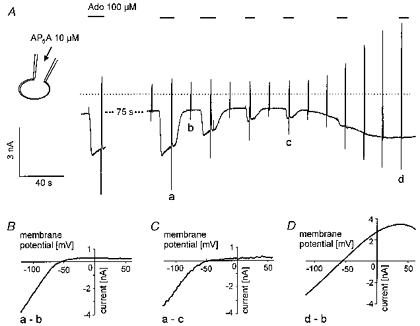
A, recording of membrane current as in previous figures. Ado (100 μm) was applied as indicated. Pipette filling solution was supplemented with 10 μm AP5A at standard [ATP]i of 2 mm. B-D, difference I–V curves of Ado-activated (control) IK(ACh) (B), inhibited IK(ACh) (C) and current activated by intracellular AP5A (D). Letters correspond to those in A.
Effects of APnA are mimicked by intracellular AMP
Recent studies of intracellular actions of APnA using isolated (inside-out) membrane patches have identified this class of compounds as potent inhibitors of IK(ATP) in pancreatic β-cells and cardiac myocytes (Ripoll et al. 1996; Jovanovic et al. 1997). Our observation to the contrary could mean that the stimulatory action is not seen in isolated membrane patches because upon extracellular application or slow intracellular loading these compounds in an intact myocyte are metabolized to yield an activator of KATP channels. The most potent physiological stimulating ligands of cardiac-type KATP channels are nucleotide diphosphates (NDP) such as ADP or UDP (e.g. Lederer & Nichols, 1989). If ADP (10–100 μm) was included in the pipette solution, neither activation of IK(ATP) nor inhibition of IK(ACh) were observed in six of six myocytes dialysed for a minimum of 8 min and a maximum of ∼24 min. Intracellularly NDPs are in equilibrium with tri- and monophosphates via the adenylate kinase reaction. If ADP from the pipette slowly diffuses into the cell, it can be converted via this reaction to yield AMP and ATP, resulting in an equilibrium concentration of the nucleotide diphosphate, which might be substantially lower than the nominal concentration in the pipette solution. On the other hand, this equilibrium in the presence of ATP can be shifted to yield ADP by including AMP in the pipette solution. As AMP is the first product of enzymatic hydrolysis of APnA (Mateo et al. 1997), formation of ADP via this pathway could be the mechanism underlying activation of IK(ATP) by intracellular APnA. This hypothesis was tested by supplementing the pipette filling solution with 100 μm AMP (at standard [ATP]i of 2 mm). A representative experiment is illustrated in Fig. 9. As with APnA in the pipette filling solution (see Fig. 8), dialysis of the cell with this solution resulted in activation of IK(ATP) and inhibition of IK(ACh). Again, both effects were fully reversed by 10 μm Gli. Comparable results were obtained in a total of eight myocytes.
Figure 9. Internal loading with AMP mimics the effects of APnA.
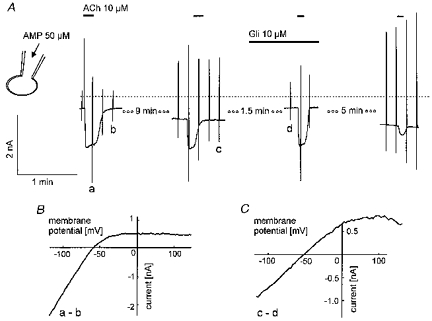
A, sections of membrane current recording as in previous figures. ACh (10 μm) and Gli were superfused as indicated. B, difference current-voltage relations as indicated by the labelling in A.
Inhibition of IK(ACh) occurs downstream of the receptor
This hypothesis of activation of IK(ATP) by ADP does not explain the inhibitory effects of extra- and intracellular APnA, metabolic inhibition, and intracellular AMP on IK(ACh). To further delineate the site of action within the muscarinic signalling pathway we tested whether IK(ACh) when irreversibly activated by intracelluar GTP-γ-S is sensitive to any of the experimental conditions listed above. In Fig. 10A a representative response of a cell to loading with GTP-γ-S is depicted. Repetitive exposure to ACh (10 μm) resulted in irreversible activation of IK(ACh) to a level of about 1.3 nA more negative than the initial background current. This level of IK(ACh) activation, confirmed by the strongly inward rectifying I–Vcurve (Fig. 10B), was maintained for another 15 min. Stable activation of IK(ACh) by intracellular GTP-γ-S was confirmed in a total of five cells. A comparable protocol was used for the response illustrated in Fig. 11. The pipette filling solution again contained 500 μm GTP-γ-S but was additionally supplemented with 50 μm AP6A. The compound was included in the patch clamp pipette rather than superfused extracellularly. As shown in Fig. 8, there was a tendency for the two effects to occur sequentially, inhibition of IK(ACh) preceding activation of IK(ATP). The separation of the effects on the two different currents permits an unambiguous identification of the individual currents and their contributions to total current. The current level activated by the GTP analogue was not maintained, but IK(ACh) gradually decreased to approximately the level of the initial holding current within about 4 min (Fig. 11A). This complete inhibition of IK(ACh) confirmed by the difference I–Vcurve (Fig. 11B) was followed by activation of IK(ATP), as revealed by the change of the whole-cell I–Vcharacteristics (Fig. 11C).
Figure 10. Persistent activation of IK(ACh) by intracellular GTP-γ-S.
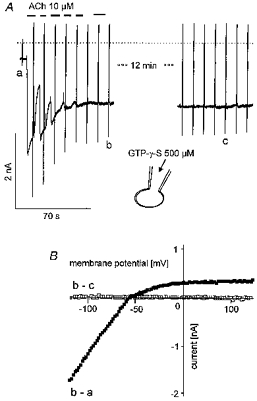
Pipette filling solution was supplemented with the GTP analogue (500 μm). A, continuous recording of membrane current, interrupted by a period of time of 12 min as indicated. The beginning of the trace corresponds to less than 10 s after starting the whole-cell mode by breaking the patch under the tip of the pipette. ACh (10 μm) was applied as indicated. B, difference I–V curves of GTP-γ-S-activated current (b - a). The data labelled b - c indicate that no change in the I–V characteristics of the cell occurred between the points of time in A representing about 13 min.
Figure 11. Inhibition of GTP-γ-S-activated IK(ACh) and activation of IK(ATP) by intracellular AP6A.
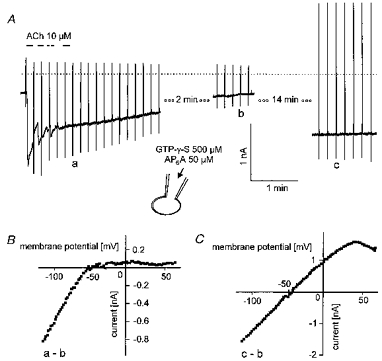
Pipette solution was supplemented with the GTP analogue (500 μm) and AP6A (50 μm). The beginning of the current trace in A corresponds to less than 10 s after breaking the membrane under the tip of the recording pipette. B, difference I–V curves as indicated by the labelling in A.
This clearly demonstrates that IK(ACh) induced by irreversibly activating the G protein is susceptible to inhibition by the putative changes in intracellular concentrations of nucleotide phosphates induced by APnA. Thus it is likely that inhibition of IK(ACh) by APnA or its metabolite(s) occurs downstream of G protein activation, presumably at the GIRK1-GIRK4 channel complex.
DISCUSSION
Actions of APnA in perfused hearts
In isolated atria and ventricular muscle strips APnA exert effects which by common pharmacological criteria are undoubtedly mediated via A1 receptors (Hoyle et al. 1996; Vahlensick et al. 1996). In addition to cardiac actions mediated by myocyte A1 receptors Rubino & Burnstock (1996) described an inhibition of calcitonin gene-related peptide release from capsaicin-sensitive nerve endings in isolated atria. From its sensitivity to an A1-selective antagonist, this was also identified as being mediated by A1 receptors. In this regard our data demonstrating inhibition of APnA-induced bradycardia by the A1 antagonist CPX in Langendorff perfused hearts, seem to be confirmatory. The additional observation in the present study of a strong enhancement of the bradycardia induced by Dipy, however, suggests that Ado itself might be the active compound. As, to our knowledge, there are no data in the literature indicating whether or not APnA are substrates of this transport mechanism, this finding does not unequivocally exclude the possibility that the diadenosine compounds could act as A1 agonists. The hypothesis that APnA actions in the system under study are mediated by Ado is supported by the finding that only a fraction of the initial concentration of APnA is detected in the effluent of perfused hearts, which however does contain Ado at concentrations high enough to activate IK(ACh). The final proof that APnA have no direct A1-agonistic action comes from the complete failure of these compounds to activate the muscarinic signalling pathway in isolated atrial myocytes. Conversion of APnA to Ado in the perfused heart, as demonstrated indirectly by the experiments using either CPX or dipyridamole, and directly by the HPLC assay, is in line with a recent study on coronary effects of AP4A (Nakae et al. 1997). These authors came to a similar conclusion, namely that the decrease in coronary resistance by this compound is mediated by Ado. At present no information is available regarding the identity and location of APnA cleaving enzymes in the heart. In adrenomedullary vascular endothelial cells an ectoenzyme has been described (ectodiadenosine polyphosphate hydrolase), which catalyses the hydrolysis of APnA to AMP and adenosine-5′-(n - 1)-phosphate (Mateo et al. 1997). Both, AMP and An - 1P can be further metabolized via the ectonucleotidase system to yield Ado. whether the putative enzyme in the heart is identical to the one described is unknown at present, as is its location on endothelial cells and/or myocytes.
Cellular actions of APnA
APnA failed to cause any measurable activation of IK(ACh), which represents a major target of A1 receptors in supraventricular myocytes. In the presence of APnA for periods > 3 min a current was activated which was identified as IK(ATP). Simultaneously IK(ACh) induced by activation of M2 receptors or A1 receptors or GTP-γ-S was inhibited. As both effects were observed upon extracellular application of the compounds as well as during intracellular loading via the patch clamp pipette, we suggest an intracellular site of action. This requires that the compounds, when applied extracellularly, have to be taken up by a mechanism yet to be identified. Due to the highly charged nature of the compounds, their cellular uptake via passive diffusion can be safely excluded. In preliminary experiments Dipy did not prevent the effects mediated by extracellular APnA (data not shown), suggesting that the Dipy-sensitive Ado transporter is not involved in internalizing the compounds. Moreover, Ado at a concentration of 1 mm superfused for > 10 min did not mimic the effects of APnA on IK(ATP) (not shown). This excludes the possibility that Ado, which might be produced extracellularly is accumulated by the cells and directly or indirectly contributes to the intracellular actions of APnA. This can be excluded also on technical grounds, since a cell under study was exposed to a continuous rapid flow of solution which should prevent accumulation of molecules produced on the surface of that cell. In summary, the striking similarity of effects induced by extra- and intracellular application of APnA on atrial membrane currents clearly favour (an) intracellular site(s) of action. For the identification of the pathway of internalization further experiments are required.
Activation of IK(ATP)
Identification of the APnA-activated current as IK(ATP) is based on (i) sensitivity to Gli at submicromolar concentrations and (ii) identity of the voltage dependence of this current with the voltage dependence of the current activated by metabolic inhibition. It should be noted that Gli, particularly if used at concentrations ≥ 10 μm, is not a perfectly selective drug but blocks various other phenomena such as cardiac cAMP-dependent and stretch-activated Cl− currents (Tominaga et al. 1995; Sakaguchi et al. 1997). At concentrations ≥ 1 μm a partial inhibition of atrial background and Ado-induced IK(ACh) has been described (Song et al. 1996). This could not be confirmed in the present study. Even at 10 μm no inhibitory effect on IK(ACh) was found. Inhibition of the APnA-activated current by almost 60 % in the presence of 100 nm Gli can be considered as a safe criterion for the identification. Moreover, the voltage-dependent properties, i.e. behaviour as a background current without obvious voltage-dependent gating, reversal at EK, and the weak inward rectification represent properties characteristic of IK(ATP), as confirmed by the comparison with the DNP-activated current.
We suggest that intracellularly the diadenosine polyphosphates are hydrolysed to yield AMP. In chromaffin cells a cytosolic enzyme activity degrading AP4A and AP5A with Km values of around 10 μm has been described previously (Rodriguez del Castillo et al. 1988). No information is available at present as to whether this enzyme is expressed in cardiac myocytes. The major evidence from the present study is indirect and comes from the finding that the effects of APnA on IK(ACh) and IK(ATP) can be mimicked by supplementing the pipette filling solution with AMP at a concentration as low as 50 μm, which itself is unlikely to have an major effect on either of the two K+ currents (e.g. Elvir-Mairena et al. 1996). AMP in turn, is a substrate of the adenylate kinase equilibrium reaction:
The equilibrium constant of this reaction among other factors depends on the absolute concentrations of nucleotides and Mg2+. It is not possible therefore to give a reliable estimate of the concentration of ADP for given values of [AMP] and [ATP] in the pipette solution. Modulation of KATP channels via this reaction by AMP in the presence of ATP has recently been demonstrated in isolated inside-out membrane patches from guinea-pig ventricular myocytes (Elvir-Mairena et al. 1996). These authors found substantial activation of KATP channels by 100 μm AMP in the presence of ATP in the millimolar range, which qualitatively is in line with our whole-cell data. In that publication evidence for a membrane association of this enzyme was provided, leading to the interesting hypothesis that it might be involved in local regulation of KATP channel activity.
Based on results from experiments on isolated inside-out membrane patches from cardiac myocytes and pancreatic β-cells, various APnA at micromolar concentrations have been identified as potent intracellular inhibitors of KATP channels. Activation of IK(ATP) by APnA seems to be in contradiction to these findings. We suggest that this apparent discrepancy reflects the different experimental conditions, namely recording from isolated (inside-out) patches vs. whole-cell recording. During exposure of the cytosolic side of an inside-out patch to APnA, or any other compound, its concentration can be considered as clamped. Proteins or binding sites sensitive to that compound will encounter a stable concentration - unless the compound represents a substrate of an efficient membrane-bound enzymatic mechanism catalysing its chemical modification (see Elvir-Mairena et al. 1996). Under the whole-cell recording conditions of the present study the effective intracellular concentration is not necessarily clamped. APnA are slowly entering the cell, either through the orifice of the recording pipette or by transmembrane transport. If, as our data suggest, these compounds are subject to intracellular metabolic pathways yielding an activator of KATP channels such as ADP, their genuine blocking actions would be completely irrelevant under such conditions. Previously APnA have been described as potent inhibitors of nucleoside and nucleotide kinases (Bone et al. 1986). Our intepretation of how IK(ATP) is activated strongly relies on the activity of adenylate kinase which catalyses the conversion of AMP/ATP to ADP. Again, if APnA entry is balanced by hydrolysis, inhibition of adenylate kinase would be of little importance.
Inhibition of IK(ACh)
Under all conditions that caused activation of IK(ATP) an inhibition of IK(ACh) was recorded which was independent of the mode of activation (M2 receptors, A1 receptors, GTP-γ-S). The apparent coupling of inhibition of IK(ACh) to activation of IK(ATP) could reflect a phenomenon related to the flux of K+ ions rather than a genuine inhibition of KACh channels. Such a mechanism, where flux through one type of K+ channel limits the flux through a different channel due to local effects on EK, has been discussed previously (McHugh & Beech, 1995). However, since in a number of cells loaded with APnA both effects were recorded sequentially, i.e. inhibition of IK(ACh) before activation of IK(ATP), this seems rather unlikely in the present case. In experiments using DNP, however, separation of the two effects was never observed, which might reflect a more rapid and dramatic decrease of the [ATP]/[ADP] ratio. We found, however, that in myocytes treated with antisense oligodeoxynucleotides directed against the cardiac sulphonylurea receptor SUR2A, in a fraction of cells DNP completely failed to activate whole-cell IK(ATP) but still caused inhibition of IK(ACh) (Brandts et al. 1998). This clearly suggests that activation of IK(ATP) and inhibition of IK(ACh) reflect two effects which may have a common cause but which are independent of each other.
Whereas our hypothesis that ADP is the compound responsible for mediating activation of IK(ATP) by APnA is in line with the current literature, we have no conclusive explanation at present for the inhibition of IK(ACh). A conceivable mechanism would be a reduction of the [GTP]/[GDP] ratio, since intracellular nucleotide phosphates are subject to common equlibrium reactions. Whether such a mechanism acting on the level of the coupling G protein can account for the complete inhibition of IK(ACh) by APnA, AMP, and DNP at present cannot be answered. The reversal of the inhibition of IK(ACh) by Gli, which was seen under all conditions which caused activation of IK(ATP)/inhibition of IK(ACh), would hardly be explained by a ‘metabolic effect’ acting on the [GTP]/[GDP] ratio. The molecular target of Gli at the KATP channel is the sulphonylurea receptor (SUR2A, Sakura et al. 1995). There is however no evidence for any relation of SURs to the Kir3.x subunits of KACh channels. Attempts to further delineate the mechanism underlying the adverse effects on the two different ion channel systems in inside-out patches has so far failed. Both channels are present at a high density in the membrane of atrial cells. Because of superimposed multiple openings of both types of channels, an analysis of the behaviour of one channel type, e.g. upon application of ADP to the internal face of the membrane, so far has been unsuccessful. Thus, for studying the mechanism(s) underlying the inhibition of IK(ACh) a system is necessary in which activity of KATP channels is eliminated by non-pharmacological tools, either by heterologous expression of the muscarinic signalling pathway in a cell lacking intrinsic KATP channels or antisense knockout of KATP channel subunits in native myocytes.
Acknowledgments
The excellent technical assistance of Mrs Bing Liu is gratefully acknowledged. This work was supported by the Hans und Gertie Fischer Stiftung and the Deutsche Forschungsgemeinschaft.
References
- Bone R, Cheng YC, Wolfenden R. Inhibition of adenosine and thymidylate kinases by bisubstrate analogs. Journal of Biological Chemistry. 1986;261:16410–16413. [PubMed] [Google Scholar]
- Brady PA, Alekseev AE, Terzic A. Operative condition-dependent response of cardiac ATP-sensitive K+ channels toward sulfonylureas. Circulation Research. 1998;82:272–278. doi: 10.1161/01.res.82.2.272. [DOI] [PubMed] [Google Scholar]
- Brandts B, Bender K, Pott L. SUR2A antisense removes sensitivity of IK(ATP) to metabolic inhibition but not to cromakalim in guinea-pig atrial myocytes. Pflügers Archiv. 1998;435:R96. [Google Scholar]
- Bünemann M, Brandts B, Liliom K, Pott L, Tseng J-L, Desiderio GM, Sun G, Miller D, Tigyi G. A novel receptor with high affinity for lysosphingomyelin and sphingosine 1-phosphate in atrial myocytes. EMBO Journal. 1996a;15:5527–5534. [PMC free article] [PubMed] [Google Scholar]
- Bünemann M, Brandts B, Pott L. Downregulation of muscarinic M2 receptors linked to K+ current in cultured guinea-pig atrial myocytes. The Journal of Physiology. 1996b;492:351–362. doi: 10.1113/jphysiol.1996.sp021497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bünemann M, Brandts B, Pott L. In vivo downregulation of M2 receptors revealed by measurement of muscarinic K+ current in cultured guinea-pig atrial myocytes. The Journal of Physiology. 1997;501:549–554. doi: 10.1111/j.1469-7793.1997.549bm.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bünemann M, Pott L. Downregulation of A1 adenosine receptors coupled to muscarinic K+ current in cultured guinea-pig atrial myocytes. The Journal of Physiology. 1995;482:81–92. doi: 10.1113/jphysiol.1995.sp020501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busse R, Ogilvie A, Pohl U. Vasomotor activity of diadenosine triphosphate and diadenosine tetraphosphte in isolated arteries. American Journal of Physiology. 1988;254:828–832. doi: 10.1152/ajpheart.1988.254.5.H828. [DOI] [PubMed] [Google Scholar]
- Dascal N. Signalling via the G protein-activated K+ channels. Cellular Signalling. 1997;9:551–573. doi: 10.1016/s0898-6568(97)00095-8. [DOI] [PubMed] [Google Scholar]
- Elvir-Mairena JR, Jovanovic A, Gomez LA, Alekseev AE, Terzic A. Reversal of the ATP-liganded state of ATP-sensitive K+ channels by adenylate kinase activity. Journal of Biological Chemistry. 1996;271:31903–31908. doi: 10.1074/jbc.271.50.31903. [DOI] [PubMed] [Google Scholar]
- Fosset M, De Weille JR, Green RD, Schmid-Antomarchi H, Lazdunski M. Antidiabetic sulfonylureas control action potential properties in heart cells via high affinity receptors that are linked to ATP-dependent K+ channels. Journal of Biological Chemistry. 1988;263:7933–7936. [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hildermann RH, Martin M, Zimmermann JK, Pivorun EB. Identification of a unique membrane receptor for adenosine 5′,5’‘’-P1,P4-tetraphosphate. Journal of Biological Chemistry. 1991;266:6915–6918. [PubMed] [Google Scholar]
- Hoyle CHV, Ziganshin AU, Pintor J, Burnstock G. The activation of P1- and P2-purinoceptors in the guinea-pig left atrium by diadenosine polyphosphates. British Journal of Pharmacology. 1996;118:1294–1300. doi: 10.1111/j.1476-5381.1996.tb15536.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CL, Slesinger PA, Casey PJ, Jan YN, Jan LY. Evidence that direct binding of Gβγ to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 1995;15:1133–1143. doi: 10.1016/0896-6273(95)90101-9. [DOI] [PubMed] [Google Scholar]
- Jovanovic A, Alekseev AE, Terzic A. Intracellular diadenosine polyphosphates — A novel family of inhibitory ligands of the ATP-sensitive K+ channel. Biochemical Pharmacology. 1997;54:219–225. doi: 10.1016/s0006-2952(97)00262-1. [DOI] [PubMed] [Google Scholar]
- Kaibara M, Nakajima T, Irisawa H, Giles W. Regulation of spontaneous opening of muscarinic K+ channels in rabbit atrium. The Journal of Physiology. 1991;433:589–613. doi: 10.1113/jphysiol.1991.sp018445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kisselev LL, Justesen J, Wolfson AD, Frolova LY. Diadenosine oligophosphates (ApnA), a novel class of signalling molecules? FEBS Letters. 1998;427:157–163. doi: 10.1016/s0014-5793(98)00420-7. [DOI] [PubMed] [Google Scholar]
- Kurachi Y. G protein regulation of cardiac muscarinic potassium channel. American Journal of Physiology. 1995a;269:C821–830. doi: 10.1152/ajpcell.1995.269.4.C821. [DOI] [PubMed] [Google Scholar]
- Kurachi Y. Adenosine and adenine nucleotide action on cardiac cellular excitation. In: Sperelakis N, editor. Physiology and Pathophysiology of the Heart. 3. Dordrecht: Kluwer Academic Publishers; 1995b. pp. 789–800. [Google Scholar]
- Lederer WJ, Nichols CG. Nucleotide modulation of the activity of rat heart ATP-sensitive K+ channels in isolated membrane patches. The Journal of Physiology. 1989;419:193–211. doi: 10.1113/jphysiol.1989.sp017869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHugh D, Beech DJ. Inhibition of delayed rectifier K+-current by levcromakalim in singel intestinal smooth muscle cells: effects of cations and dependence on K+-flux. British Journal of Pharmacology. 1995;114:391–399. doi: 10.1111/j.1476-5381.1995.tb13239.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mateo J, Miras-Portugal MT, Rotllan P. Ectoenzymatic hydrolysis of diadenosine polyphosphates by cultured adrenomedullary vascular endothelial cells. American Journal of Physiology. 1997;273:C918–927. doi: 10.1152/ajpcell.1997.273.3.C918. [DOI] [PubMed] [Google Scholar]
- Nakae I, Takahashi M, Takaoka A, Liu Q, Matsumoto T, Amano M, Sekine A, Nakjima H, Kinoshita M. Coronary effects of diadenosine tetraphosphate resemble those of adenosine in anesthetized pigs: involvement of ATP-sensitive potassium channels. Journal of Cardiovascular Pharmacology. 1997;28:124–133. doi: 10.1097/00005344-199607000-00019. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- Ogilvie A, Blasius R, Schulze-Lohoff E, Sterzel RB. Adenine dinucleotides: a novel class of signalling molecules. Journal of Autonomic Pharmacology. 1996;16:325–328. doi: 10.1111/j.1474-8673.1996.tb00045.x. [DOI] [PubMed] [Google Scholar]
- Pohl U, Ogilvie A, Lamontagne D, Busse R. Potent effects of AP3A and AP4A on coronary resistance and autacoid release of intact rabbit hearts. American Journal of Physiology. 1991;260:1692–1697. doi: 10.1152/ajpheart.1991.260.5.H1692. [DOI] [PubMed] [Google Scholar]
- Pott L, Bünemann M. Rate of activation of muscarinic K+ current in atrial myocytes by adenosine is limited by density of A1 receptors. Pflügers Archiv. 1994;426:R126. [Google Scholar]
- Ripoll C, Martin F, Rovira JM, Pintor J, Miras-Portugal MT, Soria B. Diadenosine polyphosphates — A novel class of glucose-induced intracellular messengers in the pancreatic β-cell. Diabetes. 1996;45:1431–1434. doi: 10.2337/diab.45.10.1431. [DOI] [PubMed] [Google Scholar]
- Rodriguez del Castillo A, Torres M, Delicado EG, Miras-Portugal MT. Subcellular distribution studies of diadenosine polyphosphates — Ap4A and Ap5A — in bovine adrenal medulla: presence in chromaffin granules. Journal of Neurochemistry. 1988;51:1696–1703. doi: 10.1111/j.1471-4159.1988.tb01147.x. [DOI] [PubMed] [Google Scholar]
- Rubino A, Burnstock G. Possible role of diadenosine polyphosphates as modulators of cardiac sensory-motor neurotransmission in guinea-pigs. The Journal of Physiology. 1996;495:515–523. doi: 10.1113/jphysiol.1996.sp021611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi M, Matsuura H, Ehara T. Swelling-induced Cl− current in guinea-pig atrial myocytes: inhibition by glibenclamide. The Journal of Physiology. 1997;505:41–52. doi: 10.1111/j.1469-7793.1997.041bc.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakura H, Ämmlälä C, Smith PH, Gribble FM, Ashcroft FM. Cloning and functional expression of the cDNA encoding a novel ATP-sensitive potassium channel subunit expressed in pancreatic β-cells, brain, heart and skeletal muscle. FEBS Letters. 1995;377:338–344. doi: 10.1016/0014-5793(95)01369-5. [DOI] [PubMed] [Google Scholar]
- Song Y, Srinivas M, Belardinelli L. Nonspecific inhibition of adenosine-activated K+ current by glibenclamide in guinea pig atrial myocytes. American Journal of Physiology. 1996;40:H2430–2437. doi: 10.1152/ajpheart.1996.271.6.H2430. [DOI] [PubMed] [Google Scholar]
- Takano M, Noma A. Development of muscarinic potassium current in fetal and neonatal rat heart. American Journal of Physiology. 1997;41:H1188–1195. doi: 10.1152/ajpheart.1997.272.3.H1188. [DOI] [PubMed] [Google Scholar]
- Tepel M, Bachmann J, Schlüter H, Zidek W. Diadenosine polyphosphates increase cytosolic: Calcium and attenuate angiotensin-II-induced changes of calcium in vascular smooth muscle cells. Journal of Vascular Research. 1996;33:132–138. doi: 10.1159/000159141. [DOI] [PubMed] [Google Scholar]
- Tominaga M, Horie M, Sasayama S, Okada Y. Glibenclamide, an ATP-sensitive K+ channel blocker, inhibits cardiac cAMP-activated CI-conductance. Circulation Research. 1995;77:417–423. doi: 10.1161/01.res.77.2.417. [DOI] [PubMed] [Google Scholar]
- Vahlensick U, Boknik P, Knapp J, Linck B, Müller FU, Neumann J, Herzig S, Schlüter H, Zidek W, Deng MC, Scheld HH, Schmitz W. Negative chronotropic and inotropic effects exerted by diadenosine hexaphosphate (AP6A) via A1-adenosine receptors. British Journal of Pharmacology. 1996;119:835–844. doi: 10.1111/j.1476-5381.1996.tb15748.x. [DOI] [PMC free article] [PubMed] [Google Scholar]