

Abstract
The involvement of ryanodine-sensitive Ca2+ release (RsCR) in bradykinin (Bk)-induced Ca2+ release, capacitative Ca2+ entry (CCE) and nitric oxide synthase (NOS) activation was assessed in freshly isolated bovine coronary artery endothelial cells.
Using deconvolution microscopy fura-2 was found throughout the whole cytosol, while the cell membrane impermeable dye FFP-18 was exclusively in the cell membrane. Thus, perinuclear ([Ca2+]pn) and subplasmalemmal Ca2+ concentration ([Ca2+]sp) were monitored using fura-2 and FFP-18.
Inhibition of Na+−Ca2+ exchange by lowering extracellular Na+ concentration augmented the Bk-induced [Ca2+]pn signal in Ca2+-free solution. This effect was abolished when RsCR was prevented with 25 μmmu;mol l−1 ryanodine, while inhibition of RsCR had no effect on Bk-induced increase in [Ca2+]pn without inhibition of Na+−Ca2+ exchange.
Initiating RsCR by 200 nmol l−1 ryanodine increased [Ca2+]sp, while [Ca2+]pn remained constant. However, when Na+−Ca2+ exchange was prevented, ryanodine was also able to elevate [Ca2+]pn.
Blockage of RsCR diminished Ca2+ extrusion in response to stimulation with Bk in normal Na+-containing solution.
Inhibition of RsCR blunted Bk-activated CCE, while inhibition of Na+−Ca2+ exchange during stimulation enhanced CCE.
Although direct activation of RsCR failed to activate NOS, inhibition of RsCR diminished the effect of ATP and Bk on NOS, while the effect of thapsigargin remained unchanged.
These data suggest that during stimulation subplasmalemmal RsCR occurs, which contributes to the activities of CCE and NOS. Thus, the function of the subplasmalemmal Ca2+ control unit must be extended as a regulator for CCE and NOS.
In endothelial cells an increase in intracellular free Ca2+ concentration is an important mediator in the formation of vasoactive compounds, such as nitric oxide and endothelin (for review see Graier et al. 1994b). Agonists activate phospholipase C which synthesizes inositol-1,4,5 trisphosphate (InsP3) and evokes Ca2+ release from InsP3-sensitive Ca2+ stores (Freay et al. 1989). Depletion of Ca2+ stores triggers activation of capacitative Ca2+ entry (CCE; Putney, 1986; Putney, 1990; Schilling et al. 1992), possibly mediated via tyrosine kinase (Fleming et al. 1996) and/or cytochrome P450 epoxygenase-derived metabolites (Graier et al. 1995; Hoebel et al. 1997).
Besides InsP3-mediated Ca2+ release and CCE, the involvement of a Ca2+-induced Ca2+ release mechanism (ryanodine-sensitive Ca2+ release; RsCR) has been proposed to contribute to endothelial Ca2+ signalling. Although binding sites for ryanodine have been demonstrated on the endoplasmic reticulum membrane in endothelial cells (Lesh et al. 1993; Graier et al. 1998), our knowledge of the importance of RsCR for Ca2+ signalling and endothelial cell function is limited. While there is evidence that RsCR occurs during agonist stimulation (Mozhayeva, 1996), direct stimulation of RsCR by ryanodine failed to evoke Ca2+ transients; instead ryanodine slowly increased the intracellular Ca2+ concentration (Wang et al. 1995). A similar slow release of Ca2+ was described for caffeine, a non-specific activator of Ca2+-induced Ca2+ release, in rabbit aortic endothelial cells (Wang et al. 1995). However, caffeine evoked rapid Ca2+ transients in human (Ziegelstein et al. 1994) and porcine endothelial cells (Graier et al. 1994a). In contrast to these reports indicating an involvement of RsCR in endothelial Ca2+ transients, several authors have failed to find its contribution to CCE (Graier et al. 1994a; Ziegelstein et al. 1994). Recently, we described the generation of localized Ca2+ gradients in the subplasmalemmal area and suggested RsCR may contribute to subplasmalemmal Ca2+ elevation (Graier et al. 1998). Although there is increasing evidence that localized Ca2+ signalling is important for the generation of vasoactive compounds, such as prostacyclin and nitric oxide (Lückhoff et al. 1988), very little is known about the contribution of RsCR to agonist-induced stimulation of nitric oxide synthase (NOS).
In the present study we used endothelial cells freshly isolated from bovine left circumflex coronary arteries to investigate the involvement of RsCR in agonist-induced intracellular Ca2+ release, CCE, and activation of NOS. Our data indicate that, during stimulation with agonists, RsCR occurs mainly in the subplasmalemmal area. Hence, although RsCR may not stimulate CCE per se, it contributes to activity of CCE and stimulation of NOS.
METHODS
Materials
Cell culture chemicals were purchased from Life Technologies, Vienna, Austria and fetal calf serum (FCS, premium quality) was from PAA Laboratories, Linz, Austria. Petri dishes were obtained from Corning, Vienna, Austria. Fura-2 AM and fura dextran potassium salt (MW 70 000) were from Molecular Probes, Leiden, The Netherlands. FFP-18 and ryanodine were purchased from Calbiochem, Vienna, Austria. Hepes acid was from Amresco, Vienna, Austria and buffer salts were purchased from Merck, Vienna, Austria. All other materials were from Sigma, Vienna, Austria. The human umbilical vein endothelial cell line EA.hy 926 was a generous gift from Dr Cora-Jean S. Edgell, Pathology Department, University of North Carolina, NC, USA (Edgell et al. 1983).
Cell isolation and culture
Endothelial cells were isolated from bovine left circumflex coronary arteries as described previously (Graier et al. 1992; Graier et al. 1995). Cattle were killed by a shot in the head in the local slaughterhouse. After they were exsanguinated, the hearts were extracted and put on ice for transport to the laboratory. For cell isolation, vessels were incubated at 37°C in Dulbecco's minimum essential medium (DMEM) containing 200 U ml−1 collagenase (type II), trypsin inhibitor (soybean type I; 1 mg ml−1), minimum essential medium (MEM) dilutions (v/v) of essential amino acids (2 %), MEM non-essential amino acids (1 %) and MEM (1 %) vitamins (Life Technologies, Vienna, Austria). After 15 min incubation, parts of the incubation buffer were aspirated for cell culture followed by an additional 20 min for experiments with freshly isolated cells. Endothelial cells were cultured up to passage 2 in Opti-MEM containing 2 % (v/v) fetal calf serum. Cell culture purity was tested by typical cobblestone morphology and the lack of smooth muscle α-actin. Cells from the EA.hy 926 cell line (passage 63 and higher) were grown in DMEM containing 10 % fetal calf serum, 4.5 mg l−1 D-glucose and a 1 % (w/v) hypoxanthine- aminopterine-thymidine mixture (HAT).
Ca2+ measurement
Intracellular free Ca2+ concentration was determined in single endothelial cells using microfluorometry (Graier et al. 1995). As described previously (Graier et al. 1998), different Ca2+-sensitive dyes were used to monitor perinuclear free Ca2+ concentration ([Ca2+]pn; fura-2) and the subplasmalemmal Ca2+ concentration ([Ca2+]sp; FFP-18). For fura-2 loading, the cells were incubated in the dark with 2 μmmu;mol l−1 fura-2 AM in DMEM. After 45 min at room temperature, cells were centrifuged, washed twice and resuspended in Hepes-buffered solution containing (mmol l−1): 145 NaCl, 5 KCl, 1 MgCl2, 2.5 CaCl2, 10 Hepes acid, pH = 7.4 (Hepes-buffered solution; HBS). The cell membrane impermeable dye FFP-18 was loaded using the laser-generated stress wave loading technique (Graier et al. 1998). Briefly, approximately 106 cells were suspended in 350 μl Phenol Red-free MEM containing 2 μmmu;mol l−1 FFP-18 and transferred into a well of a microtitre plate. After the well had been sealed with 1.25 mm aluminum foil, laser-induced stress waves were generated by 3 to 5 shots of a Nd:YAG laser (70 mJ) to the foil, focused 0.5 cm behind the foil. After an equilibration period of 10 to 30 min in the dark, cells were centrifuged and resuspended in HBS. For Ca2+ measurements Ca2+-free HBS was used containing (mmol l−1): either 145 NaCl, 5 KCl, 1 MgCl2, 0.01 EGTA, 10 Hepes-acid, pH = 7.4 (i.e. control buffer) or 19 NaCl, 5 KCl, 1 MgCl2, 126 choline chloride, 0.01 EGTA, 10 Hepes acid, pH = 7.4 (i.e. low Na+ solution). For measurement of Ca2+ extrusion, cells were resuspended in Ca2+-free HBS containing 1 μmmu;mol l−1 fura dextran potassium salt (MW 70 000).
Data acquisition
Single cell [Ca2+]pn was recorded in a microfluorometer, which excited cells with alternating 360 and 380 nm light, while emission was detected at 510 nm using a modified photon counting technique (Sturek et al. 1991; Graier et al. 1995). Counts were converted to an analog signal by an optical processor and the fluorescence intensity for each excitation and/or emission wavelength was acquired by a personal computer running AxoBASIC® 1.0 (Axon Instruments, Foster City, CA, USA). For measurements of [Ca2+]sp, fluorescence of FFP-18 at 335-365 nm excitation and 500 nm emission was monitored in suspended cells every 0.2 s in a spectrofluorometer (Hitachi F-4500). Extrusion of Ca2+ was measured with fura dextran by monitoring extracellular Ca2+ concentration at 340 and 364 nm excitation and 500 nm emission.
Data analysis
Due to the reported difficulties in the calibration techniques for intracellular Ca2+ concentration (Sturek et al. 1991; Graier et al. 1995; Wang et al. 1995), Ca2+ concentrations are expressed in ratio units (ratio F360/F380 for fura-2; ratio F335/F365 for FFP-18 and ratio F340/F364 for fura dextran).
Digital confocal microscopy
Images were obtained with a scientific-grade CCD camera using conventional lamp illumination. Out-of-focus fluorescence was calculated with a computer algorithm using the three-dimensional point spread function for each microscope objective (Waters & Brown, 1996). The instrument setup included a liquid-cooled CCD camera (Quantix, Photometrics, Munich, Germany) on an inverted microscope (Nikon Eclipse TE 300, Nikon, Vienna, Austria) with z-stage motor controlled by the Ludl-Z-stage control box (Ludl, Inc., Fairfield Imaging, Turnbridge Wells, UK) in association with Image Pro 3.0 software (Media Cybernetics, Fairfield Imagining, Turnbridge Wells, UK). The camera head was cooled to −40°C and the gain level was 2. Images were collected with a CFI Plan Fluor × 100 oil immersion objective (NA 1.3; 0.068 μmmu;m pixel−1, Nikon, Vienna, Austria) and the distance between images in the z scan was 0.3 μmmu;m. Images were collected with Volume Scan® (VayTek, Inc., Fairfield Imaging, Turnbridge Wells, UK) and out-of-focus fluorescence was calculated with Micro Tome® deconvolution software (VayTek, Inc., Fairfield Imaging, Turnbridge Wells, UK) using the advanced constrained iterative deconvolution algorithm (5 iterations). These deconvolution procedures removed out-of-focus fluorescence, thus producing ‘digital confocal’ images.
Three-dimensional reconstruction of data
The deconvolved images were transformed into 8-bit images to conduct three-dimensional reconstruction with the volume rendering program VoxBlast®, which utilizes a back propagation-weighted average algorithm (VayTek, Inc., Fairfield Imaging, Turnbridge Wells, UK).
Measurements of NOS activity
Activity of NOS was determined by measuring the conversion of [3H]-L-arginine to [3H]-L-citrulline as described previously (Graier et al. 1996). Briefly, cells were cultured in 6-well plastic dishes. Experiments were performed in HBS and the extracellular Na+ concentration ([Na+]o) indicated. After equilibrating the cells in HBS containing the solvent (0.5 % DMSO) or ryanodine for 5 min, [3H]-L-arginine (106 c.p.m. dish−1) was added together with the compound to be tested. After 15 min, incubation buffer was aspirated and 1 ml chilled 0.01 mol l−1 HCl was added. After 1 h at 4°C, a 100 μl aliquot was saved (i.e. incorporated radioactivity) and to the remaining 900 μl, 100 μl of 200 mmol l−1 sodium acetate buffer containing 10 mmol l−1 L-citrulline (pH = 13.0) was added. From this mixture, 900 μl were placed on a Dowex AG50W-X8 and [3H]-L-citrulline was purified by washing with 1 ml water. After considering the column recovery for each column and a dilution correction, activity of NOS was calculated as the percentage of [3H]-L-citrulline radioactivity within the total incorporated radioactivity. In the presence of 100 μmmu;mol l−1 L-GN-nitroarginine, which reduced basal [3H]-L-citrulline formation by about 40–50 %, no increase in the conversion of [3H]-L-arginine to [3H]-L-citrulline with agonists was found. The L-GN-nitroarginine resistant formation of [3H]-L-citrulline under each condition was subtracted to demonstrate NOS-dependent formation of [3H]-L-citrulline.
Statistics
All data points represent the mean ±s.e.m. Data evaluation was performed by analysis of variance (ANOVA) and statistical significance was estimated by Scheffé‘s post hoc test. The level of significance was defined as P < 0.05.
RESULTS
Contribution of RsCR to [Ca2+]pn elevation
In the absence of extracellular Ca2+, lowering [Na+]o from 145 to 19 mmol l−1 increased the Bk-induced elevation of [Ca2+]pn by 112 %, while resting [Ca2+]pn remained unchanged (Fig. 1). Under low [Na+]o conditions, the mobilization of [Ca2+]pn following the addition of 2.5 mmol l−1 extracellular Ca2+ (a marker for CCE) was augmented by 55 % in cells prestimulated by Bk (Fig. 1). Accordingly, Bk-induced Mn2+ quench was increased by 23 % when cells were stimulated with 100 nmol l−1 Bk under low [Na+]o conditions (data not shown). Similar findings were obtained in the EA.hy 926 cell line using 10 μmmu;mol l−1 histamine (data not shown).
Figure 1. Effect of low extracellular [Na+]o on agonist-induced changes of [Ca2+]pn in endothelial cells freshly isolated from bovine left circumflex arteries.
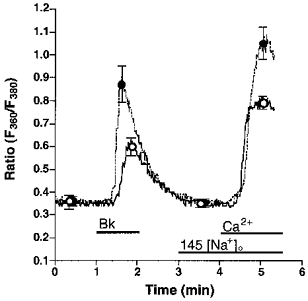
Freshly isolated endothelial cells were loaded with fura-2 to measure [Ca2+]pn. In Ca2+-free HBS containing 145 (145 [Na+]o, ○, continuous line) or 19 (19 [Na+]o, •, dashed line) mmol l−1 Na+, cells were stimulated for 1 min with 100 nmol l−1 Bk followed by an addition of 2.5 mmol l−1 Ca2+ as indicated. In experiments using 19 mmol l−1 Na+-containing solution, [Na+]o was changed to 145 mmol l−1 at time 3 min. Points represent means ±s.e.m. (n= 22).
To investigate the involvement of RsCR in the Bk-induced Ca2+ signalling, Bk-initiated Ca2+ release and CCE in the absence and presence of 25 μmmu;mol l−1 ryanodine at normal (i.e. 145 mmol l−1) [Na+]o were compared. There was no difference in the increase in [Ca2+]pn to 100 nmol l−1 Bk in the Ca2+-free HBS in the absence and presence of ryanodine, while CCE activity was reduced by 60 % in the presence of ryanodine (Fig. 2A). Similar results were obtained in Mn2+ quench experiments. Thus, in the presence of ryanodine Bk-induced Mn2+ quench was reduced by 78 % (7.3 ± 2.2 to 1.6 ± 0.2 % decrease of initial fluorescence at 360 nm excitation min−1; n= 4, P < 0.05). In contrast, addition of ryanodine to Bk-prestimulated cells immediately prior to the addition of Ca2+ had no effect on CCE activity (data not shown).
Figure 2. Effect of 25 μmmu;mol l−1 ryanodine on Bk-induced changes of [Ca2+]pn in single freshly isolated bovine left circumflex endothelial cells in 145 (panel A) and 19 (panel B) mmol l−1 Na+-containing solution.
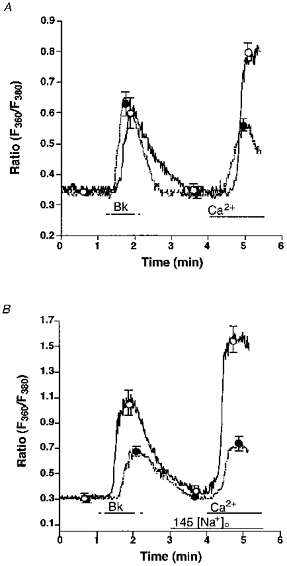
Freshly isolated endothelial cells were loaded with fura-2 to measure [Ca2+]pn. Panel A, in Ca2+-free HBS containing 145 mmol l−1 Na+ without (control, ○, continuous line) or with 25 μmmu;mol l−1 ryanodine (•, dashed line) endothelial cells were stimulated with 100 nmol l−1 Bk followed by the addition of 2.5 mmol l−1 Ca2+ as indicated. Panel B, in Ca2+-free HBS containing 19 mmol l−1 Na+ without (control, ○, continuous line) or with 25 μmmu;mol l−1 ryanodine (•, dashed line) cells were stimulated with 100 nmol l−1 Bk followed by the addition of 2.5 mmol l−1 Ca2+. As indicated, [Na+]o was increased to 145 mmol l−1. Points represent means ±s.e.m. (panel A, n= 36; panel B, n= 32).
When Na+-Ca2+ exchange was prevented by lowering [Na+]o to 19 mmol l−1, 25 μmmu;mol l−1 ryanodine diminished the effect of Bk on [Ca2+]pn in Ca2+-free solution by 51 % (Fig. 2B). Moreover, CCE stimulated by Bk was diminished by 77 % when ryanodine was present (Fig. 2B).
Figure 3 summarizes the data obtained in the above protocols in terms of Bk-induced intracellular Ca2+ release (Fig. 3A) and CCE (Fig. 3B).
Figure 3.
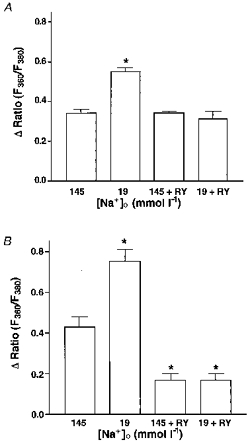
Summary of the results on the effect of Bk on intracellular Ca2+ release (A) and CCE (B) in normal and low Na+-containing solution in the absence and presence of ryanodine.
Measurement of RsCR in endothelial cells
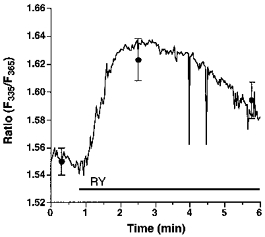
Endothelial RsCR was further investigated using 200 nmol l−1 ryanodine in Ca2+-free HBS. As shown in Fig. 4, there was no detectable increase in [Ca2+]pn to 200 nmol l−1 ryanodine in 145 mmol l−1 Na+-containing buffer. In contrast, under low [Na+]o conditions 200 nmol l−1 ryanodine evoked a small, but significant, increase in [Ca2+]pn (Fig. 4). In cells stimulated with 200 nmol l−1 ryanodine in 19 mmol l−1[Na+]o, elevation of [Ca2+]pn following the external addition of 2.5 mmol l−1 Ca2+ was not significantly different from that observed using 145 mmol l−1 Na+-containing solution. This increase in [Ca2+]pn was also similar to that measured in resting cells (data not shown). Furthermore, 200 nmol l−1 ryanodine failed to initiate Mn2+ entry in either 19 or 145 mmol l−1 Na+-containing solution (data not shown).
Figure 4. Prevention of Na+-Ca2+ exchange by lowering [Na+]o unmasked ryanodine-induced Ca2+ release.
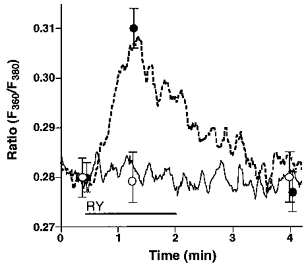
Single endothelial cells were loaded with fura-2 to measure [Ca2+]pn. As indicated, cells were stimulated with 200 nmol l−1 ryanodine (RY) in Ca2+-free HBS containing 145 (RY, ○, continuous line) or 19 mmol l−1 Na+ (RY, •, dashed line). Each point represents the mean ±s.e.m. (n= 24).
To verify whether, in 145 mmol l−1 Na+-containing solution, ryanodine induces an elevation of [Ca2+]sp which is insulated from [Ca2+]pn, experiments were conducted using cells that were loaded with FFP-18. This is a fura-2 analogue which selectively monitors [Ca2+]sp (Etter et al. 1996; Graier et al. 1998). As shown in Fig. 5, dye distribution of FFP-18 markedly differed from that of fura-2. Both dyes were loaded in freshly isolated bovine left circumflex coronary artery endothelial cells (panels A and B) or EA.hy 926 cells (panels C and D) using the laser-pulse wave loading technique (Graier et al. 1998). FFP-18 fluorescence was mainly found at the edge of the cells (i.e. plasma membrane; Fig. 5, panels A and C). By contrast, fura-2 was distributed throughout the cytosol (Fig. 5, panels B and D). In FFP-18-loaded cells 200 nmol l−1 ryanodine induced an elevation in [Ca2+]sp in Ca2+-free solution containing 145 mmol l−1 Na+ (Fig. 6). In the presence of 2.5 mmol l−1 extracellular Ca2+, ryanodine (200 nmol l−1) yielded a similar elevation in [Ca2+]sp to that observed in Ca2+-free HBS (data not shown).
Figure 5. Distribution of FFP-18 and fura-2 in the bovine left circumflex coronary artery endothelial cells and in EA.hy 926 cells.
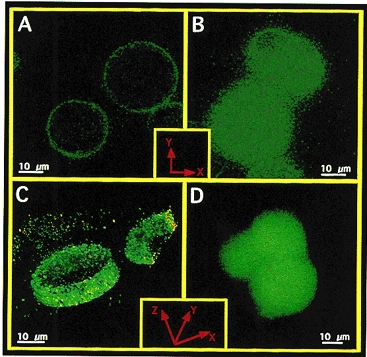
Freshly isolated bovine left circumflex coronary artery endothelial cells (panels A and B) and cultured EA.hy 926 (panels C and D) were loaded with FFP-18 (panels A and C) or fura-2 free acid (panels B and D) using laser-stress wave loading technique (Graier et al. 1998), equilibrated for 10 min at room temperature, washed three times in HBS and placed in a glass-bottomed chamber. Fluorescence images were collected using a × 100 objective (NA 1.3) with an excitation of 340 ± 5 nm and emission of 510 ± 10 nm for both dyes (5 to 10 s exposure time). Panels A and B: 2-D image of a slice in the middle depth of freshly isolated bovine endothelial cells. Panels C and D: 3-D reconstructed image of EA.hy 926 cells. Images were collected throughout the whole cell with 0.3 μmmu;m interslice distance, and out-of-focus fluorescence was removed using the advanced constrained iterative algorithm (MicroTome, Vaytek, Turnbridge Wells, UK). 3-D reconstruction was performed with VoxBlast (Vaytek, Turnbridge Wells, UK). In the case of FFP-18 (panel C) just 10 slices at the cell equator are shown, while panel D (fura-2) shows the whole cell. Identical distribution of fura-2 was found when fura-2 was loaded conventionally using 2 μmmu;mol l−1 fura-2 AM over 30 min.
Figure 6. Ryanodine induced subplasmalemmal Ca2+ increase in bovine coronary endothelial cells monitored by FFP-18.
Endothelial cells freshly isolated from bovine circumflex coronary artery were loaded with FFP-18 using laser-generated stress waves to monitor changes in [Ca2+]sp. After 10 min equilibration time at room temperature, cells were washed twice, resuspended in Ca2+-free HBS and placed into a stirred cuvette. Changes in [Ca2+]sp were monitored at 335 and 365 nm excitation and 500 nm emission. Each point represents the mean ±s.e.m. (n= 14).
Next we tested whether in the presence of 145 mmol l−1 Na+, RsCR is linked to Ca2+ extrusion. Here, extrusion of Ca2+ was monitored in cells stimulated with Bk in Ca2+-free HBS. Inhibition of RsCR with 25 μmmu;mol l−1 ryanodine diminished Ca2+ extrusion in response to a stimulation with 100 nmol l−1 Bk by 66 % (Fig. 7).
Figure 7. Inhibition of ryanodine-sensitive Ca2+ release attenuates Ca2+ extrusion in response to bradykinin.
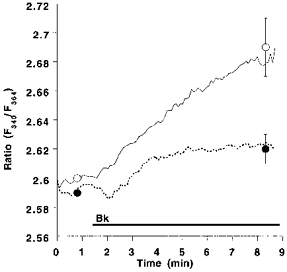
Suspended cultured endothelial cells (passage 2; 7 × 107 cells ml−1) were stimulated with 100 nmol l−1 Bk in the absence (control, ○, continuous line) or presence (•, dashed line) of 25 μmmu;mol l−1 ryanodine in Ca2+-free HBS containing 1 μmmu;mol l−1 fura-2 dextran. The Ca2+ extrusion was monitored at 340 and 364 nm excitation and 500 nm emission. Each point represents the mean ±s.e.m. (n= 6).
Correlation of RsCR with activity of CCE
The data shown above indicate that RsCR contributes to activity of CCE (Fig. 3B). Next, the (cumulative) amplitude of Bk-induced Ca2+ transients after (repetitive) stimulation taken as an index of Ca2+ store depletion, was correlated with the magnitude of CCE. Under control conditions (i.e. 145 mmol l−1[Na+]o), activity of CCE increased stepwise with the number of repeats of the Bk stimulation (open circles; Fig. 8). In contrast, in low Na+ solution, CCE was already maximal after the first stimulation with Bk and did not increase further after repetitive stimulation with Bk (filled circles; Fig. 8). Inhibition of RsCR with 25 μmmu;mol l−1 ryanodine blunted CCE under normal and low [Na+]o conditions and prevented increases in CCE, induced by repetitive stimulation with Bk (squares; Fig. 8).
Figure 8. Correlation of Ca2+ release and CCE on repetitive stimulation with Bk in normal and low [Na+] conditions in the absence and presence of ryanodine.
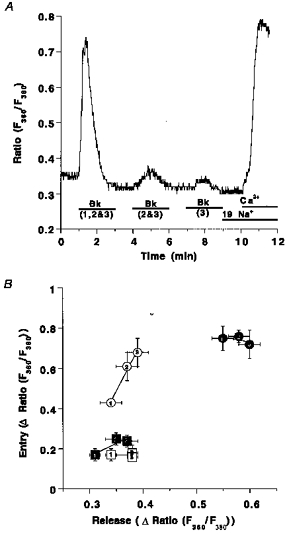
Panel A shows the experimental protocol: fura-2-loaded endothelial cells freshly isolated from bovine circumflex arteries were stimulated with 100 nmol l−1 Bk for 2 min either at time point 1 min (1), at time points 1 and 4 min (2) or at time points 1, 4 and 7 min (3). At time 9 min [Na+]o was set to 19 mmol l−1 and 2.5 mmol l−1 Ca2+ was added at time 10 min. Panel B, elevation of [Ca2+]pn to Bk at time 1 min (1), or the total sum of the increases of [Ca2+]pn upon two (2: 1st peak plus 2nd peak) or three (3: 1st peak plus 2nd peak plus 3rd peak) stimulations is shown on the x-axis. The increase in [Ca2+]pn to the addition of Ca2+ is shown on the y-axis. Bk stimulation was performed in Ca2+-free HBS containing 145 mmol l−1 Na+ (○), 19 mmol l−1 Na+ (•), 145 mmol l−1 Na+ and 25 μmmu;mol l−1 ryanodine (□) and 19 mmol l−1 Na+ plus 25 μmmu;mol l−1 ryanodine (▪). Each point represents the mean ±s.e.m. (n= 12).
Importance of RsCR for NOS activity
To test whether RsCR itself stimulates NOS, cultured endothelial cells from bovine left circumflex coronary artery (passage 2) were stimulated for 15 min with 20, 200 and 500 nmol l−1 ryanodine in the presence of 2.5 mmol l−1 Ca2+ under normal and low [Na+]o conditions. Ryanodine, in all concentrations tested, failed to stimulate NOS (Fig. 9A). However, inhibition of RsCR with 5 μmmu;mol l−1 ryanodine diminished NOS activation by Bk and ATP (Fig. 9B). The inhibitory effect of ryanodine on autacoid-induced NOS activation was more pronounced at lower agonist concentrations (i.e. 10 μmmu;mol l−1 ATP, 10 nmol l−1 Bk), and was less effective for the highest concentrations tested (i.e. 100 μmmu;mol l−1 ATP, 100 nmol l−1 Bk). In contrast, thapsigargin-induced activation of NOS was not affected by inhibition of RsCR (Fig. 9B). Similar findings were obtained in the human umbilical vein endothelial cell-derived cell line EA.hy 926 using 10 and 100 μmmu;mol l−1 ADP and 10 and 100 μmmu;mol l−1 histamine (data not shown).
Figure 9. Effect of ryanodine-induced RsCR on NOS activity (A) and changes in autacoid-induced NOS activation by inhibition of RsCR with a high concentration of ryanodine (B).
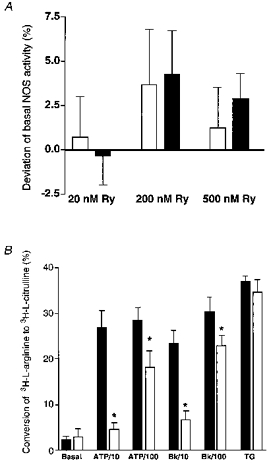
Cultured endothelial cells from the bovine circumflex coronary artery (passage 2) were stimulated with 20, 200 and 500 nmol l−1 ryanodine (panel A), or with ATP (10 and 100 μmmu;mol l−1), Bk (10 and 100 nmol l−1) and 2 μmmu;mol l−1 thapsigargin (TG) in the absence or presence of 5 μmmu;mol l−1 ryanodine (panel B) for 15 min in HBS. Columns represent the percentage deviation from basal NOS activity (panel A) or the percentage of conversion of [3H]-L-arginine to [3H]-l-citrulline (panel B). Each column represents the mean ±s.e.m. (A, n= 4; B, n= 5). *P < 0.05 vs. absence of ryanodine.
DISCUSSION
Role of Na+-Ca2+ exchange and RsCR in intracellular Ca2+ signalling
Impairment of Na+−Ca2+ exchange by lowering [Na+]o increased the Bk-induced elevation in [Ca2+]pn. This suggests that during Bk stimulation a large amount of the Ca2+ released is extruded via the Na+−Ca2+ exchange in endothelial cells. This is in agreement with the proposed involvement of Na+−Ca2+ exchange in endothelial cell Ca2+ handling during agonist stimulation (Sage et al. 1991).
In contrast to the prevention of Na+−Ca2+ exchange activity, inhibition of RsCR by 25 μmmu;mol l−1 ryanodine did not affect Bk-induced mobilization of [Ca2+]pn in 145 mmol l−1 Na+-containing buffer. Hence, under these conditions RsCR does not contribute to Bk-induced elevation of [Ca2+]pn. However, under low [Na+]o conditions inhibition of RsCR attenuated the ability of Bk to increase [Ca2+]pn, suggesting that under these conditions RsCR contributes to the observed elevation of [Ca2+]pn.
In spite of the existence of ryanodine receptors in endothelial cells (Lesh et al. 1993; Graier et al. 1998), direct stimulation of RsCR by 200 nmol l−1 ryanodine failed to elevate [Ca2+]pn under control conditions. These findings are consistent with the report of Wang et al. (1995) who observed that ryanodine did not induce Ca2+ transients. Our findings that inhibition of Na+−Ca2+ exchange unmasked ryanodine-induced Ca2+ release suggest that activation of RsCR occurs in the subplasmalemmal compartment and that the Ca2+ released by RsCR is extruded by the Na+−Ca2+ exchange without increasing [Ca2+]pn. This hypothesis is further supported by results obtained with FFP-18. In contrast to fura-2, which is found in the whole cytosol, FFP-18 has been shown to accumulate only in the plasma membrane and the nuclear membrane in smooth muscle cells when introduced into the cell from a patch pipette (Etter et al. 1996). In human neutrophils loaded for 1 to 3 h with the membrane permeable ester FFP-18 AM, FFP-18 was found mainly in the plasma membrane (Davies & Hallett, 1996). In agreement with these findings, when cells were loaded with FFP-18 using a laser-generated stress wave loading technique (Graier et al. 1998) FFP-18 was found mainly in the plasma membrane. Using FFP-18, ryanodine-induced subplasmalemmal Ca2+ release could be monitored even under control conditions (i.e. 145 mmol l−1[Na+]o). Hence, in 145 mmol l−1 Na+-containing solution, inhibition of RsCR blunted the amount of Ca2+ extruded during stimulation with Bk. These data suggest that during stimulation of endothelial cells RsCR increases [Ca2+]sp by vectorial Ca2+ release towards plasmalemmal Na+-Ca2+ exchange proteins without increasing [Ca2+]pn. A similar functional organization of the plasmalemmal Na+−Ca2+ exchange with sarcoplasmic reticulum Ca2+ release was described in smooth muscle cells (Moore et al. 1993), a cell type where subplasmalemmal Ca2+ handling is also insulated from [Ca2+]pn (van Breemen et al. 1986; Sturek et al. 1992; Fay, 1995; Nelson et al. 1995; Yamaguchi et al. 1995).
Role of RsCR for CCE activity
The activity of CCE has been shown to increase progressively with enhanced Ca2+ store depletion (Hofer et al. 1998). Recently, we have shown that inhibition of Na+−Ca2+ exchange during autacoid stimulation enhanced depletion of Ca2+ stores due to Ca2+−induced Ca2+ release initiated by Ca2+ normally extruded by Na+−Ca2+ exchange (Graier et al. 1998). In agreement with these reports CCE was increased when cells were stimulated under low [Na+]o conditions in our experiments, a phenomenon possibly linked to the pronounced depletion of intracellular Ca2+ stores by RsCR when Na+-Ca2+ exchange activity was prevented. Consistent with this explanation, inhibition of RsCR diminished CCE under normal and low [Na+]o conditions. Moreover, ryanodine uncoupled the correlation of intracellular Ca2+ release and activity of CCE. These data may indicate that RsCR contributes to the depletion of Ca2+ stores and hence activation of CCE. Although this explanation seems attractive, the contribution of caffeine- and/or ryanodine-sensitive compartments of the endoplasmic reticulum in CCE activation in endothelial cells was excluded very recently (Sasajima et al. 1997). Hence, several authors have failed to measure CCE in response to activation of RsCR by either caffeine (Graier et al. 1994; Ziegelstein et al. 1994; Wang et al. 1995) or ryanodine (Wang et al. 1995). Consistent with these reports, direct stimulation of RsCR by 200 nmol l−1 ryanodine did not evoke CCE in this study. In agreement with these findings, we have previously reported that ryanodine failed to activate microsomal cytochrome P450 epoxygenase (Hoebel et al. 1997), an enzyme thought to be involved in the regulation of CCE (Graier et al. 1995). Therefore, a direct involvement of RsCR in the activation of CCE seems unlikely.
In endothelial cells stimulated by agonists, the amount of Ca2+ entering the cell depends strictly on the driving force provided by membrane hyperpolarization due to activation of Ca2+-activated K+ channels (Colden-Stanfield et al. 1987; Busse et al. 1988). Since we have shown that RsCR preferably occurs in the subplasmalemmal region, one might expect that RsCR may activate Ca2+-activated K+ channels to promote membrane hyperpolarization and thus increase the driving force for Ca2+ influx. This is consistent with the finding that caffeine, an activator of RsCR, induced Ca2+-sensitive K+ current in freshly isolated rabbit aortic endothelial cells (Rusko et al. 1995). Hence, caffeine has been shown to elevate CCE in activated endothelial cells (Corda et al. 1995). Therefore, the reduction of CCE activity by inhibition of RsCR might be due to a reduction of agonist-induced membrane hyperpolarization. However, a more detailed investigation on the contribution of RsCR on CCE is necessary to determine the exact mechanism by which RsCR affects CCE activity.
Role of RsCR in autacoid-induced NOS activation
There is little known about the involvement of RsCR in the activation of NOS. In rat aortic rings inhibition of RsCR attenuated endothelium-dependent relaxation in response to caffeine (Hatano et al. 1995). In isolated rabbit arterial rings ryanodine induced nitric oxide-mediated relaxation (Hutcheson & Griffith, 1997), while in rat lung and pulmonary arteries ryanodine failed to induce nitric oxide release (Muramatsu et al. 1996). In agreement with the latter findings, ryanodine at a concentration where it activates RsCR (20, 200 and 500 nmol l−1) failed to activate NOS in the present study. However, inhibition of RsCR diminished activation of NOS, evoked either by ADP, ATP, Bk or histamine. These findings suggest that the partial inhibition of CCE by ryanodine accounts for the reduced NOS activation to autacoids. Since the inhibitory properties of ryanodine were diminished with increased agonist concentration, one might speculate that RsCR is more important for regulation of NOS under submaximal and/or physiological stimulation. Under supramaximal stimulation the strong InsP3-mediated Ca2+ release and/or the maximal CCE may compensate for the lack of RsCR.
Recently, a subplasmalemmal Ca2+ control unit (SCCU) was introduced to explain the control of subplasmalemmal Ca2+ gradients under submaximal stimulation (Graier et al. 1998). The data presented here extend the function of the SCCU as a regulator of activation of NOS, which is localized in cavaeoli of the plasma membrane (García-Cardena et al. 1996).
In conclusion, our data provide evidence that during autacoid stimulation RsCR occurs in endothelial cells. Ca2+ is vectorially released towards plasmalemmal Na+−Ca2+ exchange proteins for rapid extrusion without any impact on [Ca2+]pn. However, RsCR elevates [Ca2+]sp and contributes to the activity of CCE, while RsCR alone does not activate CCE. Finally, the subplasmalemmal Ca2+ release by RsCR is important for activation of NOS under submaximal stimulation. These data extend the function of the SCCU not only to control [Ca2+]sp but to the regulation of NOS activity.
Acknowledgments
The excellent technical assistance from Mrs Beatrix Petschar is highly appreciated. We thank Drs Michael Sturek and Jean-Vivien Mombouli for critically reviewing this manuscript. This work was supported from the Austrian Science Fund (P1 2341-MED; SFB 714).
References
- Busse R, Fichter H, Lückhoff A, Kohlhardt M. Hyperpolarization and increased free calcium in acetylcholine-stimulated endothelial cell. American Journal of Physiology. 1988;255:H965–969. doi: 10.1152/ajpheart.1988.255.4.H965. [DOI] [PubMed] [Google Scholar]
- Colden-Stanfield M, Schilling WP, Ritchie AK, Eskin SG, Navarro LT, Kunze DL. Bradykinin-induced increases in cytosolic free calcium and ionic currents in bovine aortic endothelial cells. Circulation Research. 1987;61:632–640. doi: 10.1161/01.res.61.5.632. [DOI] [PubMed] [Google Scholar]
- Corda S, Spurgeon HA, Lakatta EG, Capogrossi MC, Ziegelstein R. Endoplasmic reticulum Ca2+ depletion unmasks a caffeine-induced Ca2+ influx in human aortic endothelial cells. Circulation Research. 1995;77:927–935. doi: 10.1161/01.res.77.5.927. [DOI] [PubMed] [Google Scholar]
- Davies EV, Hallett MB. Near membrane Ca2+ changes resulting from store release in neutrophils: detection by FFP-18. Cell Calcium. 1996;19:355–362. doi: 10.1016/s0143-4160(96)90076-7. 10.1016/S0143-4160(96)90076-7. [DOI] [PubMed] [Google Scholar]
- Edgell CJS, McDonald CC, Graham JB. Permanent cell line expressing factor VIII related antigen established byhybridization. Proceedings of the National Academy of Sciences of the USA. 1983;80:3734–3737. doi: 10.1073/pnas.80.12.3734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etter EF, Minta A, Poenie M, Fay FS. Near-membrane [Ca2+] transients resolved using the Ca2+ indicator FFP-18. Proceedings of the National Academy of Sciences of the USA. 1996;93:5368–5373. doi: 10.1073/pnas.93.11.5368. 10.1073/pnas.93.11.5368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay FS. Calcium sparks in vascular smooth muscle: relaxation regulators. Science. 1995;270:588–589. doi: 10.1126/science.270.5236.588. [DOI] [PubMed] [Google Scholar]
- Fleming I, Fisslthaler B, Busse R. Interdependence of calcium signaling and protein tyrosine phosphorylation in human endothelial cells. Journal of Biological Chemistry. 1996;271:11009–11015. doi: 10.1074/jbc.271.18.11009. 10.1074/jbc.271.18.11009. [DOI] [PubMed] [Google Scholar]
- Freay A, Johns A, Adams DJ, Ryan US, Ivan Breemen C. Bradykinin and inositol 1,4,5-trisphosphate-stimulated calcium release from intracellular stores in cultured bovine endothelial cells. Pflügers Archiv. 1989;414:377–384. doi: 10.1007/BF00585046. [DOI] [PubMed] [Google Scholar]
- García-Cardena G, Oh P, Liu J, Schnitzer JE, Sessa WC. Targeting of nitric oxide synthase to endothelial cell caveolae via palmitoylation: Implication for nitric oxide signaling. Proceedings of the National Academy of Sciences of the USA. 1996;93:6448–6453. doi: 10.1073/pnas.93.13.6448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Groschner K, Schmidt K, Kukovetz WR. SK&F 96365 inhibits histamine-induced formation of endothelium-derived relaxing factor in human endothelial cells. Biochemical and Biophysical Research Communications. 1992;186:1539–1545. doi: 10.1016/s0006-291x(05)81582-7. [DOI] [PubMed] [Google Scholar]
- Graier WF, Paltauf-Doburzynska J, Hill H, Fleischhacker B, Hoebel BG, Kostner GM, Sturek M. Submaximal stimulation of porcine endothelial cells causes focal Ca 2+ elevation beneath the cell membrane. The Journal of Physiology. 1998;506:109–125. doi: 10.1111/j.1469-7793.1998.109bx.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Bowles DK, Sturek M. Heterogeneity of caffeine and bradykinin-sensitive stores in vascular endothelial cells. Biochemical Journal. 1994a;300:637–641. doi: 10.1042/bj3000637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Kukovetz WR, Kostner GM. High-D-glucose-induced changes in endothelial Ca2+/EDRF signaling is due to generation of superoxide anions. Diabetes. 1996;45:1386–1395. doi: 10.2337/diab.45.10.1386. [DOI] [PubMed] [Google Scholar]
- Graier WF, Simecek S, Sturek M. Cytochrome P450 mono-oxygenase-regulated signalling of endothelial Ca 2+ entry in human and bovine endothelial cells. The Journal of Physiology. 1995;482:259–274. doi: 10.1113/jphysiol.1995.sp020515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graier WF, Sturek M, Kukovetz WR. Ca2+ regulation and endothelial vascular function. Endothelium. 1994b;1:223–236. [Google Scholar]
- Hatano Y, Mizumoto K, Yoshiyama T, Yamamoto M, Iranami H. Endothelium-dependent and -independent vasodilation of isolated rat aorta induced by caffeine. American Journal of Physiology. 1995;269:H1679–1684. doi: 10.1152/ajpheart.1995.269.5.H1679. [DOI] [PubMed] [Google Scholar]
- Hoebel BG, Kostner GM, Graier WF. Activation of microsomal cytochrome P450 mono-oxygenase by Ca2+ store depletion and its contribution to Ca2+ entry in porcine aortic endothelial cells. British Journal of Pharmacology. 1997;121:1579–1588. doi: 10.1038/sj.bjp.0701304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: a study using simultaneous measurements of ICRAC and intraluminal [Ca2+] Journal of Cellular Biology. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutcheson IR, Griffith TM. Central role of intracellular calcium stores in acute flow- and agonist-evoked endothelial nitric oxide release. British Journal of Pharmacology. 1997;122:117–125. doi: 10.1038/sj.bjp.0701340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesh RE, Marks AR, Somlyo AV, Fleischer S, Somlyo AP. Anti-ryanodine receptor antibody binding sites in vascular and endocardial endothelium. Circulation Research. 1993;72:481–488. doi: 10.1161/01.res.72.2.481. [DOI] [PubMed] [Google Scholar]
- Lückhoff A, Pohl U, Mülsch A, Busse R. Differential role of extra- and intracellular calcium in the release of EDRF and prostacyclin from cultured endothelial cells. British Journal of Pharmacology. 1988;95:189–186. doi: 10.1111/j.1476-5381.1988.tb16564.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore EDW, Etter EF, Philipson KD, Carrington WA, Fogarty KE, Lifshitz LM, Fay FS. Coupling of the Na+/Ca2+ exchanger, Na+/K+ pump and sacroplasmic reticulum in smooth muscle. Nature. 1993;365:657–660. doi: 10.1038/365657a0. [DOI] [PubMed] [Google Scholar]
- Mozhayeva MG. [Ca2+]i elevation evoked by Ca2+ readdition to the medium after agonist-induced Ca2+ release can involve both IP3-, and ryanodine-sensitive Ca2+ release. Pflügers Archiv. 1996;433:180–187. doi: 10.1007/s004240050265. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Tyler RC, Rodman DM, McMurtry IF. Thapsigargin stimulates increased NO activity in hypoxic hypertensive rat lung and pulmonary arteries. Journal of Applied Physiology. 1996;80:1336–1344. doi: 10.1152/jappl.1996.80.4.1336. [DOI] [PubMed] [Google Scholar]
- Nelson MT, Cheng H, Rubart M, Santana LF, Bonev AD, Knot HJ, Lederer WJ. Relaxation of arterial smooth muscle by calcium sparks. Science. 1995;270:633–637. doi: 10.1126/science.270.5236.633. [DOI] [PubMed] [Google Scholar]
- Putney JW. A model for receptor-regulated calcium-entry. Cell Calcium. 1986;7:1–12. doi: 10.1016/0143-4160(86)90026-6. [DOI] [PubMed] [Google Scholar]
- Putney JW. Capacitative calcium entry revisited. Cell Calcium. 1990;11:611–624. doi: 10.1016/0143-4160(90)90016-n. [DOI] [PubMed] [Google Scholar]
- Rusko J, Van Slooten G, Adams DJ. Caffeine-evoked, calcium-sensitive membrane currents in rabbit aortic endothelial cells. British Journal of Pharmacology. 1995;115:133–141. doi: 10.1111/j.1476-5381.1995.tb16330.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage SO, van Breemen C, Cannell MB. Sodium-calcium exchange in cultured bovine pulmonary artery endothelial cells. The Journal of Physiology. 1991;440:569–580. doi: 10.1113/jphysiol.1991.sp018725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasajima H, Wang X, van Breemen C. Fractional Ca2+ release from the endoplasmic reticulum activates Ca2+ entry in freshly isolated rabbit aortic endothelial cells. Biochemical and Biophysical Research Communications. 1997;241:471–475. doi: 10.1006/bbrc.1997.7844. [DOI] [PubMed] [Google Scholar]
- Schilling WP, Cabello OA, Rajan L. Depletion of the inositol 1,4,5-trisphosphate-sensitive intracellular Ca2+ store in vascular endothelial cells activates the agonist-sensitive Ca2+-influx pathway. Biochemical Journal. 1992;284:521–530. doi: 10.1042/bj2840521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturek M, Kunda K, Hu Q. Sarcoplasmic reticulum buffering of myoplasmic calcium in bovine coronary artery smooth muscle. The Journal of Physiology. 1992;451:25–48. doi: 10.1113/jphysiol.1992.sp019152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturek M, Smith P, Stehno-Bittle L. In vitro models of vascular endothelial cell calcium regulation. In: Sperelakis N, Kuriyama H, editors. Ion Channels of Vascular Smooth Muscle Cells and Endothelial Cells. New York: Elsevier; 1991. pp. 349–364. [Google Scholar]
- van Breemen C, Lukeman S, Leijten P, Yamamoto H, Loutzenhiser R. The role of superficial SR in modulating force development induced by Ca2+ entry into artery smooth muscle. Journal of Cardiovascular Pharmacology. 1986;8(suppl. 8):S111–S116. doi: 10.1097/00005344-198600088-00023. [DOI] [PubMed] [Google Scholar]
- Wang X, Lau F, Li L, Yoshikawa A, van Breemen C. Acetylcholine-sensitive intracellular Ca2+ store in fresh endothelial cells and evidence for ryanodine receptors. Circulation Research. 1995;77:37–42. doi: 10.1161/01.res.77.1.37. [DOI] [PubMed] [Google Scholar]
- Waters D, Brown C. New high-resolution 3-D microscope - avoids damage to live samples. Biophotonics International. 1996;9/10:40–44. [Google Scholar]
- Yamaguchi H, Kajita J, Madison JM. Isoproterenol increases peripheral [Ca2+]i and decreases inner [Ca2+]i in single airway smooth muscle cells. American Journal of Physiology. 1995;268:C771–779. doi: 10.1152/ajpcell.1995.268.3.C771. [DOI] [PubMed] [Google Scholar]
- Ziegelstein RC, Spurgeon HA, Pili R, Passaniti A, Cheng L, Corda S, Laketta EG, Capogrossi MC. A functional ryanodine-sensitive intracellular Ca2+ store is present in vascular endothelial cells. Circulation Research. 1994;74:151–156. doi: 10.1161/01.res.74.1.151. [DOI] [PubMed] [Google Scholar]