

Abstract
Ca2+ release flux across the sarcoplasmic reticulum (SR) during cardiac excitation-contraction coupling was investigated using a novel fluorescence method. Under whole-cell voltage-clamp conditions, rat ventricular myocytes were dialysed with a high concentration of EGTA (4.0 mm, 150 nm free Ca2+), to minimize the residence time of released Ca2+ in the cytoplasm, and a low-affinity, fast Ca2+ indicator, Oregon Green 488 BAPTA-5N (OG-5N; 1.0 mm, Kd ≈ 31 μm), to optimize the detection of localized high [Ca2+] in release site microdomains. Confocal microscopy was employed to resolve intracellular [Ca2+] at high spatial and temporal resolution.
Analytical and numerical analyses indicated that, under conditions of high EGTA concentration, the free [Ca2+] change is the sum of two terms: one major term proportional to the SR release flux/Ca2+ influx, and the other reflecting the running integral of the released Ca2+.
Indeed, the OG-5N transients in EGTA-containing cells consisted of a prominent spike followed by a small pedestal. The OG-5N spike closely resembled the first derivative (d[Ca2+]/dt) of the conventional Ca2+ transient (with no EGTA), and mimicked the model-derived SR Ca2+ release function reported previously. In SR Ca2+-depleted cells, the OG-5N transient also closely followed the waveform of L-type Ca2+ current (ICa). Using ICa as a known source of Ca2+ influx, SR flux can be calibrated in vivo by a linear extrapolation of the ICa-elicited OG-5N signal.
The OG-5N image signal was localized to discrete release sites at the Z-line level of sarcomeres, indicating that the local OG-5N spike arises from ‘Ca2+ spikes’ at transverse (T) tubule-SR junctions (due to the imbalance between calcium ions entering the cytosol and the buffer molecules).
Both peak SR release flux and total amount of released Ca2+ exhibited a bell-shaped voltage dependence. The temporal pattern of SR release also varied with membrane voltage: Ca2+ release was most synchronized and produced maximal peak release flux (4.2 mm s−1) at 0 mV; in contrast, maximal total release occurred at −20 mV (71 versus 61 μm at 0 mV), but the localized release signals were partially asynchronous. Since the maximal conventional [Ca2+] transient and contraction were elicited at 0 mV, it appears that not only the amount of Ca2+ released, but also the synchronization among release sites affects the whole-cell Ca2+ transient and the Ca2+-myofilament interaction.
Release of Ca2+ from internal stores is a common pathway of intracellular signal transduction in many types of excitable and non-excitable cells, including neurons (e.g. Shmigol et al. 1995), oocytes (e.g. Lechleiter & Clapham, 1992), endothelial cells (Ziegelstein et al. 1994) and cells in plant root hairs (Ehrhardt et al. 1996). In mammalian ventricular myocytes, Ca2+ release from the sarcoplasmic reticulum (SR) constitutes a crucial step in the excitation-contraction (E–C) coupling cascade. This process is triggered by ‘cross-talk’ of single L-type Ca2+ channels to their adjacent SR Ca2+ release channels or ryanodine receptors via the Ca2+-induced Ca2+ release (CICR) mechanism (Fabiato, 1985; Stern, 1992a; Wier et al. 1994; Cannell et al. 1994, 1995; López-López et al. 1994, 1995; Sham et al. 1995; Adachi-Akahane et al. 1996), and contributes up to 90% of the cytosolic [Ca2+] ([Ca2+]cyto) transient (Wier et al. 1994; Cheng et al. 1994). Determination of the flux of Ca2+ across the SR membrane (Jsr) is thus essential to mechanistic studies of local and global Ca2+ signalling in the heart.
The conventional [Ca2+]cyto transient is the result of several Ca2+ fluxes, predominantly Jsr, and Ca2+ removal processes (e.g. SR Ca2+-ATPase, endogenous buffers, etc.). The Jsr in skeletal muscle cells has been estimated from measurements of [Ca2+]cyto transients by explicitly accounting for Ca2+ removal mechanisms as well as properties of the indicator employed (Melzer et al. 1986; Brum et al. 1988; see Ríos & Pizarro, 1991, for review). Applying this generic Ca2+ removal model to cardiac cells, Sipido & Wier (1991) have deduced the Jsr of E–C coupling indirectly from the conventional [Ca2+]cyto transient. Their studies showed that the time course of Jsr differs markedly from that of the [Ca2+]cyto transient per se, but resembles the time derivative (d[Ca2+]cyto/dt) of the rising phase of the transient, and that the peak release flux has a bell-shaped voltage dependence with maximum of 3–5 mm s−1 at 0 mV. It was also emphasized that it is difficult to determine Jsr unequivocally by this method, due to the lack of precise information on intracellular Ca2+ handling (e.g. SR Ca2+ pump). In particular, it is dubious whether there is a steady unidirectional SR efflux during the late phase of a depolarization pulse (when the SR net flux is very small). This problem was later solved by fitting of the declining phase of the [Ca2+]cyto transient for estimations of SR pump parameters (Balke et al. 1994).
The recent discovery of ‘Ca2+ sparks’ and their characterization as the elementary SR Ca2+ release events (Cheng et al. 1993; López-López et al. 1994, 1995; Cannell et al. 1994, 1995; Cheng et al. 1995) provides another means to quantify Jsr, i.e. a ‘digital’ SR Ca2+ release function can be constructed by counting discrete spark events in confocal images (Cannell et al. 1995; López-López et al. 1995; Klein et al. 1996). Visualization of these release events further permits study of SR Ca2+ release in both spatial and temporal dimensions. Unfortunately, a prerequisite of this ‘digital’ measurement is that these sparks should not be too frequent, to prevent overlap either spatially or temporally. To date, Ca2+ sparks are discernible only over a very narrow voltage range around the foot of L-type channel activation (≤ −35 mV) (Cannell et al. 1995; Santana et al. 1996); during larger depolarizations, resolution of Ca2+ sparks requires the application of L-type Ca2+ channel blockers to reduce either the single channel conductance (e.g. Cd2+; Cannell et al. 1994) or the availability of the channel (e.g. verapmil or D600; Cheng et al. 1995; López-López et al. 1995).
In the present study, we developed a new fluorescence method to measure directly Jsr in confocal images without partial inhibition of L-type Ca2+ channels. Our approach made use of a combination of a fast, low-affinity indicator, Oregon Green 488 BAPTA-5N (OG-5N), and a high concentration of a slow, non-fluorescent Ca2+ chelator, EGTA. The conditions were chosen to minimize the residence time of free released Ca2+ in the cytoplasm and to optimize the detection of high [Ca2+] in the vicinity of localized release sites. Under our experimental conditions, the fluorescence transient signal consists of a prominent spike directly proportional to Jsr and a small component reflecting the time integral of Jsr (i.e. the total amount of Ca2+ released). This novel technique affords characterization of local and global SR Ca2+ release function over the entire physiological voltage range. A novel finding employing this method is that the extent of synchronization among release sites, as well as the total amount of release, is an important, heretofore unrecognized determinant of the peak [Ca2+]cyto transient and contraction amplitude.
METHODS
Cell preparation
Ventricular cardiac myocytes were isolated from adult Sprague-Dawley rats (2–3 months old; weight, 225–300 g) using standard enzymatic techniques, as described previously (Spurgeon et al. 1990). Briefly, following anaesthesia (sodium pentobarbital, 100 mg kg−1 injected i.p.), the heart was removed from the chest and perfused retrogradely via the aorta using the Langendorff method and collagenase (Worthington Type II, 1 mg ml−1). Following this perfusion procedure, single cells were shaken loose from the heart and stored in Hepes-buffered solution containing (mm): 137 NaCl, 5.4 KCl, 1.2 MgCl2, 1 NaH2PO4, 1 CaCl2, 20 glucose and 20 Hepes (pH 7.4).
Confocal microscopy
Myocytes were placed on the stage of a Zeiss LSM-410 inverted confocal microscope (Carl Zeiss, Inc., Germany) and excited by the 488 nm line of an argon laser. All image data were taken in the line-scanning mode, with the scan line usually oriented along the long axis of the myocyte, while the cell nucleus was excluded from the line. Each image consisted of 512 linescans (2.09 ms per linescan) and each line consisted of 512 pixels spaced at 0.104 μm intervals. The microscope objective was a Zeiss Plan-Neofluar × 40 oil immersion lens (NA, 1.3), and the confocal pinhole was set to render spatial resolutions of 0.4 μm in the horizontal plane and 0.9 μm in the axial direction, except in experiments shown in Fig. 5, in which the pinhole diameter was enlarged to render a 2-fold increase in focus depth. Image processing, data analysis and presentation were made using IDL software (Research Systems, Boulder, CO, USA).
Figure 5. Typical OG-5N transient evoked by Ca2+ influx through L-type channels.
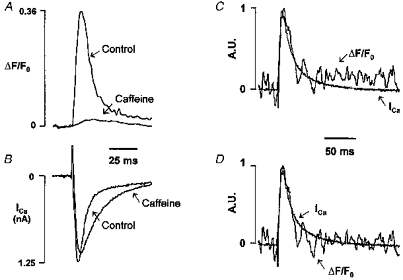
A and B, OG-5N transients (A) and ICa (B) at 0 mV before (Control) and after 10 mm caffeine. The OG-5N signal in the presence of caffeine was averaged from 5 consecutive recordings. C, OG-5N transient (noisy trace) obtained in the presence of caffeine was scaled and superimposed on the inverted ICa (A.U., arbitrary units). D, same as in C except that the integral component of the OG-5N transient was subtracted (α = 6.0 s−1). See the text for the algorithm.
Simultaneous electrophysiological recording of ICa and confocal imaging
The whole-cell patch-clamp technique was employed in conjunction with confocal imaging. Patch pipette filling solutions of designated EGTA concentrations (0, 2, 4 or 10 mm) were made by mixing, in appropriate proportions, the following two solutions: an EGTA-free solution containing (mm): 120 CsCl, 1.5 MgCl2, 5 MgATP, 10 NaCl, 10 TEACl, and 20 Hepes (pH 7.2 adjusted with CsOH); and an EGTA-containing solution containing (mm): 100 CsCl, 1.5 MgCl2, 5 MgATP, 10 NaCl, 10 TEACl, 10 EGTA, 5 CaCl2, and 20 Hepes (pH 7.2 adjusted with CsOH). The reduction of CsCl in the latter solution was to compensate for the osmolarity change due to EGTA and CaCl2, and the additional CsOH required for pH adjustment. Assuming a Kd value of 150 nm for EGTA at pH 7.2, the free [Ca2+] in the EGTA-containing solutions was 150 nm. The Ca2+ indicator Oregon Green 488 BAPTA-5N (OG-5N) hexapotassium salt (Molecular Probes, lot 5561, 1 mm) was directly dissolved in the pipette solution and then backfilled into glass microelectrodes with resistances of 1.5–2.5 MΩ. An Axopatch 200B patch-clamp amplifier (Axon Instruments) was used for electrophysiological recordings. Following the establishment of voltage control of the cell, the local cell perfusion solution was switched to a recording solution at room temperature (22–24°C) containing (mm): 137 NaCl, 5.4 CsCl, 1.2 MgCl2, 1 NaH2PO4, 2 CaCl2, 20 glucose, 0.01 TTX, and 20 Hepes (pH 7.4). After a period of ∼ 5 min to allow adequate intracellular dialysis, the L-type Ca2+ current and SR Ca2+ release were activated by a series of 200 ms depolarizations from a holding potential of −60 mV to test pulses ranging from −50 to +60 mV. The magnitude of the Ca2+ current was indexed by the difference between the peak inward current and the current at the end of the 200 ms pulse. The voltage pulses were applied every 10 or 15 s and confocal images were acquired simultaneously; the exact timing of voltage pulse was marked on a second image channel by a 2 ms flash from a light-emitting diode.
In some experiments, Cd2+ (100 μm) or caffeine (10 mm) was used to block L-type channels or to empty the SR of Ca2+, respectively. In our preliminary experiments, we varied the EGTA concentration while maintaining free Ca2+ at 150 nm and OG-5N concentrations constant in order to determine the optimal conditions for resolving the Jsr-related transient (see Results). Similar results were obtained at 4 and 10 mm EGTA, albeit the signal-to-noise ratio of image data was noticeably degraded at the higher EGTA concentration. In the presence of 2 mm EGTA, however, residual cell contractions were still visible following the voltage pulse in some cells, indicating that the global [Ca2+]cyto was not sufficiently suppressed. We therefore used 4 mm EGTA mixed with 1 mm OG-5N in all experiments, except those presented in Fig. 2.
Figure 2. Conventional [Ca2+]cyto transient measured with OG-5N (1 mm) in the absence of EGTA.
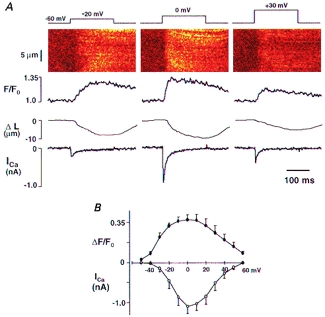
A, simultaneous recording of confocal image, cell length and ICa in a representative rat ventricular myocyte. From top to bottom: voltage-clamp protocol, confocal linescan image of OG-5N fluorescence, spatially averaged OG-5N fluorescence transient normalized by its prestimulus level (F/F0), shortening of cell length (ΔL) and ICa. Columns from left to right show results recorded on depolarization to −20, 0 and +30 mV. B, average peak fluorescence increases (ΔF/F0) and peak ICa from 6 cells plotted against membrane voltage.
In vitro properties of the Ca2+ indicator OG-5N
In the present study, the low-affinity Ca2+ indicator OG-5N was used to measure Ca2+ release flux as well as conventional [Ca2+]cyto transients. An in vitro calibration curve for OG-5N fluorescence intensity as a function of free [Ca2+] was determined on-stage using solutions containing free [Ca2+] of 1 μm to 1 mm (Ca2+ buffer kit no. 3, Molecular Probes) and 1 mm Mg2+. As shown in Fig. 1, the OG-5N signal was well described by the following equation:
| (1) |
where Kd denotes the dissociation constant, and Fmax and Fmin the fluorescence intensity at saturating and zero [Ca2+], respectively. This fitting yielded a Kd of 31.4 μm and an Fmax/Fmin of 34, comparable with values reported by Molecular Probes (Kd ≈ 20 μm, Fmax/Fmin ≈ 44). Nearly identical results were obtained in the absence of Mg2+ (data not shown). Utilizing the pseudo-ratio method described previously (Cheng et al. 1993), the absolute [Ca2+] in cell experiments can be estimated from the fold increase in OG-5N fluorescence intensity (R = F/F0, where F0 is the prestimulus value of F), the in vitro measured Kd and Rmax (Rmax = Fmax/Fmin), and an assumed resting [Ca2+] ([Ca2+]0):
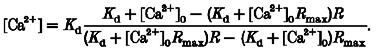 |
(2) |
Figure 1. Semi-logarithmic plot of OG-5N fluorescence intensity as a function of free [Ca2+].
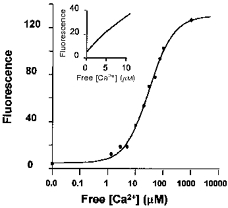
The smooth curve is the best fit of the data using eqn (1) described in Methods, yielding a Kd of 31.4 μm and Fmax/Fmin of 34. Inset shows OG-5N fluorescence intensity over a range of Ca2+ concentrations from 0 to 10 μm on a linear scale. A calcium buffer kit for low-affinity indicators was used with the addition of 1 mm MgCl2 and 20 μm OG-5N dye.
Conversion of Jsr between different units
In this report, Jsr is expressed in three interconvertible units. The raw data are given by the dimensionless ΔF/F0 of the OG-5N signal. The OG-5N signal was calibrated by determining the conversion factor between ΔF/F0 and ICa (in the absence of SR Ca2+ release; 41.7 nA per 1 unit ΔF/F0, see Results). From this, the SR release fluxes are presented as the amount of equivalent Ca2+ current (Isr), having units of nanoamps. To compare our results with those of Sipido & Wier (1991), we further employed the same conversion procedure suggested by these authors. In brief, all cells were assumed to be of the scaled geometry of a standard cell of dimensions 150 μm long, 35 μm wide and 7.5 μm thick. The volume (V) of a given cell was estimated from the membrane capacitance (Cm) of the cell, standard cell volume (Vs) and standard membrane capacitance (Cm,s, 167 pF) according to the formula:
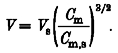 |
(3) |
The following equation was then applied:
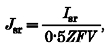 |
(4) |
where the number 0.5 refers to the fraction of cell volume accessible to Ca2+, Z is the valence of the calcium ion, and F is the Faraday constant. The final Jsr would thus possess units of millimolar per second. Accordingly, the total amount of Ca2+ released, or the integral of Jsr, is reported in either ΔF/F0 or micromolar units.
Statistics
Data are reported as means ± s.e.m. Student's t test or paired t test was applied, when appropriate, to determine differences between mean values. A P value of < 0.05 was taken as the statistical significance level.
RESULTS
Detection of Ca2+ flux associated with a ‘Ca2+ spike’: the strategy
Injection of Ca2+ into a buffer-containing medium, such as the cytosol, would produce a surge in free [Ca2+], dubbed a ‘Ca2+ spike’, which arises from a kinetic imbalance between calcium ions and the buffer. This phenomenon has been documented experimentally in the context of rapid photorelease of Ca2+ from caged compounds (Escobar et al. 1995). We therefore proposed that tracking ‘Ca2+ spikes’ during cardiac E–C coupling may afford a novel means for direct measurement of the SR Ca2+ release flux. To provide the tracking mechanism, we devised a combination of a fast Ca2+ indicator such as OG-5N with a slow Ca2+ chelator such as EGTA. Intuitively, the inclusion of a slow buffer as the Ca2+ scavenger would reduce the residence time of released free Ca2+, so that the Ca2+ spike would be shaped to follow closely the waveform of the injecting flux, but without being abolished altogether. In quantifying the relationship between the magnitude of Ca2+ flux and the amplitude of the resultant Ca2+ spike, our analytical results (see Appendix) revealed that in the presence of a high concentration of the slow buffer EGTA, changes in free [Ca2+] (Δ[Ca2+]) are composed of two terms, a major spike term proportional to the input Ca2+ flux, and a minor one proportional to its time integral:
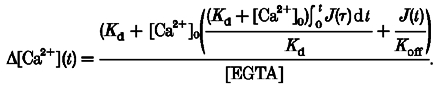 |
(5) |
The choice of a linear, fast Ca2+ indicator such as OG-5N is crucial to detection of the EGTA-shaped Ca2+ spikes. The high Kd value of OG-5N favours the detection of high [Ca2+]cyto in the microdomains of release sites and yields a good linear relation between fluorescence intensity and Ca2+ concentration (inset of Fig. 1). The linearity not only is a convenient feature for interpretation of the experimental data but also eliminates errors in measuring non-uniform [Ca2+]cyto (Wier et al. 1987; Cheng et al. 1993; Cannell et al. 1994) due to dye saturation and non-linearity (Yue & Wier, 1985). Moreover, a higher Kd usually implicates a faster Ca2+ dissociation rate; a fast off-rate is advantageous for tracking a rapid onset and decline of local Ca2+ release, so that no deconvolution is necessary to restore the true kinetics of the signal. In this respect, a closely related low-affinity dye, Calcium Orange-5N (Kd = 53 μm), has been shown to be superior to fluo-3 (Kd = 0.75 μm) in tracking the rapid decline of Ca2+ pulses generated by photolysis of caged Ca2+ (fluorescence relaxation time, 143 μs versus 5.9 ms) (Escobar et al. 1995). The dye OG-5N has the extra advantage of a much higher Fmax/Fmin than Calcium Orange-5N (3.4), an important feature for achieving better signal contrast and signal-to-noise ratio, and thereby optimizing the spatiotemporal resolution of a weak image signal. Another similar dye, Calcium Green-5N (Kd ≈ 14 μm, Fmax/Fmin ≈ 38, technical information from Molecular Probes) has been used successfully in detecting intrasarcomeric [Ca2+]cyto gradients in skeletal muscle cells (Escobar et al. 1994).
[Ca2+]cyto transients measured with OG-5N in the absence of EGTA
Before experimentally testing the idea that OG-5N combined with EGTA can track the SR release flux, we first measured conventional [Ca2+]cyto transients in rat ventricular myocytes using OG-5N in the absence of EGTA. Figure 2A shows results obtained in a representative cell with 1 mm OG-5N dialysed via the patch pipette. Each column shows, from top to bottom, membrane voltage, confocal linescan image, spatially averaged OG-5N fluorescence (F/F0), shortening of cell length (ΔL) and simultaneously recorded ICa. Depolarization to 0 mV (middle column) elicited the maximal amplitudes of the OG-5N fluorescence transient and cell contraction. The OG-5N transient reached a peak F/F0 value of 1.37 ± 0.05 in 46.3 ± 4.8 ms and declined with a half-decay time (t1/2) of 192 ± 5.1 ms (n = 5). Using eqn (2) and assuming Kd = 31.4 μmand Fmax/Fmin = 34 for OG-5N (Fig. 1), and a resting [Ca2+]cyto of 150 nm, this 37% increase in fluorescence intensity corresponds to a peak [Ca2+]cyto of 566 nm, which is within the range of the values reported previously in cardiac myocytes (0.4–1.5 μm; see Bers, 1991, for review). Figure 2B shows the bell-shaped curve for peak ΔF/F0 as a function of membrane voltage, mirrored by the current-voltage plot for ICa. These measurements using OG-5N are thus in agreement with results in the literature obtained using other indicators (Cannell et al. 1987; Beuckelmann & Wier, 1988; Cleemann & Morad, 1991).
OG-5N transients in the presence of EGTA
When 4 mm EGTA (2 mm total Ca2+ and 150 nm free Ca2+) was co-dialysed with the Ca2+ indicator, cell contraction was completely inhibited (data not shown). The most striking observation, however, was that the configuration of the OG-5N fluorescence transient (Fig. 3, F/F0) was distinctly different from that in the absence of EGTA. The OG-5N transient elicited by depolarization to middle-range voltages (e.g. −20, 0 and +20 mV) exhibited a prominent initial spike followed by a small, sustained pedestal that lasted beyond the depolarization pulse. At extreme voltages (e.g. −40 or +40 mV), the OG-5N transient displayed a flat waveform. The OG-5N spike at 0 mV had a time to peak (Tpeak) of 13.1 ± 1.3 ms and a t1/2 of 10.5 ± 0.46 ms (n = 7), which was abbreviated 18-fold compared with the t1/2 of the conventional transient in the absence of EGTA. It is particularly noteworthy that this brisk OG-5N spike is reminiscent of the previously reported SR Ca2+ release function (Fig. 8 in Sipido & Wier, 1991). This observation supports the idea that the OG-5N spike in the presence of EGTA is related to Ca2+ fluxes (mainly Jsr) into the cytosol-OG-5N-EGTA medium, rather than to the conventional, global [Ca2+]cyto transient.
Figure 3. OG-5N fluorescence transients in the presence of EGTA.
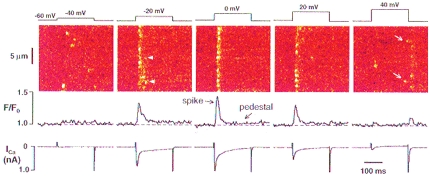
The experimental conditions were the same as in Fig. 2, except that 4 mm EGTA (with 2 mm CaCl2 to retain 150 nm free Ca2+) was included in the pipette filling solution and the trace for cell length change was omitted because EGTA totally suppressed cell contraction. Results from left to right were obtained at −40, −20, 0, +20 and +40 mV. Note that the OG-5N signal exhibits a brisk inital ‘spike’ component and a ‘pedestal’ component, except at extreme voltages (see Fig. 6 for further analysis). Arrowheads in the image at −20 mV mark probable events of repetitive activation at the same release sites. Arrows at +40 mV mark release events late into the depolarization pulse. Note also release events accompanying repolarization at +20 and +40 mV.
In contrast, the pedestal component of the OG-5N transient, which reflects a quasi-steady-state increase in global [Ca2+]cyto, displayed an extremely slow relaxation, during the sustained depolarization and after repolarization (Fig. 3, see also Fig. 5). This indicates that the rate of cytosolic Ca2+ removal by the SR and sarcolemmal Ca2+ transport mechanisms was negligible under these experimental conditions, presumably because of the large quantity of Ca2+ buffer in the cytosol and the small elevation in [Ca2+]cyto. (As we will demonstrate later, this condition further simplifies the detection of SR unidirectional release flux during E–C coupling.) The pedestal level at 0 mV was only 0.020 ± 0.004 ΔF/F0 units or Δ[Ca2+]cyto of 22 nm (see eqn (2) and Fig. 5), indicating that global elevation in [Ca2+]cyto had been damped by a factor of ∼ 20 by EGTA.
Comparison of OG-5N spike and d[Ca2+]cyto/dt of the conventional [Ca2+]cyto transient
During the rising phase of a conventional [Ca2+]cyto transient, the rate of increase in [Ca2+]cyto or d[Ca2+]cyto/dt provides a good predictor of Jsr (Sipido & Wier, 1991). To further evaluate the nature of the OG-5N spike, it is therefore instructive to compare waveforms of the OG-5N signal in the presence of EGTA and d[Ca2+]cyto/dt of the conventional transient. Figure 4A shows typical OG-5N transients at 0 mV with (red) and without (black) EGTA, and the dF/dt, which approximates d[Ca2+]cyto/dt, in the EGTA-free cell (green). All traces were normalized to the same peak and baseline. It is clear that general aspects of waveform for the OG-5N spike (in EGTA-containing cells) and d[Ca2+]cyto/dt (in EGTA-free cells) are very similar, as evidenced by similar ascending and descending rates and a nearly identical Tpeak. Figure 4B shows that over a wide range of membrane potentials, Tpeak curves for OG-5N spike and d[Ca2+]cyto/dt overlapped each other, both exhibiting a U-shaped voltage dependence centred around 0 mV. However, T90 for the OG-5N spike (time from onset to 90% decay towards the pedestal level) was consistently higher than that for d[Ca2+]cyto/dt (time from onset to 90% decay towards the baseline level; Fig. 4C). The latter result is not unexpected, because when Ca2+ removal outweighs release, d[Ca2+]cyto/dt eventually becomes negative during the descending phase of the [Ca2+]cyto transient and therefore deviates from a true SR Ca2+ release function. The kinetic similarities between the OG-5N spike and d[Ca2+]cyto/dt in the early phase of the [Ca2+]cyto transient further support the notion that the spike component of the OG-5N transient reports the SR Ca2+ release function under these conditions.
Figure 4. Waveform of the OG-5N transient in the presence of EGTA compared with the waveform of dF/dt of the OG-5N transient in an EGTA-free cell.
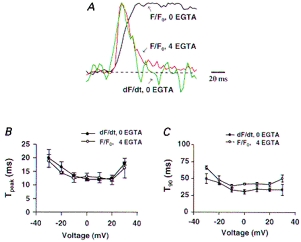
A, typical conventional [Ca2+]cyto transient elicited by depolarization to 0 mV and its first derivative, shown in red and green, respectively. The black trace shows the OG-5N transient in another cell in the presence of 4 mm EGTA. Dashed line indicates the baseline level. B and C, kinetic parameters of the OG-5N transient (with 4 mm EGTA) and dF/dt (with no EGTA). In B, Tpeak refers to time to peak. In C, T90 refers to time from peak to 90% decay to pedestal (for OG-5N transient with EGTA) or to baseline (for dF/dt, indicated by the dashed line in A).
OG-5N transient produced by Ca2+ influx via ICa
To test rigorously the idea that the OG-5N spike tracks Jsr and to quantify the relation between the magnitude of the OG-5N spike and the magnitude of Jsr, we examined the OG-5N response to a known source of Ca2+ flux, i.e. ICa injected into the cytosol. In these experiments, SR Ca2+ stores were depleted by 10 mm caffeine applied continuously in the perfusion solution. To maintain an adequate signal-to-noise ratio for resolving the ICa-induced OG-5N transient, the focus depth was enlarged 2-fold (to enhance photon collection efficiency) and traces obtained with the same voltage protocol were signal averaged. The ICa after SR depletion (0.78 ± 0.14 nA, n = 5) was slightly, but not significantly, smaller than under control conditions (0.92 ± 0.19 nA, n = 5), and elicited a tiny (< 2%) increase in OG-5N signal (ΔF/F0 = 0.017 ± 0.003, n = 5), which was only 6% of the peak OG-5N signal prior to caffeine application (0.30 ± 0.05, n = 5) (Fig. 5A). Concomitantly, the time constant of the fast component of L-type Ca2+ channel inactivation, which is sensitive to Ca2+ release from the SR (Sham et al. 1995; Adachi-Akahane et al. 1996; Sham, 1997), was increased by 2.4-fold, from 6.7 ± 0.64 ms under control conditions to 15.9 ± 2.9 ms (n = 5, P < 0.05) in the presence of caffeine (Fig. 5B). Figure 5C shows representative results for the overlay of the inverted ICa (smooth trace) and the OG-5N transient (noisy trace) in the presence of caffeine. The optically recorded OG-5N transient closely tracks the waveform of the electrophysiologically recorded ICa; after subtraction of a component related to the integral of ICa (see below), the two traces almost perfectly overlap each other (Fig. 5D). Addition of 100 μm Cd2+ completely abolished both ICa and the OG-5N transients (data not shown), confirming the ICa origin of the OG-5N signal.
Several important conclusions can be drawn from these observations. (1) OG-5N combined with EGTA does indeed track Ca2+fluxes into the cytosol. The OG-5N signal appears to be linearly related to the magnitude of Ca2+ flux. (2) Absolute calibration of the optical signal of release flux during E–C coupling can be made by a linear extrapolation of results obtained from ICa (see Discussion). The conversion factor measured in five cells was, on average, 41.7 ± 5.7 nA per unit increment of ΔF/F0. (3) ICa contributes little to the OG-5N signal when the SR is intact. This is, in part, because ICa inactivation would be hastened by SR Ca2+ release, contributing even smaller total flux than was observed with the SR empty.
Separation of the OG-5N signal into the flux- and integral-related components
The above theoretical and experimental results indicate that the OG-5N transient in the presence of EGTA can be subdivided into two components: a prominent spike directly related to Ca2+ flux (fr) and a small component representing the running integral of the flux (fs). That is,
| (6) |
Considering eqn (5) and taking the OG-5N signal as a linear measurement of instantaneous [Ca2+]cyto, we have:
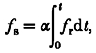 |
(7) |
where:
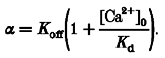 |
(8) |
Thus, α is a constant determined mainly by the dissociation constant, Kd, and off-rate, Koff, of EGTA, but appears to be independent of EGTA concentration in the high EGTA limit. Experimentally, the single parameter α was determined by fitting fs to the pedestal level after repolarization from 0 mV, when Jsr, ICa and thereby fr are expected to be zero. The same α value was subsequently applied to all traces obtained in the same cell, including the repolarization-elicited releases following strong depolarization (see below). Strictly speaking, sources of Ca2+ fluxes for fr should include input fluxes from sources other than Jsr, such as ICa (Fig. 5) and, at strong depolarizations, possibly the reverse mode Na+-Ca2+ exchange (Beuckelmann & Wier, 1989; Crespo et al. 1990). However, in most situations, these sarcolemmal influxes are small relative to Jsr (Fig. 5; Wier, 1990; Sipido & Wier, 1991; Cleemann & Morad, 1991), so we will use fr and Jsr interchangeably unless otherwise indicated. Similarly, Ca2+ clearance during the pulse by transporting mechanisms, which represents a negative unidirectional Ca2+ flux, is neglected because it is extremely slow (Fig. 3).
Voltage dependence of SR release: peak flux, total release and temporal pattern
Figure 6A shows traces of OG-5N transients obtained in the same cell at −40, −20, 0, +20, +40 and +60 mV. Applying the algorithm described above, the value of α was determined to be 5.5 s−1 in this cell (varying from 4.8 to 6.7 s−1 in 5 other cells). The dissected running integral component, fs, is shown as the dashed lines in Fig. 6A, and the concurrent SR release component, fr, is shown in Fig. 6B. The SR release was detectable at −40 mV, reached a maximum at 0 mV, and then decreased with further depolarization until virtually vanishing at +60 mV. For test potentials beyond +20 mV, repolarization also activated SR Ca2+ release, in agreement with previous observations that the tail ICa of repolarization triggers a tail [Ca2+]cyto transient (Cannell et al. 1987; Beuckelmann & Wier, 1988; Cleemann & Morad, 1991). The maximal ΔF/F0 of the OG-5N spike was 0.25 ± 0.04 (n = 7), which represents a Ca2+ flux equivalent to a Ca2+ current of 10.5 nA. Given the measured cell membrane capacitance (Cm = 111 ± 16.9 pF, n = 7), and using the conversion procedure described in Methods, this translated into a peak Jsr of 4.6 mm s−1 or 4.2 mm s−1 after subtracting ICa flux. This value of peak Jsr is thus highly comparable to that inferred from the [Ca2+]cyto transient (3–5 mm s−1 in guinea-pig heart cells at 0 mV; Sipido & Wier, 1991; Balke et al. 1994). It is important to note that the time integral of the release (fs at pedestal) reached its maximum of 0.023 ΔF/F0 units or 70.5 μm at −20 mV rather than at 0 mV (0.020 ΔF/F0 units or 61.3 μm). The values for maximal cumulative SR Ca2+ release at 0 mV are also similar to those reported previously (Sipido & Wier, 1991; Balke et al. 1994).
Figure 6. Time course and voltage dependence of SR Ca2+ release.
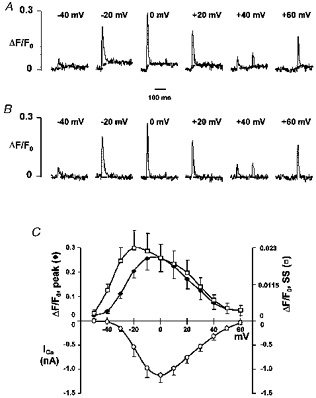
A, OG-5N transients measured at −40, −20, 0, 20, 40 and 60 mV (from left to right) in a different cell from that in Fig. 3. The dashed lines represent the integral component fs. B, release functions fr or Jsr were obtained by subtracting fs from the original records using the algorithm described in the text (α = 5.5 s−1) (horizontal dashed lines indicate the zero level). C, average voltage dependence of peak release, integral of release (SS) and peak ICa from the same cells. The total release and the peak Jsr curves were scaled so that they matched at 0 mV.
We next compared the voltage dependence of peak Jsr, of the total SR release, and of the peak ICa (Fig. 6C). Curves for peak Jsr and total release were scaled and aligned vertically at 0 mV. Although both peak and total SR release were bell shaped, they were notably different, with the largest discrepancies occurring at negative potentials. For example, the maximum for total release was observed at −20 mV rather than at 0 mV at which Jsr peaked; the total releases at −30 mV and at 0 mV were about the same but the peak Jsr at −30 mV was 45% less than that at 0 mV. These results indicate that the peak release does not always reflect the total release. Moreover, it is noteworthy that the curves for both peak and total release were shifted leftwards in relation to the peak ICa-V curve, in agreement with previous results (Cannell et al. 1987; Cleemann & Morad, 1991; Wier et al. 1994).
We also examined the temporal spreading of the SR activation at different voltages. At −40 mV, discrete release events were sporadic and occurred at any time during the pulse. Spatial averaging resulted in a small, flat OG-5N transient (Fig. 3). However, release events at middle-range voltages (−20 to +20 mV) were clustered to the early part of the pulse, and were mostly synchronized at around 0 mV (Figs 3 and 7), conferring upon the OG-5N transient the distinctive spike-like appearance. Sporadic, late release events were also seen during depolarization to very positive potentials (arrowheads in Fig. 3). Tail releases, whether large or small, were all highly synchronized and, in the accompanying line plots, had rather narrow durations. This temporal pattern of SR release provides a good explanation for the aforementioned discrepancy in the voltage dependence of total SR Ca2+ release and peak SR release flux. Evidently, the more scattered the release (e.g. at negative potentials), the smaller the peak flux, and after integration, the larger the total amount of released Ca2+ relative to the peak flux.
Figure 7. Visualization of TT-SR diadic release events during a large Jsr.
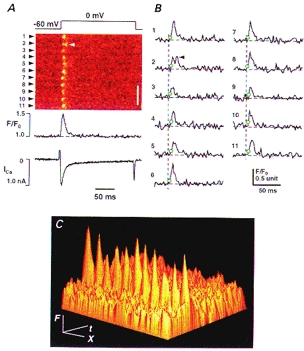
A, confocal images showing discrete TT-SR release events at 11 sarcomeres (arrowheads), release function (with the integral component subtracted) and ICa. Depolarization voltage was 0 mV (top). Arrowhead in the image shows what probably represents a reopening subsequent the first opening. Scale bar: 5 μm. B, release function at individual TT-SR sites. The dashed lines show the onset of depolarization and the open circles indicate the activation of individual TT-SR sites. C, data in A are shown as a 3-D plot of fluorescence (F), time (t) and space (X). Upon depolarization, the OG-5N fluorescence peaks sharply at the level of the Z-lines, but no detectable increase is observed at centre regions between Z-lines, locations from which the SR Ca2+ release apparatus is probably excluded.
Measuring ‘Ca2+ spikes’ at T tubule-SR diadic junctions
In addition to providing direct measurements of whole-cell Jsr and its integral, the present method provides detailed information on spatial and local characteristics of the SR release function. With EGTA limiting the spatial diffusion of the released Ca2+, the OG-5N signal was highly localized to regular, discrete sites, which were clearly discernible, even at the largest SR release (e.g. at 0 mV; Figs 3 and 7). When the scanline was placed along the longitudinal axis of the cell, the interval between neighbouring sites was about 1.8 μm (Figs 3 and 7), roughly the slack sarcomere length, indicating that these sites probably correspond to T tubule (TT)-SR diadic junctions at the Z-lines of the sarcomeres. The intrasarcomeric gradients are more clearly illustrated in Fig. 7C where fluorescence intensity (F) is portrayed as a 3-dimensional plot of time (t) and space (X). This observation substantiates our conclusion that in the presence of EGTA, the OG-5N spike directly reflects the TT-SR junctional ‘Ca2+ spikes’ associated with local release, and is consistent with the fact that the elementary SR Ca2+ release events, Ca2+ sparks, occur at the TT-SR junctional regions only (Shacklock et al. 1995; Cheng et al. 1996). Hence, it is natural for the OG-5N spike to be linearly related to the Jsr. (Note that putative discrete OG-5N spikes due to single L-type Ca2+ channel currents in SR Ca2+-depleted cells were too faint to be discerned by imaging (data not shown).) With the improved spatial resolution of release sites afforded by the present method, the timing and the time course of individual TT-SR diadic release (Jsr) can be readily resolved. Figure 7B shows an example of TT-SR response to depolarization to 0 mV. The onset of local releases (marked by open circles) was as fast as the time resolution of the scanning (2.1 ms or 1 linescan for labels 7 and 8). On average, the TT-SR release had a Tpeak of 8.78 ± 0.78 ms and a T90 of 27.6 ± 1.33 ms (n = 11 sarcomeres). Usually, a given TT-SR junction could be activated only once (within the space-time resolution limit of the present method) during a test pulse. Doublet activation events at a single TT-SR junction were only occasionally observed (arrowheads in Figs 3 and 7), suggesting that individual release sites become refractory after local release. The localized refraction could be explained by inactivation (Fabiato, 1985) or ‘adaptation’ (Györke & Fill, 1993) of the release channels (see also Sham et al. 1998), but is unlikely to be due to local depletion of SR Ca2+ stores (because pharmacologically modified ‘Ca2+ sparks’ can last several seconds) (Cheng et al. 1993; Xiao et al. 1997).
DISCUSSION
Tracking ‘Ca2+ spikes’ by OG-5N in combination with EGTA
Utilizing a combination of OG-5N and EGTA, we have, for the first time, directly measured the SR Ca2+ release function underlying cardiac E–C coupling. Specifically, the OG-5N transients in the presence of EGTA were composed of two inter-related yet separable components: a major spike component directly related to Ca2+ fluxes into the cytosol, predominantly Jsr, and a minor component (< 10% of peak) related to the running integral of the fluxes. Dissection of these components required the fitting of OG-5N transients to eqns (6) and (7) (to determine the constant α), but no specific knowledge of intracellular Ca2+ handling was necessary. Absolute calibration of Jsr made use of linear extrapolation of the response of the OG-5N-EGTA- endogenous buffer system to a known Ca2+ source, the ICa.
The experimental evidence that the OG-5N spike (with intact SR) reflects SR Ca2+ release flux is compelling. Firstly, the OG-5N spike has a unique time course that resembles d[Ca2+]cyto/dt of the conventional transient in the rising phase (Fig. 4), a rough estimator of Jsr, and mimicks the reported SR Ca2+ release function (Sipido & Wier, 1991; Balke et al. 1994). Other salient features of the directly resolved Jsr, including its magnitude and voltage dependence, are also in close agreement with those inferred from deconvolution of the [Ca2+]cyto transient (Sipido & Wier, 1991; Balke et al. 1994). Secondly, in SR Ca2+-depleted cells, the residual OG-5N transient (< 2% increase in OG-5N signal) due to ICa alone tracked the time course of the electrophysiologically recorded ICa (Fig. 5). The most direct evidence to support our conclusion, however, comes from confocal imaging. Regardless of the magnitude of SR release, the OG-5N transients consisted of discrete, localized OG-5N spikes reflecting TT-SR diadic ‘Ca2+ spikes’. As a result, the OG-5N-EGTA transient reflects the spatial and temporal summation of Ca2+ spikes at individual release sites, as well as the minute change in background [Ca2+]cyto due to the continuous Ca2+ flux.
Although TT-SR Ca2+ spikes are closely related to Ca2+ sparks (measured with fluo-3), they are conceptually different. According to model simulation, the amplitude of Ca2+ sparks in the absence of EGTA depends on not only the Ca2+ source strength, but also the duration of release; its rate of decay after termination of release is determined by diffusion of Ca2+, the mobility and kinetics of the indicator, and the exchange of Ca2+ between the indicator and endogenous buffers (Smith et al. 1998). The spikes in this study, however, are related to the local release flux underlying these Ca2+ sparks; collectively, they provide a measurement of the release function underlying the conventional [Ca2+]cyto transients (arising from the summation of the elemental Ca2+ sparks). For example, the whole-cell Ca2+ spike at 0 mV lasts about 20 ms (at 50% peak level), whereas solitary sparks have an even longer duration (typical Tpeak ≈ 15 ms and t1/2 ≈ 20 ms in ventricular myocytes) (Cheng et al. 1993; López-López et al. 1995; Xiao et al. 1997). Depending on the degree of TT-SR activation, a TT-SR junctional Ca2+ spike may or may not represent a single Ca2+ spark event. Future experimental studies are required to define the number of Ca2+ sparks involved during a junctional spike under various circumstances.
Numerical simulation of the OG-5N-EGTA- endogenous buffer system
An analytical approach detailed in the Appendix provides a theoretical basis for our utilization of OG-5N-EGTA to track Ca2+ fluxes into the cytosol. To further elucidate how OG-5N-EGTA works, we resorted to numerical simulation using realistic buffer kinetics and concentrations. In the simplest case, we assumed that Ca2+ flux of square waveform was uniformly injected into a homogeneous medium containing OG-5N, EGTA and various buffers of the characteristics of endogenous Ca2+-binding sites (Fig. 8). After subtracting the running integral component (α = 6.2 s−1), the initial overshoot in OG-5N signal or OG-5N spike reproduces well the waveform of Ca2+ flux, with an ‘on’ and ‘off’ response time of less than 1 ms (measured at 50% peak level). More importantly, the relationships between the amplitude of the OG-5N spike, the steady-state OG-5N signal and the strength of Ca2+ injection are almost linear over a wide range of Ca2+ flux (0–30 mm s−1, corresponding to peak ΔF/F0 values up to 0.8; Fig. 8B). These numerical results suggest that the ‘high, slow buffer concentration limit’ used for derivation of eqn (8) is valid under our experimental conditions. In addition, it is justifiable to use linear extrapolation of results obtained with small flux to calibrate larger flux, as we did in this study (see Results). Owing to these linear relationships, qualitatively similar results would be expected even if the source Ca2+ is spatially non-uniform, as occurs within cells, provided that the local release flux is below that required to produce local saturation of buffers.
Figure 8. The OG-5N Ca2+ spike in the presence of a high concentration of a slow Ca2+ buffer: numerical simulation.
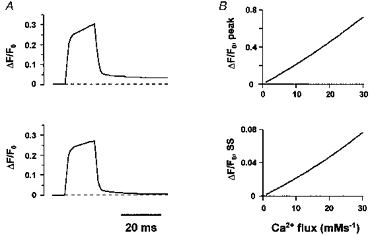
The following buffer species were included in our simulation: EGTA, 4000 μm, Kon = 20 μm−1 s−1, Koff = 3 s−1; OG-5N, 1000 μm, Kon = 323 μm−1 s−1, Koff = 10 000 s−1; calmodulin, 24 μm, Kon = 100 μm−1 s−1, Koff = 38 s−1; troponin C, 70 μm, Kon = 39 μm−1 s−1, Koff = 20 s−1; SR membrane binding sites, 47 μm, Kon = 115 μm−1 s−1, Koff = 100 s−1; sarcolemmal membrane binding sites, 1124 μm, Kon = 115 μm−1 s−1, Koff = 115 000 s−1. The initial resting [Ca2+] was 150 nm. The dissociation constants for EGTA are based on its Kd (150 nm at pH7.2), the measured value of α in cells (≈ 6 s−1) and eqn (8). The dissociation rate for OG-5N was chosen in reflection of its Kd value (31 μm) and the result of Escobar et al. (1995) on a similar dye. The protein and membrane Ca2+-binding parameters are integrated from data in the literature (Sipido & Wier, 1991; Weir et al. 1994; Langer & Peskoff, 1996). A, waveforms of Ca2+ flux (15 ms square wave, 12.5 mm s−1) for the raw OG-5N transient (top), and the OG-5N transient after removal of the running integral (α = 6.2 s−1; bottom). The on- and off-response time is 0.8 ms measured at 50% peak level. The small creeping component seen after subtraction of the integral component reflects non-equilibrium attribution of injected Ca2+ among various buffers (during injection) or interchange of Ca2+ between EGTA and OG-5N (after termination of injection). Increasing EGTA concentration further suppressed the creeping component, and reduced the spike amplitude, but only slightly affected the value of α (data not shown). B, peak OG-5N spike (top) and steady-state pedestal OG-5N fluorescence (bottom) as a function of the magnitude of Ca2+ flux.
Does EGTA alter Jsr under physiological conditions?
Within a TT-SR diadic junction, main Ca2+ signalling routes are thought to proceed from L-type Ca2+ influx to activation of the release channels; released Ca2+ reciprocally inactivates the L-type channels, and release channels interact with each other, all occurring at nanoscopic and microscopic scales. The inclusion of EGTA at concentrations used in the present study is, however, not expected to disturb, to a significant extent, any of these intermolecular Ca2+ signalling processes. In studies using millimolar concentrations of fura-2, it has been found that the SR release remains intact despite the fact that the global [Ca2+]cyto transient is markedly reduced and cell contraction is totally abolished (Adachi-Akahane et al. 1996). Recently, Sham (1997) provided more direct evidence that intracellular dialysis of up to 10 mm EGTA has little effect on the release flux, as manifested by little change in the release-dependent L-type Ca2+ channel inactivation (Sham, 1997). A similar result was observed in the present study (data not shown). Consistent with these observations, it has been illustrated theoretically that millimolar EGTA is impotent with respect to chelation of Ca2+ in the vicinity of permeation pores of Ca2+ channels (Stern, 1992a, b). We therefore may conclude that in the present study EGTA does not alter the Jsr. Indeed, the absolute SR flux determined here is not different from the value obtained in the absence of excessive exogenous buffers (Sipido & Wier, 1991; Balke et al. 1994; Wier et al. 1994). Nevertheless, it should be cautioned that supramicrometre Ca2+ communications (e.g. possible interaction between adjacent TT-SR diads, Ca2+ waves) or global Ca2+-mediated effects (e.g. cell contraction) could be retarded or abolished by millimolar EGTA. Another limitation imposed by EGTA is that a long interpulse interval (≥ 10 s) is necessary to ensure repletion of the SR store (as judged by a large, stable SR release in response to repetitive depolarizations to 0 mV; data not shown).
Temporal synchronization of SR release and its physiological significance
Both confocal images and line plots of Jsr revealed that the degree of synchronization of SR Ca2+ release is distinctly voltage dependent. The most synchronized release occurs at around 0 mV, as reflected by the fastest Tpeak and the narrowest T90 of Jsr, as well as the fact that discrete TT-SR spikes clustered to the beginning of the pulse. At the negative voltage end, however, release events were activated more asynchronously, resulting in a scattered TT-SR activation and a flat Jsr. Interestingly, the inactivation time constants of ICa exhibit similar U-shaped voltage dependencies, with the fastest ICa observed at around 0 mV (Sham, 1997). This suggests that the SR Ca2+ release kinetics are tightly controlled by the activation and inactivation kinetics of L-type channels. This interpretation is in agreement with the observation that, in the presence of L-type channel blockers, activation of Ca2+ sparks at all voltages follows the waveform of L-type current in the presence of verapmil (López-López et al. 1995). However, local refractoriness after release, as shown in Fig. 7, and possibly SR Ca2+ depletion may provide additional mechanisms to accentuate the temporal localization of large Jsr.
A novel concept arising from the present results is that the synchronization among SR Ca2+ release sites may be of vital physiological significance in determining the effectiveness of released Ca2+ to activate the contractile machinery. For example, the maximal cell contraction, Jsr and [Ca2+]cyto transient occur at 0 mV, at which the releases occur synchronously (Sipido & Wier, 1991; Balke et al. 1994; this study); in contrast, total SR Ca2+ release peaks around −20 mV (Fig. 6C), but the release is relatively unsynchronized. In addition, cells contract twice as vigorously at 0 mV as at −30 mV (data not shown), with about the same amount of released Ca2+ (Fig. 6). We speculate that substantial modulation of cardiac contractility can occur simply by varying the degree of synchronization of SR Ca2+ release.
Comparison with previous work
In general, the direct measurement of Jsr using the OG-5N-EGTA fluorescence method is an improvement upon the Ca2+ removal modelling method to estimate Jsr (Melzer et al. 1986; Brum et al. 1988; Sipido & Wier, 1991). As long as the high, slow buffer concentration condition is valid, it does not require cell-type specific knowledge of intrinsic Ca2+ transporting and buffering mechanisms. In addition, because the ‘differentiation’ implicit in the calculation of release flux is done by the buffer system, before the addition of photon shot noise, the signal-to-noise ratio is more favourable than in methods that determine release flux numerically from fluorescence signals that reflect [Ca2+]cyto. Possible errors due to saturation and non-linearity of the Ca2+ indicator (Yue & Wier, 1985) have essentially been circumvented, and the interpretation of the experimental data is rather straightforward. The present method also extends the ‘digital’ Ca2+ spark-counting method (Cheng et al. 1993; Cannell et al. 1995; López-López et al. 1995; Klein et al. 1996) by providing continuous, absolute measurement of Jsr while maintaining a submicrometre spatial resolution of release origin, even at large values of Jsr. In contrast to the spark-counting method, this approach is also applicable when unitary properties of Ca2+ sparks are subjected to modulation by physiological and pharmacological mechanisms (e.g. Cheng et al. 1993; Xiao et al. 1997; Song et al. 1997). In skeletal muscle, the [Ca2+]cyto transient in the presence of EGTA (15 mm) has the same general shape as the Ca2+ release waveform but without an apparent integral component (Ríos & Pizarro, 1991). The present results clearly show both release- and integral-related components. Our theoretical reasoning further suggests that the relative magnitude of these components (the value α) would be insensitive to EGTA concentration, provided it is sufficiently large. The reason for the absence of an integral component under the experimental conditions of Ríos & Pizarro (1991) is unclear. Transient, large intrasarcomere [Ca2+]cyto gradients have been observed previously in both skeletal (Escobar et al. 1994) and cardiac muscle cells (Isenberg et al. 1996). In the presence of EGTA, which serves mainly as a free-Ca2+ scavenger rather than as a Ca2+ diffusion facilitator, these gradients appeared to be further steepened under our experimental conditions (Figs 3 and 7), as evidenced by the lack of cellular contraction despite large TT-SR junctional Ca2+ spikes.
Similar to the previous studies (Cleemann & Morad, 1991; Wier et al. 1994), a leftwards shift of the SR Ca2+ release as a function of voltage in relation to the ICa-V curve was observed in the present study (Fig. 6C). This indicates that the efficacy of L-type Ca2+ influxes as the trigger of release decreases as membrane potential is increasingly positive. This phenomenon can now be explained on the basis that: (1) SR release is controlled locally by single L-type channel current (iCa) (Niggli & Lederer, 1990; Stern 1992a; Wier et al. 1994; Cannel et al. 1995; López-López et al. 1995; Sham et al. 1995; Sham, 1997); (2) the electrochemical driving force and therefore the amplitude of iCa decrease monotonically with depolarization towards the reversal potential; and importantly, (3) SR activation is a non-linear (square) function of iCa (Santana et al. 1996).
The intermolecular Ca2+ signalling at TT-SR diadic junctions has been the main focus of several recent theoretical studies (Langer & Peskoff, 1996; Soeller & Cannell, 1997). It seems that a thorough understanding of cardiac E–C coupling would ultimately rely on resolution of local Ca2+ signalling on submicrometre and submillisecond scales. In this regard, the present method of visualization of Ca2+ spikes associated with TT-SR release may provide a stepping stone towards achieving this challenging goal. Moreover, since mobilization of internal Ca2+ through ryanodine receptors or inositol trisphosphate receptors is a ubiquitous signalling process, this method may have general applications for studies in many other types of cell.
Acknowledgments
The authors acknowledge the expert technical support from Dr Harold Spurgeon and Bruce Ziman. H. C. would like to thank Dr J. L. Vergara for discussion on the use of low-affinity dye. This work was supported by NIH Intramural Research Program (H. C., M. D. S. and E. G. L.) and extramural grants (HL-52652, J. S. K. S.) and AHA Established Investigator Award (J. S. K. S.).
APPENDIX
An analytical derivation of the behaviour of Ca2+ under the high EGTA conditions was carried out with the assistance of the symbolic computer mathematics package Macsyma (Macsyma Corp., Arlington, MA, USA). We began with the basic equation of Ca2+ balance in the presence of EGTA, including a factor b + 1 for effective volume due to other buffers treated in the rapid buffering approximation, which is the worst case for the error due to flux on and off the fast buffers (see Discussion for a more realistic numerical simulation of fast buffer effects). The free Ca2+ concentration is ca(t), the source flux into the cell is J(t), total EGTA is egt,and [Ca2+] : [EGTA] is egca(t):
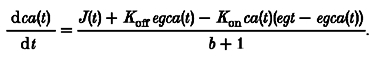 |
(A1) |
Net Ca2+ balance is expressed by the following, where bb(t) is Ca2+ bound to fast buffers (including Oregon Green), and ig(t) is the running integral of J(t):
| (A2) |
where the starting equilibrium values are given by:
| (A3) |
and
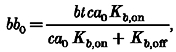 |
(A4) |
where Kb,on and Kb,off are association and dissociation rate constants for the fast buffers. Using eqn (A2) to replace egca(t) in eqn (A1), and defining δ as the deviation of [Ca2+] from resting:
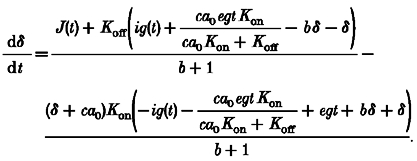 |
(A5) |
Equation (A5) cannot be solved analytically because of the quadratic appearance of δ together with the unknown functions J(t) and ig(t). However, it is intuitively obvious (and can be proven mathematically) that the excursion of [Ca2+] from equilibrium, δ, can be made as small as desired by using a sufficiently large concentration of EGTA, which will also permit the total amount of Ca2+ entering, ig(t), to be treated as a (relatively) small quantity. Therefore, in the high EGTA conditions, we can linearize the equation in δ, J and ig, obtaining, after dropping constants which are relatively small in the high EGTA conditions, a linear differential equation with constant coefficients:
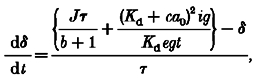 |
(A6) |
where:
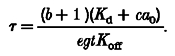 |
(A7) |
This can be recognized as the equation of a single-compartment system with a source given by the quantity in curly brackets, and transient (mono-exponential) response time τ. For times and rates of change of J(t) long compared with this transient response time (which itself becomes arbitrarily short in the high EGTA conditions), δ will be given by the steady-state response, which is found by setting the right-hand side of eqn (A6) to zero. After replacing δ and ig by their definitions, we find that the deviation of [Ca2+] from equilibrium is given by eqn (A8), identical to eqn (5) in the Results, confirming that it is the sum of two terms, one proportional to the Ca2+ flux/release function J(t)) and the other to its time integral:
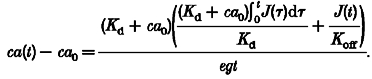 |
(A8) |
References
- Adachi-Akahane S, Cleemann L, Morad M. Cross-signaling between L-type Ca2+ channels and ryanodine receptors in rat ventricular myocytes. Journal of General Physiology. 1996;108:435–454. doi: 10.1085/jgp.108.5.435. 10.1085/jgp.108.5.435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balke CW, Egan TM, Wier WG. Processes that remove calcium from the cytoplasm during excitation-contraction coupling in intact rat heart cells. The Journal of Physiology. 1994;474:447–462. doi: 10.1113/jphysiol.1994.sp020036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. Boston, USA: Kluwer Academic Publishers; 1991. [Google Scholar]
- Beuckelmann DJ, Wier WG. Mechanism of release of calcium from sarcoplasmic reticulum of guinea-pig cardiac cells. The Journal of Physiology. 1988;405:233–255. doi: 10.1113/jphysiol.1988.sp017331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beuckelmann DJ, Wier WG. Sodium-calcium exchange in guinea-pig cardiac cells: exchange current and changes in intracellular Ca2+ The Journal of Physiology. 1989;414:499–520. doi: 10.1113/jphysiol.1989.sp017700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brum G, Ríosiacute;os E, Stefani E. Effects of extracellular calcium on calcium movements of excitation-contraction coupling in frog skeletal muscle fibres. The Journal of Physiology. 1988;398:441–473. doi: 10.1113/jphysiol.1988.sp017052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Berlin JR, Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. Spatial non-uniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Biophysical Journal. 1994;67:1942–1956. doi: 10.1016/S0006-3495(94)80677-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannell MB, Cheng H, Lederer WJ. The control of calcium release in heart muscle. Science. 1995;268:1045–1049. doi: 10.1126/science.7754384. [DOI] [PubMed] [Google Scholar]
- Cheng H, Cannell MB, Lederer WJ. Propagation of excitation-contraction coupling into ventricular myocytes. Pflügers Archiv. 1994;428:415–417. doi: 10.1007/BF00724526. [DOI] [PubMed] [Google Scholar]
- Cheng H, Cannell MB, Lederer WJ. Partial inhibition of Ca2+ current by methoxyverapamil (D600) reveals spatial nonuniformities in [Ca2+]i during excitation-contraction coupling in cardiac myocytes. Circulation Research. 1995;76:236–241. doi: 10.1161/01.res.76.2.236. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- Cheng H, Lederer MR, Lederer WJ, Cannell MB. Calcium sparks and [Ca2+]i waves in cardiac myocytes. American Journal of Physiology. 1996;270:C148–159. doi: 10.1152/ajpcell.1996.270.1.C148. [DOI] [PubMed] [Google Scholar]
- Cleemann L, Morad M. Role of Ca2+ channel in cardiac excitation-contraction coupling in the rat: evidence from Ca2+ transients and contraction. The Journal of Physiology. 1991;432:283–312. doi: 10.1113/jphysiol.1991.sp018385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crespo LM, Grantham CJ, Cannell MB. Kinetics, stoichiometry and role of the Na-Ca exchange mechanism in isolated cardiac myocytes. Nature. 1990;345:618–621. doi: 10.1038/345618a0. [DOI] [PubMed] [Google Scholar]
- Ehrhardt DW, Wais R, Long SR. Calcium spiking in plant root hairs responding to Rhizobium nodulation signals. Cell. 1996;85:673–681. doi: 10.1016/s0092-8674(00)81234-9. [DOI] [PubMed] [Google Scholar]
- Escobar AL, Cifuentes F, Vergara JL. Detection of Ca2+-transients elicited by flash photolysis of DM-nitrophen with a fast calcium indicator. FEBS Letters. 1995;364:335–338. doi: 10.1016/0014-5793(95)00425-9. [DOI] [PubMed] [Google Scholar]
- Escobar AL, Monck JR, Fernandez JM, Vergara JL. Localization of the site of Ca2+ release at the level of a single sarcomere in skeletal muscle fibres. Nature. 1994;367:739–741. doi: 10.1038/367739a0. [DOI] [PubMed] [Google Scholar]
- Fabiato A. Time and calcium dependence of activation and inactivation of calcium-induced release of calcium from the sarcoplasmic reticulum of a skinned canine cardiac Purkinje cell. Journal of General Physiology. 1985;85:247–289. doi: 10.1085/jgp.85.2.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gyyörkeouml;rke S, Fill M. Ryanodine receptor adaptation: control mechanism of Ca2+-induced Ca2+ release in heart. Science. 1993;260:807–809. doi: 10.1126/science.8387229. [DOI] [PubMed] [Google Scholar]
- Isenberg G, Etter EF, Wendt-Gallitelli MF, Schiefer A, Carrington WA, Tuft RA, Fay FS. Intrasarcomere [Ca2+] gradients in ventricular myocytes revealed by high speed digital imaging microscopy. Proceedings of the National Academy of Sciences of the USA. 1996;93:5413–5418. doi: 10.1073/pnas.93.11.5413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein MG, Cheng H, Santana LF, Jiang YH, Lederer WJ, Schneider MF. Two mechanisms of quantized calcium release in skeletal muscle. Nature. 1996;379:455–458. doi: 10.1038/379455a0. [DOI] [PubMed] [Google Scholar]
- Langer GA, Peskoff A. Calcium concentration and movement in the diadic cleft space of the cardiac ventricular cell. Biophysical Journal. 1996;70:1169–1182. doi: 10.1016/S0006-3495(96)79677-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lechleiter JD, Clapham DE. Molecular mechanisms of intracellular calcium excitability in X. laevis oocytes. Cell. 1992;69:283–294. doi: 10.1016/0092-8674(92)90409-6. [DOI] [PubMed] [Google Scholar]
- López-López JR, Shacklock PS, Balke CW, Wier WG. Local, stochastic release of Ca2+ in voltage-clamped rat heart cells: visualization with confocal microscopy. The Journal of Physiology. 1994;480:21–29. doi: 10.1113/jphysiol.1994.sp020337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-López JR, Shacklock PS, Balke CW, Wier WG. Local calcium transients triggered by single L-type calcium channel currents in cardiac cells. Science. 1995;268:1042–1045. doi: 10.1126/science.7754383. [DOI] [PubMed] [Google Scholar]
- Melzer W, Ríosiacute;os E, Schneider MF. The removal of myoplasmic free calcium following calcium release in frog skeletal muscle. The Journal of Physiology. 1986;372:261–292. doi: 10.1113/jphysiol.1986.sp016008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niggli E, Lederer WJ. Voltage-independent calcium release in heart muscle. Science. 1990;250:565–568. doi: 10.1126/science.2173135. [DOI] [PubMed] [Google Scholar]
- Ríosiacute;os E, Pizarro G. Voltage sensor of excitation-contraction coupling in skeletal muscle. Physiological Reviews. 1991;71:849–908. doi: 10.1152/physrev.1991.71.3.849. [DOI] [PubMed] [Google Scholar]
- Santana LF, Cheng H, Gomez AM, Cannell MB, Lederer WJ. Relation between the sarcolemmal Ca2+ current and Ca2+ sparks and local control theories for cardiac excitation-contraction coupling. Circulation Research. 1996;78:166–171. doi: 10.1161/01.res.78.1.166. [DOI] [PubMed] [Google Scholar]
- Shacklock PS, Wier WG, Balke CW. Local Ca2+ transients (Ca2+ sparks) originate at transverse tubules in rat heart cells. The Journal of Physiology. 1995;487:601–608. doi: 10.1113/jphysiol.1995.sp020903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham JS. Ca2+ release-induced inactivation of Ca2+ current in rat ventricular myocytes: evidence for local Ca2+ signalling. The Journal of Physiology. 1997;500:285–295. doi: 10.1113/jphysiol.1997.sp022020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham JS, Cleemann L, Morad M. Functional coupling of Ca2+ channels and ryanodine receptors in cardiac myocytes. Proceedings of the National Academy of Sciences of the USA. 1995;92:121–125. doi: 10.1073/pnas.92.1.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham JS, Song LS, Chen Y, Deng LH, Stern MD, Lakatta EG, Cheng H. Termination of Ca2+ release by local inactivation of ryanodine receptors in cardiac myocytes. Proceedings of the National Academy of Sciences of the USA. 1998 doi: 10.1073/pnas.95.25.15096. (in the Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A, Verkhratsky A, Isenberg G. Calcium-induced calcium release in rat sensory neurons. The Journal of Physiology. 1995;489:627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipido KR, Wier WG. Flux of Ca2+ across the sarcoplasmic reticulum of guinea-pig cardiac cells during excitation-contraction coupling. The Journal of Physiology. 1991;435:605–630. doi: 10.1113/jphysiol.1991.sp018528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GD, Keizer JE, Stern MD, Lederer WJ, Cheng H. A simple numerical model of calcium spark formation and detection in cardiac myocytes. Biophysical Journal. 1998;75:15–32. doi: 10.1016/S0006-3495(98)77491-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soeller C, Cannell MB. Numerical simulation of local calcium movements during L-type calcium channel gating in the cardiac diad. Biophysical Journal. 1997;73:97–111. doi: 10.1016/S0006-3495(97)78051-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song LS, Stern MD, Lakatta EG, Cheng H. Partial depletion of sarcoplasmic reticulum calcium does not prevent calcium sparks in rat ventricular myocytes. The Journal of Physiology. 1997;505:665–675. doi: 10.1111/j.1469-7793.1997.665ba.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spurgeon HA, Stern MD, Baartz G, Raffaeli S, Hansford RG, Talo A, Lakatta EG, Capogrossi MC. Simultaneous measurement of Ca2+, contraction, and potential in cardiac myocytes. American Journal of Physiology. 1990;258:H574–586. doi: 10.1152/ajpheart.1990.258.2.H574. [DOI] [PubMed] [Google Scholar]
- Stern MD. Theory of excitation-contraction coupling in cardiac muscle. Biophysical Journal. 1992a;63:497–517. doi: 10.1016/S0006-3495(92)81615-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern MD. Buffering of calcium in the vicinity of a channel pore. Cell Calcium. 1992b;13:183–192. doi: 10.1016/0143-4160(92)90046-u. [DOI] [PubMed] [Google Scholar]
- Wier WG. Cytoplasmic [Ca2+] in mammalian ventricle: dynamic control by cellular processes. Annual Review of Physiology. 1990;52:467–485. doi: 10.1146/annurev.ph.52.030190.002343. [DOI] [PubMed] [Google Scholar]
- Wier WG, Cannell MB, Berlin JR, Marban E, Lederer WJ. Cellular and subcellular heterogeneity of [Ca2+]i in single heart cells revealed by fura-2. Science. 1987;235:325–328. doi: 10.1126/science.3798114. [DOI] [PubMed] [Google Scholar]
- Wier WG, Egan TM, Lópezoacute;pez-Lópezoacute;pez JR, Balke CW. Local control of excitation-contraction coupling in rat heart cells. The Journal of Physiology. 1994;474:463–471. doi: 10.1113/jphysiol.1994.sp020037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao RP, Valdivia HH, Bogdanov K, Valdivia C, Lakatta EG, Cheng H. The immunophilin FK506-binding protein modulates Ca2+ release channel closure in rat heart. The Journal of Physiology. 1997;500:343–354. doi: 10.1113/jphysiol.1997.sp022025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yue DT, Wier WG. Estimation of intracellular [Ca2+] by nonlinear indicators. A quantitative analysis. Biophysical Journal. 1985;48:533–537. doi: 10.1016/S0006-3495(85)83810-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziegelstein RC, Spurgeon HA, Pili R, Passaniti A, Cheng L, Corda S, Lakatta EG, Capogrossi MC. A functional ryanodine-sensitive intracellular Ca2+ store is present in vascular endothelial cells. Circulation Research. 1994;74:151–156. doi: 10.1161/01.res.74.1.151. [DOI] [PubMed] [Google Scholar]