

Abstract
The presynaptic calcium current (IpCa) was recorded from the calyx of Held in rat brainstem slices using the whole-cell patch clamp technique.
Tetanic activation of IpCa by 1 ms depolarizing voltage steps markedly enhanced the amplitude of IpCa. Using a paired pulse protocol, the second (test) response was facilitated with inter-pulse intervals of less than 100 ms. The facilitation was greater at shorter intervals and was maximal (about 20 %) at intervals of 5–10 ms.
When the test pulse duration was extended, the facilitation was revealed as an increased rate of IpCa activation. From the current-voltage relationship measured at 1 ms from onset, facilitation could be described by a shift in the half-activation voltage of about −4 mV.
IpCa facilitation was not attenuated when guanosine-5′-O-(3-thiotriphosphate) (GTPγS) or guanosine-5′-O-(2-thiodiphosphate) (GDPβS) was included in the patch pipette, suggesting that G-proteins are not involved in this phenomenon.
On reducing [Ca2+]o, the magnitude of facilitation diminished proportionally to the amplitude of IpCa. Replacement of [Ca2+]o by Ba2+ or Na+, or buffering of [Ca2+]i with EGTA or BAPTA attenuated IpCa facilitation.
We conclude that repetitive presynaptic activity can facilitate the presynaptic Ca2+ current through a Ca2+-dependent mechanism. This mechanism would be complementary to the action of residual Ca2+ on the exocytotic machinery in producing activity-dependent facilitation of synaptic responses.
An activity-dependent rise in intracellular calcium plays a crucial role in triggering transmitter release and intracellular signals linked to ion channel modulation, gene expression and induction of synaptic plasticity (for reviews see Bliss & Collingridge, 1993; Milner et al. 1998). Voltage-gated Ca2+ channels are important routes for the entry of Ca2+ into excitable cells and their open probability is closely linked to neuronal excitability. Many Ca2+ channel subtypes are modulated by phosphorylation through protein kinase activity or via membrane delimited mechanisms involving GTP binding proteins (G-proteins) (for review see Dolphin, 1996). Although the functional consequences of Ca2+ channel modulation are not fully understood, modulation of presynaptic Ca2+ currents (IpCa) can modulate synaptic transmission, for example by G-protein-coupled receptors (Takahashi et al. 1996, 1998).
Activity-dependent accumulation of presynaptic intra-cellular calcium is thought to contribute to synaptic facilitation (Katz & Miledi, 1968). Direct recordings from the squid giant synapse showed that the presynaptic Ca2+ current did not change during repetitive activation (Charlton et al. 1982), so the mechanism of facilitation has been wholly attributed to the exocytotic process downstream of Ca2+ influx. At the calyx of Held, however, activity-dependent changes in presynaptic calcium currents have been observed (Forsythe et al. 1998), with repetitive presynaptic stimulation producing post-tetanic depression of excitatory postsynaptic currents (EPSCs), due to accumulating inactivation of presynaptic Ca2+ currents (IpCa). Surprisingly, the inactivation was preceded by a transient facilitation of IpCa.
The aim of the present study is to characterize the facilitation of IpCa at this giant presynaptic terminal. This synapse forms on the cell bodies of principal neurones in the medial nucleus of the trapezoid body (MNTB) and exocytosis is predominantly triggered by P-type Ca2+ channels (Forsythe et al. 1998; Iwasaki & Takahashi, 1998). Our results indicate that the facilitation of this P-type IpCa is dependent on [Ca2+]i acting on the Ca2+ channel itself. In this respect, this novel mechanism is complementary to the residual calcium hypothesis for synaptic facilitation, which has previously been implicated as a direct effect of Ca2+ on exocytosis (for review see Zucker, 1996).
METHODS
Brainstem slices were prepared from Lister Hooded or Wistar rats, 9–17 days old, killed by decapitation following published methods (Barnes-Davies & Forsythe, 1995). For most experiments, animals older than 10 days were used to minimize developmental changes of Ca2+ channel types (Iwasaki & Takahashi, 1998). In brief, the brain was rapidly removed into cold (< 4°C) low sodium artificial cerebrospinal fluid (ACSF; in which NaCl was substituted by 250 mm sucrose). Transverse brainstem slices (125–150 μm thick) containing the MNTB were cut and transferred into an incubation chamber containing normal ACSF at 37°C. After 1 h incubation, slices were stored at 24–26°C. The normal ACSF consisted of (mm): NaCl, 125; KCl, 2.5; NaHCO3, 26; glucose, 10; NaH2 PO4, 1.25; CaCl2, 2; MgCl2, 1; myo-inositol, 3; sodium pyruvate, 2; ascorbic acid, 0.5 (pH 7.4 when saturated with 95 % O2 and 5 % CO2).
Experiments were carried out at 24–26°C. One slice was transferred into an experimental chamber, mounted onto a fixed-stage Axioskop microscope (Zeiss) and continuously perfused with ACSF at a rate of 1 ml min−1. Presynaptic terminals surrounding MNTB neurones were visualized using a × 63 water immersion objective and differential interference contrast optics. Whole-cell voltage-clamp recordings of presynaptic currents were made from the calyx of Held (Forsythe, 1994; Borst et al. 1995; Takahashi et al. 1996) with patch electrodes pulled from standard walled glass capillaries (1.5 mm outer diameter, Clark Electromedical, Pangbourne, UK) having a resistance of 4–8 MΩ when filled with an internal solution of (mm): CsCl, 110; Hepes, 40; EGTA, 0.5; sodium phosphocreatine, 12; tetraethylammonium (TEA)-Cl, 10; ATP, 2; GTP, 0.5 (pH 7.2 with CsOH). To isolate Ca2+ currents TEA-Cl (10 mm) and tetrodotoxin (TTX, 1 μm) were added to the ACSF. Stable recordings (for up to 2 h) were obtained using Olympus ONU 31-P ultrasound micromanipulators mounted on the fixed stage. The cell capacitance of presynaptic terminals was 10–22 pF. The series resistance ranged from 7 to 20 MΩ and was compensated by > 80 %. An Axopatch 200A or 200B amplifier was used for patch-clamp recordings. For analysis, pCLAMP 6.03 software suite was used with data sampled at 20 kHz on the Digidata 1200 A/D converter (Axon Instruments) and filtered at 5–10 kHz (8-pole Bessel filter). To assess whether possible voltage-clamp errors may affect leak currents in paired pulse experiments, the test pulse leak currents were estimated with a −10 mV voltage step from the holding potential of −80 mV following a conditioning voltage step to −70 mV (which did not generate Ca2+ currents) in one protocol and to −10 mV (which did generate Ca2+ currents) in another protocol. No difference was found in the magnitude of leak currents between these protocols, suggesting that the Ca2+ conductance activated by the conditioning pulse had no effect on the test pulse leak current. In subsequent experiments leak currents were generated using +10 mV steps and 5 or 10 traces averaged and scaled appropriately for subtraction from the +70 mV test pulse potential (P/n protocol). The current-voltage relationship for the test IpCa was fitted by a modified Boltzmann equation: IpCa = Gmax (Vm–Vr)/1 + exp((V1/2–Vm)/k), where Vm, Vr, V1/2 and k respectively correspond to the membrane potential, reversal potential, half-activation voltage and the slope. Data values are presented as means ± s.e.m.
The following pharmacological agents were added to the perfusate: 1-5-isoquinolinylsulphonyl)-2-methylpiperazine (H-7) and baclofen; and the following agents were added to the internal patch pipette solution: 1-(N,O-bis[5-isoquinolinesulphonyl]-N-methyl-L-tyrosyl)-4-phenylpiperazine (KN-62), calyculin A, deltamethrin, GTPγS and GDPβS. All compounds were obtained from Sigma except for H-7, KN-62, deltamethrin and calyculin A, which were obtained from Calbiochem-Novabiochem. Ca2+ channels were rendered Na+ permeable by removing external divalent cations and adding 1 mm EGTA to ACSF (Forsythe et al. 1998). The concentration of Na+ in ACSF was reduced to 75 mm by substitution with N-methyl-D-glucamine chloride.
RESULTS
Facilitation of presynaptic calcium currents
Presynaptic Ca2+ currents (IpCa) were evoked in a calyx by a brief depolarizing pulse (1 ms). The amplitude of IpCa was stable when stimulated at 0.1 Hz but underwent a dramatic enhancement during tetanic stimulation (100 Hz), followed by a gradual decline due to inactivation of IpCa (Fig. 1A, see also Forsythe et al. 1998). To characterize the IpCa facilitation in isolation from inactivation, we used a paired pulse protocol. When a pair of brief depolarizing pulses were given at a short interval, the second (test) IpCa was clearly facilitated relative to the first (Fig. 1B). The magnitude of facilitation with a 5 ms inter-pulse interval was 21.0 ± 1.8 % (n = 6), which was similar to that at 10 ms. For longer inter-pulse intervals the magnitude of the facilitation declined and no facilitation was observed with intervals longer than 100 ms. For comparative purposes, the 10 ms inter-pulse interval was used as standard in further studies; the mean facilitation was 19.7 ± 1.2 % (n = 12), with 2 mm [Ca2+]o.
Figure 1. Presynaptic calcium currents exhibit facilitation by repetitive activation.
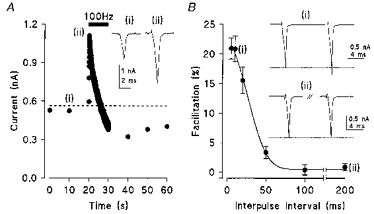
A, IpCa facilitation elicited by tetanic activation in a calyx voltage clamped at a holding potential of −70 mV. A brief depolarizing pulse (1 ms) to −20 mV was applied at 0.1 Hz (i). A dashed line indicates the baseline corresponding to the mean value of 6 control events. A tetanic stimulus (100 Hz, 10 s) was delivered as indicated by the bar (ii). IpCa increased for the first 15 events, and then declined. B, paired pulse facilitation of IpCa. Calcium currents were generated by a pair of 1 ms depolarizing voltage steps to −10 mV from a holding potential of −80 mV. Mean magnitude of the facilitation (± s.e.m., n = 6, except for the 200 ms time point where n = 4) is plotted against the inter-pulse interval. Paired pulse sample traces are averages of 10–20 events here and in subsequent figures.
Facilitation of IpCa results from accelerated activation kinetics
Calcium current facilitation could in principle arise from an increase in the number of channels opened by a given depolarization, or an acceleration in the kinetics of channel activation. To determine which of these factors are responsible for the IpCa facilitation, longer depolarizing pulses (5–10 ms duration from −80 mV to −10 mV) were applied either in the absence or presence of a 1 ms conditioning prepulse applied 10 ms prior to the test pulse (Fig. 2A). As shown superimposed in Fig. 2B, when IpCa was preceded by a conditioning prepulse (*), it exhibited a faster rate of rise than that measured without a prepulse. In contrast, no change was observed in the maximum current amplitude (measured at 4.9 ms from the test pulse onset; 100 ± 5 %, n = 3). The current-voltage relationship for the test IpCa (measured at 1 ms after the pulse onset, dashed line in Fig. 2A) showed a clear shift in the half-activation voltage of −4.1 mV (V1/2 = −6.7 mV in control and −10.8 mV in facilitation, see Methods) when conditioned by a prepulse (Fig. 2B). An intense hyperpolarization (−80 mV to −160 mV for 2.5 or 5 ms, n = 4) applied between the conditioning and test pulses failed to attenuate the facilitation (not shown), suggesting that the shift in the activation curve is not simply due to a voltage-dependent Markov process.
Figure 2. Acceleration of calcium current rise time by a conditioning pulse.
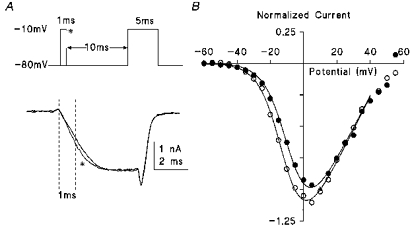
A, IpCa (lower traces) was evoked by a test pulse (5 ms duration to −10 mV) in the presence (*) or absence of a conditioning prepulse (1 ms, to −10 mV) applied 10 ms prior to the test pulse (voltage protocol indicated in upper panel). Holding potential was −80 mV. B, the current-voltage relationship measured 1 ms from the pulse onset (dashed line in A) with current normalized to the amplitude of control current at +10 mV (*). Test pulse command voltage was varied. The mean current amplitude plotted from 3 calyces is plotted in the presence (○) and absence (•) of the 1 ms conditioning pulse. The continuous lines are drawn according to a modified Boltzmann equation (see Methods). In control (no prepulse), V1/2 was −7.5 mV. The facilitation of IpCa could be explained by a hyperpolarizing shift of −4.1 mV in V1/2 (with k = 7 for control and test data).
The IpCa facilitation cannot be due to the leak subtraction error since no facilitation was observed for the test pulses to below −50 mV or above +20 mV. Also, it cannot be due to a change in potassium conductance, since this was blocked by external and internal TEA and internal Cs+ (see Methods). Furthermore, a similar magnitude of facilitation was observed in the presence of external Cs+ (1 mm, not shown), which blocks inwardly rectifying potassium and cationic currents. Finally, since the facilitation was clearly observed at the chloride equilibrium potential (0 mV), it cannot be due to the chloride current.
Facilitation is independent of G-protein activity
One possible mechanism for IpCa facilitation is the voltage-dependent relief from an endogenous G-protein-mediated inhibition of Ca2+ channels (Tsunoo et al. 1986; Ikeda, 1991; see Dolphin, 1996 for review). To test this possibility we included guanosine-5′-O-(3-thiotriphosphate) (GTPγS) in the patch pipette to fully activate G-proteins. In the presence of GTPγS (0.5 mm) the rise time of IpCa was significantly slowed (see also Takahashi et al. 1998). However, the magnitude of IpCa facilitation was similar to control (19.0 ± 2.5 %, n = 4, Fig. 3). Although a longer and larger prepulse significantly relieved the inhibitory effect of GTPγS on IpCa (10 ms to +100 mV, Y. Kajikawa & T. Takahashi, unpublished observation), the prepulse used here (1 ms to −10 mV) was too small to relieve G-protein-dependent inhibition in these experiments.
Figure 3. G-proteins are not involved in IpCa facilitation.
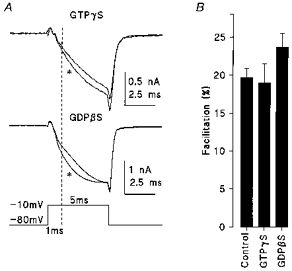
A, IpCa facilitation in the presence of GTPγS (0.5 mm, top trace) or GDPβS (3 mm, middle trace) using the same protocol as Fig. 2A. B, the mean magnitude of facilitation in control, with GTPγS or GDPβS (measured at 1 ms from the pulse onset, dashed line in A).
Since G-protein activation can be blocked by guanosine-5′-O-(2-thiodiphosphate) (GDPβS) we also examined if this might attenuate IpCa facilitation. GDPβS (3 mm) had no clear effect when included in the patch pipette, except for a marginal increase in the magnitude of IpCa facilitation (23.7 ± 1.8 %, n = 5) which was not statistically different from control (Fig. 3B). In the same recordings (in the presence of GDPβS) baclofen (10 μm) failed to suppress IpCa (not shown) providing a positive control for this drug (Takahashi et al. 1998). These results obtained using guanine nucleotide analogues suggest that G-proteins are not critically involved in IpCa facilitation.
Presynaptic calcium influx is involved in IpCa facilitation
We next tested whether Ca2+ that entered during the conditioning pulse may remain and facilitate the subsequent IpCa. When [Ca2+]o was reduced, IpCa diminished concomitantly with a reduction of IpCa facilitation (Fig. 4A). As illustrated in Fig. 4B, the magnitude of IpCa facilitation was proportional to that of the IpCa. In 0.1 mm [Ca2+]o, IpCa facilitation averaged 6.9 ± 1.4 % (n = 4, Fig. 4C), which was significantly less than control (P < 0.005). Similarly, when [Ca2+]o was totally replaced by Ba2+ (2 mm) the magnitude of IpCa facilitation was markedly reduced to 4.0 ± 1.6 % (n = 4, Fig. 4C). Furthermore, when the extracellular divalent cations were replaced by Na+, the sodium current through Ca2+ channels (Forsythe et al. 1998) no longer showed the paired-pulse facilitation (−0.7 ± 1.2 %, n = 3, Fig. 4C).
Figure 4. Calcium dependence of IpCa facilitation.
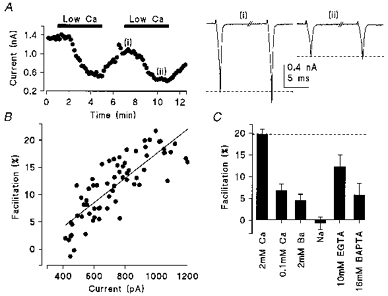
A, time plot of the amplitude of the calcium current during switched perfusion with 0.1 mm [Ca2+]o (filled bars) and 2 mm [Ca2+]o. Sample traces show paired pulse facilitation (10 ms inter-pulse interval) at 2 mm (i) and 0.1 mm [Ca2+]o (ii). The depolarizing test pulse was to −10 mV. B, relationship between the magnitude of facilitation and IpCa. The parameters are strongly correlated (r2 = 0.65) suggesting that calcium influx is involved in generating facilitation. Data points were taken from the 2–12 min time period in A, including wash-in and wash-out of 0.1 mm [Ca2+]o. C, effect of several manipulations which reduce calcium influx or intracellular calcium concentration on IpCa facilitation.
The current amplitude carried by Ba2+ or Na+ was similar or larger than that by Ca2+ (Forsythe et al. 1998). Less facilitation despite larger or comparable Ba2+ or Na+ current argues against a possibility that IpCa facilitation may result from a residual depolarization by a preceding pulse under inadequate voltage-clamp condition.
Given that Ca2+ influx is essential for the IpCa facilitation, we examined the extent to which IpCa facilitation is dependent upon [Ca2+]i. When EGTA (10 mm) was included in the patch pipette, IpCa facilitation was reduced (12.2 ± 2.8 %, n = 4; P < 0.02). More dramatically, the faster Ca2+ chelator BAPTA, at the higher concentration of 16 mm, significantly reduced IpCa facilitation to 5.9 ± 2.6 % (n = 7, P < 0.001; Fig. 4) and the negative shift in the half-activation voltage on presentation of a conditioning voltage pulse was substantially blocked (n = 4, data not shown; V1/2 = −7.4 mV in control, −8.2 mV in facilitation, see Methods). These results indicate that a rise in [Ca2+]i through Ca2+ influx is essential for the IpCa facilitation.
Kinase inhibitors and phosphatase inhibitors had no effect on IpCa facilitation
To examine further the mechanism of IpCa facilitation downstream of [Ca2+]i, we tested agents which are known to block Ca2+-dependent kinases and phosphatases. The broad spectrum kinase blocker H-7 applied from outside (200 μm) had no significant effect on IpCa facilitation (19.6 ± 3.2 %, n = 3, P > 0.98). Similarly, the Ca2+-calmodulin dependent kinase II inhibitor KN-62 (5 μm) applied intracellularly through the patch pipette had no appreciable effect (18.0 ± 2.0 %, n = 4, P > 0.53). The broad spectrum phosphatase inhibitor deltamethrin at a high concentration (100 nm) significantly reduced the magnitude of IpCa facilitation (13.0 ± 1.0, n = 4, P < 0.01). However, an inhibitor of the Ca2+-dependent phosphatase, calcineurin (calyculin A, 0.5 μm) did not attenuate the facilitation (18.3 ± 1 %, n = 4).
DISCUSSION
We have examined the presynaptic mechanism underlying short-term modulation at an excitatory synapse in the rat CNS. Presynaptic Ca2+ currents evoked by a brief depolarizing pulse underwent a robust facilitation when activated repetitively. This facilitation was due to a negative shift in the voltage-dependent activation of the Ca2+ current, resulting in an increased rate of activation at a given voltage. The mechanism underlying IpCa facilitation was independent of G-proteins, but was highly dependent upon [Ca2+]i. This result shows that the mechanism of paired pulse potentiation based on the residual calcium hypothesis can include modulation of the calcium current itself in addition to direct actions of [Ca2+]i on the exocytotic machinery.
Facilitation of somatic Ca2+ currents has been reported for many types of Ca2+ channels in a range of preparations (for review, see Dolphin, 1996). For example, protein kinase A-dependent phosphorylation of Ca2+ channels results in facilitation of Ca2+ currents in chromaffin cells (Artalejo et al. 1990). Like the P-type IpCa facilitation observed here at this auditory synapse, L-type Ca2+ current facilitation in cardiac myocytes was also Ca2+ dependent (Gurney et al. 1989) and the mechanism appeared Ca2+ specific since it was abolished by replacement of external Ca2+ by Ba2+ (Zygmund & Maylie, 1990). However, in contrast to the presynaptic IpCa facilitation, the cardiac Ca2+ current facilitation was associated with an increase in peak current amplitude with no shift in the voltage dependence of activation (Gurney et al. 1989), and chelation of intracellular Ca2+ had no effect on facilitation (Zygmunt & Maylie, 1990). Another type of Ca2+ channel facilitation is the depolarization-induced reversal of G-protein inhibition (Tsunoo et al. 1986) reported for N-type Ca2+ channels (Ikeda, 1991) and also for recombinant α1A channels (Brody et al. 1997). Although it is conceivable that IpCa is tonically suppressed by the presynaptic G-protein-coupled receptors and its relief may cause the IpCa facilitation, our results using GTPγS and GDPβS indicate that this G-protein-coupled mechanism is not involved in the IpCa facilitation. Another possibility is that facilitation may be due to recruitment of other types of Ca2+ channels. This is also unlikely since the effect of facilitation was to accelerate activation kinetics rather than enhance IpCa magnitude. We have previously demonstrated that IpCa also exhibits a Ca2+-dependent inactivation (Forsythe et al. 1998). Although both the facilitation and inactivation are dependent upon Ca2+ influx, the mechanisms underlying the processes seem quite distinct since the latter was less sensitive to [Ca2+]i buffering and had little specificity for Ca2+ over Ba2+ as the charge carrier.
Short-term potentiation of synaptic efficacy, such as paired pulse facilitation or augmentation, can result from accumulation of intracellular Ca2+ following subsequent activity (Katz & Miledi, 1968; Charlton et al. 1982). This residual Ca2+ is thought to sum with subsequent Ca2+ transients to increase the probability of transmitter release through a direct action on the exocytotic machinery. Our results indicate that raised intracellular Ca2+ can also facilitate presynaptic Ca2+ channels, thereby generating a larger Ca2+ influx. The maximal facilitation of IpCa was about 20 % in the paired pulse protocol. Assuming a power relationship of around 2 at this synapse (Barnes-Davies & Forsythe, 1995; Takahashi et al. 1996), EPSCs could be facilitated by 44 % through this IpCa facilitation mechanism alone. In the case of tetanic stimulation at 100 Hz, nearly a 2-fold increase of IpCa was observed, which could have enhanced EPSCs by as much as 4-fold. However, in normal ACSF, both the paired pulse protocol (Barnes-Davies & Forsythe, 1995) and tetanic stimulation (Forsythe et al. 1998) give rise to synaptic depression at this synapse. The overall depression or facilitation of transmitter release would be determined as the sum of all the attenuating and facilitating factors. The former includes postsynaptic receptor desensitization (Otis et al. 1996), synaptic vesicle depletion (Stevens & Tsujimoto, 1995; von Gersdorff et al. 1997) and inactivation of IpCa (Forsythe et al. 1998), whereas the latter comprises residual Ca2+ effects on IpCa facilitation as well as on the exocytotic machinery. Thus, residual calcium may exert multiple facilitatory actions within the same synaptic terminal which would function in concert to promote synaptic facilitation. Since mammalian central and peripheral synaptic transmission are largely mediated by P/Q-type Ca2+ channels (Takahashi & Momiyama, 1993; Luebke et al. 1993; Katz et al. 1996; Iwasaki & Takahashi, 1998), the Ca2+-induced facilitation of the presynaptic Ca2+ current may be a general facilitatory mechanism for activity-dependent synaptic modulation in the CNS.
The calyx of Held is a relay synapse in the binaural auditory pathway which generates an EPSC mediated by glutamate receptors that can sustain transmission at very high rates (see Trussell, 1997) while also preserving the timing information essential for its function in sound source localization. The large magnitude and rapid kinetics of the EPSCs (Barnes-Davies & Forsythe, 1995) combined with the expression of both low and high threshold potassium currents (Brew & Forsythe, 1995) are important factors in this behaviour. It is therefore not surprising that a synapse with such physiological constraints should also possess a diverse modulatory feedback, since the balance of the facilitatory and inhibitory feedback must change with the frequency of synaptic input, thereby providing a dynamic drive or filter to synaptic transmission and modifying release probabilities over the full range of physiological firing rates.
We have demonstrated that IpCa can be facilitated by prior Ca2+ influx. None of our pharmacological tests revealed the underlying mechanism downstream of Ca2+ influx, except for a possible involvement of an as yet unidentified phosphatase. If metabolic cascades are involved in the IpCa facilitation, they must be fully activated within the minimal paired pulse interval of 5 ms and their effects last up to 100 ms. Another possibility would be the Ca2+ binding to the channel pore from inside and subsequent change in gating kinetics (Ohmori & Yoshii, 1977; Yoshii et al. 1988). It is also possible that Ca2+ influx might induce Ca2+release from internal stores, thereby producing the pronounced effect. Whatever the underlying mechanism, Ca2+-dependent facilitation of presynaptic calcium current is a novel phenomenon which should be taken into account when interpreting results from experiments involving short-term and long-term synaptic modulation in the brain.
Acknowledgments
This work was supported by the International Scientific Research Programme and Grants-in-Aid for Young Scientists from the Ministry of Education of Japan, the Research for the Future Programme by the Japan Society for the Promotion of Sciences and The Wellcome Trust. I.D.F. is a Wellcome Senior Research Fellow in Basic Biomedical Sciences. We thank Margaret Barnes-Davies for comments on the manuscript.
References
- Artalejo CR, Ariano MA, Perlman RL, Fox AP. Activation of facilitation calcium channels in chromaffin cells by D1 dopamine receptors through a cAMP/protein kinase A-dependent mechanism. Nature. 1990;348:239–242. doi: 10.1038/348239a0. 10.1038/348239a0. [DOI] [PubMed] [Google Scholar]
- Barnes-Davies M, Forsythe ID. Pre- and postsynaptic glutamate receptors at a giant excitatory synapse in rat auditory brainstem slices. The Journal of Physiology. 1995;488:387–406. doi: 10.1113/jphysiol.1995.sp020974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Borst JGG, Helmchen F, Sakmann B. Pre- and postsynaptic whole-cell recordings in the medial nucleus of the trapezoid body of the rat. The Journal of Physiology. 1995;489:825–840. doi: 10.1113/jphysiol.1995.sp021095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brew HM, Forsythe ID. Two voltage-dependent K+ conductances with complementary functions in postsynaptic integration at a central auditory synapse. Journal of Neuroscience. 1995;15:8011–8022. doi: 10.1523/JNEUROSCI.15-12-08011.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brody DL, Patil PG, Mulle JG, Snutch TP, Yue DT. Bursts of action potential waveforms relieve G-protein inhibition of recombinant P/Q-type Ca2+ channels in HEK 293 cells. The Journal of Physiology. 1997;499:637–644. doi: 10.1113/jphysiol.1997.sp021956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlton MP, Smith SJ, Zucker RS. Role of presynaptic calcium ions and channels in synaptic facilitation and depression at the squid giant synapse. The Journal of Physiology. 1982;323:173–193. doi: 10.1113/jphysiol.1982.sp014067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC. Facilitation of Ca2+ current in excitable cells. Trends in Neurosciences. 1996;19:35–43. doi: 10.1016/0166-2236(96)81865-0. 10.1016/0166-2236(96)81865-0. [DOI] [PubMed] [Google Scholar]
- Forsythe ID. Direct patch recording from identified presynaptic terminals mediating glutamatergic EPSCs in the rat CNS, in vitro. The Journal of Physiology. 1994;479:381–387. doi: 10.1113/jphysiol.1994.sp020303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forsythe ID, Tsujimoto T, Barnes-Davies M, Cuttle MF, Takahashi T. Inactivation of presynaptic calcium current contributes to synaptic depression at a fast central synapse. Neuron. 1998;20:797–807. doi: 10.1016/s0896-6273(00)81017-x. 10.1016/S0896-6273(00)81017-X. [DOI] [PubMed] [Google Scholar]
- Gurney AM, Charnet P, Pye JM, Nargeot J. Augmentation of cardiac calcium current by flash photolysis of intracellular caged-Ca2+ molecules. Nature. 1989;341:65–68. doi: 10.1038/341065a0. 10.1038/341065a0. [DOI] [PubMed] [Google Scholar]
- Ikeda SR. Double-pulse calcium channel current facilitation in adult rat sympathetic neurones. The Journal of Physiology. 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwasaki S, Takahashi T. Developmental changes in calcium channel types mediating synaptic transmission in rat auditory brainstem. The Journal of Physiology. 1998;509:419–423. doi: 10.1111/j.1469-7793.1998.419bn.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz B, Miledi R. The role of calcium in neuromuscular facilitation. The Journal of Physiology. 1968;195:481–492. doi: 10.1113/jphysiol.1968.sp008469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Ferro PA, Weisz G, Uchitel OD. Calcium channels involved in synaptic transmission at the mature and regenerating mouse neuromuscular junction. The Journal of Physiology. 1996;497:687–697. doi: 10.1113/jphysiol.1996.sp021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luebke JI, Dunlap K, Turner TJ. Multiple calcium channel types control glutamatergic synaptic transmission in the hippocampus. Neuron. 1993;11:895–902. doi: 10.1016/0896-6273(93)90119-c. 10.1016/0896-6273(93)90119-C. [DOI] [PubMed] [Google Scholar]
- Milner B, Squire LR, Kandel ER. Cognitive neuroscience and the study of memory. Neuron. 1998;20:445–468. doi: 10.1016/s0896-6273(00)80987-3. 10.1016/S0896-6273(00)80987-3. [DOI] [PubMed] [Google Scholar]
- Ohmori H, Yoshii M. Surface potential reflected in both gating and permeation mechanisms of sodium and calcium channels of the tunicate egg cell membrane. The Journal of Physiology. 1977;267:429–463. doi: 10.1113/jphysiol.1977.sp011821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otis T, Zhang S, Trussell LO. Direct measurement of AMPA receptor desensitization induced by glutamatergic synaptic transmission. Journal of Neuroscience. 1996;16:7496–7504. doi: 10.1523/JNEUROSCI.16-23-07496.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens CF, Tsujimoto T. Estimates for the pool size of releasable quanta at a single central synapse and for the time required to refill the pool. Proceedings of the National Academy of Sciences of the USA. 1995;92:846–849. doi: 10.1073/pnas.92.3.846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Forsythe ID, Tsujimoto T, Barnes-Davies M, Onodera K. Presynaptic calcium current modulation by a metabotropic glutamate receptor. Science. 1996;274:594–597. doi: 10.1126/science.274.5287.594. 10.1126/science.274.5287.594. [DOI] [PubMed] [Google Scholar]
- Takahashi T, Kajikawa Y, Tsujimoto T. G-protein-coupled modulation of presynaptic calcium currents and transmitter release by a GABAB receptor. Journal of Neuroscience. 1998;18:3138–3146. doi: 10.1523/JNEUROSCI.18-09-03138.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi T, Momiyama A. Different types of calcium channels mediate central synaptic transmission. Nature. 1993;366:156–158. doi: 10.1038/366156a0. 10.1038/366156a0. [DOI] [PubMed] [Google Scholar]
- Trussell LO. Cellular mechanisms for preservation of timing in central auditory pathways. Current Opinion in Neurobiology. 1997;7:487–492. doi: 10.1016/s0959-4388(97)80027-x. 10.1016/S0959-4388(97)80027-X. [DOI] [PubMed] [Google Scholar]
- Tsunoo A, Yoshii M, Narahashi T. Block of calcium channels by enkephalin and somatostatin in neuroblastoma-glioma hybrid NG 108–15 cells. Proceedings of the National Academy of Sciences of the USA. 1986;83:9832–9836. doi: 10.1073/pnas.83.24.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- von Gersdorff H, Schneggenburger R, Weis S, Neher E. Presynaptic depression at a calyx synapse: The small contribution of metabotropic glutamate receptors. Journal of Neuroscience. 1997;17:8137–8146. doi: 10.1523/JNEUROSCI.17-21-08137.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii M, Tsunoo A, Narahashi T. Gating and permeation properties of two types of calcium channels in neuroblastoma cells. Biophysical Journal. 1988;54:885–895. doi: 10.1016/S0006-3495(88)83025-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zucker RS. Exocytosis: a molecular and physiological perspective. Neuron. 1996;17:1049–1055. doi: 10.1016/s0896-6273(00)80238-x. 10.1016/S0896-6273(00)80238-X. [DOI] [PubMed] [Google Scholar]
- Zygmunt AC, Maylie J. Stimulation-dependent facilitation of the high threshold calcium current in guinea-pig ventricular myocytes. The Journal of Physiology. 1990;428:653–671. doi: 10.1113/jphysiol.1990.sp018233. [DOI] [PMC free article] [PubMed] [Google Scholar]