

Abstract
We examined the effects of noradrenaline on steady-state intracellular pH (pHi) and the recovery of pHi from internal acid loads imposed by the NH4+ prepulse technique in hippocampal CA1 neurones acutely dissociated from adult rats.
Under nominally HCO3−-free conditions, acid extrusion was accomplished by a Na+-dependent mechanism, probably the amiloride-insensitive variant of the Na+-H+ exchanger previously characterized in both fetal and adult rat hippocampal neurones. In the presence of external HCO3−, acid extrusion appeared to be supplemented by a Na+-dependent HCO3−-Cl− exchanger, the activity of which was dependent upon the absolute level of pHi.
Noradrenaline evoked a concentration-dependent and sustained rise in steady-state pHi and increased rates of pHi recovery from imposed intracellular acid loads. The effects of noradrenaline were not dependent upon the presence of external HCO3− but were blocked by substituting external Na+ with N-methyl-D-glucamine, suggesting that noradrenaline acts to increase steady-state pHi by increasing the activity of the Na+-H+ exchanger.
The effects of noradrenaline on steady-state pHi and on rates of pHi recovery from imposed acid loads were mimicked by β1- and β2-, but not α-, adrenoceptor agonists. The β-adrenoceptor antagonist propranolol blocked the ability of noradrenaline to increase both steady-state pHi and rates of pHi recovery from acid loads.
The effects of noradrenaline on steady-state pHi and on pHi recovery rates following acid loads were not dependent on changes in [Ca2+]i. However, the effects of noradrenaline were blocked by pre-treatment with the adenylate cyclase inhibitor 2′,5′-dideoxyadenosine and the cAMP-dependent protein kinase inhibitors Rp-adenosine-3′,5′-cyclic monophosphorothioate (sodium salt; Rp-cAMPS) and N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulphonamide (H-89).
Forskolin, an activator of endogenous adenylate cyclase, and 3-isobutyl-1-methylxanthine, a phosphodiesterase inhibitor, mimicked the ability of noradrenaline to increase both steady-state pHi and rates of pHi recovery from imposed acid loads, as did Sp-cAMPS, a selective activator of cAMP-dependent protein kinase. The effect of forskolin on steady-state pHi was blocked by pre-treatment with Rp-cAMPS whereas the effect of Sp-cAMPS was enhanced by pre-treatment with the protein phosphatase inhibitor, okadaic acid.
Noradrenaline also increased steady-state pHi and rates of pHi recovery from imposed acid loads in cultured postnatal rat hippocampal neurones. In this preparation, the effects of noradrenaline were occluded by 18–24 h pre-treatment with cholera toxin.
We conclude that noradrenaline increases the activity of the Na+-H+ exchanger in rat hippocampal neurones, probably by inducing an alkaline shift in the pHi dependence of the antiport, thereby raising steady-state pHi. The effects of noradrenaline are mediated by β-adrenoceptors via a pathway which involves the α-subunit of the stimulatory G-protein Gs (Gsα), adenylate cyclase, cAMP and the subsequent activation of cAMP-dependent protein kinase which, in turn, may phosphorylate the exchange mechanism.
It is now apparent that changes in intracellular pH (pHi) can both reflect and influence neuronal function. Activation of either γ-aminobutyric acidA (GABAA) or N-methyl-D-aspartate receptors, for example, leads to falls in pHi attributable, respectively, to a net efflux of HCO3− ions across GABAA receptor-activated anion channels or to a rise in [Ca2+]i and (possibly) subsequent activation of Ca2+-H+ exchange (see Kaila, 1994; Trapp et al. 1996). Changes in pHi evoked by amino acid neurotransmitters may, in turn, represent a physiologically important facet of their mechanism of action given that neuronal ionic conductances, the activities of intracellular second messenger systems, and buffering and transport mechanisms for various intracellular ions are all sensitive to changes in pHi (e.g. Dipolo & Beaugé, 1982; Daumas & Andersen, 1993; Vignes et al. 1996; Tombaugh & Somjen, 1997).
Intracellular pH is critically dependent on the activity of pHi-regulating mechanisms. In peripheral cell types, it is well established that the activities of pHi-regulating mechanisms, including Na+-H+ and HCO3−-Cl− exchangers, are highly controlled (reviewed by Mahnensmith & Aronson, 1985; Grinstein et al. 1989; Noël & Pouysségur, 1995; Wakabayashi et al. 1997). Depending upon cell type, the activities of these exchangers can be modulated not only by changes in extracellular pH (pHo) and pHi but also by a wide variety of external stimuli, including transmitters, mitogens and hormones. Furthermore, it is clear that surface receptors for these and other extracellular agents are coupled to changes in the activities of pHi-regulating mechanisms through divergent intracellular signalling pathways. The importance of the modulation of pHi by changes in the activities of pHi-regulating mechanisms has been highlighted in studies of cell metabolism, growth and proliferation, where pHi shifts transform the functional state of cells (Busa & Nuccitelli, 1984; Mahnensmith & Aronson, 1985; Grinstein et al. 1989; Wang et al. 1997). However, in contrast to peripheral cell types, the possibility that external stimuli such as neurotransmitters may change pHi in mammalian central neurones by specifically altering the activities of pHi-regulating mechanism(s) has not been explored. In the present study, we have investigated the ability of noradrenaline to modulate the activities of the acid extrusion mechanisms present in hippocampal CA1 neurones acutely dissociated from adult rats. Not only does the hippocampus receive a dense noradrenergic innervation from the locus coeruleus (Loy et al. 1980) but also catecholamines, including noradrenaline, are known to affect the activities of pHi-regulating mechanisms in a variety of peripheral cell types (see Discussion).
Portions of this work have been presented in abstract form (Smith & Church, 1997; Brett & Church, 1998).
METHODS
Cell preparation
Acutely dissociated adult rat hippocampal CA1 neurones
Acutely dissociated adult rat hippocampal CA1 neurones were prepared using a modification of the procedure described by Mody et al. (1989). Male Wistar rats (200–240 g) were anaesthetized with 3% halothane in air, decapitated and the brains rapidly removed and placed in ice-cold (4–6°C) HCO3−/CO2-buffered saline (see below) equilibrated with 5% CO2-95% O2. Transverse hippocampal slices (450 μm) were then prepared and allowed to recover for at least 1 h at 32°C in HCO3−/CO2-buffered saline. To isolate CA1 neurones, three slices were removed from the incubation chamber and enzymatically digested for 30 min at 32°C in 2 ml of HCO3−/CO2-buffered saline containing 1.5 mg ml−1 protease type XIV (Sigma Chemical Co.). The CA1 regions were then removed under a dissecting microscope and triturated with fire-polished Pasteur pipettes in 0.5 ml of Hepes-buffered saline (see below) to which 3 mm NaHCO3 had been added in place of NaCl. The triturated suspension was deposited onto an 18 mm poly-D-lysine-coated glass coverslip mounted in a perfusion chamber so as to form the base of the chamber. Neurones were allowed to adhere to the substrate for 15 min at room temperature (18–22°C) before being loaded with fluorophore.
Cultured postnatal rat hippocampal neurones
Primary cultures of postnatal hippocampal neurones were prepared from 4- to 5-day-old Wistar rat pups as previously described (Sidky & Baimbridge, 1997). In brief, animals were anaesthetized with CO2, decapitated and the hippocampi removed. Neurones were plated at a density of 3 × 105 cells cm−2 on glass coverslips, treated with 5-fluorodeoxyuridine to arrest glial cell proliferation and were maintained in a 5% CO2 atmosphere at 36°C in serum-free, N2-supplemented Dulbecco's modified Eagle's medium (Life Technologies, Burlington, ON, Canada) containing 22 mm NaHCO3. Cultured neurones were used 7–14 days after plating in experiments in which prolonged (18–24 h) pre-incubation with cholera toxin was required.
Solutions and test compounds
The standard HCO3−/CO2-free perfusion medium contained (mm): NaCl, 136.5; KCl, 3; CaCl2, 2; NaH2PO4, 1.5; MgSO4, 1.5; d-glucose, 17.5; and Hepes, 10; titrated to pH 7.35 (at 37°C) with 10 m NaOH. In standard HCO3−/CO2-containing media, Hepes was isosmotically replaced by NaCl and solutions contained 20 mm NaHCO3, by equimolar substitution for NaCl, together with the constituents listed above (pH 7.35 after equilibration with 5% CO2-95% air). During perfusion with HCO3−-containing media, the atmosphere in the recording chamber contained 5% CO2-95% air.
Solutions containing 20 mm NH4Cl were prepared by equimolar substitution for NaCl. When external Na+ was omitted, N-methyl-D-glucamine (NMDG+) or Li+ were employed as substitutes in Hepes-buffered media; choline was employed as the substitute in HCO3−/CO2-buffered media. When external Cl− was omitted, gluconate was substituted. For Ca2+-free media, Ca2+ was omitted, [Mg2+] was increased to 3.5 mm and 200 μm EGTA was added. The pH of each solution was re-measured following every experiment.
5-(N-ethyl, N-isopropyl)-amiloride (EIPA) and 4,4′-diisothiocyanatostilbene-2,2′-disulphonic acid (DIDS) were prepared as 50 and 100 mm stock solutions, respectively, in dimethylsulphoxide (DMSO) and stored at −80°C. Forskolin, 2′,5′-dideoxyadenosine, 1′,9′-dideoxyforskolin and 3-isobutyl-1-methylxanthine (IBMX) were dissolved in DMSO to stock concentrations of 50 mm, whilst N-[2-(p-bromocinnamylamino)ethyl]-5-isoquinolinesulphonamide (H-89) was dissolved in DMSO to a stock concentration of 10 mm. Noradrenaline and other adrenoceptor agonists and antagonists were dissolved in ultrapure water (Milli-Q UF Plus, Millipore Ltd) and stored as 50 mm stock solutions with 5 mm sodium ethylenediaminetetraacetate (NaEDTA); on the day of an experiment, stock solutions were dissolved in physiological media to the desired test concentration with 0.3 mm ascorbic acid. NaEDTA and ascorbic acid were employed to delay the oxidative degradation of the compounds, especially important at 37°C (Hughes & Smith, 1978). The Sp- and Rp-isomers of adenosine-3′,5′-cyclic monophosphorothioate (Sp- and Rp-cAMPS, respectively; sodium salts) were dissolved in ultrapure water to 25 or 50 mm stock solutions. Stock solutions of cholera toxin were prepared in ultrapure water at 500 μg ml−1. Control experiments were performed with DMSO, NaEDTA and ascorbic acid at their final working concentrations and none of the agents affected steady-state pHi or rates of pHi recovery from imposed intracellular acid loads (data not shown). Perfusion lines were replaced following each experiment.
Compounds were obtained from Research Biochemicals International with the exceptions of cholera toxin, terbutaline and DIDS (Sigma Chemical Co.); 2′,5′-dideoxyadenosine and H-89 (Biomol Research Laboratories Inc., Plymouth Meeting, PA, USA); Rp- and Sp-cAMPS, Na+ salts (Biolog Life Science Institute, La Jolla, CA, USA); and 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein acetoxymethyl ester (BCECF-AM) and fura-2 AM (Molecular Probes Inc.).
Recording techniques
BCECF and fura-2 were employed to estimate pHi and [Ca2+]i, respectively. In studies where information was required on the effects of an experimental manoeuvre on both [Ca2+]i and pHi, measurements of [Ca2+]i and pHi were performed separately in parallel experiments conducted on neurones isolated from the same hippocampus. Neurones were loaded with either 5 μm fura-2 AM for 30 min at 37°C or 2 μm BCECF-AM for 15 min at room temperature and were then superfused at a rate of 2.4 ml min−1 for 15 min with the initial experimental solution at 37°C prior to the start of an experiment. All experiments were performed at 37°C. pHi and [Ca2+]i were measured using the dual-excitation ratio method, employing a digital fluorescence microscopy system (Atto Instruments Inc., Rockville, MD, USA; Carl Zeiss Canada Ltd, Don Mills, ON, Canada). Details of the methods employed have been presented previously (Church et al. 1994; Baxter & Church, 1996). In brief, fluorescence emissions measured at 520 or 510 nm from neurones loaded with BCECF or fura-2, respectively, were obtained from one to three neuronal somata simultaneously (up to 21 neurones simultaneously when using cultured cells) and raw intensity data at each excitation wavelength (488 and 452 nm for BCECF; 334 and 380 nm for fura-2) were corrected for background fluorescence prior to calculation of the ratio. Freshly isolated neurones were chosen for study based on the morphological criteria described by Schwiening & Boron (1994), i.e. a smooth, non-granular appearance with a single major process (presumably an apical dendrite) projecting from one pole of the cell which was at least twice the length of the diameter of the cell body, and the presence of two or more smaller processes at the opposite pole. Analysis was restricted to those neurones able to retain BCECF (as judged by raw emission intensity values recorded during excitation at 452 nm) throughout the course of an experiment (see Bevensee et al. 1996). In order to reduce photobleaching of the fluorophores and cell damage, the intensity of the 100 W mercury arc lamp was reduced by 50% and neutral density filters were placed in the light path. The one-point high-[K+]/nigericin technique was employed to convert background-corrected BCECF emission intensity ratios (I488/I452) into pHi values as described (Baxter & Church, 1996). For the thirteen full calibration experiments utilized in analysing all data, the values of pKa (-log of the dissociation constant of the fluorophore), Rn(min) (the minimum obtainable value for the normalized ratio) and Rn(max) (the maximum obtainable value for the normalized ratio) were (mean ±s.e.m.) 7.17 ± 0.03, 0.45 ± 0.03 and 2.03 ± 0.04, respectively. Values for Rn(min) and Rn(max) were derived from non-linear least-squares regression fits to background-subtracted ratio versus pH data which, in turn, were obtained in full calibration experiments (see Baxter & Church, 1996). Periodically, brief fluctuations in the incident radiation from the arc lamp produced variations in emission intensities. In order to smooth the graphical representation of the pHiversus time records, a moving average (period = 3) was applied to all records shown (see Baxter & Church, 1996). Calibration of the fura-2 signal was not attempted and the effects of experimental manoeuvres on [Ca2+]i are presented as changes in background-corrected I334/I380 ratio values.
Experimental procedures and data analysis
The effects of changes in perfusate composition and pharmacological treatments were examined on both steady-state pHi and on rates of pHi recovery from internal acid loads imposed by the NH4+ prepulse technique. In each experiment in which rates of pHi recovery were examined, two consecutive intracellular acid loads were imposed, the first being employed to calculate control rates of pHi recovery for a given neurone and the second being performed under the influence of a pharmacological or other treatment. The mean percentage difference between rates of pHi recovery following two consecutive acid loads imposed in the absence of a test treatment was established in control experiments. In twenty-one neurones, the difference observed in the overall rate of pHi recovery between the second and first acid loads was a 3 ± 22% (mean ±s.d.) increase. Therefore, in any given experiment, rates of pHi recovery under the influence of a test treatment were considered to be different from control rates of pHi recovery only if they displayed a mean overall increase greater than 47% or a mean overall decrease greater than 41% (i.e. a mean ± 2 s.d. difference from the overall control rate of pHi recovery established in the same cell). Only neurones which exceeded these criteria were considered to have responded to a test treatment and only data from these neurones underwent further analysis.
Control rates of pHi recovery were compared with rates of pHi recovery under a test condition at the same absolute values of pHi. At each corresponding absolute value of pHi, the percentage difference between the control rate of pHi recovery and the rate of pHi recovery under the influence of the test treatment was determined. The mean of the resultant percentage differences was then calculated and employed to describe the overall effect of the test treatment on the rate of pHi recovery. In addition, a formal statistical comparison was made between rates of pHi recovery (evaluated at 0.05 pHi unit increments from the point of maximum acidification) under control and test conditions. For a given absolute value of pHi, control rates of pHi recovery and rates of pHi recovery under a given test condition were grouped separately and Student's paired two-tailed t test was employed to assess statistical significance. Net acid efflux in nominally HCO3−-free media was calculated as the product of the measured rate of recovery of pHi (dpHi/dt) from an imposed acid load at a given pHi value and the intrinsic intracellular buffering power (βi) at the same pHi value. Values for βi were calculated from the equation:
which was derived by Bevensee et al. (1996) in acutely dissociated rat hippocampal CA1 neurones.
Results are reported as means ±s.e.m. with the accompanying n value referring either to the number of neurones from which data were obtained (acutely dissociated cells) or, in cultured neurones, to the number of cell populations (i.e. number of coverslips) examined. Statistical comparisons were performed using Student's two-tailed t test, paired or unpaired as appropriate, with a 95% confidence limit.
RESULTS
Characterization of acid extrusion mechanisms
Acid extrusion from adult rat hippocampal CA1 neurones is reported to be governed by an amiloride-insensitive Na+-H+ exchanger and a Na+-dependent HCO3−-Cl− exchanger (Schwiening & Boron, 1994; Bevensee et al. 1996). Initially, we performed a limited series of experiments to assess whether these mechanisms contribute to acid extrusion in our preparation of acutely dissociated adult rat hippocampal CA1 neurones.
In nominally HCO3−-free medium, resting pHi was 7.29 ± 0.01 (n = 439). The values of resting pHi had a broad range (pH 6.6–7.9) and, in agreement with Bevensee et al. (1996), their distribution was best fitted with the sum of two Gaussian distributions with means at pHi = 6.91 ± 0.01 and 7.43 ± 0.01 (Fig. 1A). Application of the amiloride analogue EIPA (50 μm), a pharmacological inhibitor of Na+-H+ exchange in a wide variety of cell types, did not alter resting pHi (n = 3) and had no effect on rates of pHi recovery from imposed intracellular acid loads (n = 4; data not shown). However, the removal of external Na+ (substitution with NMDG+) evoked a rapid 0.30 ± 0.03 pH unit fall in pHi (n = 15; Fig. 2A) and blocked the recovery of pHi from intracellular acid loads (n = 5; Fig. 2B), suggesting that a Na+-dependent acid extrusion mechanism contributes to the maintenance of steady-state pHi under HCO3−-free conditions. As shown in Fig. 2C, the replacement of external Na+ with Li+ was marked by an initial acidification but pHi recovered in the continued absence of Na+ (n = 4; also see Baxter & Church, 1996). In addition, when Na+-free, Li+-substituted medium was applied at the point of maximum acidification following an NH4+ prepulse, pHi recovery still occurred (n = 6; Fig. 2D). The results indicate that acid extrusion under nominally HCO3−-free conditions is governed by a Na+-dependent mechanism that can also transport Li+ in exchange for internal protons. This mechanism is likely to be the amiloride-insensitive variant of the Na+-H+ exchanger previously characterized in detail by Schwiening & Boron (1994) and Bevensee et al. (1996).
Figure 1. Distribution of steady-state pHi.
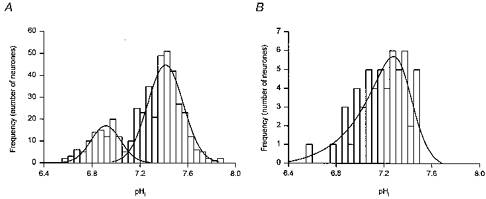
A, a frequency histogram (bin width = 0.05 pH units) of steady-state pHi values for 439 acutely dissociated adult rat hippocampal CA1 neurones during perfusion with pH 7.35 Hepes-buffered medium. The distribution was fitted best with the sum of two Gaussian distributions with means at pHi = 6.91 ± 0.01 and 7.43 ± 0.01. B, a frequency histogram (bin width = 0.05 pH units) of steady-state pHi values for 55 acutely dissociated adult rat hippocampal CA1 neurones during perfusion with pH 7.35 HCO3−/CO2-buffered medium. The distribution was negatively skewed and was fitted best with an asymmetric logistic function with a modal value at pHi = 7.28.
Figure 2. Steady-state pHi and acid extrusion in the absence of HCO3−.
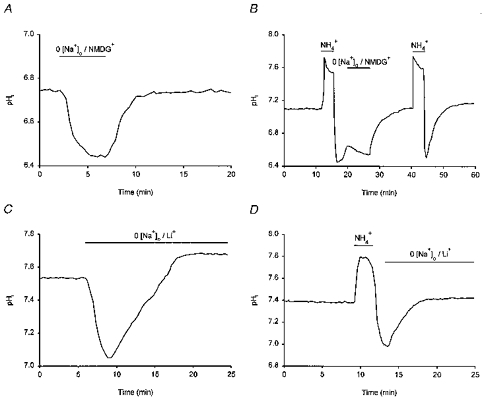
A, the removal of external Na+ (NMDG+ substitution) for the period indicated by the bar above the trace caused pHi to fall by ∼0.3 pH units. pHi recovered when external Na+ was reintroduced. B, following the first NH4+-induced internal acid load, the removal of external Na+ (replacement with NMDG+) reversibly interrupted pHi recovery. A second acid load was then performed and pHi recovery was allowed to take place in the continued presence of external Na+. C, the replacement of external Na+ with Li+ evoked a transient intracellular acidification followed by a recovery of pHi despite the continued absence of external Na+. D, pHi recovery from an imposed intracellular acid load could take place in the absence of external Na+ when Li+ was employed as the substitute cation. Each record was obtained from a different neurone under HCO3−/CO2-free, Hepes-buffered conditions at a pHo of 7.35.
In HCO3−/CO2-buffered saline, resting pHi was 7.20 ± 0.03 (n = 55). As was the case under HCO3−-free conditions, values of resting pHi under HCO3−-containing conditions had a broad range (pH 6.6–7.5) although the distribution of steady-state pHi values in the presence of HCO3− was uni-modal and best fitted by a negatively skewed asymmetric logistic function with a modal value of pHi 7.28 (Fig. 1B). Initially, we explored the effect on steady-state pHi of the transition from a HCO3−/CO2-free medium at pH 7.35 to a medium buffered with HCO3−/CO2 at the same pHo. Upon exposure to HCO3−/CO2-buffered medium, an increase in pHi typically occurred, the magnitude of which was dependent upon the initial resting level of pHi in Hepes-buffered medium (Fig. 3A). The results, which are in agreement with those of Schwiening & Boron (1994) and Bevensee et al. (1996) in the same cell type, indicate that HCO3−-dependent mechanism(s) can contribute to the maintenance of steady-state pHi in adult rat hippocampal CA1 neurones. This possibility was further suggested by the fact that, in six additional neurones with an initial resting pHi < 7.3 in Hepes-buffered medium, the increase in pHi expected upon exposure to HCO3−/CO2-buffered medium was blocked by 300 μm DIDS (Fig. 3B); applied in the nominal absence of HCO3−/CO2, 200–300 μm DIDS failed to affect steady-state pHi (n = 8; data not shown). In neurones with resting pHi < 7.3, replacing external Cl− with gluconate under HCO3−/CO2-buffered conditions evoked a pHi increase of 0.15 ± 0.02 pH units (n = 8; Fig. 3C). In turn, the 0 [Cl−]o-induced alkalinization was blocked by 300 μm DIDS (n = 5; Fig. 3C), consistent with its mediation by a carrier coupling HCO3− and Cl− fluxes. Finally, removal of Na+ from the perfusion medium in the presence of HCO3− caused a 0.35 ± 0.10 pH unit fall in pHi (n = 3), similar to the change observed under HCO3−-free conditions, and blocked pHi recovery from imposed intracellular acid loads (n = 3; data not shown).
Figure 3. Steady-state pHi in the presence of HCO3−.
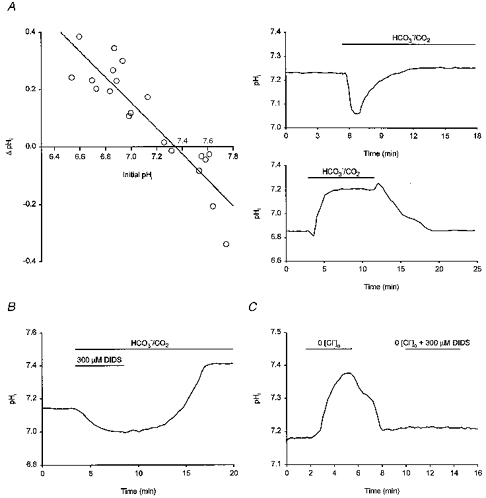
A, to the left, the change in steady-state pHi evoked by the addition of HCO3− at a constant pHo (7.35) is plotted against initial pHi in the absence of HCO3− (n = 20). The line shown is a linear least-squares best fit to the data points indicated. To the right are records showing the effect of introducing HCO3− on pHi in two different neurones with initial pHi values in Hepes-buffered medium of ∼7.25 (upper record) and ∼6.85 (lower record). In both cases, pHo was 7.35 throughout. B, in a different neurone with an initial pHi in Hepes-buffered medium of ∼7.1, the introduction of a HCO3−/CO2-buffered medium containing 300 μm DIDS did not evoke an increase in pHi. An intracellular alkalinization of ∼0.25 pH units above the resting level occurred upon the removal of DIDS from the HCO3−/CO2-buffered medium. Extracellular pH was 7.35 throughout. C, in a fourth neurone, the removal of external Cl− in the presence of HCO3− evoked an ∼0.2 pH unit internal alkalinization which was abolished by the co-application of 300 μm DIDS.
The above findings are entirely consistent with the literature concerning acid extrusion in both cultured fetal (Raley-Susman et al. 1991; Baxter & Church, 1996) and acutely dissociated adult CA1 (Schwiening & Boron, 1994; Bevensee et al. 1996) rat hippocampal neurones. They indicate that acid extrusion in our preparation of adult rat CA1 neurones under HCO3−-free conditions is mediated by an amiloride-insensitive Na+-H+ exchanger. Under HCO3−-containing conditions, acid extrusion appears to be supplemented by the activity of a DIDS-sensitive, Na+-dependent HCO3−-Cl− exchanger, the activity of which is dependent upon the absolute level of pHi.
Effects of noradrenaline on pHi
Steady-state pHi
Under HCO3−/CO2-buffered conditions, application of 10 μm noradrenaline evoked, after a 3–5 min delay, an increase in steady-state pHi of 0.19 ± 0.02 pH units in 6/7 neurones tested (in the remaining neurone, noradrenaline had no effect). A similar rise in steady-state pHi of 0.21 ± 0.02 pH units was observed under nominally HCO3−-free conditions in 18/20 neurones tested (Fig. 4A; P > 0.1 for difference to change in pHi evoked in the presence of HCO3−; noradrenaline was without effect in the remaining two neurones). The increase in pHi evoked by noradrenaline under Hepes-buffered conditions was concentration dependent; 5 and 20 μm noradrenaline increased steady-state pHi by 0.11 ± 0.01 (n = 4) and 0.24 ± 0.04 (n = 3) pH units, respectively. Under both HCO3−-free and HCO3−-containing conditions, the increase in pHi persisted following the washout of noradrenaline (see Fig. 4A) and for as long as stable recordings could be maintained (up to 60 min). Beneath the pHi trace in Fig. 4A are shown the background-subtracted I452 values which were employed in the measurement of pHi; the stability of the I452 values indicates that the persistent nature of the rise in steady-state pHi evoked by noradrenaline was not an artifact produced by a decline in I452 values consequent upon a deterioration of membrane integrity (see Methods). Because the increase in steady-state pHi evoked by noradrenaline was similar in the presence or absence of HCO3−, it is unlikely to reflect primarily changes in the activities of HCO3−-dependent pHi-regulating mechanisms. However, application of 10 μm noradrenaline under HCO3−- and Na+-free (NMDG+-substituted) conditions failed to affect steady-state pHi in 8/8 neurones tested (Fig. 4B), consistent with the possibility that the increase in steady-state pHi might be mediated by an increase in the activity of the Na+-H+ exchanger. Note that, in experiments of the type illustrated in Fig. 4B, the weak base trimethylamine was employed to elevate pHi towards normal resting levels in the continued absence of external Na+.
Figure 4. The effect of noradrenaline on steady-state pHi and the recovery of pHi from imposed intracellular acid loads.
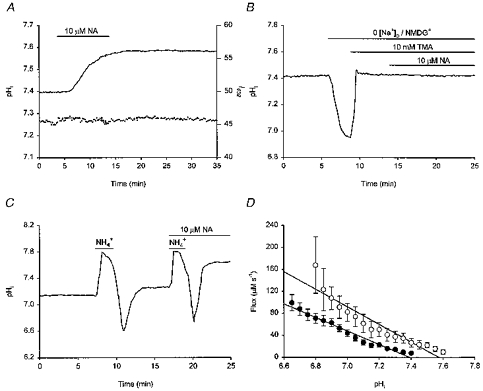
All traces were obtained under HCO3−/CO2-free, Hepes-buffered conditions. Records shown in A-C were obtained from different neurones. A, a 10 min exposure to 10 μm noradrenaline (NA) evoked, after a short delay, an increase in pHi (continuous line) which persisted following washout of the catecholamine. The background-subtracted I452 values which were employed in the measurement of pHi are shown beneath the pHi trace (see Results). B, the Na+ dependence of the noradrenaline-evoked intracellular alkalinization was examined by substituting NMDG+ for external Na+. This produced a fall in pHi which was returned towards the normal resting value by the addition of 10 mm trimethylamine (TMA). Under these conditions, noradrenaline failed to elicit an increase in steady-state pHi. C, following the first NH4+-induced intracellular acid load, pHi was allowed to recover. A second acid load was then performed in the presence of 10 μm noradrenaline, which increased the rate of pHi recovery from the imposed internal acidification. pHi recovered to a higher steady-state level in the presence than in the absence of noradrenaline. D, the pHi dependence of net acid extrusion in the presence (○) and absence (•) of 10 μm noradrenaline. Continuous lines represent the least-squares linear regression fits to the data points indicated for each experimental condition. Data points were obtained from 15 experiments of the type illustrated in C; where missing, standard error bars lie within the symbol areas. Noradrenaline significantly (P < 0.05) increased H+ efflux at each absolute value of pHi and shifted the pHi dependence of acid extrusion to the right.
pHi recovery from internal acid loads
Noradrenaline (10 μm) increased the rate of pHi recovery from an imposed acid load in 5/5 neurones tested in the presence of HCO3− and in 15/17 neurones tested under HCO3−-free conditions; overall rates of pHi recovery were increased by 193 ± 28% (n = 5) and 161 ± 20% (n = 15; Fig. 4C), respectively (P > 0.1). In the remaining two neurones tested under HCO3−-free conditions, 10 μm noradrenaline evoked a 21% decrease and an 11% increase in the overall rate of pHi recovery. In fifteen paired experiments of the type shown in Fig. 4C, which were conducted under Hepes-buffered conditions, pHi recoveries from imposed acid loads under the influence of 10 μm noradrenaline were compared with control pHi recoveries at the same absolute values of pHi. The resulting plots of the pHi dependence of net acid efflux before and after noradrenaline application are presented in Fig. 4D. Noradrenaline (10 μm) significantly increased H+ efflux (P < 0.05 at all absolute values of pHi) and shifted the pHi dependence of acid extrusion by ∼0.2 pH units in the alkaline direction, suggesting that noradrenaline activates Na+-H+ exchange by changing the cytoplasmic pH sensitivity of the antiport (see Grinstein et al. 1989; Wakabayashi et al. 1997). As was the case for the effect of noradrenaline on steady-state pHi (see above), the effect of noradrenaline to increase rates of pHi recovery from internal acid loads required the presence of external Na+. Thus, in five neurones examined under HCO3−/CO2-free conditions in which the recovery of pHi from an imposed acid load took place in the presence of 10 μm noradrenaline (see Fig. 4C), removal of external Na+ (NMDG+ substitution) blocked pHi recovery in a manner identical to that observed in the absence of noradrenaline (see Fig. 2B).
As the noradrenaline-evoked increase in steady-state pHi was abolished under external Na+-free conditions (Fig. 4B), it is unlikely to reflect alterations in intrinsic intracellular buffering power (βi). In addition, as an indication of the apparent intracellular buffering power, we quantified the increase in pHi caused by exposure to 20 mm NH4Cl by taking the difference between the steady-state pHi immediately prior to the application of NH4+ and the maximum pHi immediately after its application. Under nominally HCO3−-free conditions, application of NH4+ evoked a 0.51 ± 0.04 pH unit rise under both control conditions and in the presence of 10 μm noradrenaline (n = 17 in each case). As the alkaline shift evoked by NH4+ was similar in the presence or absence of noradrenaline, changes in βi are unlikely to underlie the marked increases in acid extrusion rates evoked by the neurotransmitter (also see Bevensee et al. 1996). In this regard, it has previously been found that changes in pHi evoked by adrenoceptor agonists in a variety of peripheral cell types do not reflect changes in βi or background acid loading rates (e.g. Guo et al. 1992; Lagadic-Gossmann & Vaughan-Jones, 1993).
In summary, the results indicate that the rise in steady-state pHi evoked by noradrenaline in adult rat hippocampal CA1 neurones is mediated by an increase in the activity of the Na+-H+ exchanger. Consequently, subsequent experiments were performed under nominally HCO3−-free, Hepes-buffered conditions.
Pharmacology of the pHi response to noradrenaline
The adrenergic receptor subtypes mediating the effects of noradrenaline on steady-state pHi and on acid extrusion following intracellular acid loads were determined by employing receptor subtype-selective agonists and antagonists. β-Adrenoceptor agonists (isoprenaline, dobutamine and terbutaline) were applied in the presence of the full α-adrenoceptor antagonist, phentolamine (1–10 μm), whereas media containing the α-adrenoceptor agonist 6-fluoro-noradrenaline also contained the non-selective β-adrenoceptor antagonist, propranolol (10 μm).
The full α-adrenoceptor agonist, 6-fluoro-noradrenaline (10 μm), had no effect on steady-state pHi in five neurones tested (data not shown) and failed to alter the pHi dependence of acid extrusion following imposed acid loads in 9/9 neurones examined (Fig. 5A and B). In contrast, the full β-adrenoceptor agonist isoprenaline (10 μm) mimicked the effects of noradrenaline, increasing both resting pHi by 0.15 ± 0.03 pH units in 6/7 neurones tested and overall rates of pHi recovery from imposed acid loads by 145 ± 12% in 14/16 neurones examined. Steady-state pHi in the remaining neurone was not affected by 10 μm isoprenaline whereas in the two remaining cells in which pHi recovery rates were examined, isoprenaline increased the overall rates by 5 and 31%. In fourteen paired experiments of the type shown in Fig. 5C, 10 μm isoprenaline significantly increased acid extrusion following imposed acid loads at all absolute levels of pHi and shifted the pHi dependence of acid extrusion by ∼0.15 pH units in the alkaline direction (Fig. 5D).
Figure 5. Pharmacology of the pHi response to noradrenaline.
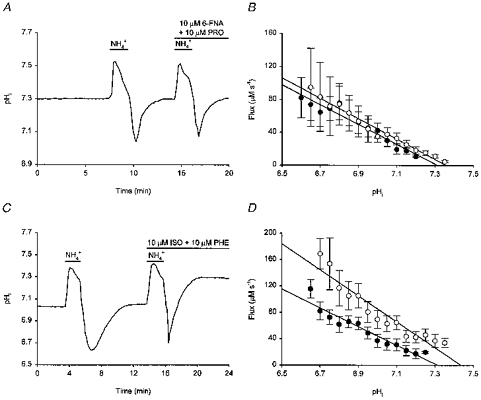
Following an initial acid load and recovery of pHi to resting levels, a second acid load was performed in the presence of the full α-adrenoceptor agonist 6-fluoro-noradrenaline (6-FNA, 10 μm; applied in the presence of 10 μm propranolol, PRO) (A) and in the presence of the full β-adrenoceptor agonist isoprenaline (ISO, 10 μm; applied in the presence of 10 μm phentolamine, PHE) (C). In contrast to 6-FNA, isoprenaline increased the rate of pHi recovery from the intracellular acid load and pHi recovered to a higher steady-state level than that prevailing under control conditions. To the right are shown the pHi dependencies of net acid extrusion in the presence (○) and absence (•) of 10 μm 6-FNA (B) and 10 μm isoprenaline (D). Data were obtained from 9 (in B) and 14 (in D) paired experiments of the types shown in A and C, respectively; error bars represent s.e.m. Continuous lines represent the least-squares linear regression fits to the data points indicated for each experimental condition. Acid extrusion in the presence of 6-FNA was not significantly different from control (P > 0.1 at each absolute value of pHi). In contrast, isoprenaline significantly increased acid extrusion (P < 0.05 at each absolute value of pHi).
The selective β1- and β2-adrenoceptor agonists dobutamine and terbutaline, respectively, also mimicked the effects of noradrenaline on pHi. Experiments with dobutamine were performed in the presence of 10 μm ICI 118,551, a highly selective β2-adrenoceptor antagonist, as well as phentolamine to further ensure the selectivity of the compound for β1-adrenoceptors. Dobutamine (1 μm) increased steady-state pHi by 0.18 ± 0.03 pH units in 6/7 neurones examined (Fig. 6A; dobutamine had no effect on the remaining neurone) and increased the overall rate of pHi recovery from internal acid loads by 134 ± 10% in 6/7 neurones tested (Fig. 6B; in the remaining neurone, the overall rate of pHi recovery was increased by 16%). Similarly, terbutaline (1 μm, tested in the presence of phentolamine) increased steady-state pHi by 0.29 ± 0.06 pH units (n = 5/5; Fig. 6C) and, in 10/10 neurones, increased the overall rate of pHi recovery from imposed acid loads by 165 ± 11% (Fig. 6D). The effect of terbutaline to increase steady-state pHi was concentration dependent; 0.5 and 10 μm terbutaline increased steady-state pHi by 0.11 ± 0.03 (n = 3) and 0.52 ± 0.04 pH units (n = 5), respectively (Fig. 6C). Both dobutamine (1 μm) and terbutaline (1 μm) shifted the pHi dependence of acid extrusion (derived from experiments of the types shown in Fig. 6B and D, respectively) to the right by ∼0.2 pH units (not illustrated).
Figure 6. The pHi response to noradrenaline is mimicked by β1- and β2-adrenoceptor agonists.
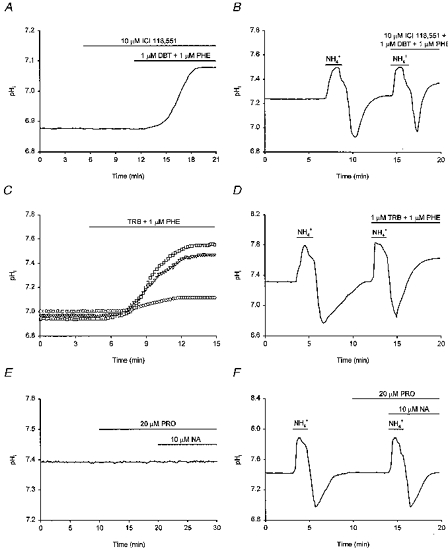
A, exposure to 1 μm dobutamine (DBT), a selective β1-adrenoceptor agonist, increased resting pHi. Dobutamine was applied with 1 μm phentolamine (PHE) in the presence of 10 μm ICI 118,551 (a selective β2-adrenoceptor antagonist). B, the neurone underwent an initial acid load and, following the recovery of pHi, a second acid load was performed in the presence of 1 μm dobutamine, 1 μm phentolamine and 10 μm ICI 118,551. The rate of pHi recovery was increased in the presence of dobutamine. C, representative records from three different neurones, each with a similar resting pHi, are shown to illustrate the concentration dependence of the effect of the selective β2-adrenoceptor agonist, terbutaline (TRB), on steady-state pHi. Terbutaline was applied at 0.5 (○), 1 (▿) and 10 μm (□), in the presence of 1 μm phentolamine in each case. D, terbutaline (1 μm; applied in the presence of 1 μm phentolamine) increased the rate of pHi recovery from an imposed acid load. E, applied in the presence of the β-adrenoceptor antagonist propranolol (PRO, 20 μm), 10 μm noradrenaline (NA) failed to increase resting pHi. F, an initial intracellular acid load was imposed under control conditions. Following the recovery of pHi to resting values, 20 μm propranolol was applied and a second acid load was then performed in the combined presence of 10 μm noradrenaline and 20 μm propranolol. Under these conditions, noradrenaline failed to increase the rate of pHi recovery from the imposed acid load (compare with Fig. 4C). Each record shown in A-F was obtained from a different neurone.
The results indicate that the effects of noradrenaline on steady-state pHi and on acid extrusion following imposed acid loads are mediated by β-adrenoceptors. Consistent with this possibility, application of 10 μm noradrenaline in the presence of the full β-adrenoceptor antagonist propranolol (20 μm) did not evoke a rise in steady-state pHi in 4/4 neurones tested (Fig. 6E) and failed to affect rates of pHi recovery from acid loads in 7/7 neurones examined (Fig. 6F). Concomitantly, propranolol abolished the noradrenaline-evoked alkaline shift in the pHi dependence of acid extrusion (not illustrated).
It has been suggested that the bimodal distribution of resting pHi values observed in adult rat hippocampal CA1 neurones in the absence of HCO3− (see Fig. 1A) may reflect different functional states of the Na+-H+ exchanger (Bevensee et al. 1996). We therefore examined whether exposure to noradrenaline or β-adrenoceptor agonists affected the distribution of steady-state pHi values. As illustrated in Fig. 7A, the distribution of steady-state pHi values for fifty neurones prior to exposure to noradrenaline or a β-adrenoceptor agonist was fitted best by the sum of two Gaussian distributions, with means at pHi 6.93 ± 0.03 and 7.44 ± 0.02. Upon exposure to noradrenaline, isoprenaline, dobutamine or terbutaline, pHi alkalinized to a new steady-state level and the distribution of pHi values was now fitted best by a single Gaussian distribution with a mean at pHi = 7.54 ± 0.03 (Fig. 7B).
Figure 7. Effect of β-adrenoceptor activation on the distribution of steady-state pHi values.
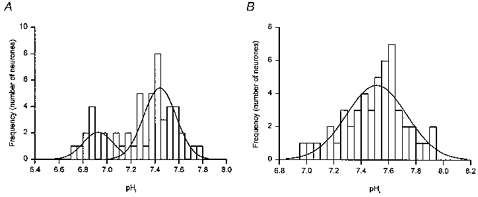
Distributions of steady-state pHi from 50 neurones prior to (A) and following (B) the addition of β-adrenoceptor agonists under HCO3−/CO2-free, Hepes-buffered conditions. A, under control conditions the frequency distribution (bin width = 0.05 pH units) was fitted best with the sum of two Gaussian distributions with means at pHi values of 6.93 ± 0.03 and 7.44 ± 0.02 (compare with Fig. 1A). B, steady-state pHi values measured following the addition of noradrenaline, isoprenaline, dobutamine or terbutaline were pooled and the frequency distribution was fitted best with a single Gaussian distribution with a mean at a pHi of 7.54 ± 0.03.
Intracellular mechanisms mediating the effects of β-adrenoceptor stimulation on pHi
In some systems, Na+-H+ exchange activity and steady-state pHi can be modulated by changes in [Ca2+]i (see Mahnensmith & Aronson, 1985; Wakabayashi et al. 1997). In addition, β-adrenoceptor activation or manoeuvres which act to increase the intracellular concentration of adenosine-3′,5′-cyclic monophosphate ([cAMP]i) can enhance Ca2+ currents in some cell types (e.g. Chetkovich et al. 1991). To investigate whether changes in [Ca2+]i might contribute to the effects of β-adrenoceptor activation on pHi in hippocampal neurones, the effects of noradrenaline and isoprenaline on steady-state pHi and on rates of pHi recovery following imposed acid loads were examined under external Ca2+-free conditions. Exposure to Ca2+-free medium caused an increase in steady-state pHi of 0.16 ± 0.04 pH units (n = 19; see Fig. 8A and B). Once pHi had stabilized at the new resting level, 10 μm noradrenaline evoked a further increase in pHi of 0.18 ± 0.03 pH units in 12/15 cells tested (Fig. 8A; three cells showed no response) whereas 10 μm isoprenaline (tested in the presence of 10 μm phentolamine) evoked an increase in pHi of 0.18 ± 0.02 pH units (n = 4/4; not illustrated). Noradrenaline (10 μm) and isoprenaline (10 μm) also increased overall rates of pHi recovery from acid loads imposed under Ca2+-free conditions by 145 ± 9% (n = 6/7; Fig. 8B; in the remaining neurone, the overall rate of pHi recovery was increased by 8%) and 160 ± 12% (n = 6/6), respectively. The increases in steady-state pHi and rates of pHi recovery evoked by noradrenaline and isoprenaline in the absence of external Ca2+ were not statistically different from the corresponding changes observed in the presence of the cation. Thus, the effects of β-adrenoceptor activation on pHi are not dependent upon Ca2+ influx. In addition, isoprenaline 10 μm had no effect on [Ca2+]i when applied in the absence of external Ca2+ (n = 5/5; Fig. 8C). We therefore conclude that β-adrenoceptor agonists act in a Ca2+-independent manner to increase acid extrusion and thereby increase steady-state pHi.
Figure 8. The pHi response to noradrenaline is not dependent on changes in [Ca2+]i.
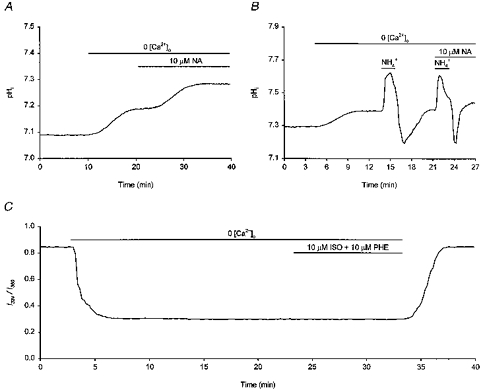
A, exposure to a Ca2+-free medium evoked an ∼0.1 pH unit rise in resting pHi. Once pHi had stabilized at the new resting level, application of 10 μm noradrenaline (NA) elicited a further rise in pHi. B, in a different neurone, removal of external Ca2+ again elicited a rise in steady-state pHi. An initial intracellular acid load was then imposed and, following the recovery of pHi, a second acid load was performed in the presence of 10 μm noradrenaline. Noradrenaline increased the rate of pHi recovery in the absence of external Ca2+. C, in a third neurone loaded with fura-2, exposure to Ca2+-free medium caused a decrease in the I334/I380 ratio value. Application of 10 μm isoprenaline (ISO, applied in the presence of 10 μm phentolamine (PHE)) did not evoke a change in the I334/I380 ratio value. In contrast, like noradrenaline (see B), 10 μm isoprenaline increased steady-state pHi under Ca2+-free conditions (see text).
The occupation of β-adrenoceptors by noradrenaline is linked classically to the activation of adenylate cyclase and a subsequent increase in [cAMP]i. We therefore examined whether the effects of noradrenaline on steady-state pHi and on rates of pHi recovery from imposed acid loads could be mimicked and attenuated, respectively, by activators and inhibitors of adenylate cyclase. Applied alone, 25 μm forskolin mimicked the actions of noradrenaline, producing a sustained increase in steady-state pHi of 0.23 ± 0.03 pH units in 16/17 neurones examined (Fig. 9A) and increasing the overall rate of pHi recovery from acid loads by 169 ± 18% (n = 5/5; Fig. 9B). The steady-state pHi of the remaining neurone was unaffected by forskolin. Exposure to 25 μm forskolin under external Ca2+-free conditions also evoked a sustained increase in steady-state pHi of 0.20 ± 0.06 pH units (n = 4/4; not illustrated), a result not significantly different from that obtained in the presence of external Ca2+. An inactive analogue of forskolin, 1′,9′-dideoxyforskolin (25 μm), failed to influence steady-state pHi (n = 5/5) or rates of pHi recovery from imposed intracellular acid loads (n = 7/7). The effects of forskolin on steady-state pHi and on rates of pHi recovery from imposed acid loads were mimicked by the phosphodiesterase inhibitor IBMX which, like forskolin, acts to raise [cAMP]i. Applied at 200 μm, IBMX evoked an increase in steady-state pHi of 0.20 ± 0.03 pH units (n = 12/12; Fig. 9C) and increased the overall rate of pHi recovery by 142 ± 13% (n = 14/15; Fig. 9D; the remaining neurone was not affected). In a manner analogous to noradrenaline and β-adrenoceptor agonists, both forskolin (25 μm; Fig. 9E) and IBMX (200 μm; not illustrated) shifted the pHi dependence of acid extrusion in the alkaline direction. Conversely, the effects of noradrenaline on both steady-state pHi and rates of pHi recovery from imposed acid loads were attenuated by pre-treatment with the adenylate cyclase inhibitor 2′,5′-dideoxyadenosine. Applied alone, 100 μm 2′,5′-dideoxyadenosine did not markedly affect steady-state pHi (n = 9/9). However, in cells pre-treated for 5–15 min with 100–200 μm 2′,5′-dideoxyadenosine, noradrenaline (10 μm) failed to affect steady-state pHi (n = 6/6; not illustrated) and did not increase rates of pHi recovery from imposed acid loads (n = 7/7; Fig. 9F). Finally, the ability of forskolin to increase steady-state pHi was occluded by prior exposure to noradrenaline. Applied following a 15 min period of perfusion with 20 μm noradrenaline, which of itself increased pHi by 0.23 ± 0.06 pH units (n = 5), 25 μm forskolin increased steady-state pHi by only a further 0.02 ± 0.01 pH units. The results suggest that the effects of noradrenaline on pHi are probably mediated via a signal transduction pathway which involves the activation of adenylate cyclase and the subsequent production of cAMP.
Figure 9. Involvement of cAMP in the pHi response to noradrenaline.
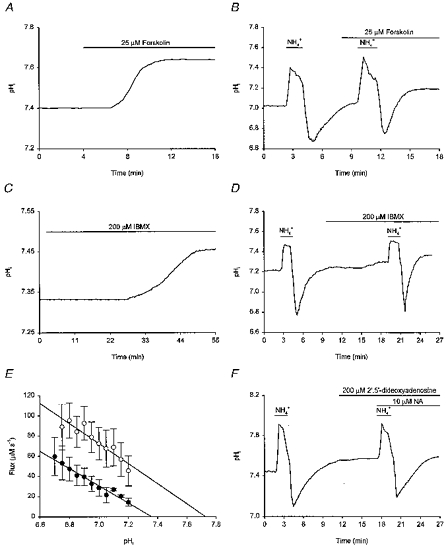
Records in A-D and F were obtained from different neurones. A, application of 25 μm forskolin evoked, after a short delay, a rise in steady-state pHi. B, following an initial intracellular acid load, pHi was allowed to recover. Forskolin (25 μm) was then applied and a second acid load was imposed. The rate of pHi recovery was increased in the presence of forskolin and pHi recovered to a new higher steady-state level. C, the phosphodiesterase inhibitor IBMX (200 μm) increased steady-state pHi. D, an initial intracellular acid load was performed and, after pHi had recovered, IBMX (200 μm) was applied. The rate of pHi recovery following a second acid load was increased by IBMX and pHi recovered to a new higher steady-state level. E, the pHi dependence of net acid extrusion in the presence (○) and absence (•) of 25 μm forskolin. Continuous lines represent the least-squares linear regression fits to the data points indicated for each experimental condition. Data points were obtained from 5 experiments of the type illustrated in B; error bars represent s.e.m. Forskolin increased H+ efflux (P < 0.05 at each absolute value of pHi except pHi 6.75, where P = 0.23) and shifted the pHi dependence of acid extrusion to the right. F, following pre-treatment of the neurone with 200 μm 2′,5′-dideoxyadenosine, 10 μm noradrenaline failed to increase the rate of pHi recovery from an imposed intracellular acid load.
In the next series of experiments we examined the involvement of cAMP-dependent protein kinase (protein kinase A; PKA) in the pHi response to β-adrenoceptor activation. Pre-treatment of neurones with the PKA inhibitors Rp-cAMPS (50 μm; Fig. 10A) and H-89 (10 μm; not illustrated) abolished the ability of 10 μm noradrenaline to increase steady-state pHi (n = 5/5 and n = 10/10, respectively). In addition, both compounds inhibited the effect of 10 μm noradrenaline to increase rates of pHi recovery following imposed acid loads (n = 4/4 and n = 5/5 for Rp-cAMPS (Fig. 10B) and H-89 (not illustrated), respectively). Conversely, a selective activator of PKA, Sp-cAMPS, mimicked the effects of noradrenaline by evoking a concentration-dependent rise in steady-state pHi. Thus, applied at 5, 10, 25 and 40 μm, Sp-cAMPS caused steady-state pHi to rise by 0.10 ± 0.01 (n = 6/8), 0.15 ± 0.01 (n = 11/13), 0.19 ± 0.01 (n = 11/13) and 0.20 ± 0.02 (n = 2/2) pH units, respectively (Fig. 10C); in the remaining neurones, Sp-cAMPS was without effect. The increase in steady-state pHi evoked by 25 μm Sp-cAMPS under external Ca2+-free conditions (a 0.20 ± 0.03 pH unit increase, n = 3/3) was not statistically different from that evoked in the presence of Ca2+. Sp-cAMPS (25 μm) also increased the overall rate of pHi recovery from imposed acid loads by 168 ± 24% in 9/10 neurones examined (Fig. 10D; in the remaining neurone, 25 μm Sp-cAMPS increased the overall rate of pHi recovery by 33%) and shifted the pHi dependence of acid extrusion in an alkaline direction by ∼0.2 pH units (Fig. 10E). Finally, we examined whether inhibition of PKA could attenuate the ability of forskolin to increase steady-state pHi. Applied following a 3–5 min period of pre-perfusion with 50 μm Rp-cAMPS, forskolin (25 μm) increased steady-state pHi by 0.06 ± 0.04 pH units (n = 10/10; not shown), a change which was significantly smaller than that observed in the absence of Rp-cAMPS (P < 0.005).
Figure 10. Involvement of cAMP-dependent protein kinase.
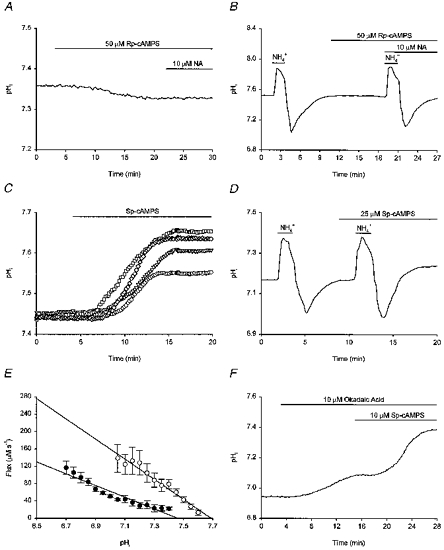
Records in A-F were obtained from different neurones. A, following pre-treatment with 50 μm Rp-cAMPS for ∼20 min, 10 μm noradrenaline (NA) failed to increase steady-state pHi. B, an initial intracellular acid load was performed and, following the recovery of pHi, 50 μm Rp-cAMPS was applied. A second acid load was then performed in the presence of 10 μm noradrenaline. Noradrenaline failed to increase the rate of pHi recovery from the imposed acid load following pre-treatment with Rp-cAMPS. C, Sp-cAMPS evoked a concentration-dependent rise in steady-state pHi. Shown are the effects of 5 (○), 10 (▿), 25 (⋄) and 40 μm Sp-cAMPS (□), applied to four different neurones with similar resting pHi values prior to the application of the compound. D, two intracellular acid loads were performed, the second in the presence of 25 μm Sp-cAMPS. The rate of pHi recovery was increased by Sp-cAMPS and pHi recovered to a new higher steady-state level. E, the pHi dependence of net acid extrusion in the presence (○) and absence (•) of 25 μm Sp-cAMPS. Continuous lines represent the least-squares linear regression fits to the data points indicated for each experimental condition. Data points were obtained from 9 experiments of the type illustrated in D; error bars represent s.e.m. Sp-cAMPS significantly (P < 0.05) increased H+ efflux at each absolute value of pHi and shifted the pHi dependence of acid extrusion to the right. F, application of 10 μm okadaic acid evoked a rise in resting pHi. Sp-cAMPS (10 μm) was then applied and caused resting pHi to increase to a greater extent than was observed in the absence of okadaic acid (see Results).
The above results lead us to conclude that the effects of noradrenaline on steady-state pHi and on acid extrusion following internal acid loads involve the activation of PKA by cAMP. The level of phosphorylation of acid extruding exchangers (and thus their activities) may result from an equilibrium between kinase and phosphatase activities (see Bianchini et al. 1991; Sardet et al. 1991). The possibility that phosphorylation of the Na+-H+ exchanger (or of an ancillary protein; see Discussion) might be involved in the pHi response to PKA activation was suggested by experiments with the protein phosphatase inhibitor, okadaic acid. In HCO3−-free medium containing 10 μm okadaic acid (Na+ salt), which of itself produced a 0.18 ± 0.05 pH unit rise in pHi, 10 μm Sp-cAMPS evoked a 0.36 ± 0.10 pH unit rise in pHi in 4/4 neurones tested (Fig. 10F). This increase was significantly greater (P < 0.01) than the rise in pHi evoked by 10 μm Sp-cAMPS in the absence of okadaic acid. We therefore suggest that a phosphorylation step may be involved in the effects of β-adrenoceptor activation on pHi.
β-Adrenoceptors are coupled to adenylate cyclase activation through the α-subunit of the stimulatory G protein, Gs. If Gsα participates in the noradrenaline-evoked increase in Na+-H+ exchange activity, cholera toxin-catalysed ADP-ribosylation of Gsα should activate Na+-H+ exchange as if stimulated by noradrenaline and should not only increase steady-state pHi but also occlude the effects of noradrenaline to increase steady-state pHi and rates of pHi recovery from acid loads. We therefore examined whether β-adrenoceptor-mediated regulation of acid extrusion proceeds through a pathway involving Gsα by pre-treating cultured postnatal rat hippocampal neurones with cholera toxin. Resting pHi was 7.33 ± 0.03 in six neuronal cultures pre-treated with 500 ng ml−1 cholera toxin for 18–24 h, a value significantly higher than that measured in six sister cultures which had not been pre-treated with the toxin (7.18 ± 0.04). In addition, in neurones pre-treated with cholera toxin, 10 μm noradrenaline failed to increase steady-state pHi (n = 3/3; Fig. 11B) and did not affect the rate of pHi recovery from imposed intracellular acid loads (n = 3/3; Fig. 11D). In contrast, in sister cultures which had not been pre-treated with cholera toxin, 10 μm noradrenaline increased resting pHi by 0.24 ± 0.03 pH units (n = 3/3; Fig. 11A) and increased the overall rate of pHi recovery from imposed acid loads by 126 ± 9% (n = 3/3; Fig. 11C). We therefore conclude that the effects of noradrenaline on pHi are dependent upon the activation of Gsα.
Figure 11. Pre-treatment with cholera toxin occludes the pHi response to noradrenaline.
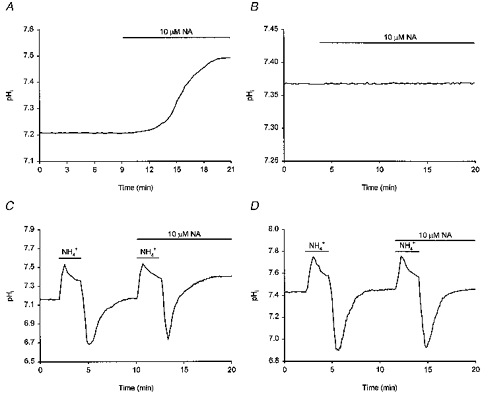
A, noradrenaline (NA, 10 μm) was applied to a population of cultured postnatal rat hippocampal neurones and, after a short delay, pHi increased to a new steady-state level (compare with Fig. 4A, the same experiment conducted in an acutely dissociated adult neurone). The trace represents the mean of data obtained from 18 neurones simultaneously. B, a sister culture to that employed in the experiment shown in A was pre-treated with 500 ng ml−1 cholera toxin for 18 h. Noradrenaline (10 μm) failed to increase steady-state pHi. The trace represents the mean of data obtained from 21 neurones simultaneously. C, two consecutive intracellular acid loads were imposed on a third population of cultured postnatal hippocampal neurones, the second in the presence of 10 μm noradrenaline. The rate of pHi recovery was increased by noradrenaline (compare with Fig. 4C, the same experiment conducted in an acutely dissociated adult neurone). The trace represents the mean of data obtained from 5 neurones simultaneously. D, a sister culture to that employed in the experiment shown in C was pre-treated with 500 ng ml−1 cholera toxin for 24 h. The overall rate of pHi recovery from the second acid load conducted in the presence of 10 μm noradrenaline was comparable to the overall rate of pHi recovery observed following the first acid load conducted in its absence. The trace represents the mean of data obtained from 10 neurones simultaneously.
DISCUSSION
In acutely dissociated adult rat hippocampal CA1 neurones, noradrenaline evoked a concentration-dependent increase in steady-state pHi by increasing the activity of a Na+-dependent, HCO3−-independent acid extrusion mechanism, probably the amiloride-insensitive variant of the Na+-H+ exchanger which has been extensively characterized in both fetal and adult rat hippocampal neurones (Raley-Susman et al. 1991; Schwiening & Boron, 1994; Baxter & Church, 1996; Bevensee et al. 1996). We found no evidence to suggest that the effect of noradrenaline to increase steady-state pHi reflected changes in the activities of HCO3−-dependent pHi-regulating mechanisms. Nevertheless, it remains to be determined whether noradrenaline might modulate equally the activities of both HCO3−-dependent acid loading mechanisms (e.g. Na+-independent HCO3−-Cl− exchange) and HCO3−-dependent acid extruding mechanisms (e.g. Na+-dependent HCO3−-Cl− exchange), such that the resultant steady-state pHi changes are no different from those found in the absence of HCO3− (see Ganz et al. 1989).
The effect of noradrenaline to increase both steady-state pHi and net acid extrusion following imposed acid loads was mediated by β-adrenoceptors and proceeded via a pathway which probably involved a cholera toxin-sensitive G protein-coupled activation of adenylate cyclase and the subsequent stimulation of cAMP-dependent protein kinase. The evidence for this pathway is as follows. Firstly, the effects of noradrenaline on steady-state pHi and acid efflux were mimicked by selective β1- and β2-adrenoceptor agonists, but not by a full α-adrenoceptor agonist, and were blocked by a full β-adrenoceptor antagonist. Secondly, incubation with cholera toxin occluded the effects of noradrenaline to increase both steady-state pHi and rates of pHi recovery following imposed acid loads. Thirdly, the effects of noradrenaline on both steady-state pHi and acid extrusion were blocked by the adenylate cyclase inhibitor 2′,5′-dideoxyadenosine and were mimicked by forskolin and IBMX. Finally, pre-treatment with PKA inhibitors (Rp-cAMPS and H-89) blocked the effects of noradrenaline on steady-state pHi and on rates of pHi recovery from acid loads whereas an activator of PKA (Sp-cAMPS) mimicked the effects of noradrenaline on both parameters. In contrast to other cell types, we found no evidence to suggest either that β-adrenoceptor-mediated activation of Na+-H+ exchange in rat hippocampal neurones is independent of receptor coupling to Gs (Dhanasekaran et al. 1994; Voyno-Yasenetskaya et al. 1994) or that β-adrenoceptor stimulation regulates Na+-H+ exchange independently from a signalling cascade which involves cAMP (see Ganz et al. 1990). In addition, the regulatory pathway characterized in the present study differs from that described in other neuronal preparations. Thus, Na+-H+ exchange activity in rat brain synaptosomes is modulated by internal Ca2+ and not by protein kinases A or C (Sánchez-Armass et al. 1994). In contrast, in cerebellar Purkinje cells, Na+-H+ exchange is activated by protein kinase C (Gaillard & Dupont, 1990; the effect of modulating the cAMP/PKA pathway was not examined).
In peripheral cell types, it is well established that the activities of Na+-H+ exchangers are regulated by a wide variety of external stimuli that act via surface receptors linked to diverse intracellular signal transduction cascades. Furthermore, the response of Na+-H+ exchangers to a given stimulus appears highly dependent not only on the particular isoform of the exchanger being studied but also on the cell type or cell line in which the particular isoform is expressed (see Wakabayashi et al. 1997). Indeed, the adrenoceptor subtype and intracellular pathway which couples activation of the receptor to the Na+-H+ exchanger in rat hippocampal neurones appear to differ from those involved in many other preparations. Thus, in the majority of cell types studied to date, α-adrenoceptors mediate the stimulatory effects of adrenergic receptor agonists on Na+-H+ exchange. In cardiac myocytes, for example, α1-adrenoceptor stimulation increases steady-state pHi through protein kinase C-mediated activation of Na+-H+ exchange whereas β-adrenoceptor agonists either fail to influence exchange activity or inhibit it (e.g. Guo et al. 1992; Wallert & Fröhlich, 1992; Lagadic-Gossman & Vaughan-Jones, 1993). Furthermore, elevating [cAMP]i or activating PKA either has no effect on or inhibits Na+-H+ exchange (NHE) activity in most cell types studied (e.g. Wu & Vaughan-Jones, 1994; Kurashima et al. 1997), although there are some exceptions (reviewed by Wakabayashi et al. 1997; see also Kandasamy et al. 1995). Perhaps the best-characterized example of a cAMP-mediated upregulation of Na+-H+ exchange activity is seen in the case of the β-NHE isoform, which can be activated both by β-adrenoceptor agonists and by direct elevation of [cAMP]i (see Noël & Pouysségur, 1995). Interestingly, β-NHE contains two potential consensus sites for phosphorylation by PKA in its C-terminal cytoplasmic domain (Borgese et al. 1992). Although it is unclear precisely which NHE isoform participates in pHi regulation in rat hippocampal CA1 neurones, it is tempting to speculate that it might also possess an intrinsic capability to respond to PKA.
Activation of Na+-H+ exchange by mitogens, hormones and other external agents occurs through an increased affinity of the allosteric internal modifier site on the exchanger for protons and/or an increase in the maximum velocity of transport (see Mahnensmith & Aronson, 1985; Noël & Pouysségur, 1995). The former process, which may reflect a conformational change in the transport protein produced by phosphorylation, results in an alkaline shift in the pHi dependence of the exchanger. The possibility that a similar mechanism might underlie the effects of β-adrenoceptor activation observed in the present study is suggested by the rightward shifts in the pHi dependence of acid extrusion evoked by noradrenaline (Fig. 4D), β-adrenoceptor agonists (Fig. 5D), forskolin (Fig. 9E) and Sp-cAMPS (Fig. 10E). Furthermore, the ability of the protein phosphatase inhibitor okadaic acid to augment the increase in pHi evoked by a PKA activator (Fig. 10F) suggests that, as in other cell types (see Bianchini et al. 1991; Sardet et al. 1991; Wakabayashi et al. 1997), a phosphorylation event might be involved in modifying the mechanistic properties of the exchange mechanism in hippocampal neurones. However, we have no evidence to indicate whether activation of Na+-H+ exchange by PKA involves direct phosphorylation of the exchange protein itself or of an associated regulatory protein whose phosphorylation state influences the former's activity (see Sardet et al. 1991; Lin & Barber, 1996; Yun et al. 1997; Hall et al. 1998).
The functional significance of the present findings remains unclear. Nevertheless, the magnitude of the effect of noradrenaline on steady-state pHi is consistent with the possibility that, as in peripheral cell types, adrenoceptor-mediated modulation of the activity of neuronal acid extrusion mechanisms may represent a physiologically relevant transmembrane signalling pathway. In this regard, a number of speculative possibilities exist. Firstly, as noted in the Introduction, neuronal ionic conductances (Daumas & Andersen, 1993; Tombaugh & Somjen, 1997) and the activities of intracellular enzymes (e.g. Vignes et al. 1996) are sensitive to shifts in pHi, as are the activities of transport and buffering mechanisms for various ions (e.g. Zucker, 1981; Dipolo & Beaugé, 1982; Sidky & Baimbridge, 1997). Although the net functional result of changes in pHi on all of these processes is impossible to predict, β-adrenoceptor-mediated changes in steady-state pHi might contribute to at least some of the complex effects of β-adrenoceptor stimulation or activation of the cAMP/PKA signalling cascade on hippocampal neuronal function. The possible participation of increases in pHi in some of the more persistent effects of β-adrenoceptor activation or [cAMP]i elevation, such as increases in excitatory synaptic strength (Dunwiddie et al. 1992; Raman et al. 1996) and facilitation of the induction of long-term potentiation (Thomas et al. 1996; Bolshakov et al. 1997), is especially attractive given that changes in pHi are known to affect processes such as gene expression, protein synthesis and cytoskeletal reorganization in peripheral cell types (reviewed by Busa & Nuccitelli, 1984; Grinstein et al. 1989). Secondly, the increase in acid efflux evoked by β-adrenoceptor activation may provide a mechanism to rapidly alleviate intracellular acid loads occasioned by neuronal activity and the application of neurotransmitters such as glutamate (see Introduction). Thirdly, changes in pHi consequent upon β-adrenoceptor activation will inevitably affect the pH of the microenvironment. The changes in pHo may, in turn, affect not only the activities of pHi-regulating mechanisms in adjacent cells but also neuronal excitability, given the established sensitivity of neuronal voltage- and ligand-activated ion channels to changes in pHo (e.g. Vyklick´y et al. 1990; Tombaugh & Somjen, 1996). Finally, in light of the fact that activation of Na+-H+ exchange contributes to neuronal death following metabolic inhibition (see Vornov et al. 1996), it is possible that β-adrenoceptor-mediated stimulation of acid extrusion participates in β-adrenoceptor-mediated potentiation of ischaemia-induced neuronal damage (e.g. Shibata et al. 1992).
In summary, noradrenaline increases steady-state pHi in acutely dissociated adult rat hippocampal CA1 neurones. The effect, which is mediated by β-adrenoceptors, is probably due to activation of the acid-extruding Na+-H+ exchanger via a pathway involving Gs, cAMP and PKA. The exact relationship between PKA activation and stimulation of the transport mechanism, however, remains to be determined. In many peripheral cell types, the activities of Na+-H+ exchangers can be controlled independently by multiple external stimuli acting via different signalling pathways (see Borgese et al. 1992; Kandasamy et al. 1995; Noël & Pouysségur, 1995; Wakabayashi et al. 1997). Given that pHi is an important determinant of neuronal function, it will be of interest to examine whether additional external stimuli and intracellular signal transduction cascades might be involved in the regulation of the activities of the acid extrusion mechanisms present in rat hippocampal neurones.
Acknowledgments
We are grateful to Ms S. Atmadja for the preparation and maintenance of the neuronal cultures. Financial support was provided by an operating grant to J. C. from the Medical Research Council of Canada.
References
- Baxter KA, Church J. Characterization of acid extrusion mechanisms in cultured fetal rat hippocampal neurones. The Journal of Physiology. 1996;493:457–470. doi: 10.1113/jphysiol.1996.sp021396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevensee MO, Cummins TR, Haddad GG, Boron WF, Boyarsky G. pH regulation in single CA1 neurons acutely isolated from the hippocampi of immature and mature rats. The Journal of Physiology. 1996;494:315–328. doi: 10.1113/jphysiol.1996.sp021494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bianchini L, Woodside M, Sardet C, Pouysségur J, Takai A, Grinstein S. Okadaic acid, a phosphatase inhibitor, induces activation and phosphorylation of the Na+/H+ antiport. Journal of Biological Chemistry. 1991;266:15406–15413. [PubMed] [Google Scholar]
- Bolshakov VY, Golan H, Kandel ER, Siegelbaum SA. Recruitment of new sites of synaptic transmission during the cAMP-dependent late phase of LTP at CA3-CA1 synapses in the hippocampus. Neuron. 1997;19:635–651. doi: 10.1016/s0896-6273(00)80377-3. [DOI] [PubMed] [Google Scholar]
- Borgese F, Sardet C, Cappadoro M, Pouysségur J, Motais R. Cloning and expression of a cAMP-activated Na+/H+ exchanger: Evidence that the cytoplasmic domain mediates hormonal regulation. Proceedings of the National Academy of Sciences of the USA. 1992;89:6765–6769. doi: 10.1073/pnas.89.15.6765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brett CL, Church J. Modulation of the activity of intracellular pH regulating mechanisms in rat hippocampal CA1 neurones: The cAMP/PKA pathway. Canadian Journal of Physiology and Pharmacology. 1998;76:Aii. [Google Scholar]
- Busa WB, Nuccitelli R. Metabolic regulation via intracellular pH. American Journal of Physiology. 1984;246:R409–438. doi: 10.1152/ajpregu.1984.246.4.R409. [DOI] [PubMed] [Google Scholar]
- Chetkovich DM, Gray R, Johnston D, Sweatt JD. N-methyl-D-aspartate receptor activation increases cAMP levels and voltage-gated Ca2+ channel activity in area CA1 of hippocampus. Proceedings of the National Academy of Sciences of the USA. 1991;88:6467–6471. doi: 10.1073/pnas.88.15.6467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Church J, Fletcher EJ, Abdel-Hamid K, MacDonald JF. Loperamide blocks high-voltage-activated calcium channels and N-methyl-D-aspartate-evoked responses in rat and mouse cultured hippocampal pyramidal neurons. Molecular Pharmacology. 1994;45:747–757. [PubMed] [Google Scholar]
- Daumas P, Andersen OS. Proton block of rat brain sodium channels. Journal of General Physiology. 1993;101:27–43. doi: 10.1085/jgp.101.1.27. 10.1085/jgp.101.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhanasekaran N, Vara Prasad MVVS, Wadsworth SJ, Dermott JM, Van Rossum G. Protein kinase C-dependent and -independent activation of Na+/H+ exchanger by Gα12 class of G proteins. Journal of Biological Chemistry. 1994;269:11802–11806. [PubMed] [Google Scholar]
- Dipolo R, Beaugé L. The effect of pH on Ca2+ extrusion mechanisms in dialyzed squid axons. Biochimica et Biophysica Acta. 1982;688:237–245. doi: 10.1016/0005-2736(82)90599-5. [DOI] [PubMed] [Google Scholar]
- Dunwiddie TV, Taylor M, Heginbotham LR, Proctor WR. Long-term increases in excitability in the CA1 region of rat hippocampus induced by β-adrenergic stimulation: Possible mediation by cAMP. Journal of Neuroscience. 1992;12:506–517. doi: 10.1523/JNEUROSCI.12-02-00506.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaillard S, Dupont J-L. Ionic control of intracellular pH in rat cerebellar Purkinje cells maintained in culture. The Journal of Physiology. 1990;425:71–83. doi: 10.1113/jphysiol.1990.sp018093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganz MB, Boyarsky G, Sterzel RB, Boron WF. Arginine vasopressin enhances pHi regulation in the presence of HCO3− by stimulating three acid-base transport systems. Nature. 1989;337:648–650. doi: 10.1038/337648a0. 10.1038/337648a0. [DOI] [PubMed] [Google Scholar]
- Ganz MB, Pachter JA, Barber DL. Multiple receptors coupled to adenylate cyclase regulate Na-H exchange independent of cAMP. Journal of Biological Chemistry. 1990;265:8989–8992. [PubMed] [Google Scholar]
- Grinstein S, Rotin D, Mason MJ. Na+/H+ exchange and growth factor-induced cytosolic pH changes. Role in cellular proliferation. Biochimica et Biophysica Acta. 1989;988:73–97. doi: 10.1016/0304-4157(89)90004-x. [DOI] [PubMed] [Google Scholar]
- Guo H, Wasserstrom JA, Rosenthal JE. Effect of catecholamines on intracellular pH in sheep cardiac Purkinje fibres. The Journal of Physiology. 1992;458:289–306. doi: 10.1113/jphysiol.1992.sp019418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall RA, Premont RT, Chow C-W, Blitzer JT, Pitcher JA, Claing A, Stoffel RH, Barak LS, Shenolikar S, Weinman EJ, Grinstein S, Lefkowitz RJ. The β2-adrenergic receptor interacts with the Na+/H+-exchanger regulatory factor to control Na+/H+ exchange. Nature. 1998;392:626–630. doi: 10.1038/33458. 10.1038/33458. [DOI] [PubMed] [Google Scholar]
- Hughes IE, Smith JA. The stability of noradrenaline in physiological saline solutions. Journal of Pharmacy and Pharmacology. 1978;30:124–126. doi: 10.1111/j.2042-7158.1978.tb13179.x. [DOI] [PubMed] [Google Scholar]
- Kaila K. Ionic basis of GABAA receptor channel function in the nervous system. Progress in Neurobiology. 1994;42:489–537. doi: 10.1016/0301-0082(94)90049-3. 10.1016/0301-0082(94)90049-3. [DOI] [PubMed] [Google Scholar]
- Kandasamy RA, Yu FH, Harris R, Boucher A, Hanrahan JW, Orlowski J. Plasma membrane Na+/H+ exchanger isoforms (NHE-1, -2, and -3) are differentially responsive to second messenger agonists of the protein kinase A and C pathways. Journal of Biological Chemistry. 1995;270:29209–29216. doi: 10.1074/jbc.270.49.29209. 10.1074/jbc.270.49.29209. [DOI] [PubMed] [Google Scholar]
- Kurashima K, Yu FH, Cabado AG, Szabó EZ, Grinstein S, Orlowski J. Identification of sites required for downregulation of Na+/H+ exchanger NHE3 activity by cAMP-dependent protein kinase. Journal of Biological Chemistry. 1997;272:28672–28679. doi: 10.1074/jbc.272.45.28672. 10.1074/jbc.272.45.28672. [DOI] [PubMed] [Google Scholar]
- Lagadic-Gossmann D, Vaughan-Jones RD. Coupling of dual acid extrusion in the guinea-pig isolated ventricular myocyte to α1- and β-adrenoceptors. The Journal of Physiology. 1993;464:49–73. doi: 10.1113/jphysiol.1993.sp019624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin X, Barber DL. A calcineurin homologous protein inhibits GTPase-stimulated Na-H exchange. Proceedings of the National Academy of Sciences of the USA. 1996;93:12631–12636. doi: 10.1073/pnas.93.22.12631. 10.1073/pnas.93.22.12631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loy R, Koziell DA, Lindsey JD, Moore RY. Noradrenergic innervation of the adult rat hippocampal formation. Journal of Comparative Neurology. 1980;189:699–710. doi: 10.1002/cne.901890406. [DOI] [PubMed] [Google Scholar]
- Mahnensmith RL, Aronson PS. The plasma membrane sodium-hydrogen exchanger and its role in physiological and pathophysiological processes. Circulation Research. 1985;56:773–788. doi: 10.1161/01.res.56.6.773. [DOI] [PubMed] [Google Scholar]
- Mody I, Salter MW, MacDonald JF. Whole-cell voltage-clamp recordings in granule cells acutely isolated from hippocampal slices of adult or aged rats. Neuroscience Letters. 1989;96:70–75. doi: 10.1016/0304-3940(89)90245-0. 10.1016/0304-3940(89)90245-0. [DOI] [PubMed] [Google Scholar]
- Noël J, Pouysségur J. Hormonal regulation, pharmacology, and membrane sorting of vertebrate Na+/H+ exchanger isoforms. American Journal of Physiology. 1995;268:C283–296. doi: 10.1152/ajpcell.1995.268.2.C283. [DOI] [PubMed] [Google Scholar]
- Raley-Susman KM, Cragoe EJ, Jr, Sapolsky RM, Kopito RR. Regulation of intracellular pH in cultured hippocampal neurons by an amiloride-insensitive Na+/H+ exchanger. Journal of Biological Chemistry. 1991;266:2739–2745. [PubMed] [Google Scholar]
- Raman IM, Tong G, Jahr CE. β-Adrenergic regulation of synaptic NMDA receptors by cAMP-dependent protein kinase. Neuron. 1996;16:415–421. doi: 10.1016/s0896-6273(00)80059-8. 10.1016/S0896-6273(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Sánchez-Armass S, Martínez-Zaguilán R, Martínez GM, Gillies RJ. Regulation of pH in rat brain synaptosomes. I. Role of sodium, bicarbonate, and potassium. Journal of Neurophysiology. 1994;71:2236–2248. doi: 10.1152/jn.1994.71.6.2236. [DOI] [PubMed] [Google Scholar]
- Sardet C, Fafournoux P, Pouysségur J. α-Thrombin, epidermal growth factor, and okadaic acid activate the Na+/H+ exchanger, NHE-1, by phosphorylating a set of common sites. Journal of Biological Chemistry. 1991;266:19166–19171. [PubMed] [Google Scholar]
- Schwiening CJ, Boron WF. Regulation of intracellular pH in pyramidal neurones from the rat hippocampus by Na+-dependent Cl−-HCO3− exchange. The Journal of Physiology. 1994;475:59–67. doi: 10.1113/jphysiol.1994.sp020049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata S, Kodama K, Tominaga K, Ueki S, Watanabe S. Assessment of the role of adrenoceptor function in ischemia-induced impairment of 2-deoxyglucose uptake and CA1 field potential in rat hippocampal slices. European Journal of Pharmacology. 1992;221:255–260. doi: 10.1016/0014-2999(92)90710-l. 10.1016/0014-2999(92)90710-L. [DOI] [PubMed] [Google Scholar]
- Sidky AO, Baimbridge KG. Calcium homeostatic mechanisms operating in cultured postnatal rat hippocampal neurones following flash photolysis of nitrophenyl-EGTA. The Journal of Physiology. 1997;504:579–590. doi: 10.1111/j.1469-7793.1997.579bd.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith G, Church J. Modulation of the activity of the Na+-H+ exchanger by noradrenaline in acutely dissociated adult rat hippocampal CA1 neurones. The Journal of Physiology. 1997;501:150–151P. doi: 10.1111/j.1469-7793.1998.487be.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas MJ, Moody TD, Makhinson M, O'Dell TJ. Activity-dependent β-adrenergic modulation of low frequency stimulation induced LTP in the hippocampal CA1 region. Neuron. 1996;17:475–482. doi: 10.1016/s0896-6273(00)80179-8. 10.1016/S0896-6273(00)80179-8. [DOI] [PubMed] [Google Scholar]
- Tombaugh GC, Somjen GG. Effects of extracellular pH on voltage-gated Na+, K+ and Ca2+ currents in isolated rat CA1 neurons. The Journal of Physiology. 1996;493:719–732. doi: 10.1113/jphysiol.1996.sp021417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tombaugh GC, Somjen GG. Differential sensitivity to intracellular pH among high- and low-threshold Ca2+ currents in isolated rat CA1 neurons. Journal of Neurophysiology. 1997;77:639–653. doi: 10.1152/jn.1997.77.2.639. [DOI] [PubMed] [Google Scholar]
- Trapp S, Lückermann M, Kaila K, Ballanyi K. Acidosis of hippocampal neurones mediated by a plasmalemmal Ca2+/H+ pump. NeuroReport. 1996;7:2000–2004. doi: 10.1097/00001756-199608120-00029. [DOI] [PubMed] [Google Scholar]
- Vignes M, Blanc E, Guiramand J, Gonzalez E, Sassetti I, Récasens M. A modulation of glutamate-induced phosphoinositide breakdown by intracellular pH changes. Neuropharmacology. 1996;35:1595–1604. doi: 10.1016/s0028-3908(96)00102-5. 10.1016/S0028-3908(96)00102-5. [DOI] [PubMed] [Google Scholar]
- Vornov JJ, Thomas AG, Jo D. Protective effects of extracellular acidosis and blockade of sodium/hydrogen ion exchange during recovery from metabolic inhibition in neuronal tissue culture. Journal of Neurochemistry. 1996;67:2379–2389. doi: 10.1046/j.1471-4159.1996.67062379.x. [DOI] [PubMed] [Google Scholar]
- Voyno-Yasenetskaya T, Conklin BR, Gilbert RL, Hooley R, Bourne HR, Barber DL. Gα13 stimulates Na-H exchange. Journal of Biological Chemistry. 1994;269:4721–4724. [PubMed] [Google Scholar]
- Vyklick´y L, Vlachová V, Krusek J. The effect of external pH changes on responses to excitatory amino acids in mouse hippocampal neurones. The Journal of Physiology. 1990;430:497–517. doi: 10.1113/jphysiol.1990.sp018304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakabayashi S, Shigekawa M, Pouysségur J. Molecular physiology of vertebrate Na+/H+ exchangers. Physiological Reviews. 1997;77:51–74. doi: 10.1152/physrev.1997.77.1.51. [DOI] [PubMed] [Google Scholar]
- Wallert MA, Fröhlich O. α1-Adrenergic stimulation of Na-H exchange in cardiac myocytes. American Journal of Physiology. 1992;263:C1096–1102. doi: 10.1152/ajpcell.1992.263.5.C1096. [DOI] [PubMed] [Google Scholar]
- Wang H, Singh D, Fliegel L. The Na+/H+ antiporter potentiates growth and retinoic acid-induced differentiation of P19 embryonal carcinoma cells. Journal of Biological Chemistry. 1997;272:26545–26549. doi: 10.1074/jbc.272.42.26545. [DOI] [PubMed] [Google Scholar]
- Wu M-L, Vaughan-Jones RD. Effect of metabolic inhibitors and second messengers upon Na+-H+ exchange in the sheep cardiac Purkinje fibre. The Journal of Physiology. 1994;478:301–313. doi: 10.1113/jphysiol.1994.sp020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun CHC, Oh S, Zizak M, Steplock D, Tsao S, Tse C-M, Weinman EJ, Donowitz M. cAMP-mediated inhibition of the epithelial brush border Na+/H+ exchanger, NHE3, requires an associated regulatory protein. Proceedings of the National Academy of Sciences of the USA. 1997;94:3010–3015. doi: 10.1073/pnas.94.7.3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Cytoplasmic alkalization reduces calcium buffering in molluscan central neurons. Brain Research. 1981;225:155–170. doi: 10.1016/0006-8993(81)90325-5. [DOI] [PubMed] [Google Scholar]