

Abstract
Using perforated-patch, whole cell recording, we investigated the membrane mechanisms underlying O2 chemosensitivity in neonatal rat adrenomedullary chromaffin cells (AMC) bathed in extracellular solution containing tetrodotoxin (TTX; 0.5–1 μm), with or without blockers of calcium entry.
Under voltage clamp, low PO2 (0–15 mmHg) caused a graded and reversible suppression in macroscopic outward K+ current. The suppression during anoxia (PO2 = 0 mmHg) was ∼35% (voltage step from −60 to +30 mV) and was due to a combination of several factors: (i) suppression of a cadmium-sensitive, Ca2+-dependent K+ current, IK(CaO2); (ii) suppression of a Ca2+-insensitive, delayed rectifier type K+ current, IK(VO2); (iii) activation of a glibenclamide- (and Ca2+)-sensitive current, IK(ATP).
During normoxia (PO2 = 150 mmHg), application of pinacidil (100 μm), an ATP-sensitive potassium channel (KATP) activator, increased outward current density by 45.0 ± 7.0 pA pF−1 (step from −60 to + 30 mV), whereas the KATP blocker glibenclamide (50 μm) caused only a small suppression by 6.3 ± 4.0 pA pF−1. In contrast, during anoxia the presence of glibenclamide resulted in a substantial reduction in outward current density by 24.9 ± 7.9 pA pF−1, which far exceeded that seen in its absence. Thus, activation of IK(ATP) by anoxia appears to reduce the overall K+ current suppression attributable to the combined effects of IK(CaO2) and IK(VO2).
Pharmacological tests revealed that IK(CaO2) was carried predominantly by maxi-K+ or BK potassium channels, sensitive to 50–100 nm iberiotoxin; this current also accounted for the major portion (∼60%) of the anoxic suppression of outward current. Tetraethylammonium (TEA; 10–20 mm) blocked all of the anoxia-sensitive K+ currents recorded under voltage clamp, i.e. IK(CaO2), IK(VO2) and IK(ATP).
Under current clamp, anoxia depolarized neonatal AMC by 10–15 mV from a resting potential of ∼-55 mV. At least part of this depolarization persisted in the presence of either TEA, Cd2+, 4-aminopyridine or charybdotoxin, suggesting the presence of anoxia-sensitive mechanisms additionalto those revealed under voltage clamp. In Na+/Ca2+-free solutions, the membrane hyperpolarized, though at least a portion of the anoxia-induced depolarization persisted.
In the presence of glibenclamide, the anoxia-induced depolarization increased significantly to ∼25 mV, suggesting that activation of KATP channels may function to attenuate the anoxia-induced depolarization or receptor potential.
Adrenomedullary chromaffin cells (AMC) mediate the elevation in plasma catecholamine (CA) that occurs when animals are exposed to stressors, e.g. acute hypoxia. In the neonatal rat, this CA surge is vital for the animal's ability to survive hypoxic stress associated with the transition to extrauterine life, but occurs through a ‘non-neurogenic’ mechanism that is present prior to the onset of mature sympathetic innervation (Seidler & Slotkin, 1985). We recently reported that rat AMC possess a developmentally regulated oxygen sensing mechanism, since in the majority of cells derived from neonatal (postnatal (P) day 1-P3), but not juvenile (P13-P21) animals, acute hypoxia caused suppression of the outward K+ current, membrane depolarization and CA secretion (Thompson et al. 1997). These responses appear qualitatively similar to those of prototypic O2 chemoreceptors, i.e. type 1 cells of the carotid body (Buckler & Vaughan-Jones, 1994; Gonzalez et al. 1994; Peers & Buckler, 1995; Lopez-Barneo, 1996; Jackson & Nurse, 1997) and interestingly, both cell types derive from a similar lineage, the sympathoadrenal branch of the neural crest. However, hypoxia is known to modulate differentially several types of K+ channels and cause either membrane depolarization or hyperpolarization in a number of other cell types (Haddad & Jiang, 1997), including arterial myocytes (Post et al. 1992), pulmonary neuroepithelial bodies (Youngson et al. 1993), PC12 cells (Zhu et al. 1996) and central neurons (Jiang et al. 1994).
The K+ channel subtypes that are inhibited by hypoxia in carotid body type 1 cells include large conductance Ca2+-dependent K+, or maxi-K+ channels (Wyatt & Peers, 1995) and voltage-independent, small conductance K+‘leak’ channels (Buckler, 1997) in the rat, and Ca2+-independent, slow-inactivating, delayed rectifier type K+ channels in the rabbit (Lopez-Lopez et al. 1989). In PC12 cells, a cell line derived from adrenal chromaffin cells, hypoxia inhibits a slow-inactivating, delayed rectifier type K+ channel which mediates membrane depolarization (Zhu et al. 1996). However, in these cells hypoxia also appears to activate a large conductance Ca2+-dependent K+ channel (Conforti & Millhorn, 1997). In central neurons, hypoxia activates a glibenclamide-sensitive ATP-sensitive K+ current, IK(ATP), which is thought to play a protective role in low oxygen conditions by inducing hyperpolarization and preventing action potential generation (Jiang et al. 1994). These results suggest that hypoxia can modulate multiple K+ channels in different tissues, and that the effect on a particular channel (i.e. closure vs. opening) may be both species and cell-type dependent.
In our initial study (Thompson et al. 1997), it was unclear which K+ channel types mediate hypoxic chemosensitivity in neonatal rat AMC. This is of additional interest, since, as discussed above, hypoxia inhibits K+ channels with different calcium sensitivities in two cell types, carotid body type 1 and PC12 cells, that are related developmentally to AMC. Furthermore, in a recent study KATP channels were presumed to play a crucial role in hypoxia-induced responses in adult rat chromaffin cells (Mochizuki-Oda et al. 1997). In the present study we used perforated-patch, whole cell recording and pharmacological tools to characterize the types of O2-sensitive K+ currents in neonatal rat AMC and to investigate whether these currents can account for the hypoxia-induced membrane depolarization or receptor potential (Thompson et al. 1997). Preliminary results of some of these findings were reported in a recent abstract (Thompson & Nurse, 1997).
METHODS
Cell culture
Pregnant or lactating Wistar rats and pups (Charles River, Quebec, Canada) were housed in our animal facility under a 12 h light-12 h dark cycle. All animal handling and tissue removal conformed to guidelines established by the Canadian Council on Animal Care. Primary cultures enriched in dissociated adrenomedullary chromaffin cells (AMC) were prepared as previously described (Thompson et al. 1997). Adrenal glands were dissected from neonatal rats (i.e. postnatal (P) day P1-P2) that were rendered unconscious by a blow to the head and killed by decapitation. Most of the surrounding cortex was trimmed and discarded. The remaining (medullary) tissue was dissociated by incubation (at 37°C) in an enzymatic solution containing v/v 0.1% trypsin, 0.1% collagenase (Gibco) and 0.01% deoxyribonuclease (Millipore). After 1 h incubation, most of the enzyme was removed and the remainder inactivated by addition of growth medium consisting of F-12 nutrient medium (Gibco) supplemented with 10% v/v fetal calf serum (Gibco), 80 U l−1 insulin (Sigma), 0.6% v/v glucose, 2 mm glutamine, 1% v/v penicillin-streptomycin (Gibco) and 0.01% v/v dexamethasone (Sigma). Tissue was then triturated using a Pasteur pipette, and the final cell suspension was pre-plated for 1–24 h on a collagen-coated culture dish to remove most of the cortical cells. The non-adherent AMC were then plated onto the central region of Nunclon culture dishes that had been previously coated with a thin layer of Matrigel (Collaborative Research, Bedford, MA, USA). The cells were grown at 37°C in a humidified atmosphere of 95% air, 5% CO2 for 3–72 h before they were used in patch clamp experiments.
Electrophysiology
All voltage and current clamp data were obtained using the perforated-patch configuration of the whole cell technique as previously described (Thompson et al. 1997). The seal resistance was typically 2–10 GΩ, and most (∼75%) of the series resistance (range, 12–50 MΩ) was compensated in voltage clamp experiments; voltage errors due to series resistance were minimal due to the high input resistance (∼2 GΩ) of the cells. Junction potentials were typically 2–5 mV in the standard bathing solution, and were cancelled prior to seal formation. The pipette solution for perforated-patch recording contained (mm): potassium gluconate, 105; KCl, 30; NaCl, 5; CaCl2, 0.1; Hepes, 10; at pH 7.2, plus nystatin (450 μg ml−1). The standard bathing solution for both voltage clamp and current clamp experiments consisted of (mm): NaCl, 135; KCl, 5; CaCl2, 2; MgCl2, 2; glucose, 10; Hepes, 10; tetrodotoxin (TTX), 0.0005–0.001; pH adjusted to 7.4 with NaOH. In experiments where a Ca2+-free bathing solution was required, CaCl2 (2 mm) was replaced with equimolar MgCl2 and 1 mm EGTA. In some experiments, 200 μm CdCl2 was added to block Ca2+ currents and, indirectly, Ca2+-dependent K+ currents. In experiments requiring a Na+-free solution, Na+ was replaced with equimolar N-methyl-D-glucamine (NMDG) and the pH was adjusted with HCl.
All recordings were obtained at room temperature (20–23°C) with an Axopatch-1D amplifier equipped with a 500 MΩ head stage feedback resistor (Axon Instruments). Records were digitized with a Digidata 1200 computer interface (Axon) and stored on hard disk in an IBM-compatible computer using pCLAMP software version 6.0.3 (Axon). In the majority of experiments anoxia was used as the low PO2 stimulus. Anoxia was generated by bubbling 100% N2 into the perfusion reservoir in the presence of the O2 scavenger sodium dithionite; the pH was adjusted to 7.4 with NaOH. For hypoxic solutions, 100% N2 was bubbled in the absence of dithionite. Measurements of PO2 were obtained with an O2 microelectrode (Diamond Electro-Tech Inc., Ann Arbor, MI, USA) placed near the recording site. During calibration, 0 mmHg (anoxia) was the designated PO2 of a solution equilibrated with 100% N2 in the presence of 1 mm sodium dithionite. The solution in the recording chamber (volume, 750 μl-1 ml) was exchanged by perfusion under gravity and simultaneous removal by suction at a rate of 5–6 ml min−1.
The effects of anoxia and/or hypoxia on voltage-activated currents were examined by comparing peak currents from the average of four records at each step potential, over the range of −50 to +50 mV (10 mV increments) from a holding potential of −60 mV. Records were taken before (control), during, and after (recovery) application of the stimulus, and the averages were obtained either during data collection (on-line), or during subsequent analysis (off-line). All measurements of membrane potential were obtained under current clamp in zero current (I = 0) mode. Membrane capacitance (pF) was obtained by first integrating the capacitative transient elicited by a hyperpolarizing voltage step from −60 to −100 mV, then dividing by the magnitude of the step. Currents (pA) or current densities (pA pF−1) were compared using either paired or independent Student's t tests, with the level of significance set at P < 0.05. Voltage clamp current traces are shown in the text with ‘leak currents’ unsubtracted; current or current density vs. voltage (I–V) plots are leak subtracted. All data are presented as means ±s.e.m.
Drugs
In order to block various types of K+ currents (as indicated in the text) the following drugs were added directly to the perfusion fluid: tetraethylammonium (TEA), glibenclamide and 4-aminopyridine (4-AP), obtained from Sigma; and iberiotoxin (IbTx) and charybdotoxin (ChTx) obtained from Alomone Laboratories (Jerusalem, Israel). In some experiments the KATP channel activator pinacidil, a gift of Dr Jan Huizinga, was used. Additionally, in some experiments cadmium at 200 μm (CdCl2) was used to block Ca2+ entry and, indirectly, Ca2+-dependent K+ currents. To block voltage-dependent Na+ currents, tetrodotoxin (TTX; Sigma) was added to the bathing solution.
RESULTS
Neonatal rat adrenomedullary chromaffin cells (AMC) were first tested for hypoxic sensitivity between 3 h and 3 days in culture, using the perforated-patch technique for whole cell recording. As in our previous report (Thompson et al. 1997), low PO2 caused a reversible suppression of outward current (Figs 1 and 2) and/or membrane depolarization (e.g. Fig. 7) in > 80% of cells tested (n = 68/78). As shown in Fig. 1, the magnitude of the effect was graded, with the maximum current suppression occurring in anoxia (PO2 = 0 mmHg). During a voltage step from −60 to +30 mV, the mean (±s.e.m.) percentage suppression was 12.2 ± 0.02% during hypoxia (PO2, ∼15 mmHg) compared with 43.6 ± 0.08% in anoxia, for the same group of five cells examined at both PO2 levels. Thus, in the experiments reported below, an anoxic stimulus was routinely used in order to optimize the cell response. O2-sensitive AMC were observed after acute cell isolation (i.e. 3 h in vitro) and in short-term cultures (1–3 days), suggesting that the hypoxia-sensing mechanism was expressed in vivo, before the cells were brought into culture.
Figure 1. PO2-dependent suppression of outward current in neonatal rat adrenal chromaffin cells (AMC).
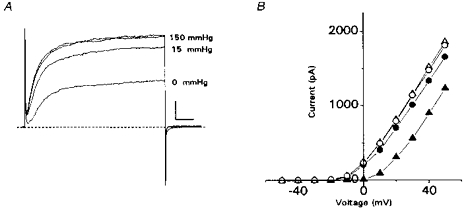
A, leak-unsubtracted outward currents recorded from a cell exposed sequentially to normoxia (PO2 = 150 mmHg), hypoxia (PO2∼15 mmHg), normoxia again (not shown), anoxia (PO2 = 0 mmHg) and finally normoxia (wash). All traces were obtained during voltage steps from −60 to +30 mV, and each trace is the average of 4 records; the 2 top traces are superimposed initial (control) and final (wash) recordings in normoxia. Horizontal scale represents 10 ms and vertical scale 200 pA. B, current vs. voltage plots for the cell in A. Records were taken at 10 mV voltage increments from a holding potential of −60 mV. Symbols are as follows: control, ○; hypoxia, •; anoxia, ▴; wash, ▵. The anoxic stimulus was applied after the cell had recovered from the hypoxic stimulus (not shown). Wash represents recovery after anoxia. Note the magnitude of outward current suppression is PO2 dependent. Similar results were obtained in 4 other cells exposed to normoxia, hypoxia and anoxia.
Figure 2. Effect of anoxia on Ca2+-dependent and Ca2+-independent outward currents in neonatal rat AMC.
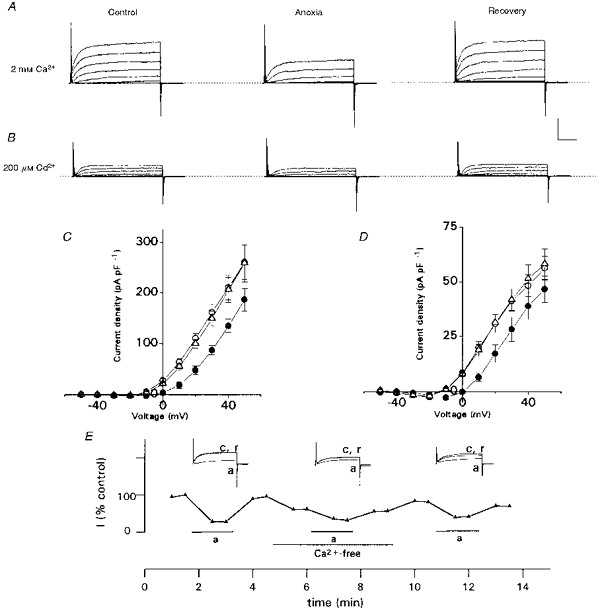
A, leak-unsubtracted outward current traces recorded from a cell in normal (2 mm) Ca2+, before (left; control), during (middle) and after (right; recovery) exposure to anoxia. Traces shown are for voltage steps from a holding potential of −60 to +50 mV in 10 mV increments; the top trace in each record represents the step to +50 mV. Note anoxia reversibly suppresses outward current. B, the same cell (as in A) exposed to a similar protocol, except that Ca2+-dependent currents were blocked by inclusion of 200 μm Cd2+ in the bathing solution. Note anoxia still had a suppressive effect on the residual Ca2+-independent outward current. In A and B, the vertical scale represents 500 pA and horizontal scale represents 10 ms. C and D, current density vs. voltage plots for 10 representative cells, including the one in A. Mean current density (± s.e.m.) is shown for cells recorded in 2 mm Ca2+ (C) and 200 μm Cd2+ (D). The current density measured during anoxia was significantly different from control (P < 0.05) at all voltage steps between −10 and +50 mV in C, or −10 and +30 mV in D, and recovery was complete at each step. Control, ○; anoxia, •; recovery, ▵. E, time course of anoxic inhibition of outward currents (step to +40 mV) in the presence and absence (Ca2+-free) of extracellular Ca2+. Note that the cell responded to a third anoxic stimulus after return to normal Ca2+-containing medium. Outward current is expressed as a percentage of the maximum recorded in normal Ca2+. c, a and r refer to control, anoxia and recovery, respectively.
Figure 7. Effects of anoxia and manipulation of the extracellular fluid on membrane potential of neonatal AMC.
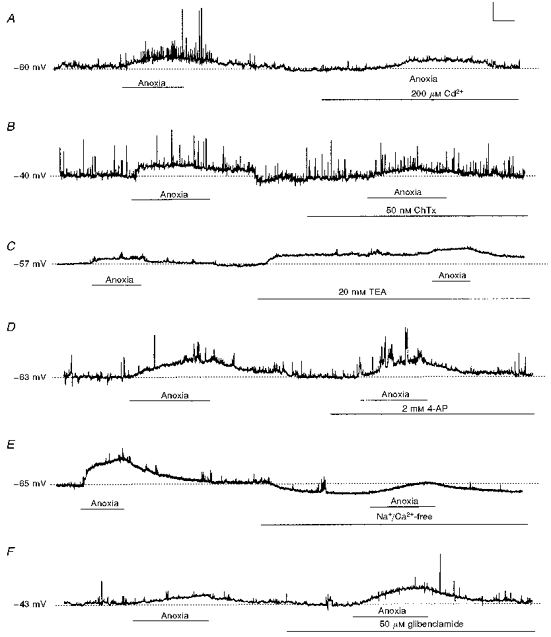
Recordings under current clamp were obtained in the continuous presence of TTX (0.5–1 μm). A, anoxia depolarized AMC, though addition of Cd2+ alone did not; further, Cd2+ did not prevent the anoxia-induced depolarization, but abolished spike activity (compare right with left portions of trace). B, direct blockade of BK currents with 50 nm ChTx did not significantly alter membrane potential, nor prevent the anoxia-induced depolarization (compare left and right portions of trace). C, addition of TEA (20 mm) caused a slight membrane depolarization (right portion of trace), but did not block the anoxia-induced depolarization. D, addition of 4-AP did not depolarize nor prevent anoxia-induced depolarization of neonatal AMC. E, effects of perfusion with a Na+/Ca2+-free (NMDG+ was substituted for Na+; Ca2+ was replaced with 2 mm Mg2+ and 1 mm EGTA) bathing solution. Exposure to this solution hyperpolarized AMC and reduced, but did not block, the anoxia-induced depolarization. F, blockade of KATP channels with 50 μm glibenclamide did not affect resting membrane potential, nor did it prevent the anoxia-induced depolarization. However, in the presence of glibenclamide the anoxia-induced depolarization was significantly (P < 0.05) enhanced. Vertical scale bars represent 20 mV in A-E and 15 mV in F; horizontal bar represents 25 s.
To allow comparisons among cells of different sizes, current density (pA pF−1) was determined by dividing the steady-state outward current (at 45 ms; step from −60 mV) by the whole cell capacitance (range, 4–10 pF; see Methods). Under voltage clamp, the mean (±s.e.m.) outward current density during normoxia (PO2 = 150 mmHg) was 115 ± 8.24 pA pF−1 (n = 48) for a voltage step from −60 to +30 mV. Exposure to anoxia caused a significant reduction in outward current density to 74.25 ± 5.96 pA pF−1 (n = 48; P < 0.01); after reperfusion with control (normoxic) solution, the current density returned to 100.8 ± 8.8 pA pF−1 (n = 48), a value not significantly different from control (P > 0.2). The anoxia-sensitive component of outward current (IK(O2)) comprised a mean of 34.5 ± 0.03% (n = 48) of the total outward K+ current recorded in normoxia for the voltage step from −60 to +30 mV. By first testing for the presence of this anoxia- sensitive K+ current, O2-sensitive AMC were identified, thereby allowing the K+ current subtype(s) that mediate O2 chemosensitivity to be investigated in greater detail. In the voltage clamp experiments reported below, cells that failed to show > 90% recovery of the control current, after exposure to anoxia or pharmacological agents (with the exception of the poorly reversible, Ca2+-dependent K+ channel blocker, iberiotoxin), were excluded.
Anoxia suppresses both Ca2+-dependent and Ca2+-independent K+ currents in neonatal AMC
Closure of a variety of K+ channels, including both Ca2+-dependent and Ca2+-independent subtypes, mediates hypoxic chemosensitivity in carotid body type 1 cells (Lopez-Lopez et al. 1989; Peers, 1990; Gonzalez et al. 1994; Buckler, 1997) and in PC12 cells (Conforti & Millhorn, 1997). To test whether both subtypes participate in the anoxic suppression of K+ current in neonatal AMC, cells were studied in either Ca2+-free or Cd2+-containing (200 μm) bathing solutions. These conditions have been shown to inhibit Ca2+-dependent K+ currents in adult rat AMC (Neely & Lingle, 1992) and neonatal rat type 1 cells (Peers, 1990). Blockade of Ca2+ entry by addition of 200 μm Cd2+ to the bathing solution resulted in a reduction in outward current by 64.3 ± 0.06% relative to control (n = 10; step from −60 to +30 mV); an example is shown in Fig. 2A and B (compare left traces). This reduction was not significantly different from that seen in Ca2+-free medium, where the corresponding suppression was 60.1 ± 0.08% (n = 12). Thus, as in adult rat AMC (Neely & Lingle, 1992), the majority (∼62%) of the outward current in neonatal, O2-sensitive AMC is Ca2+ dependent.
Since anoxia suppressed the outward current by ∼35% when Ca2+ currents were present (see above and Fig. 2A), it was of interest to determine whether the same stimulus had any effect on the residual K+ current recorded in Ca2+-free or Cd2+-containing solutions. As shown in Fig. 2B and D, anoxia caused a small but significant suppression in outward current in the presence of 200 μm Cd2+ and the effect was reversible. Also, cells exposed to Ca2+-free solutions were still capable of responding normally to another anoxic stimulus, after return to normal Ca2+ (n = 3; Fig. 2E). Experiments similar to those shown in Fig. 2B, D and E allowed a quantitative estimate of IK(VO2), i.e. the magnitude of the Ca2+-independent component of the total anoxia-sensitive current, IK(O2). This current, IK(VO2), is shown as difference current traces for the cell in Fig. 3A, lower traces, and compared with IK(O2) for the same cell in Fig. 3A, upper traces. Comparisons of current densities (pA pF−1) for groups of cells exposed to anoxia in Ca2+-free or Cd2+-containing solutions are shown in Fig. 3B and C. The difference current traces shown in Fig. 3A, where currents recorded in anoxia are subtracted from control (normoxic) currents, allow the anoxia-sensitive component to be isolated in both normal Ca2+ (upper traces) and Ca2+-free (lower traces) solutions. It is evident from these traces, as well as from the current density vs. voltage plot in Fig. 3B, that the Ca2+-independent component, IK(VO2), represents only a small fraction of the total IK(O2). For a voltage step from −60 to +30 mV, IK(VO2) represents 39.1 ± 0.1% (n = 10) of IK(O2) in Ca2+-free, and 26.1 ± 0.05% (n = 12) in Cd2+-containing solutions. This implies that the remaining ∼65% of IK(O2) must be Ca2+ dependent, i.e. IK(CaO2). Both of these O2-sensitive K+ currents were observed in the majority of cells tested (n = 21/22). A comparison of the absolute K+ current densities in normoxia and anoxia, as well as the difference K+ current density, is shown in Fig. 3C for cells recorded in normal Ca2+, Ca2+-free and Cd2+-containing solutions. Note that the anoxia-sensitive, or difference K+ current density, is similar in Ca2+-free and Cd2+-containing solutions (i.e. IK(VO2)).
Figure 3. Comparison of anoxia-sensitive difference currents in neonatal chromaffin cells.
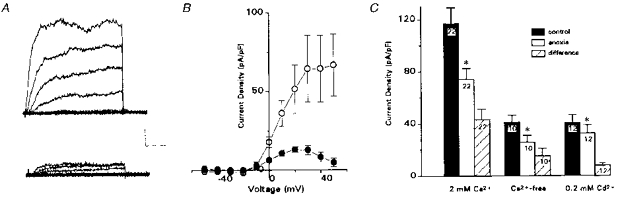
A, anoxia-sensitive difference currents, obtained by subtracting current traces recorded during anoxia from corresponding ones in normoxia, shown in normal Ca2+-containing (upper traces) and Ca2+- free (lower traces) solutions. Subtracted traces are for 10 mV incremental steps between −50 and +30 mV; holding potential was −60 mV. Note the larger component of the anoxia-sensitive difference current is Ca2+ sensitive; lower traces represent the Ca2+-independent O2-sensitive current, IK(VO2). Vertical scale bar represents 100 pA, horizontal scale bar represents 10 ms. B, current density vs. voltage plots for 6 representative cells, showing the total (mean ± s.e.m.) anoxia-sensitive component, IK(O2) (○) and IK(VO2) (•; recorded in 200 μm Cd2+). C, comparison of mean (± s.e.m.) outward current density at +30 mV, for all cells investigated in normal Ca2+ (2 mm), Ca2+-free and Cd2+-containing (200 μm) bathing solutions. * Significantly different from control group (P < 0.05). The mean current density (at +30 mV) during anoxia in the presence of Cd2+, or in Ca2+-free solutions, was significantly different from that in the presence of 2 mm Ca2+ (P < 0.01).
Neonatal AMC possess anoxia-sensitive KATP currents
KATP channels have been implicated in hypoxic chemosensitivity of adult rat AMC, based on the observation that openers of these channels prevented the hypoxia-induced rise in intracellular Ca2+ (Mochizuki-Oda et al. 1997). In addition, activation of KATP channels during hypoxia is hypothesized to play a protective role in some central neurons (Jiang et al. 1994). If similar channels were present in neonatal rat AMC, then their activation by anoxia would tend to oppose or reduce the inhibition of outward current resulting from closure of the Ca2+-dependent and Ca2+-independent channels described above. To determine if O2-sensitive AMC possess KATP channels we used both pinacidil (a KATP activator) and glibenclamide (a KATP blocker; Ashcroft & Ashcroft, 1990). In Fig. 4Aa and Ab, exposure of AMC to 100 μm pinacidil resulted in a reversible augmentation of outward current and current density. For a voltage step from −60 to +30 mV, the outward current density was increased from the control value of 128.3 ± 19.5 pA pF−1 to 173.3 ± 16.6 pA pF−1 in the presence of 100 μm pinacidil (n = 5; P < 0.05); after washout of pinacidil the current density recovered to 132.7 ± 25.8 pA pF−1. These results suggest that O2-sensitive AMC possess KATP channels.
Figure 4. Comparison of the effects of KATP channel modulators on K+ current and K+ current density recorded during normoxia or anoxia in neonatal AMC.
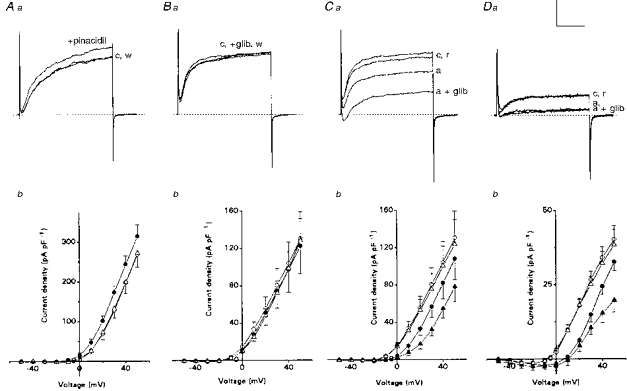
All current records are shown for cells studied under voltage clamp, and representative traces are shown for the voltage step from −60 to +30 mV. Aa and b: in normoxia, effects of pinacidil (100 μm), an activator of IK(ATP), on outward currents and current density. Note that sequential application of pinacidil (+pinacidil, •), increases outward current above control (c, ○), and the effect was reversible after washout (w, ▵). Mean (± s.e.m.) K+ current density is shown for a group of 5 cells, indicating that pinacidil significantly increased K+ current density relative to control at all voltage steps between 0 and +50 mV (P < 0.05). Ba and b: current records in normoxia for cells (n = 9) treated with control saline (c, ○), 50 μm glibenclamide (+glib, •), a blocker of IK(ATP), and after washout (w, ▵) of this drug. Mean (± s.e.m.) K+ current density vs. voltage plot for 9 cells shows that glibenclamide did not significantly affect outward current density. Ca and b: effect of simultaneous exposure to anoxia and glibenclamide on outward current for the same 9 cells as in B. Cells were sequentially exposed to normoxia/control (c, ○), anoxia (a, •), anoxia plus glibenclamide (a + glib, ▴) and recovery (r, ▵). Note that the inhibitory effect of anoxia (a) on the outward current was greatly exaggerated in the presence of glibenclamide. Current density in anoxia was significantly less than normoxic control at all voltage steps between 0 and +50 mV (P < 0.01); also, current density in anoxia plus glibenclamide was significantly less than anoxia alone, between −20 and +30 mV (P < 0.05). Da and b: outward current and current density recorded in the presence of Cd2+ (200 μm), to block Ca2+-dependent K+ currents. Note that anoxia (a) reversibly suppressed the outward current, but addition of 50 μm glibenclamide (a + glib) had no additional suppressive effect during anoxia (compare Ca), suggesting that IK(ATP) is Ca2+ dependent. Mean (± s.e.m.) K+ current density vs. voltage plot is for a group of 7 cells and shows that anoxia significantly (P < 0.01) suppressed outward currents in the presence of Cd2+ at all potentials between −10 and +50 mV. In the presence of glibenclamide, the additional suppression of outward current by anoxia was absent at all test potentials below +40 mV (P > 0.05). Symbols are the same as in C. This suggests IK(ATP) is Ca2+ sensitive. Vertical scale represents 400 pA in A, B and C and 100 pA in D. Horizontal scale represents 30 ms.
We then tested whether activity of these KATP channels was regulated by O2 tension, using the specific KATP channel blocker, glibenclamide (50 μm). Figure 4Ba and Bb shows the effect of glibenclamide on outward K+ currents recorded during normoxia (PO2 = 150 mmHg). In most (6/9) cells there was no detectable effect of glibenclamide (Fig. 4B a), though in the remaining (3/9) cells, a small, reversible suppression in K+ current was observed (not shown). Pooled data from the nine cells revealed that for a voltage step to +30 mV, the mean current density for the glibenclamide-sensitive component was 6.3 ± 4.0 pA pF−1 (range, 0 to 25.2 pA pF−1), corresponding to 4.9 ± 0.8% of the total outward current density. Plots of current density vs. voltage under normoxic conditions are shown in Fig. 4Bb for these nine cells before, during and after glibenclamide; overall, the latter had negligible effect over the voltage range −30 to +50 mV.
In contrast, exposure to anoxia had a profound effect on glibenclamide sensitivity. For example, in Fig. 4Ca, anoxia alone produced the usual suppression in K+ current, but when combined with glibenclamide the suppression was much more dramatic (compare same cell in Fig. 4Ba), and the effect was completely reversible. Pooled data from nine cells revealed that for a voltage step from −60 to +30 mV, the outward current density was reduced from 80.7 ± 17.8 pA pF−1 in normoxia to 57.0 ± 11.6 pA pF−1 in anoxia alone, and this was further reduced to 32.0 ± 6.1 pA pF−1 in anoxia plus glibenclamide (significantly different from anoxia alone; P < 0.01). The mean outward current density after reperfusion with control (normoxic) solution was 75.5 ± 16.7 pA pF−1, a value not significantly different from the initial control response. Current density vs. voltage plots are shown in Fig. 4Cb for these cells, which were exposed sequentially to normoxia, anoxia, anoxia plus glibenclamide, and then returned to normoxia. These data indicate that for a step from −60 to +30 mV, the anoxia-sensitive component of outward current IK(O2) comprised 29.4 ± 0.1% (n = 9) of the control (normoxic) K+ current in the absence of glibenclamide, compared with 60.3 ± 0.1% (n = 9) in its presence (difference significant; P < 0.05). Measurements of difference current density allowed a direct comparison of the magnitude of the glibenclamide-sensitive component in normoxia and anoxia. For a step to +30 mV, the glibenclamide-sensitive component during anoxia was 24.9 ± 7.9 pA pF−1 (n = 9), a value significantly different (P < 0.05) from that seen in normoxia (6.3 ± 4.0 pA pF−1; n = 9).
Are anoxia-sensitive KATP channels in neonatal AMC also Ca2+ sensitive?
In central neurons, the O2-sensitive KATP channels are Ca2+ sensitive (Jiang et al. 1994). To investigate whether the same is true for KATP channels in neonatal AMC, the effect of glibenclamide on O2 sensitivity was tested in Cd2+-containing solutions, which block Ca2+ entry. As shown in Fig. 4Da and b, the presence of glibenclamide (50 μm) had no additional effect on the outward current recorded during anoxia over the voltage range −30 to +30 mV when Cd2+ was present. Under these conditions, the glibenclamide-sensitive component was 6.6 ± 4.9 pA pF−1 (n = 7) in normoxia vs. 10.0 ± 3.3 pA pF−1 (n = 7) in anoxia for a voltage step from −60 to +30 mV (difference not significant; P > 0.1). This contrasts with the results reported above in normal Ca2+ (Cd2+-free) solutions (Fig. 4Ca and Cb), where glibenclamide significantly reduced the K+ current recorded in anoxia. Current density vs. voltage plots are compared in Fig. 4Db for these seven cells in the continuous presence of Cd2+-containing solutions, during exposure to normoxia (○), anoxia alone (•), anoxia plus glibenclamide (▴) and finally normoxia again (▵). However, at voltage steps > +40 mV there is an apparent activation of IK(ATP) by anoxia (Fig. 4Db), even in the presence of Cd2+. The underlying mechanism is unclear, but may involve interactions of divalent cations (eg. Cd2+ or Mg2+) with the KATP channel (Ashcroft & Ashcroft, 1990).
Pharmacology of IK(CaO2) and IK(VO2)
Since the predominant Ca2+-dependent outward current in adult AMC is carried by large conductance maxi-K+ or BK potassium channels (Neely & Lingle, 1992), which are known to be inhibited by hypoxia in carotid body type 1 cells (Wyatt & Peers, 1995), it was of interest to determine if IK(CaO2) was also carried by BK channels. To test this we used the specific blocker, iberiotoxin (IbTx), to inhibit the large conductance BK channels (Galvez et al. 1990). Exposure of neonatal AMC to IbTx (50–100 nm) resulted in a suppression of the outward current, suggesting the presence of BK channels (Fig. 5A). The IbTx-sensitive component consisted of 47.6 ± 0.04% (n = 4) of the overall outward current, for a voltage step from −60 to +30 mV. This is slightly less than the values (∼62%) reported above for the magnitude of the Ca2+-dependent K+ currents recorded in Ca2+-free or Cd2+-containing solutions, and is probably due to the persistence of small-conductance Ca2+-dependent K+ channels, which are insensitive to IbTx (Park, 1994). Exposure of neonatal AMC to anoxia in the presence of IbTx (Fig. 5A, middle trace) caused a further suppression in outward current. Plots of current density vs. voltage indicate that the suppression in IbTx-containing medium occurred at all voltage steps between −10 and +50 mV, and the effect of anoxia was reversible (Fig. 5B). Generally, washout of the effects of IbTx on the total K+ current was incomplete, even after reperfusion for 10–15 min with control solution. The contribution of both the IbTx-sensitive and IbTx-insensitive K+ currents to the total anoxia-sensitive IK(O2) was determined from the difference current measurements for the voltage step to +30 mV. The IbTx- insensitive portion of IK(O2) represented 42.2 ± 0.2% (n = 4) of the total IK(O2), and the remaining 57.8 ± 0.2% was IbTx sensitive. Since the IbTx-insensitive component of IK(O2) is similar to that obtained above for the Ca2+-independent portion of IK(O2), i.e. IK(VO2), obtained in Ca2+-free (∼39%) and Cd2+-containing (∼28%) solutions, it appears that IK(CaO2) is carried almost exclusively by IbTx-sensitive BK channels. To investigate further the pharmacology of anoxia-sensitive K+ currents, the non-specific K+ channel blocker, TEA, was used.
Figure 5. Effects of iberiotoxin (IbTx) and tetraethylammonium (TEA) on anoxia-sensitive currents in neonatal AMC.
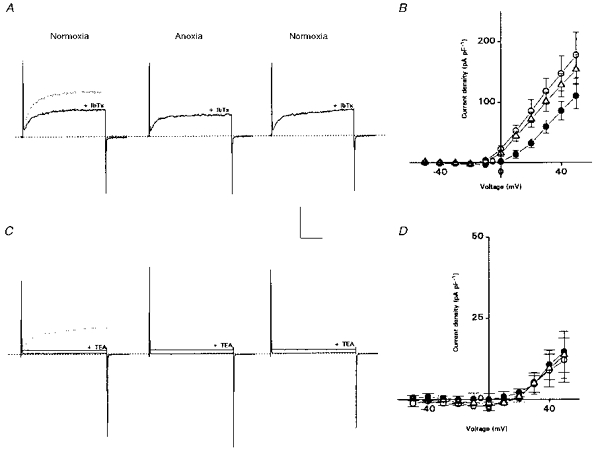
A, in normoxic conditions, IbTx (100 nm) suppressed outward current at +30 mV (left traces); upper trace is control record before IbTx. In the presence of IbTx, anoxia still suppressed outward current reversibly, though washout of the effects of IbTx was incomplete (not shown). B, mean current density (± s.e.m.) vs. voltage plots for 4 cells, in the presence of IbTx. Symbols are same as in Fig. 1. Anoxia significantly suppressed outward current at all voltage steps between 0 and +50 mV (P < 0.05). C, effects of TEA on outward currents. Addition of TEA (20 mm) suppressed ∼96% of outward current (left traces; upper dotted trace is control current for step to +30 mV). Anoxia had no additional effect on outward current in the presence of TEA (middle traces; only steps to +30 and +50 mV shown). D, mean current density vs. voltage plots for a group of 6 cells with similar initial densities; TEA was present throughout. Vertical and horizontal scale bars represent 200 pA and 20 ms, respectively, for both A and C.
Figure 5C (left traces) shows that 20 mm TEA reduced substantially the outward current in a cell that was previously identified as anoxia sensitive. In nine such cells, 10–20 mm TEA caused a mean suppression of outward current by 95.7 ± 1.4% of control, as measured during a voltage step from −60 to +30 mV. However, in the presence of 10–20 mm TEA, anoxia had no additional effect on the residual K+ currents in eight out of nine cells tested (e.g. Fig. 5C (middle traces) and D). Since the cell shown in Fig. 5C contained both IK(CaO2) and IK(VO2) components of outward current (not shown), it appears that both components are sensitive to 10–20 mm TEA. Figure 5D also indicates that the remaining outward current seen at more depolarized potentials (> 20 mV) is anoxia insensitive. Since anoxia activates IK(ATP) in these cells, the data also suggest that IK(ATP) is TEA sensitive.
O2 sensitivity in AMC with different types of BK currents
In adult rat adrenal chromaffin cells two types of BK currents have been described. One is a non-inactivating current (IBK(s); in ∼9% of adult AMC) and the other is a slowly inactivating current (IBK(i); in ∼75% of adult AMC); the remaining ∼15% of cells express both currents (Solaro et al. 1995). Since hypoxic suppression of outward currents was not observed in all neonatal AMC (∼80% respond), the question arises whether O2 chemosensitivity was restricted to a specific population of BK-expressing cells (i.e. BKi or BKs). To identify BKi- and BKs-expressing cells, a voltage-clamp protocol reported to enhance BK currents in these cells was applied (Solaro et al. 1995). Cells were held at −70 mV and stepped to 0 mV for 50 ms to load them with Ca2+, and then immediately stepped to +80 mV for 500 ms to identify the type of BK current (see Fig. 6). Out of seventeen cells examined, twelve contained predominantly BKs currents, i.e. they showed no inactivation of outward current during the 500 ms step to +80 mV (e.g. Fig. 6Aa, upper trace). The remaining five cells contained a slowly inactivating component of outward current, presumably due to the presence of IBK(i) (e.g. Fig. 6Ba, upper trace). It was found that both cells containing BKs (10/12 cells) and BKi (4/5 cells) responded to hypoxia with a suppression of outward current (Fig. 6Ab and Bb, respectively). These data suggest that O2 sensitivity can occur in neonatal rat AMC which express either IBK(s) or IBK(i) currents.
Figure 6. Chromaffin cells expressing either sustained or inactivating BK currents are anoxia sensitive.
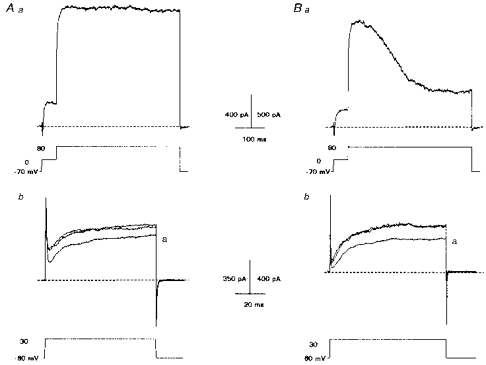
Aa and Ba, protocol used to identify AMC that expressed a sustained BK (A) or inactivating BK (B) current. Cells were initially held at −70 mV and briefly stepped to 0 mV for 50 ms (lower traces), before a final step to +80 mV for 500 ms. Both cells contained outward current that was reversibly suppressed by anoxia (a) as shown in Ab (for cell in Aa) and Bb (for cell in Ba). Traces shown for voltage step from −60 to +30 mV.
Do IK(CaO2) or IK(VO2) mediate receptor potential in AMC during anoxia?
We previously reported that neonatal AMC depolarize during hypoxia, and that this depolarization was associated with a conductance decrease (Thompson et al. 1997). These observations are consistent with the closure of, for example, K+ channels as a general mechanism for hypoxia-induced depolarization in O2-chemoreceptive cells (Gonzalez et al. 1994). However, given a resting potential in neonatal AMC of ∼-60 mV, it is unclear whether inhibition of the O2-sensitive currents described above, i.e. IK(CaO2) and IK(VO2), forms the basis of the initial membrane depolarization or receptor potential during hypoxia. To test whether inhibition of IK(CaO2) and/or IK(VO2) may contribute to the initial depolarization, neonatal AMC were exposed to solutions containing Cd2+ (200 μm) and/or TEA (20 mm), while the membrane potential was monitored using perforated-patch recording in current clamp mode.
As illustrated in Fig. 7A and B (left portion of trace), exposure of neonatal AMC to anoxia caused a significant (and reversible) depolarization from a mean resting potential of −57.6 ± 1.9 to −46.3 ± 1.8 mV (n = 11; P < 0.01). However, subsequent exposure to 200 μm Cd2+ had no effect on membrane potential (−58.3 ± 2.6 mV after Cd2+, n = 6; see Fig. 7A, right portion of trace), nor did it prevent the anoxia-induced depolarization (mean membrane potential during anoxia in the presence of Cd2+ =−49.0 ± 2.5 mV, n = 6; difference significant from Cd2+ control, P < 0.01). Since voltage-dependent Na+ channels were blocked in these experiments with 0.5 μm TTX, the recorded spike activity in Fig. 7 was probably due to Ca2+ entry, and this was abolished in the presence of Cd2+ (see Fig. 7A; note spike activity in left but not right portions of the trace). These results suggest that IK(CaO2) does not contribute significantly to the initial depolarization (or receptor potential) in neonatal AMC during anoxia, nor to the resting membrane potential. Confirmation of this point was obtained using 50 nm charybdotoxin (ChTx) to block the large conductance BK channels that mediate IK(CaO2). For these experiments ChTx was used (instead of IbTx) since it is a much faster blocker of BK channels (Galvez et al. 1990), and therefore the effects of direct BK channel inhibition could be more easily assessed. In Fig. 7B, 50 nm ChTx failed to depolarize neonatal AMC, and also failed to prevent the anoxia-induced depolarization, even in this cell which had a relatively low initial resting potential (−40 mV). This result was seen in all cells tested (n = 3).
In contrast to the above results, the more general K+ channel blocker TEA (10–20 mm) depolarized neonatal AMC from −57.6 ± 1.9 to −48.1 ± 5.2 mV (n = 5; difference significant, P < 0.01; see Fig. 7C). However, even in the presence of 20 mm TEA, exposure to anoxia caused a further significant depolarization to −40.8 ± 4.5 mV (P < 0.01), and the effect was reversible (Fig. 7C; right portion of trace). Additionally, when TEA and Cd2+ were applied together, the membrane depolarized, but the depolarizing effects of anoxia still persisted in the presence of these drugs (n = 3; not shown). These data suggest that though some TEA-sensitive K+ channels are open at rest, they are not responsible for the anoxia-induced depolarization.
Blockade of other K+ channel subtypes with 2 mm 4-AP did not alter resting membrane potential of neonatal AMC (n = 4; mean membrane potential was −59.7 ± 6.4 before, and −57.7 ± 5.6 mV after 4-AP). Further, in the presence of 2 mm 4-AP, anoxia still depolarized AMC from −59.7 ± 6.4 to −46.0 ± 5.4 mV (n = 4), an effect similar to that seen in the absence of 4-AP, where the membrane potential depolarized to −47.7 ± 6.1 mV (n = 4; see Fig. 7D). These observations suggest there is a distinct O2-sensing mechanism in neonatal AMC that controls at least part of the initial depolarization or receptor potential during anoxia, and that is insensitive to conventional blockers of voltage-dependent K+ channels.
Does a cationic current contribute to O2 sensitivity in rat AMC?
In guinea-pig AMC, a cationic current that was activated by hypoxia was recently described (Inoue et al. 1998). Since during voltage-clamp studies in Cd2+-containing (or Ca2+-free) solutions we often observed a slight enhancement of inward current during hypoxia (e.g. Fig. 2D and 4Db), we wondered whether a similar cationic current may contribute to the receptor potential in rat AMC. To test this possibility we examined the effects of anoxia on AMC under current clamp, while perfusing a Na+/Ca2+-free bathing solution. Interestingly, under these conditions anoxia-sensitive AMC hyperpolarized to −63.1 ± 9.2 mV (n = 4) from the control membrane potential of −52.6 ± 6.5 mV (P < 0.05; Fig. 7E). Since this hyperpolarization was also observed in Na+-free solutions that contained Ca2+ (not shown), it appears that a substantial Na+ leak current contributes to the resting membrane potential of AMC. Application of an anoxic stimulus in the presence of a Na+/Ca2+-free bathing solution revealed that AMC depolarized from −63.1 ± 9.2 to −51.6 ± 9.4 mV (n = 4; Fig. 7E). Thus, the anoxia-induced depolarization was reduced, but not abolished, in the absence of extracellular Na+ and Ca2+. The mean depolarization in the control solution was 24.2 ± 2.6 mV (n = 4) compared with 11.5 ± 2.5 mV for the same cells studied in the absence of external Na+ and Ca2+ (e.g. Fig. 7E; P < 0.01).
Activation of KATP attenuates the anoxia-induced depolarization
We also investigated whether KATP channels were active at the resting potential of O2-sensitive AMC and could therefore influence the magnitude of the receptor potential. Perfusion with extracellular solution containing the KATP channel blocker, glibenclamide (50 μm), had no significant effect (P > 0.2) on membrane potential (Fig. 7F); the mean resting potential was −52.6 ± 3.2 mV before, and −54.0 ± 3.5 mV after glibenclamide for a group of eleven cells that were O2 sensitive. Thus, KATP channels do not appear to contribute significantly to the resting potential of neonatal AMC during normoxia.
To determine if activation of IK(ATP) by anoxia (see above) functions to attenuate the receptor potential, the anoxia-induced depolarization was compared in the presence and absence of glibenclamide (50 μm). In the absence of glibenclamide, AMC depolarized by an average of 16.6 ± 2.5 mV (n = 11) during anoxia. However, this depolarization was significantly (P < 0.01) enhanced when anoxia was applied in the presence of glibenclamide (mean = 23.3 ± 3.1 mV; n = 11; e.g. Fig. 7F). Since IK(ATP) also appears to be TEA sensitive (see above), it was surprising that the anoxia-induced depolarization was not similarly enhanced in the presence of TEA. Although the reasons for this are unclear, the two drugs block IK(ATP) via different mechanisms and, unlike TEA, glibenclamide had no effect on resting membrane potential (compare Fig. 7C with F). These data suggest that, although IK(ATP) does not appear to contribute to the resting potential of AMC during normoxia, its activation by anoxia may serve to reduce the magnitude of the receptor potential.
DISCUSSION
The goal of the present study was to investigate the type(s) of voltage-activated K+ currents that mediate O2 chemosensitivity in neonatal rat adrenal chromaffin cells (AMC), and to determine whether these currents also contribute to the genesis of the hypoxia-induced depolarization or receptor potential (see Thompson et al. 1997). These hypoxia-sensing mechanisms appear to mediate the vital catecholamine surge that enables the neonate to survive the hypoxic stress associated with the transition to extrauterine life (Seidler & Slotkin, 1985; Slotkin & Seidler, 1988; Thompson et al. 1997). Our results from voltage-clamp studies indicated that the suppression of voltage-dependent outward K+ current by anoxia is the net result of the differential modulation of several K+ currents and comprises: (i) anoxic suppression of the large conductance Ca2+-dependent K+ current, IK(CaO2); (ii) anoxic suppression of a Ca2+-independent K+ current, IK(VO2); and (iii) anoxic activation of a glibenclamide-sensitive K+ current, IK(ATP). Additionally, results from current clamp recordings indicated that alternative mechanisms to these voltage-dependent and anoxia-sensitive K+ currents must be invoked to account for the genesis of the receptor potential seen during exposure of neonatal rat AMC to anoxia.
Anoxic suppression of outward current in neonatal AMC results primarily from the inhibition of large conductance, Ca2+-dependent K+ channels, i.e. BK or maxi-K+ channels, which mediate IK(CaO2) in this study. Though we cannot formally rule out an indirect effect due to anoxic suppression of Ca2+ currents (see Lopez-Barneo, 1996), in preliminary studies there was no consistent effect of anoxia on Ca2+ currents (R. J. Thompson & C. A. Nurse, unpublished observations), and we previously showed that entry of extracellular calcium and catecholamine secretion was enhanced in these cells by hypoxia (Thompson et al. 1997). When BK currents were eliminated, by using Ca2+-free solutions or by the addition of either Cd2+ or iberiotoxin (IbTx), a smaller Ca2+-independent component (IK(VO2)) amounting to ∼35% of the total anoxia-sensitive outward current (IK(O2)) persisted. Since TEA (10–20 mm) abolished almost all of the outward current, and anoxia had a negligible effect on the residual currents when TEA was present, it appears that both IK(CaO2) and IK(VO2) are TEA sensitive. It is noteworthy that in PC12 cells, a cell line with an adrenomedullary chromaffin cell origin, hypoxia suppresses a Ca2+-independent and TEA-sensitive outward current (Zhu et al. 1996), raising the possibility that it is similar to IK(VO2) investigated in the present study. In contrast, however, hypoxia was reported to stimulate large conductance, Ca2+-dependent K+ channels in PC12 cells, though the pharmacology of this current was not reported (Conforti & Millhorn, 1997). It is possible that this anoxia-activated current is similar to IK(ATP) described here (see below) and in central neurons (Haddad & Jiang, 1997).
Anoxic activation of KATP currents
In addition to IK(CaO2) and IK(VO2), we observed a third voltage dependent, Ca2+-sensitive K+ current, IK(ATP), which was regulated by PO2. The presence of this current was inferred from experiments where the total outward current was enhanced by pinacidil (100 μm), a KATP channel activator, and where the anoxic suppression of outward current was augmented by glibenclamide (50 μm), a KATP blocker (see Ashcroft & Ashcroft, 1990). Unlike the suppressive effects of anoxia on the other two currents, IK(ATP) was activated by anoxia in these cells, suggesting more KATP channels became available or were opened at low PO2. Since this augmentation of KATP by anoxia was absent in a Cd2+- or TEA-containing bathing solution (but persisted in the presence of the BK channel blocker, ChTx; not shown), it appears that IK(ATP) is both Ca2+ and TEA sensitive. Similar results have been reported for O2-regulated KATP channels described in central neurons (Jiang et al. 1994; Haddad & Jiang, 1997). Taken together, these results support the conclusion that anoxia augments KATP channels, but attenuates both Ca2+-dependent (IK(CaO2)) and delayed rectifier type (IK(VO2)) K+ currents in neonatal AMC.
Interestingly, the augmentation of KATP by anoxia observed in this study is in contrast with a recent suggestion that hypoxia inhibits KATP currents in adult rat chromaffin cells (Mochizuki-Oda et al. 1997). This interpretation was based on the finding that cromakalim, an activator of KATP channel currents, prevented the hypoxia-induced rise in intracellular Ca2+; however, a direct inhibitory effect of hypoxia on KATP currents was not demonstrated. It is also noteworthy that Mochizuki-Oda et al. (1997) observed hypoxic chemosensitivity in adult rat chromaffin cells, whereas we did not (Thompson et al. 1997; see also Mojet et al. 1997). It is possible that use of longer-term cultures (up to 7 days) by Mochizuki-Oda et al. (1997), and the resulting longer period of denervation in vitro, resulted in recovery of hypoxic sensing in these cells (see Slotkin & Seidler, 1988).
Possible functional role of anoxic modulation of K+ currents in neonatal AMC
In the present study, inhibition of Ca2+-dependent K+ (BK) channels was the major cause of the anoxic suppression of outward current in neonatal AMC. Similar BK channels are thought to participate in hypoxic chemosensitivity in rat carotid body type 1 cells (Wyatt & Peers, 1995; Jackson & Nurse, 1997), and are generally accepted to play important roles in action potential repolarization (Pancrazio et al. 1994), and in the ability of adult rat AMC to fire repetitive action potentials (Solaro et al. 1995). Thus, hypoxic inhibition of IK(CaO2) may result in broadening of the action potential, leading to the observed rise in intracellular Ca2+ and catecholamine release (Thompson et al. 1997; see also Mojet et al. 1997). Additionally, hypoxic inhibition of the delayed rectifier type K+ current, IK(VO2) may also contribute to action potential broadening.
On the other hand, anoxia was found to activate a Ca2+- and glibenclamide-sensitive KATP current in neonatal AMC. This activation tended to counteract or blunt the depolarizing effects of anoxia. Interestingly, in catecholaminergic neurons of the substantia nigra, hypoxic activation of Ca2+- and glibenclamide-sensitive KATP channels leads to membrane hyperpolarization, which is presumed to serve a protective role during metabolic stress associated with hypoxia and ischaemia (Jiang et al. 1994; Haddad & Jiang, 1997). Conceivably, hypoxia-sensitive KATP channels in AMC may play a similar role during periods of prolonged neonatal hypoxia, e.g. during birth or repetitive apnoeic events. Activation of KATP channels by the concomitant fall in PO2 and ATP, and the increase in intracellular Ca2+, could limit membrane depolarization and prevent CA depletion from AMC, allowing them to maintain an influence on respiratory and cardiovascular physiology during subsequent hypoxic events (see Seidler & Slotkin, 1985; Slotkin & Seidler, 1988).
Origin of the receptor potential
In this study blockers of voltage-dependent, O2-sensitive currents did not prevent the anoxia-induced depolarization in neonatal AMC. For example, direct inhibition of BK currents by charybdotoxin (50 nm) did not induce depolarization of neonatal AMC, suggesting that most of these channels are closed at the cell's resting potential (∼-58 mV). Further, the presence of the relatively non-specific K+ channel blocker TEA, which blocked all anoxia-sensitive K+ channels in voltage clamp studies, also failed to prevent the anoxia-induced depolarization. However, TEA itself caused a slight depolarization, possibly due to closure of K+ channels that were open at rest.
During exposure of AMC to anoxia, an apparent activation of a small inward current was frequently observed in voltage clamp studies when most of the outward current was blocked (e.g. Fig. 2D). This current was not studied in detail and its origin remains uncertain. On the one hand, it may result simply from inhibition of the residual outward current; alternatively, it may be due to activation of a cationic current, perhaps similar to the one recently described in hypoxia-sensitive guinea-pig AMC (Inoue et al. 1998). In the present study, the anoxia-induced depolarization of rat AMC persisted in the absence of extracellular Na+ and Ca2+, though the magnitude of the depolarization was decreased. Thus, activation of an inward cationic current by anoxia does not appear to be responsible for the receptor potential. Furthermore, these experiments suggest that the genesis of the receptor potential does not arise from the inhibition of a Na+-K+-ATPase, since under Na+-free conditions this electrogenic pump is likely to be inactive.
In summary, it appears that chromaffin cells have evolved complex mechanisms for sensing O2 and regulating catecholamine secretion in the perinatal period. These mechanisms involve the differential modulation of several K+ channel subtypes. Additional work, aided by single channel analysis, is required to elucidate the molecular mechanisms by which these channels are regulated by low PO2, and the basis of the hypoxia-induced depolarization or receptor potential.
Acknowledgments
We thank Cathy Vollmer for technical assistance in preparing and maintaining the cultures and Dr Jan Huizinga for providing pinacidil. This work was supported by an operating and equipment grant from the Natural Sciences and Engineering Research Council of Canada (NSERC). In addition R. J. T. is supported by an NSERC postgraduate scholarship.
References
- Ashcroft SJH, Ashcroft FM. Properties and functions of ATP-sensitive K- channels. Cell Signalling. 1990;2:197–214. doi: 10.1016/0898-6568(90)90048-f. 10.1016/0898-6568(90)90048-F. [DOI] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type 1 cells. The Journal of Physiology. 1997;450:33–61. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type 1 cells. The Journal of Physiology. 1994;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conforti L, Millhorn DE. Selective inhibition of slow-inactivating voltage-dependent K+ channel in rat PC12 cells by hypoxia. The Journal of Physiology. 1997;502:293–305. doi: 10.1111/j.1469-7793.1997.293bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galvez A, Gimenez-Gallego G, Reuben G, Roy-Contancin L, Fingenbaum P, Kaczorowski GJ, Garcia ML. Purification and characterization of a unique, potent, peptidyl probe for the high conductance calcium activated potassium channel from venom of the scorpion. Buthus tamulus. Journal of Biological Chemistry. 1990;265:11083–11090. [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: from natural stimuli to sensory discharges. Physiological Reviews. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Haddad GG, Jiang C. O2-sensing mechanisms in excitable cells: role of plasma membrane K+ channels. Annual Review of Physiology. 1997;59:23–43. doi: 10.1146/annurev.physiol.59.1.23. 10.1146/annurev.physiol.59.1.23. [DOI] [PubMed] [Google Scholar]
- Inoue M, Fujishiro N, Imanaga I. Hypoxia and cyanide induce depolarization and catecholamine release in dispersed guinea-pig chromaffin cells. The Journal of Physiology. 1998;507:807–818. doi: 10.1111/j.1469-7793.1998.807bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson A, Nurse CA. Dopaminergic properties of cultured rat carotid body chemoreceptors grown in normoxic and hypoxic environments. Journal of Neurochemistry. 1997;69:645–654. doi: 10.1046/j.1471-4159.1997.69020645.x. [DOI] [PubMed] [Google Scholar]
- Jiang C, Sigworth FJ, Haddad GG. Oxygen deprivation activates an ATP-inhibitable K+ channel in substantia nigra neurons. Journal of Neuroscience. 1994;14:5590–5602. doi: 10.1523/JNEUROSCI.14-09-05590.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Barneo J. Oxygen-sensing by ion channels and regulation of cellular functions. Trends in Neurosciences. 1996;19:435–440. doi: 10.1016/0166-2236(96)10050-3. [DOI] [PubMed] [Google Scholar]
- Lopez-Lopez J, Gonzalez C, Urena J, Lopez-Barneo J. Low PO2 selectively inhibits K channel activity in chemoreceptor cells of the mammalian carotid body. Journal of General Physiology. 1989;93:1001–1015. doi: 10.1085/jgp.93.5.1001. 10.1085/jgp.93.5.1001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mochizuki-Oda N, Takeuchi Y, Matsumura K, Oosawa Y, Watanabe Y. Hypoxia-induced catecholamine release and intracellular Ca2+ increase via suppression of K+ channels in cultured rat adrenal chromaffin cells. Journal of Neurochemistry. 1997;69:377–387. doi: 10.1046/j.1471-4159.1997.69010377.x. [DOI] [PubMed] [Google Scholar]
- Mojet MH, Mills E, Duchen MR. Hypoxia-induced catecholamine secretion in isolated newborn rat adrenal chromaffin cells in mimicked by inhibition of mitochondrial respiration. The Journal of Physiology. 1997;504:175–189. doi: 10.1111/j.1469-7793.1997.175bf.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neely A, Lingle CJ. Two components of calcium-activated potassium current in rat adrenal chromaffin cells. The Journal of Physiology. 1992;453:97–131. doi: 10.1113/jphysiol.1992.sp019220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pancrazio JJ, Johnson PA, Lynch C., III A major role for calcium-dependent potassium current in action potential repolarization in adrenal chromaffin cells. Brain Research. 1994;668:246–251. doi: 10.1016/0006-8993(94)90531-2. 10.1016/0006-8993(94)90531-2. [DOI] [PubMed] [Google Scholar]
- Park Y. Ion selectivity and gating of small conductance Ca2+-activated K+ channels in cultured rat adrenal chromaffin cells. The Journal of Physiology. 1994;481:555–570. doi: 10.1113/jphysiol.1994.sp020463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peers C. Hypoxic suppression of K+ currents in type 1 carotid body cells: selective effect on the Ca2+-activated K+ current. Neuroscience Letters. 1990;119:253–356. doi: 10.1016/0304-3940(90)90846-2. 10.1016/0304-3940(90)90846-2. [DOI] [PubMed] [Google Scholar]
- Peers C, Buckler KJ. Transduction of chemostimuli by the type 1 carotid body cell. Journal of Membrane Biology. 1995;144:1–9. doi: 10.1007/BF00238411. [DOI] [PubMed] [Google Scholar]
- Post JM, Hume JR, Archer SL, Weir EK. Direct role for potassium channel inhibition in hypoxic pulmonary vasoconstriction. American Journal of Physiology. 1992;262:C882–890. doi: 10.1152/ajpcell.1992.262.4.C882. [DOI] [PubMed] [Google Scholar]
- Seidler FJ, Slotkin TA. Adrenomedullary function in the neonatal rat: responses to acute hypoxia. The Journal of Physiology. 1985;385:1–16. doi: 10.1113/jphysiol.1985.sp015536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. Adrenomedullary catecholamine release in the fetus and newborn: secretory mechanisms and their role in stress and survival. Journal of Developmental Physiology. 1988;10:1–16. [PubMed] [Google Scholar]
- Solaro CR, Prakriya M, Ding JP, Lingle CJ. Inactivating and noninactivating Ca2+- and voltage-dependent K+ current in rat adrenal chromaffin cells. Journal of Neuroscience. 1995;15:6110–6123. doi: 10.1523/JNEUROSCI.15-09-06110.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Jackson A, Nurse CA. Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. The Journal of Physiology. 1997;498:503–510. doi: 10.1113/jphysiol.1997.sp021876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson RJ, Nurse CA. Hypoxia-sensitive K+ channels in rat adrenal chromaffin cells. Society for Neuroscience Abstracts. 1997;23:1746. [Google Scholar]
- Wyatt CN, Peers C. Ca2+-activated K+ channels in isolated type 1 cells of the neonatal rat carotid body. The Journal of Physiology. 1995;483:599–565. doi: 10.1113/jphysiol.1995.sp020606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngson C, Nurse CA, Yeger H, Cutz E. Oxygen sensing in airway chemoreceptors. Nature. 1993;365:153–155. doi: 10.1038/365153a0. 10.1038/365153a0. [DOI] [PubMed] [Google Scholar]
- Zhu WH, Conforti L, Czyzyk-Kreska MF, Millhorn DE. Membrane depolarization in PC-12 cells during hypoxia is regulated by an O2-sensitive K+ current. American Journal of Physiology. 1996;271:C658–665. doi: 10.1152/ajpcell.1996.271.2.C658. [DOI] [PubMed] [Google Scholar]