

Abstract
In primary cultured human glioblastoma cells extracellular application of ATP triggered elevation in cytoplasmic calcium concentration ([Ca2+]i) mediated entirely by generation of inositol 1,4,5-trisphosphate (InsP3)-dependent Ca2+ release from endoplasmic reticulum Ca2+ stores followed by the activation of store-operated Ca2+ entry into the cells.
Sensitivity of P2Y purinoceptors to extracellular ATP was regulated by extracellular Ca2+: in Ca2+-free extracellular solution the threshold concentration of ATP that induced an increase in [Ca2+]i was reduced by one order of magnitude.
Activation of Ca2+ release and store-operated Ca2+ entry was dissociated: low concentrations of ATP induced substantial Ca2+ release without activation of Ca2+ entry; activation of the latter required higher ATP concentrations.
Mitochondria participated in buffering Ca2+ loads that resulted from store-operated Ca2+ influx; in contrast Ca2+ released from intracellular stores was not accumulated by the mitochondrial depot.
We conclude that ATP-induced Ca2+ responses are governed by several pathways with different sensitivities to the agonist. This enables cells to respond either with pure Ca2+ release from intracellular stores (at low ATP concentrations) or (at high ATP concentrations) the response is amplified by plasmalemmal Ca2+ influx. Store-operated Ca2+ entry increases mitochondrial Ca2+ content providing a link between cellular activation and mitochondrial function.
In electrically non-excitable cells activation of numerous plasmalemmal receptors triggers a cytoplasmic calcium signal by utilizing two distinct, but tightly coupled pathways. The initial phase of cytoplasmic calcium concentration ([Ca2+]i) elevation is driven by the inositol 1,4,5-trisphosphate (InsP3)-mediated opening of Ca2+ channels present in the membrane of endoplasmic reticulum Ca2+ stores. Flow of Ca2+ through these channels empties the stores of releasable Ca2+. Empty stores trigger the opening of plasmalemmal Ca2+ channels, thus inducing ‘store-operated’ Ca2+ influx into the cell (Berridge, 1993, 1995; Parekh & Penner, 1997). Recent investigations of rat basophilic leukaemia cells demonstrated a non-linear relation between InsP3-induced Ca2+ release from the internal stores and activation of store-operated Ca2+ entry: the latter was triggered by InsP3 at concentrations much higher than those required for activation of Ca2+ release (Parekh et al. 1997). Ca2+ entering the cell via the store-operated pathway does not simply replenish the internal stores: at least in some types of non-excitable cell, it is taken up by mitochondria, which then redistribute to different cellular compartments (Hoth et al. 1997).
Glial cells are the non-excitable elements of the nervous system which are known to express a wide variety of receptors to neurotransmitters and hormones. The majority of these receptors are linked to generation of cytoplasmic Ca2+ signals through the InsP3-mediated intracellular signalling cascade (Verkhratsky & Kettenmann, 1996; Verkhratsky et al. 1998). Despite the fact that the store-operated Ca2+ entry pathway is operational in various types of glial cells, the precise relations between internal Ca2+ stores and plasmalemmal Ca2+ channels remain unexplored. In the present study we examined the interrelations between intracellular Ca2+ release, Ca2+ entry and mitochondrial Ca2+ buffering in primary cultured human glioblastoma cells. The latter are tumour cells of glial origin which form the most frequent and aggressive types of primary brain neoplasm. The majority of our current knowledge of glioma physiology is based on experiments on cell lines derived from different sorts of tumour tissue (for review see Brismar, 1995). Yet glioma cell lines, though being highly homogeneous, can only be regarded as a rough model of primary tumours, as they are highly influenced by culture conditions, number of passages, etc. The first attempts to investigate the basic physiological properties of primary human gliomas have only very recently been undertaken. These experiments yielded the basic characteristics of ion channels and receptor subtypes present in human neoplastic glial cells (Patt et al. 1996; Labrakakis et al. 1997). In particular these experiments demonstrated that several types of neurotransmitters, namely glutamate, GABA, ATP, histamine and bradykinin, trigger [Ca2+]i elevation in primary cultures of human gliomas of different grades (Weydt et al. 1997). As the mechanisms of Ca2+ signalling in primary cultured glioma cells have not hitherto been described in detail we investigated the main routes for [Ca2+]i metabolism in glioblastoma cells following stimulation of the purinoceptors. We found that ATP-induced Ca2+ signalling in glioblastoma cells resulted exclusively from activation of metabotropic P2Y purinoceptors. Stimulation of these receptors triggers both Ca2+ release from the internal stores and store-operated Ca2+ entry. We investigated the relations between these two processes.
METHODS
Tumours
Twelve glioblastomas of WHO (World Health Organization) grade IV (Kleihues et al. 1993) (T13/97, T14/97, T16/97, T18/97, T19/97, T20/97, T21/97, T23/97, T16/98, T17/98, T22/98, T23/98) were obtained from the Neurosurgical Department of the Clinical Centre Berlin-Buch and the Virchow-Klinikum of the Humbold University, Berlin. The use of human material was performed according to the Declaration of Helsinki and all the procedures were approved by the local ethics committee.
Primary culture of human glioma cells
After removal of blood and blood vessels, the tumour tissue was mechanically dissected, then enzymatically dissociated using incubation with 0.025 % trypsin in calcium- and magnesium-free phosphate buffered saline (Seromed, Germany) for 15 min at 37°C. After removing trypsin by centrifugation the cells were resuspended in RPMI medium (Gibco) and dissociated by mechanical trituration to obtain a suspension of single cells. Cells were plated onto poly-L-lysine-coated glass coverslips and cultured at 37°C in a humidified, 5 % CO2-95 % air incubator (Heraeus, Hanau, Germany). One day after plating, cells were washed with phosphate-buffered saline. Culture medium (RPMI) was changed twice a week. Cells were taken for experiments after different times (4-21 days) in culture.
[Ca2+]i measurements
Primary cultured glioma cells were loaded with the Ca2+ indicator by incubating glass coverslips with adhered cells in standard bath solution (for composition see Solutions and reagents) supplemented with 5 μM fura-2 acetoxymethylester (fura-2 AM) for 20 min at room temperature (21-23°C). The cells were then washed twice with physiological saline and stored in the dark for 30 min. Coverslips with loaded cells were transferred to the perfusion chamber mounted on the stage of an upright microscope (Axioskop, Zeiss). Cells were visualized using a × 20 objective lens (numerical aperture, 0.5). For fura-2 excitation, cells were illuminated with two alternating wavelengths, 340 ± 3 and 380 ± 3 nm, using a monochromator (TILL Photonics, Munich, Germany). The fluorescence emission at 530 ± 10 nm was captured by a cooled CCD camera and digitized by an image processing system (TillVision, TILL Photonics). The monochromator and CCD camera were controlled by TillVision software, which was also used for image analysis. Ratio images were collected at intervals of 0.5-5 s.
[Ca2+]i was calculated from the ratio (R) of fluorescence recorded at 340 and 380 nm excitation wavelengths using the equation of Grynkiewicz et al. (1985):
where Rmin is the fluorescence ratio of Ca2+-free fura-2 and Rmax is the ratio of Ca2+-bound fura-2. The constant KDβ was determined empirically. The system was calibrated in situ by employing a ionomycin-based intracellular calibration procedure as described previously (Usachev et al. 1993). The parameters KDβ, Rmin and Rmax characterizing the system were 552 nM, 0.2 and 1.2, respectively. All data are presented as means ±s.d., unless otherwise indicated. The statistical significance of differences between mean values was tested using Student's t test. Differences were assumed statistically significant when the error probability was < 0.05.
Electrophysiology
Membrane currents were measured using the whole-cell configuration of the patch-clamp technique (Hamill et al. 1981). Current signals were amplified with conventional electronics (EPC-9 amplifier, HEKA Electronics, Lamprecht, Germany) filtered at 3 kHz and sampled at 5 kHz by an interface connected to an IBM-compatible PC which also controls the amplifier output. The acquisition and the analysis of the data were performed by WinTida software (HEKA Electronics). Capacitive current and series resistance compensation was performed. Recording pipettes were fabricated from borosilicate glass capillaries (Hilgenberg, Malsfeld, Germany). Pipettes were filled with a solution which contained (mM): KCl, 130; CaCl2, 0.5; EGTA, 5; MgCl2, 2; and Hepes, 10. The pH was adjusted with KOH to 7.4. Pipette resistances were 3-5 MΩ.
Solutions and reagents
The standard bath solution was composed of (mM): NaCl, 150; KCl, 5.4; CaCl2, 2; MgCl2, 1; Hepes/NaOH, 10; glucose, 10; pH 7.4 adjusted with NaOH. To obtain calcium-free solution, CaCl2 was omitted, MgCl2 was increased to 2 mM and 1 mM EGTA was added. Fura-2 AM was obtained from Molecular Probes. Adenosine-5′-diphosphate (ADP), adenosine-5′-O-(2-thiodiposphate) (ADPβS), ATP, α,β-methylene-adenosine-5′-triphosphate (αβATP), 2-methylene-adenosine-5′-diphosphate (2MeSADP), uridine-5′-triphosphate (UTP) and thapsigargin were purchased from RBI. All other chemicals were obtained from Sigma.
RESULTS
ATP induces an increase in [Ca2+]i in cultured human glioblastoma cells
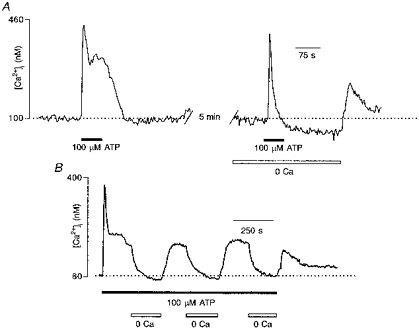
Resting [Ca2+]i in human glioma cells varied between 40 and 130 nM, and averaged 76 ± 19 nM (29 experiments, 944 cells). The majority (91 %; 25 experiments, 831 cells) of cells exposed to 100 μM ATP for 60 s exhibited a robust and transient increase in [Ca2+]i. The peak amplitudes of ATP-induced [Ca2+]i transients ranged from 100 to 1200 nM, being on average 422 ± 177 nM (756 cells). The dynamical properties of [Ca2+]i responses triggered by 100 μM ATP varied between cells. In Fig. 1A, three types of ATP-induced [Ca2+]i responses illustrate the variability in the time course of [Ca2+]i transients. In 23 % of cells, ATP induced a monophasic increase in [Ca2+]i, when [Ca2+]i after reaching the peak monotonically returned to the resting level (Fig. 1A, left trace). In most cells (66 %), ATP triggered a biphasic response, comprising an initial peak followed by a plateau phase (Fig. 1A, middle trace). Finally in 11 % of cells, the initial [Ca2+]i peak was followed by a series of [Ca2+]i oscillations (Fig. 1A, right trace).
Figure 1. Properties of ATP-induced [Ca2+]i increase in human glioblastoma cells.
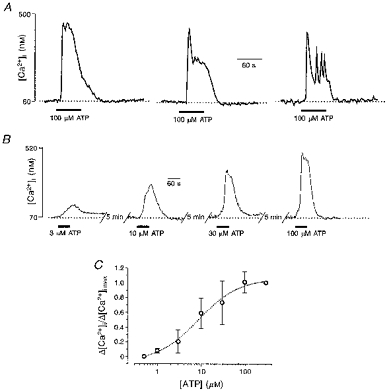
A, three types of typical [Ca2+]i response induced by extracellular application of 100 μM ATP were recorded from fura-2 AM-loaded single human glioma cells. B and C, concentration dependence of the ATP-mediated [Ca2+]i transients in cultured glioblastoma cells. B, examples of [Ca2+]i transients recorded from a fura-2 AM-loaded cell in response to 60 s applications of 3, 10, 30 and 100 μM ATP. Successive ATP applications were separated by 5 min intervals. C, ATP (0.5-300 μM)-induced [Ca2+]i transient amplitudes obtained from four different cultures (139 cells). ATP applications were made in the same way as those in B. Amplitudes are normalized to that of the maximum response to ATP (Δ[Ca2+]i,max) (here at 300 μM). The analysis yielded an apparent KD of 9 μM and a Hill coefficient of 1. Here and in subsequent figures, symbols and error bars indicate means ±s.d.
ATP induced increases in [Ca2+]i in a concentration-dependent manner, as shown in Fig. 1B and C where cells were exposed to different concentrations of ATP (0.5 − 300 μM). The threshold for ATP-induced [Ca2+]i responses was approximately 1 μM and the responses were saturated at 100 μM ATP.
Extracellular Ca2+ regulates the sensitivity of purinoceptors to ATP
ATP is a broadly recognize.d modulator of [Ca2+]i in various cell types. ATP-induced increases in [Ca2+]i occur by several distinct mechanisms including the opening of membrane channels of the P2X receptor family, as well as the triggering of Ca2+ release from InsP3-sensitive stores. Here, we have begun the analysis of the mechanisms of ATP-mediated [Ca2+]i elevation in glioma cells by determining the influence of extracellular Ca2+. Removal of Ca2+ from the extracellular solution did not affect the ability of ATP to increase [Ca2+]i. In 93.7 % of cells that were preincubated in Ca2+-free solution for 1 min, application of 100 μM ATP evoked [Ca2+]i transients (14 experiments, 372 cells; Figs 2 and 4). Surprisingly, extracellular Ca2+ appeared to modulate the concentration dependence of ATP-triggered [Ca2+]i responses. We performed experiments in which the same cells were challenged with different ATP concentrations in the presence and absence of extracellular Ca2+ (Fig. 2A). Application of 1 μM ATP in standard solution containing 2 mM Ca2+ triggered a small [Ca2+]i transient with a mean amplitude of 54 ± 11 nM (117 cells). In contrast, when the same cells were exposed to 1 μM ATP in Ca2+-free extracellular solution they responded with much larger [Ca2+]i transients that had a mean peak amplitude of 312 ± 48 nM (117 cells). Moreover, it appeared that ATP at concentrations below 1 μM was still able to trigger [Ca2+]i elevation when applied in Ca2+-free medium, whereas the same concentrations of ATP administered in Ca2+-containing solution failed to induce any change in [Ca2+]i. Therefore, we compared the ATP dose-response curves obtained from the same cells in the presence and absence of extracellular Ca2+ (Fig. 2B). We found that when cells were stimulated with ATP in Ca2+-free extracellular solution, the concentration threshold for inducing [Ca2+]i transients was approximately 0.05 μM, and the responses were saturated at 10 μM ATP. In contrast in Ca2+-containing solution, the concentration threshold was 1 μM and the responses were saturated at 100 μM ATP.
Figure 2. Extracellular Ca2+ controls the sensitivity of ATP-induced [Ca2+]i transients.

A, [Ca2+]i transients recorded from a single cell when stimulated by the same concentration of ATP applied first in standard, 2 mM Ca2+-containing extracellular solution, then in Ca2+-free solution. Note that ATP concentrations lower than 0.5 μM failed to induce [Ca2+]i responses in Ca2+-containing solutions, but when the same concentrations were applied in Ca2+-free medium [Ca2+]i transients were evoked. B, normalized amplitudes of ATP-induced [Ca2+]i transients measured in Ca2+-containing (•) and Ca2+-free (○) conditions are plotted against ATP concentration. The apparent KDs are 0.7 and 13 μM and the Hill coefficients are 1.3 and 1.1 for Ca2+-free and Ca2+-containing solutions, respectively. Data were collected from five different experiments, 117 cells.
Figure 4. ATP-induced [Ca2+]i elevation in glioblastoma cells originates primarily from thapsigargin-sensitive intracellular Ca2+ stores.
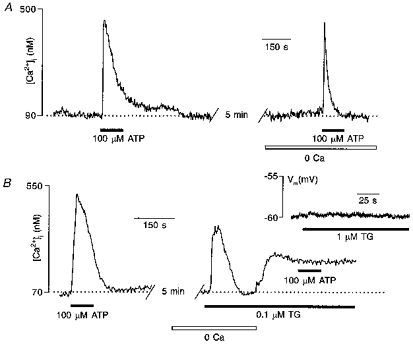
A, a single fura-2 AM-loaded glioma cell was stimulated with 100 μM ATP in standard bath solution (left trace) and after 2 min incubation in Ca2+-free extracellular medium (right trace). B, mean response of twenty-nine glioma cells to ATP in standard bath solution and after cell treatment with 100 nM TG. The inset shows that TG did not affect the membrane potential of the glioblastoma cell.
Does this phenomenon apply only to purinoceptors or is it a general feature of agonist-induced Ca2+ signals in glioblastoma cells? To test this we performed similar experiments using another neurotransmitter histamine, which is also known to trigger Ca2+ transients in human gliomas (Weydt et al. 1997). Removal of Ca2+ from the extracellular solution did not modify the concentration dependence of histamine-induced [Ca2+]i responses (3 experiments, 74 cells; data not shown).
Pharmacological characterization of purinoceptors in glioma cells
As the effects of ATP could be mediated through a wide variety of receptors (North & Barnard, 1997), we tested the efficacy of various ATP analogues in increasing [Ca2+]i in cultured human glioblastoma cells. To discriminate between ionotropic P2X and metabotropic P2Y purinoceptors we examined the effects of the following agonists (Fredholm et al. 1994; Zimmermann, 1994; North & Barnard, 1997): (i) αβATP, selective for P2X receptors, and (ii) ADP, ADPβS, UTP and 2MeSADP, known to stimulate different receptors of the P2Y family. Each cellular preparation was exposed to 100 μM of each agonist; and each agonist was applied for 60 s with a 5 min interval between applications. These experiments were performed on five different culture preparations and 164 cells were analysed. Representative examples of [Ca2+]i transients evoked by ATP analogues are illustrated in Fig. 3A, and the frequency of the [Ca2+]i response occurrence is shown in Fig. 3B. The majority of tumour cells responded with an increase in [Ca2+]i when exposed to 100 μM ADP, ADPβS and 2MeSADP. Only a minor subpopulation (∼10 %) of glioma cells demonstrated [Ca2+]i elevation during exposure to UTP. Finally, αβATP did not induce any [Ca2+]i responses in glioma cells.
Figure 3. Efficacy of different P2 purinoceptor agonists in inducing [Ca2+]i responses in human glioblastoma cells.
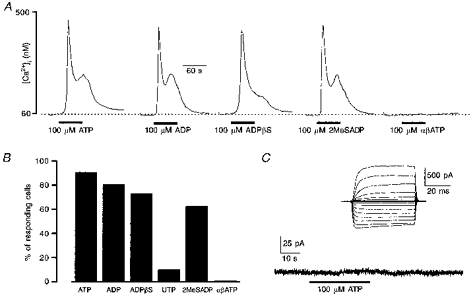
A, representative examples of [Ca2+]i transients induced by different P2 purinoceptor agonists (ATP, ADP, ADPβS, 2MeSADP and αβATP, each at 100 μM) in the same single cultured glioma cell. Applications of agonists were separated by 5 min intervals. B, percentage of glioma cells responding with a [Ca2+]i rise to P2 purinoceptor agonists. Data were collected from five different experiments, 164 cells. C, membrane current recorded from a single glioblastoma cell in whole-cell patch-clamp configuration. Extracellular application of 100 μM ATP did not evoke any changes in membrane permeability. Inset, a family of voltage-activated currents recorded from the same cell in response to de- and hyperpolarizing voltage steps, the current pattern is typical for glioblastoma cells. Holding potential was −60 mV.
Thus we conclude that human tumour cells predominantly express metabotropic P2Y purinoceptors, which are linked to generation of Ca2+ release from the intracellular InsP3-sensitive calcium pool. To substantiate these findings we also performed electrophysiological experiments monitoring membrane currents in glioblastoma cells in response to extracellular application of 100 μM ATP. In none of the thirty-two cells taken from four different preparations did ATP induce measurable changes in membrane permeability (Fig. 3C). Finally, we also checked whether depolarization of glioblastoma cells might result in [Ca2+]i elevation due to Ca2+ entry through plasmalemmal channels. In none of the 106 cells tested (4 different preparations) did extracellular application of 50 mM KCl-containing solution for 30 s induce changes in [Ca2+]i.
Activation of purinoceptors triggers Ca2+ release from intracellular stores and store-operated Ca2+ entry
As was demonstrated by agonist screening, the predominant mechanism for ATP-induced [Ca2+]i elevation in human glioma cells is the stimulation of intracellular Ca2+ release. Indeed, only those ATP analogues believed to interact with metabotropic receptors of the P2Y subfamily were able to trigger [Ca2+]i elevation in glioma cells. In contrast, none of the cells responded with a [Ca2+]i transient to the selective ionotropic (P2X) receptor agonist αβATP, and moreover, electrophysiological experiments failed to detect any ATP-induced membrane currents. This conclusion was further substantiated by comparing [Ca2+]i responses induced by ATP in standard solution containing 2 mM Ca2+ and in Ca2+-free extracellular solution. As already mentioned above, ATP was able to trigger [Ca2+]i elevation in Ca2+-free medium and, furthermore, the amplitudes of Ca2+ transients evoked by 100 μM ATP were only slightly attenuated when recorded in the absence of Ca2+ ions (e.g. Fig. 4A). The mean amplitudes of 100 μM ATP-triggered [Ca2+]i transients were 453 ± 96 nM in Ca2+-containing solution vs. 411 ± 107 nM in Ca2+-free solution (9 experiments, 194 cells). Additional evidence for the predominant role of intracellular Ca2+ release in the generation of ATP-induced [Ca2+]i elevation is derived from experiments with thapsigargin (TG), a selective and irreversible inhibitor of endoplasmic reticulum (ER) Ca2+ pumps (Thastrup et al. 1990). Exposure of glioma cells to 100 nM TG in Ca2+-free solution triggered [Ca2+]i transients due to the uncompromised Ca2+ leakage from intracellular stores (Fig. 4B, right trace). After extracellular Ca2+ was restored, the [Ca2+]i increased rapidly due to the activation of store-operated Ca2+ entry. Exposure of TG-pretreated cells to 100 μM ATP in standard, 2 mM Ca2+-containing solution failed to trigger a [Ca2+]i response in the majority of cells (Fig. 4B), only six of 147 cells (6 preparations) responded with a tiny [Ca2+]i increase. It also has to be noted that TG did not change the membrane potential of glioblastoma cells (17 cells from 3 preparations; Fig. 4B, inset). Thus, emptying the Ca2+ stores using TG prevented ATP-induced [Ca2+]i elevation in glioma cells, once more indicating that the primary source for ATP-induced [Ca2+]i elevation is release of Ca2+ from intracellular TG-sensitive stores.
Another important question was whether TG and ATP act upon the same calcium store. To address this problem we challenged glioblastoma cells in Ca2+-free solution with 100 μM ATP followed by 100 nM TG. The application of ATP caused a Ca2+ transient which apparently depleted the Ca2+ store as subsequent application of TG did not trigger a response (36 cells from 2 preparations; Fig. 8C). Similarly, when we changed the sequence and depleted the Ca2+ store with TG subsequent application of ATP was non-effective (54 cells from 3 preparations). These observations prove that TG and ATP do indeed act upon the same Ca2+ store within glioblastoma cells.
Figure 8. Relation between Ca2+ store depletion and store-operated Ca2+ entry.
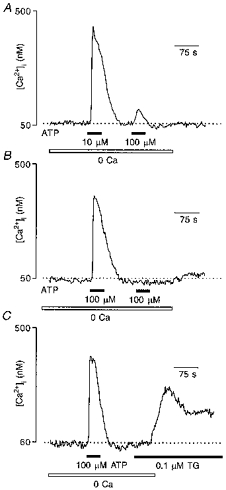
A, a cell was stimulated in Ca2+-free conditions with two different ATP concentrations, 10 μM and 100 μM, to assess the degree of store depletion produced by the lower dose. Application of 100 μM ATP after the 10 μM ATP application triggered a small [Ca2+]i transient indicating that stores were significantly depleted. Restoration of extracellular Ca2+ to 2 mM did not trigger any [Ca2+]i elevation. B, the same cell was stimulated twice more with 100 μM ATP in Ca2+-free extracellular solution. Despite the fact that the second application of ATP did not produce any measurable Ca2+ transient, restoration of Ca2+ to the bath still did not result in [Ca2+]i elevation. C, finally, the same cell was first challenged by 100 μM ATP (in Ca2+-free conditions) and then 0.1 μM TG was applied. Following stimulation with TG restoration of extracellular Ca2+ caused a large [Ca2+]i transient, indicating the activation of store-operated Ca2+ entry.
Although the peak of the ATP-induced [Ca2+]i transient was only slightly attenuated in Ca2+-free medium, the shape of the response was substantially affected. In the majority of cells, the [Ca2+]i transient induced in Ca2+-free medium was limited to the initial peak, with the plateau phase, which is frequently observed in Ca2+-containing solution, being greatly reduced (compare Figs 4A and 5A). Thus, the Ca2+ influx through the plasma membrane is significantly involved in shaping the ATP-induced [Ca2+]i signals in human glioma cells. This Ca2+ influx is likely to be associated with activation of store-operated Ca2+ entry following depletion of the intracellular stores. It is widely accepted that exhaustion of intracellular Ca2+ stores activates plasmalemmal Ca2+ channels which provide a pathway for Ca2+ influx (Penner et al. 1993; Berridge, 1995).
Figure 5. Activation of capacitative Ca2+ entry in glioblastoma cells.
A, a single cell was stimulated by 100 μM ATP in standard bath solution and in Ca2+-free extracellular medium. Restoration of extracellular Ca2+ caused a [Ca2+]i transient which slowly returned to baseline. B, mean response of fifty-one cells to a long-lasting ATP application. ATP triggered an initial peak which was followed by a plateau phase. Removal of extracellular Ca2+ eliminated the plateau phase; after restoration of Ca2+ into the extracellular medium complete recovery of the plateau was observed.
Several lines of evidence indicate that ATP triggers store-operated Ca2+ entry. First, after stimulating the cells with ATP in Ca2+-free solution, the restoration of extracellular Ca2+ caused a rapid increase of [Ca2+]i to a peak which was followed by [Ca2+]i decline to the resting level (Fig. 5A, right trace). Second, in cells exposed to 100 μM ATP for a period of several minutes, [Ca2+]i after the initial transient peak stabilized at the elevated level. Removal of extracellular Ca2+ resulted in a rapid decrease in [Ca2+]i and, vice versa, restoration of extracellular Ca2+ led to the recovery of the [Ca2+]i plateau (Fig. 5B). Third, the signal responsible for activation of Ca2+ influx was not dependent on the interval between ATP stimulation in Ca2+-free solution and restoration of extracellular Ca2+ (Fig. 6).
Figure 6. ATP-induced depletion of the intracellular Ca2+ store triggers a persistent signal associated with the activation of capacitative Ca2+ entry.
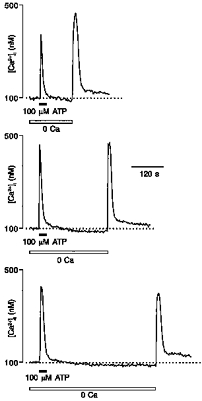
Glioma cells were stimulated with 100 μM ATP in the absence of extracellular Ca2+. Extracellular Ca2+ was then restored to 2 mM at different times after the end of ATP treatment as indicated. The results are typical of those for three independent experiments (97 cells). The [Ca2+]i rise in response to restoration of extracellular Ca2+ is unaffected by the time elapsed after the removal of ATP.
The activation of store-operated Ca2+ entry in human glioblastoma cells was characterized by a clear dissociation from Ca2+ release, similar to that demonstrated previously for other types of non-excitable cells (Parekh et al. 1997). To determine the dependence of the activation of store-operated Ca2+ entry on the concentration of ATP, cells were stimulated with ATP in Ca2+-free solution for 60 s and 3 min later, extracellular Ca2+ was restored. The amplitude of the [Ca2+]i peak following restoration of extracellular Ca2+ to 2 mM was plotted against the concentration of ATP (Fig. 7B). Store-operated Ca2+ entry was activated at much higher ATP concentrations than Ca2+ release. Despite the fact that ATP-induced [Ca2+]i release reached saturation at 10 μM ATP (when applied in Ca2+-free solution), the threshold for activation of store-operated Ca2+ entry was much higher at about 30 μM, saturation occurring at 300 μM ATP. Moreover, an increase in the amplitude of the store-operated entry-related [Ca2+]i transient reflected an increase in the probability of cells responding rather than an increase in the magnitude of the transient peak. At 30 μM ATP, only nineteen of 157 cells generated store-operated Ca2+ influx, at 100 μM this number had increased to 127 cells and at 300 μM almost all cells (154 of 157) developed Ca2+ transients in response to restoration of extracellular Ca2+. At the same time, the mean amplitude of the [Ca2+]i transient associated with store-operated Ca2+ entry when calculated only for responding cells did not differ significantly (Fig. 7C) being 73 ± 15 nM at 30 μM ATP (19 cells), 89 ± 97 nM at 100 μM (127 cells) and 96 ± 67 nM at 300 μM ATP (154 cells).
Figure 7. Dissociation between Ca2+ release and activation of capacitative Ca2+ entry.
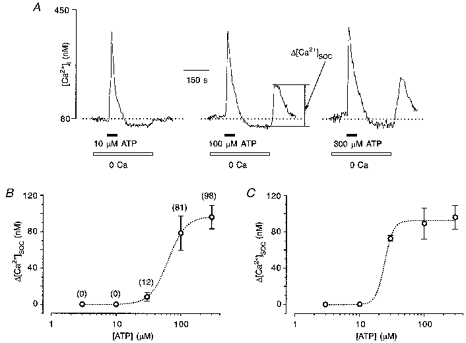
A, glioma cells were stimulated by different concentrations of ATP in the absence of extracellular Ca2+, extracellular Ca2+ was then restored to 2 mM. The amplitude of [Ca2+]i rise in response to Ca2+ restoration was determined as the amplitude of the capacitative Ca2+ entry-induced [Ca2+]i transient, Δ[Ca2+]SOC. The traces are representative for four independent experiments, 157 cells. B, from the experiments shown in A, the dose-response curve, relating ATP concentration to normalized Δ[Ca2+]SOC, was constructed. The apparent KD was 63 mM, and the Hill coefficient was 3.2. The numbers in parentheses indicate the percentage of cells which responded with [Ca2+]i transients resulting from store-operated Ca2+ entry. C, from the same experiments the mean values of Δ[Ca2+]SOC were calculated for responsive cells only. The dose-response curve has an apparent KD of 24 mM and a Hill coefficient of 6.5.
It became obvious that the degree of depletion of the intracellular Ca2+ store could not be directly assessed by simply measuring the amplitude of the ATP-induced Ca2+ transient. Thus we performed additional experiments in an attempt to ascertain how different ATP concentrations deplete the ER store of releasable Ca2+. Glioma cells in Ca2+-free solution were challenged with 10 μM ATP, followed shortly by a further application of 100 μM ATP. As demonstrated in Fig. 8A, 10 μM ATP significantly depleted intracellular Ca2+ stores because the subsequent application of 100 μM ATP induced only a small [Ca2+]i elevation. On average the amplitude of a [Ca2+]i transient evoked by 100 μM ATP was 9.5 ± 4 % (48 cells from 3 preparations) of that evoked by the conditioning application of 10 μM ATP.
Interestingly, these additional experiments revealed further important information regarding the relation between store depletion and activation of store-operated Ca2+ entry. Restoration of extracellular Ca2+ after the two applications of ATP (10 and 100 μM) resulted in a [Ca2+]i transient in approximately 70 % of cells (34 of 48 cells). We then concentrated on the ‘non-responding’ cells to determine the degree of store depletion by 100 μM ATP. In fact, when we challenged these cells with two further applications of 100 μM ATP in Ca2+-free conditions, the second of these did not produce a [Ca2+]i transient at all, indicating that the stores were depleted (Fig. 8B). But still restoration of extracellular Ca2+ did not result in a [Ca2+]i transient. Finally, one more 100 μM ATP application was made (under Ca2+-free conditions) followed by 0.1 μM TG. Application of TG per se did not induce measurable [Ca2+]i elevation, but the restoration of extracellular Ca2+ resulted in a clear [Ca2+]i elevation in all cells (Fig. 8C). Thus Ca2+ stores must be totally depleted to enable store-activated Ca2+ entry, near-maximal depletion is not effective.
Mitochondria sequester Ca2+ entering the cell via capacitative pathway
Mitochondria are known not only to be a major of source of cellular ATP but also to serve as a capacious intracellular Ca2+ pool. There is increasing evidence that mitochondrial Ca2+ stores play a dynamic role in shaping [Ca2+]i signals in many types of excitable and non-excitable cells (Werth & Thayer, 1994; Hehl et al. 1996; Babcock et al. 1997) and they are able to accumulate Ca2+ entering the cell through plasmalemmal channels. It has recently been shown that in T lymphocytes mitochondria effectively buffer the Ca2+ loads which result from the activation of store-operated Ca2+ entry (Hoth et al. 1997). To assess the possible involvement of the mitochondrial Ca2+ pool in buffering Ca2+ loads induced by ATP, we examined the ability of the mitochondrial uncoupler carbonyl cyanide chlorophenyl hydrazone (CCCP) to affect [Ca2+]i before and immediately after exposure of glioma cells to 100 μM ATP.
Application of 10 μM CCCP to unstimulated glioblastoma cells resulted in a small steady-state [Ca2+]i elevation (mean amplitude, 63 ± 13 nM; 5 experiments, 101 cells). This indicates the low amount of releasable Ca2+ stored in mitochondria under resting conditions. Washout of the ionophore resulted in a rapid restoration of [Ca2+]i to the resting level. In contrast, when CCCP was administered immediately after the end of ATP application, it evoked a much larger (mean amplitude, 137 ± 29 nM; 101 cells) and transient rise in [Ca2+]i (Fig. 9A). This demonstrates that the increase in [Ca2+]i induced by ATP was, in part, buffered by the mitochondrial pool, which thereby increased the releasable Ca2+ content of the mitochondrial store.
Figure 9. Mitochondria buffer Ca2+ loads which result from capacitative Ca2+ entry.
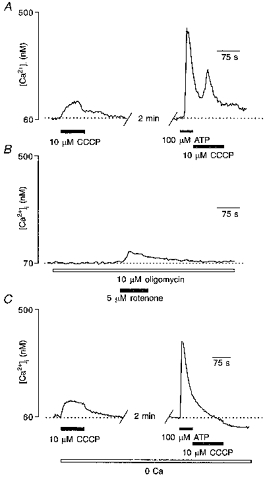
A, as shown in the left trace, application of 10 μM CCCP to the unstimulated cell triggered a small elevation in [Ca2+]i. In the same cell CCCP was applied immediately after 60 s stimulation with 100 μM ATP, triggering a large and transient elevation in [Ca2+]i. B, inhibition of ATP synthase by extracellular application of 10 μM oligomycin failed to affect [Ca2+]i. When 5 μM rotenone was added into the bath the cell responded with a small [Ca2+]i elevation. C, the same experiment as in A was repeated while incubating the cells in Ca2+-free extracellular medium. Note that CCCP did not affect [Ca2+]i when applied after cell stimulation with ATP.
As the effects on [Ca2+]i evoked by CCCP could have arisen not only from the dissipation of mitochondrial potential but also, more indirectly, from impairment of ATP production and subsequent impairment of ATP-dependent pumps, we challenged the glioblastoma cells with oligomycin, which is known to inhibit ATP synthase (Budd & Nicholls, 1996). Application of 10 μM oligomycin did not change [Ca2+]i in any cell tested (22 cells, 2 preparations, Fig. 9B;) but when 5 μM rotenone, which depolarizes the mitochondrial membrane, was added subsequently we observed a tiny [Ca2+]i elevation, similar to those induced by CCCP (mean amplitude, 46 ± 11 nM; 22 cells). Thus we may conclude that effects of CCCP on [Ca2+]i are not coupled with inhibition of ATP synthesis.
Surprisingly, when we repeated the experiment with CCCP in Ca2+-free solution, we found that CCCP applied immediately after ATP did not affect [Ca2+]i (Fig. 9C). Therefore, we conclude that mitochondria accumulate only the Ca2+ which enters the cell via the store-operated mechanism, and are unable to buffer Ca2+ that originates from the endoplasmic reticulum intracellular Ca2+ store.
DISCUSSION
Metabotropic purinoceptors in primary cultured human glioblastoma cells
In this report, we have characterized the dynamics and mechanisms of ATP-mediated [Ca2+]i signalling in human glioblastomas at the single-cell level. For the first time, we have demonstrated that human glioblastoma cells in primary culture express functional purinoceptors that control the generation of cytoplasmic Ca2+ signals. The predominant transducers of ATP-mediated Ca2+ signalling in glioma cells are metabotropic receptors of the P2Y family. The ATP-induced increases in [Ca2+]i are primarily due to Ca2+ release from intracellular stores, because the selective agonist for ionotropic P2X receptor, αβATP, failed to modulate [Ca2+]i, and because inhibition of intracellular Ca2+ accumulation by thapsigargin blocked ATP-induced [Ca2+]i responses. Direct electrophysiological measurements also failed to detect any measurable currents in response to extracellular application of ATP, once more supporting the idea that glioblastoma cells express exclusively metabotropic purinoceptors of P2Y family.
One surprising observation in the present study was the dramatic shift in the concentration dependency of ATP-induced [Ca2+]i elevation upon removal of Ca2+ ions from the extracellular medium. In Ca2+-free extracellular solution the concentration threshold for ATP-induced [Ca2+]i response was ∼10 times lower than that of ATP applied in standard bath solution. The reason for such a shift in the sensitivity of cells to ATP remains totally obscure. It is widely accepted that the activation of at least one purinoceptor subtype, the P2X7(Z) receptor, is crucially dependent upon the concentration of extracellular divalent cations, Mg2+ and Ca2+ (Cockroft & Gomperts, 1979; Ross et al. 1997). However, under our experimental conditions, we can completely rule out the possible involvement of P2X7(Z) receptors since the [Ca2+]i transients observed in Ca2+-free medium resulted entirely from intracellular Ca2+ release, and could not be linked to Ca2+ transport through the plasmalemmal pore. Another possible explanation could be the dramatic decrease in the activity of ectonucleotidases in Ca2+-deficient media resulting in deceleration of ATP metabolism. However, we think that such an explanation is highly unlikely - it is hard to believe that in standard solution ATP metabolism could have its effective concentration reduced by a factor of 10 since the cells are superfused with ATP-containing solutions at a relatively high speed. Neither are we aware of any previously published data demonstrating the possible influence of extracellular divalent cations on metabotropic P2Y purinoceptors. One possibility could be that the ATP-binding site of the receptor is modified in Ca2+-free solution, and this would lead to an increase in affinity of the receptor for ATP, however, at the moment we do not have experimental data that support such a possibility. None the less, the extracellular Ca2+ sensitivity of metabotropic purinoceptors seems to be an interesting phenomenon which clearly deserves further investigation.
Mechanisms of purinoceptor-mediated [Ca2+]i signalling: Ca2+ release and store-operated Ca2+ entry
The initial event in ATP-mediated Ca2+ signalling is associated with the activation of Ca2+ release from intracellular stores, via an InsP3-dependent pathway. The conclusion that Ca2+ release is the primary source for the increase in [Ca2+]i is supported by the following observations: (i) ATP-induced [Ca2+]i rise was preserved while stimulating cells in Ca2+-free extracellular medium, (ii) emptying of intracellular stores in the presence of thapsigargin completely abolished the ATP-triggered [Ca2+]i elevation and (iii) electrophysiology did not reveal any signs of ATP-mediated currents. However, the intracellular Ca2+ release did not appear to be the only mechanism involved in the generation of the ATP-mediated Ca2+ signal. The exhaustion of intracellular stores leads to activation of plasmalemmal Ca2+ influx that was mainly responsible for maintaining the plateau phase of the [Ca2+]i transient. We believe that the Ca2+ influx observed in glioma cells reflects the activation of store-operated calcium entry pathway, which is controlled by the filling state of intracellular Ca2+ stores. Several lines of evidence indicate that glioma cells possess such a mechanism. First, we can rule out the involvement of ATP-gated plasmalemmal channels since inhibition of ATP-driven Ca2+ release by thapsigargin also prevents the occurrence of Ca2+ influx in response to ATP application, which means that the initial Ca2+ release from the internal stores is a prerequisite for activation of the Ca2+ influx. Second, after cells were stimulated in Ca2+-free solution, which effectively depletes intracellular Ca2+ pools, restoration of Ca2+ ions to the external medium triggers a [Ca2+]i transient. This ‘restoration’[Ca2+]i transient was not affected by varying the time between ATP stimulation and restoration of external Ca2+, which indicates that the signal from the exhausted stores to the plasma membrane remains as long as the stores stay depleted. Finally, thapsigargin, which also depletes Ca2+ stores by means of unopposed leakage, also triggers a sustained [Ca2+]i elevation which is critically dependent on the extracellular Ca2+ ions, thus sharing the characteristic feature of capacitative Ca2+ influx.
Another important peculiarity of the store-operated Ca2+ influx pathway in glioma cells is the apparent dissociation from Ca2+ release. Full activation of the store-operated Ca2+ influx is observed when cells are stimulated with ATP concentrations significantly higher than necessary for producing Ca2+ release. The dependence of the degree of Ca2+ influx activation on the external ATP concentration is very steep, and moreover, an increase in [ATP]o increased the probability of activation of capacitative Ca2+ entry but had little effect on the amplitude of Ca2+-influx-associated [Ca2+]i elevation.
We observed that low ATP concentrations (10 μM) significantly deplete the ER Ca2+ store (see Fig. 8), as a subsequent application of 100 μM ATP triggers only a small [Ca2+]i elevation. Nevertheless that degree of depletion was not enough to activate store-operated Ca2+ entry. Moreover, in some cells even almost total exhaustion of the stores by 100 μM ATP did not trigger the Ca2+ influx pathway, only when the stores were completely depleted by TG did store-operated Ca2+ entry occur. Thus we can conclude that only the maximal depletion of the stores would induce activation of plasmalemmal Ca2+ entry.
A very similar dissociation between Ca2+ release and activation of store-operated Ca2+ entry has been demonstrated in rat basophilic leukaemia cells (Parekh et al. 1997). These results, however, were recently challenged by Hofer et al. (1998) who simultaneously monitored ICRAC and Ca2+ content in the ER lumen. Their experiments showed rather gradual relations between ER Ca2+ content and activation of the store-operated current. Thus the whole issue remains quite unclear.
In the present paper, for the first time, we report the phenomenon of Ca2+ release/Ca2+ entry dissociation for cells of neural origin. We demonstrated, that the ultimate condition for activation of the store-operated Ca2+ influx is the maximal depletion of the ER store, which could be achieved either with high doses of metabotropic agonist or using the non-specific agent thapsigargin. It is conceivable to speculate (in line with arguments presented by Parekh et al. 1997) that store-operated Ca2+ entry in human glioma cells is controlled by a specific set of intracellular stores with low sensitivity to InsP3, i.e. relatively high concentrations of InsP3 (which could be achieved only at high doses of the agonist) are required for activation of store-operated Ca2+ influx.
Thus, the shape of ATP-mediated Ca2+ signals in the cytoplasm of human glioblastoma cells is controlled by both intracellular Ca2+ release and store-operated Ca2+ entry. The latter is very important not only for the replenishment of intracellular Ca2+ pools, but also for creating the plateau phase of [Ca2+]i elevation in response to ATP. Long (10–20 min) exposure of glioma cells to ATP results in a [Ca2+]i response with a prominent plateau that is entirely determined by store-operated Ca2+ influx (see Fig. 5). Moreover, activation of the Ca2+ entry component could be achieved only when the cells were stimulated with high ATP concentrations, which could provide means for discriminating the strength of incoming stimuli.
Mitochondrial Ca2+ pool in glioma cells
We performed experiments to determine whether ATP-induced Ca2+ loads in glioma cells could be buffered by an additional intracellular Ca2+ pool associated with mitochondria. Mitochondrial Ca2+ buffering occurs through a uniporter which utilizes a huge negative potential across the inner mitochondrial membrane to translocate Ca2+ (Gunter & Pfeiffer, 1990; Gunter et al. 1994). In addition, mitochondria are able to release accumulated Ca2+ via Na+−Ca2+ exchange and possibly through Ca2+-permeable channels (Rizzuto et al. 1994). For many years, mitochondria were regarded as a low-affinity high-capacity Ca2+ buffering system which is activated only in pathological conditions associated with severe Ca2+ overload. However, recent experiments have demonstrated that mitochondria are able to buffer Ca2+ effectively at modest levels of [Ca2+]i (200–500 nM) and are capable of dynamically tuning [Ca2+]i transients in several types of cells (Werth & Thayer, 1994; Budd & Nicholls, 1996; Hehl et al. 1996; Wang & Thayer, 1996; Babcock et al. 1997; Hoth et al. 1997; Svichar et al. 1997).
To probe mitochondrial Ca2+ buffering in human glioma cells, we used the mitochondrial uncoupler, CCCP. In resting conditions, CCCP caused a minor steady-state elevation in [Ca2+]i, verifying that the mitochondria contained only a small amount of releasable Ca2+. We can rule out the possible involvement of ATP depletion, since inhibition of ATP synthase by oligomycin did not interfere with [Ca2+]i level. When CCCP was applied immediately after ATP (i.e. when [Ca2+]i was still elevated, 200–400 nM), a much larger, transient [Ca2+]i elevation resulted, indicating that the Ca2+ load that occurred in ATP-stimulated cells was partially buffered by mitochondria, thereby increasing the releasable Ca2+ content of mitochondrial pool. When we repeated the same experiment in Ca2+-free extracellular solution, CCCP applied during the recovery phase of the ATP-triggered [Ca2+]i transient did not result in [Ca2+]i elevation, in spite of the fact that peak amplitudes of [Ca2+]i transients in standard Ca2+-containing solution and Ca2+-free medium were not significantly different. Based on this observation, we concluded that in glioma cells, mitochondria are capable of distinguishing between routes of Ca2+ delivery into the cytoplasm: they buffer Ca2+ entering the cell through plasmalemma (via the store-operated pathway) but they are not effective in taking up Ca2+ ions released from the intracellular stores. How could such a peculiar discrimination occur? One possible explanation could lie in a non-homogeneous spatial distribution of mitochondria within glioma cells. It has been demonstrated that mitochondrial Ca2+ uptake occurs primarily when the sites of Ca2+ influx and Ca2+ uptake are situated in close proximity (Rizzuto et al. 1993; Hajnoczky et al. 1995). Provided that mitochondria are primarily located beneath the plasma membrane, they may readily sense Ca2+ entering through plasmalemmal channels, but be too far away from the ER Ca2+ release sites that are located deeper in the cell interior. Obviously such a prediction has to be proved by specially designed morphological experiments.
Nevertheless, the fact that store-operated Ca2+ entry results in an increase in mitochondrial Ca2+ could have important consequences for the function of glioma cells. It is known that an increase in mitochondrial Ca2+ concentration modulates production of ATP and is accompanied by increased levels of NADH and NADPH (Rizzuto et al. 1993; Hajnoczky et al. 1995); mitochondrial Ca2+ uptake also results in activation of pyruvate dehydrogenase, which is located in the mitochondrial matrix (Rutter et al. 1996). Therefore, an increase in mitochondrial [Ca2+] that accompanies the ATP-mediated [Ca2+]i rise could be regarded as an alternative signal transduction pathway which couples events occurring in the plasmalemma with cellular energy production.
Thus, based on the results of our observations we propose the following model describing the relations between intracellular Ca2+ stores and plasmalemmal Ca2+ entry pathway in tumour cells of glial origin (Fig. 10). The bulk of the Ca2+ underlying the generation of agonist-induced Ca2+ signals comes from ER calcium stores that are located deep within the cell and characterized by a high sensitivity to InsP3. When the cell is exposed to high concentrations of agonists another set of ER stores, possibly located beneath the plasma membrane and characterized by a relatively low sensitivity to InsP3, becomes affected. Depletion of these stores triggers plasmalemmal Ca2+ influx; Ca2+ ions delivered via this pathway are partially buffered by mitochondria, which are also located in the vicinity of Ca2+ entry sites. Obviously this model remains speculative and more experimental work is needed to prove it.
Figure 10. Possible relations between calcium stores and plasmalemmal store-operated calcium channels in glioblastoma cells.
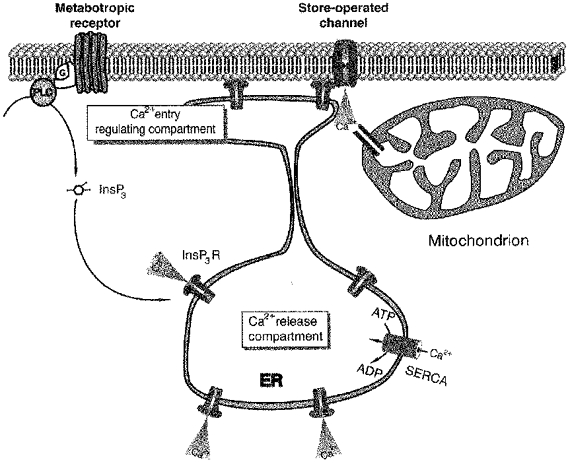
Activation of metabotropic receptors triggers Ca2+ release from the intracellular stores. On the basis of our results it is conceivable to speculate that glioma cells are endowed with two functionally distinct sets of InsP3-sensitive stores. The bulk of the Ca2+ comes from the ‘Ca2+ release compartment’ of the ER. Emptying of this compartment does not release an appropriate signal for activation of store-operated Ca2+ entry. The latter is triggered only while the other ER compartment (with presumably lower sensitivity to InsP3) is depleted; this part of the ER store (the ‘Ca2+ entry regulating compartment’) controls the store-operated Ca2+ influx. Some of the Ca2+ ions entering the cell through store-operated channels are accumulated by mitochondria, which might be located in the vicinity of Ca2+ entry sites.
Acknowledgments
This research was supported by Deutsche Forschungsgemeinschaft. The authors wish to thank Drs C. Stucky and E. Toescu for critical comments on the manuscript and G. Müller for excellent technical assistance.
References
- Babcock DF, Herrington J, Goodwin PC, Park YB, Hille B. Mitochondrial participation in the intracellular Ca2+ network. Journal of Cell Biology. 1997;136:833–844. doi: 10.1083/jcb.136.4.833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berridge MJ. Inositoltrisphosphate and calcium signalling. Nature. 1993;361:315–325. doi: 10.1038/361315a0. [DOI] [PubMed] [Google Scholar]
- Berridge MJ. Capacitative calcium entry. Biochemical Journal. 1995;312:1–12. doi: 10.1042/bj3120001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brismar T. Physiology of transformed glial cells. Glia. 1995;15:231–243. doi: 10.1002/glia.440150305. [DOI] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. Journal of Neurochemistry. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Cockroft S, Gomperts BD. ATP induces nucleotide permeability in rat mast cells. Nature. 1979;279:541–542. doi: 10.1038/279541a0. [DOI] [PubMed] [Google Scholar]
- Fredholm BB, Abbracchio MP, Burnstock G, Daly JW, Harden TK, Jacobson KA, Leff P, Williams M. Nomenclature and classification of purinoceptors. Pharmacological Reviews. 1994;46:143–156. [PMC free article] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescent properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Gunter TE, Gunter KK, Sheu S-S, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. American Journal of Physiology. 1994;267:C313–339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- Gunter TE, Pfeiffer DR. Mechanisms by which mitochondria transport calcium. American Journal of Physiology. 1990;258:C755–786. doi: 10.1152/ajpcell.1990.258.5.C755. [DOI] [PubMed] [Google Scholar]
- Hajnoczky G, Robb-Gaspers LD, Seitz MB, Thomas AP. Decoding of cytosolic calcium oscillations in the mitochondria. Cell. 1995;82:415–424. doi: 10.1016/0092-8674(95)90430-1. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Archiv. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hehl S, Golard A, Hille B. Involvement of mitochondria in intracellular calcium sequestration by rat gonadotropes. Cell Calcium. 1996;20:515–524. doi: 10.1016/s0143-4160(96)90094-9. [DOI] [PubMed] [Google Scholar]
- Hofer AM, Fasolato C, Pozzan T. Capacitative Ca2+ entry is closely linked to the filling state of internal Ca2+ stores: A study using simultaneous measurements of ICRAC and intraluminal [Ca2+] Journal of Cell Biology. 1998;140:325–334. doi: 10.1083/jcb.140.2.325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoth M, Fanger CM, Lewis RS. Mitochondrial regulation of store-operated calcium signaling in T lymphocytes. Journal of Cell Biology. 1997;137:633–648. doi: 10.1083/jcb.137.3.633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleihues P, Burger PC, Scheithauer BW. Histological Typing of Tumours of the Central Nervous System. New York: Springer; 1993. [Google Scholar]
- Labrakakis C, Patt S, Weydt P, Cervosnavarro J, Meyer R, Kettenmann H. Action potential generating cells in human glioblastomas. Journal of Neuropathology & Experimental Neurology. 1997;56:243–254. doi: 10.1097/00005072-199703000-00003. [DOI] [PubMed] [Google Scholar]
- North RA, Barnard EA. Nucleotide receptors. Current Opinion in Neurobiology. 1997;7:346–357. doi: 10.1016/s0959-4388(97)80062-1. [DOI] [PubMed] [Google Scholar]
- Parekh A, Fleig A, Penner R. The store-operated calcium current ICRAC: Nonlinear activation by InsP3 and dissociation from calcium release. Cell. 1997;89:973–980. doi: 10.1016/s0092-8674(00)80282-2. [DOI] [PubMed] [Google Scholar]
- Parekh AB, Penner R. Store depletion and calcium influx. Physiological Reviews. 1997;77:901–30. doi: 10.1152/physrev.1997.77.4.901. [DOI] [PubMed] [Google Scholar]
- Patt S, Labrakakis C, Bernstein M, Weydt P, Cervosnavarro J, Nisch G, Kettenmann H. Neuron like physiological properties of cells from human oligodendroglial tumors. Neuroscience. 1996;71:601–611. doi: 10.1016/0306-4522(95)00468-8. [DOI] [PubMed] [Google Scholar]
- Penner R, Fasolato C, Hoth M. Calcium influx and its control by calcium release. Current Opinion Neurobiology. 1993;3:368–374. doi: 10.1016/0959-4388(93)90130-q. [DOI] [PubMed] [Google Scholar]
- Rizzuto R, Bastianutto C, Brini M, Murgia M, Pozzan T. Mitochondrial Ca2+ homeostasis in intact cells. Journal of Cell Biology. 1994;126:1183–1194. doi: 10.1083/jcb.126.5.1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto R, Brini M, Murgia M, Pozzan T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighbouring mitochondria. Science. 1993;262:744–747. doi: 10.1126/science.8235595. [DOI] [PubMed] [Google Scholar]
- Ross PE, Ehring GR, Cahalan MD. Dynamics of ATP-induced calcium signaling in single mouse thymocytes. Journal of Cell Biology. 1997;138:987–998. doi: 10.1083/jcb.138.5.987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter GA, Burnett P, Rizzutto R, Brini M, Murgia M, Pozzan T, Tavare JM, Denton RM. Subcellular imaging of intramitochondrial Ca2+ with recombinant targeted aequorin: significance for the regulation of pyruvate dehydrogenase activity. Proceedings of the National Academy of Sciences of the USA. 1996;93:5489–5494. doi: 10.1073/pnas.93.11.5489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svichar N, Kostyuk P, Verkhratsky A. Mitochondria buffer Ca2+ entry but not intracellular Ca2+ release in mouse DRG neurones. Neuro Report. 1997;8:3929–3932. doi: 10.1097/00001756-199712220-00017. [DOI] [PubMed] [Google Scholar]
- Thastrup O, Cullen PJ, Drobak BK, Hanley MR, Dawson AP. Thapsigargin, a tumor promotor, discharges intracellular Ca2+ stores by specific inhibition of the endoplasmic reticulum Ca2+ ATPase. Proceedings of the National Academy of Sciences of the USA. 1990;87:2466–2470. doi: 10.1073/pnas.87.7.2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev Y, Shmigol A, Pronchuk N, Kostyuk P, Verkhrtasky A. Caffeine-induced calcium release from internal stores in rat sensory neurones. Neuroscience. 1993;57:845–859. doi: 10.1016/0306-4522(93)90029-f. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Kettenmann H. Calcium signalling in glial cells. Trends in Neurosciences. 1996;19:346–352. doi: 10.1016/0166-2236(96)10048-5. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A, Orkand RK, Kettenmann H. Glial calcium: Homeostasis and signalling function. Physiological Reviews. 1998;78:99–141. doi: 10.1152/physrev.1998.78.1.99. [DOI] [PubMed] [Google Scholar]
- Wang GJ, Thayer SA. Sequestration of glutamate-induced Ca2+ loads by mitochondria in cultured rat hippocampal neurons. Journal of Neurophysiology. 1996;76:1611–1621. doi: 10.1152/jn.1996.76.3.1611. [DOI] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurons. Journal of Neuroscience. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weydt P, Möller T, Labrakakis C, Patt S, Kettenmann H. Neuroligand-triggered calcium signaling in cultured human glioma cells. Neuroscience Letters. 1997;228:91–94. doi: 10.1016/s0304-3940(97)00366-2. [DOI] [PubMed] [Google Scholar]
- Zimmermann H. Signalling via ATP in the nervous system. Trends in Neurosciences. 1994;17:420–426. doi: 10.1016/0166-2236(94)90016-7. [DOI] [PubMed] [Google Scholar]