

Abstract
The goal of this study was to evaluate the buffer barrier hypothesis in an intact arterial smooth muscle. Specifically, we investigated the interrelationships between intracellular [Ca2+] ([Ca2+]i) homogeneity and sarcoplasmic reticulum function in swine carotid artery.
We measured focal changes in [Ca2+]i by exploiting the different characteristics of several [Ca2+]i indicators: (1) aequorin, which can detect focal increases in [Ca2+]i such as those that occur in the subplasmalemmal region ([Ca2+]pm); (2) fura-2, which is primarily a measure of mean cytoplasmic [Ca2+] ([Ca2+]c); and (3) force, which reflects increases in [Ca2+] near the contractile apparatus. We then estimated the relative degree of [Ca2+]i homogeneity with the aequorin/fura-2 ratio. Finally, we inhibited sarcoplasmic reticulum Ca2+ pumping with cyclopiazonic acid (CPA), an inhibitor of the sarco(endo)plasmic reticulum Ca2+-ATPase (SERCA).
We found that, after Ca2+ depletion, the sarcoplasmic reticulum could be partially reloaded with Ca2+ by manipulations that increased the aequorin signal relatively more than the fura-2 signal. Complete reloading required large increases in the fura-2 signal. These data suggest that increases in [Ca2+]pm (as measured with aequorin) can partially reload the sarcoplasmic reticulum, but complete reloading required increases in [Ca2+]c (as measured with fura-2). Reloading could be partially inhibited by 10 μM CPA, indicating that SERCA function was important for reloading.
In unstimulated arteries, 10 μM CPA increased the fura-2 signal without altering the aequorin signal, thereby decreasing the aequorin/fura-2 ratio. Removal of extracellular Ca2+ without CPA also reduced the aequorin/fura-2 ratio. These data suggest that resting cells have a [Ca2+] gradient with [Ca2+]pm > [Ca2+]c; this gradient is maintained by SERCA function.
CPA slowed the decline in the fura-2 signal observed when histamine stimulation was removed. This result is consistent with the concept of vectorial Ca2+ efflux in which Ca2+ pumping by SERCA reduces [Ca2+]c after stimulation.
Ca2+ depletion by prior treatment with 100 μM histamine and CPA transiently attenuated subsequent histamine-induced aequorin and fura-2 transients. The effect on contraction was smaller: a delay in contraction of approximately 10 s. These data suggest that histamine-induced Ca2+ release has at least a small role in the initial phase of contraction; however, other contractile mechanisms appear to be able to compensate for loss of Ca2+ release with only modest changes in contraction kinetics.
These data suggest that there is a complex interrelationship between smooth muscle sarcoplasmic reticulum function and [Ca2+] in at least two cytoplasmic compartments. [Ca2+]pm and [Ca2+]c can differentially regulate sarcoplasmic reticulum Ca2+ filling; and sarcoplasmic reticulum function regulates [Ca2+]pm and [Ca2+]c.
It is well accepted that contractile agonist stimulation of smooth muscle induces Ca2+ release from a non-mitochondrial intracellular store (reviewed in Rembold, 1996). This store has been identified as the sarcoplasmic reticulum. 1,4,5-Inositol trisphosphate (1,4,5-IP3) is proposed to be the primary second messenger responsible for Ca2+ release. The sarcoplasmic reticulum also contains a Ca2+ pump, termed the sarco(endo)plasmic reticulum ATPase (SERCA), which appears to be primarily responsible for Ca2+ uptake into the sarcoplasmic reticulum. These data suggest that smooth muscle contains the necessary proteins for agonist stimulated Ca2+ release. However, the relative importance of Ca2+ release in the resultant contraction has yet to be determined. Additionally, it is possible that the sarcoplasmic reticulum has role(s) in the regulation of intracellular [Ca2+] ([Ca2+]i) beyond agonist-induced Ca2+ release.
Smooth muscle sarcoplasmic reticulum is a complex three-dimensional tubular structure in the cytosol. Electron microscopy reveals the existence of a portion of the sarcoplasmic reticulum closely apposed near the plasma membrane (Devine et al. 1972). This region of close apposition may have a special role in regulation of [Ca2+]i because a number of Ca2+ transport proteins, such as the Na+-Ca2+ exchanger, are preferentially localized to this region (Villa et al. 1993; Moore et al. 1993). It is possible that Ca2+ flux through this region may be involved in Ca2+ influx or efflux. For example, Ca2+ influx across the plasma membrane may be important to refill the sarcoplasmic reticulum after Ca2+ release. Ca2+ could also flow in a reverse direction. When contractile stimuli are removed, the sarcoplasmic reticulum may pump Ca2+ into its lumen, transport it to near the plasma membrane, and release it into the subsarcolemmal cytoplasm so it can be exported into the extracellular space.
This specialized region of cytoplasm between the sarcoplasmic reticulum and the plasma membrane may have a different [Ca2+] than the remaining cytoplasm. In 1977, Van Breemen hypothesized that there are functional [Ca2+]i domains inside smooth muscle cells: (1) a contractile domain encompassing most of the cytoplasm (hereafter termed [Ca2+]c), and (2) a subplasmalemmal domain (hereafter termed [Ca2+]pm) (Van Breemen, 1977; Van Breemen et al. 1986). The latter domain includes the region where the sarcoplasmic reticulum is closely apposed to the plasma membrane. According to this hypothesis, the [Ca2+]pm may, at times, be higher than [Ca2+]i. He also hypothesized that higher [Ca2+]pm may have a role in refilling the sarcoplasmic reticulum with Ca2+. Subsequently other investigators, working in other cell types, have hypothesized the existence of subplasmalemmal Ca2+ domains (Nagai et al. 1989; Rembold, 1989; Takemura & Putney, 1989; Lederer et al. 1990; Augustine & Neher, 1992). Evidence for focal increases in [Ca2+]i was inferred by comparing fura-2-estimated [Ca2+]i with [Ca2+]pm as measured with calcium-activated K+ (KCa) channel activity (Stehno-Bittel & Sturek, 1992; Yoshikawa et al. 1996), synaptic vessel fusion (Smith & Augustine, 1988; Augustine & Neher, 1992; Brose et al. 1992), or Ca2+ dependent Cl− currents (Osipchuk et al. 1990) in isolated patch clamped cells. Unfortunately, measurement of [Ca2+]pm with fluorescent dyes has been difficult because (1) the region is very small and (2) the fluorescent dyes, per se, act as mobile Ca2+ buffers which attenuate and change the spatial characteristics of the Ca2+ domains (Neher & Augustine, 1992).
We recently developed a method to estimate whether cellular [Ca2+] is homogeneous (e.g. [Ca2+]pm=[Ca2+]c) or inhomogeneous (i.e. [Ca2+]pm > [Ca2+]c) in the cell. We measure [Ca2+]i with both aequorin and fura-2 (Rembold et al. 1995). This dual measurement allows estimation of relative [Ca2+]i homogeneity based on the characteristics of these two Ca2+ indicators (Yue & Wier, 1998). Aequorin is a Ca2+ sensitive protein that is very sensitive to increases in [Ca2+] that occur in small regions of the cell (e.g. [Ca2+]pm). In contrast, fura-2 is predominantly a measure of mean [Ca2+] (i.e. [Ca2+]c). The ratio of the observed aequorin and fura-2 signals is an index of the relative degree of [Ca2+]i homogeneity in intact swine carotid arterial tissues (Rembold et al. 1995). Increases in this ratio suggest a more inhomogeneous [Ca2+] distribution.
The goal of this study was to evaluate the physiological roles of [Ca2+]i homogeneity and sarcoplasmic reticulum function in the regulation of arterial smooth muscle [Ca2+]i and contraction. In this study we tested four hypotheses: (1) that increases in [Ca2+]pm were primarily responsible for refilling of the sarcoplasmic reticulum with Ca2+; (2) that the sarcoplasmic reticulum maintains a [Ca2+] gradient within unstimulated smooth muscle cells, specifically that basal levels of Ca2+ influx increase [Ca2+]pm more than [Ca2+]c in unstimulated smooth muscle; (3) that the sarcoplasmic reticulum is important in removal of Ca2+ from the centre of the cell; and (4) that Ca2+ release from the sarcoplasmic reticulum is crucial for the initial phase of agonist-induced contractions.
METHODS
Swine common carotid arteries were obtained from an abattoir (Smithfield Co., Smithfield, VA, USA), dissected, mounted for isometric stress measurement, and the optimal length for stress development determined (Rembold & Murphy, 1988). The physiological saline solution (PSS) for arterial tissues contained (mM): NaCl, 140; KCl, 4.7; Mops, 2; CaCl2, 1.6; MgCl2, 1.2; Na2HPO4, 1.2; EGTA, 0.02; D-glucose, 5.6; pH adjusted to 7.4 at 37°C.
Aequorin-estimated [Ca2+]
[Ca2+] was estimated in one set of tissues with the photoprotein aequorin loaded intracellularly by reversible hyperpermeabilization (Rembold & Murphy, 1988). The aequorin-derived light was collected with a photomultiplier tube and the photon count per second (L) was divided by an estimate of the total [aequorin] (Lmax). The logarithm of this ratio (logL/Lmax) depends on [Ca2+]. Light signals are reported as a change in logL/Lmax in which the resting logL/Lmax is subtracted from all subsequent logL/Lmax values. This normalization markedly decreased interexperimental variability and provided enhanced sensitivity to small changes in [Ca2+] (Gilbert et al. 1991). Aequorin light signals were calibrated in Ca2+-EGTA buffers at 37°C with [Mg2+]= 0.5 mM (Gilbert et al. 1991). We do not report estimated [Ca2+] with each experiment because assumptions introduce errors. For example, [Mg2+]i affects aequorin calibration substantially (Blinks et al. 1982). We present aequorin data as the change in logL/Lmax. See our prior publication (Rembold et al. 1995) for discussion of aequorin localization and focal aequorin depletion.
Fura-2-estimated intracellular [Ca2+]
[Ca2+] was estimated in a second set of intact swine carotid medial tissues with fura-2 loaded intracellularly by incubation in 20 μM of the acetylmethoxy ester of fura-2 (fura-2 AM) for 3–5 h at 37°C (Gilbert et al. 1991). The final loading solution was PSS containing 20 μM fura-2 AM and 250 μM neostigmine to inhibit breakdown of fura-2 AM by extracellular esterases. After loading, arterial strips were washed in PSS for 30–45 min before each experiment. Tissues were mounted isometrically and stretched to ∼1.05 of the optimal length for force development to decrease movement artifacts (Gilbert et al. 1991). The fura-2-loaded tissues were sequentially excited with 340 ± 5, 360 ± 5 and 380 ± 5 nm light with a rotating filter wheel system. The emission light was measured at 525 ± 25 mm with a photomultiplier tube. The fluorescence signals were electronically demultiplexed and the fura-2340, 360, and 380 fluorescence signals and the force signals were converted to digital signals and stored on a personal computer. After completing pharmacological manipulations, incubation in 5 mM MnCl2 lysed the cells, quenched fura-2 fluorescence and allowed measurement of background fluorescence subtracted from all fluorescence measurements. We do not report estimated [Ca2+]i with each experiment because assumptions introduce errors. For example, protein binding, extracellular leakage, and loading of fura-2 into organelles affects fura-2 calibration (Uto et al. 1991; Shuttleworth & Thompson, 1991). We present fura-2 data as a background-subtracted 340/380 ratio. See our prior publication (Rembold et al. 1995) for discussion of fura-2 localization and other artifacts of fura-2.
Estimation of [Ca2+]i homogeneity with the aequorin/fura-2 ratio
Aequorin and fura-2 detect [Ca2+] differently. Fura-2 has one Ca2+ binding site and alterations in [Ca2+] cause the fluorescence signal to vary over approximately one order of magnitude (Grynkiewicz et al. 1985). Aequorin has three Ca2+ binding sites (it is in the EF-hand superfamily of Ca2+ binding proteins) and changes in [Ca2+] regulate aequorin light emission over six orders of magnitude (Blinks et al. 1982). The slope of the aequorin calibration (∼2.5) is steeper than the slope of the fura-2 calibration (∼1). Therefore, absolute aequorin signals increase much more than fura-2 signals in response to given increases in [Ca2+]. If [Ca2+] is homogeneous, both aequorin and fura-2 give accurate representations of mean [Ca2+]; however, if [Ca2+] is not uniform, aequorin and fura-2 will report different values for mean [Ca2+].
To demonstrate this phenomenon, suppose there is a cell that has equally distributed aequorin and fura-2 in the cytoplasm. This hypothetical cell also has two regions with different [Ca2+]: (1) [Ca2+]c, the [Ca2+] in the centre of the cell, which equals 100 nM, and (2) [Ca2+]pm, the [Ca2+] near the plasma membrane, which equals 1000 nM. Within each region, [Ca2+] is uniform. Given these levels of [Ca2+] in each region, the resulting focal aequorin and fura-2 signal from each region can be predicted. The sum of the focal signals resulting from both regions can be calculated depending on the relative volume of the two regions. If the volume of the subplasmalemmal region is 10 % of the cell volume, then the measured aequorin signal would predict a mean [Ca2+] of 522 nM, the measured fura-2 signal would predict a mean [Ca2+] of 166 nM, and the actual mean [Ca2+]i would be 190 nM. Under these conditions, neither indicator was truly representative: aequorin overestimated and fura-2 underestimated true mean [Ca2+]i, a reflection of the non-linearity of the calibration curves of both indicators.
We can exploit the different predictions of mean global [Ca2+]i observed with aequorin and fura-2 as a measure of [Ca2+]i homogeneity. The ratio of the two Ca2+ indicators (aequorin/fura-2) depends on the relative volume of the two regions and the [Ca2+] within each region. In this study, we calculated the ratio of the aequorin signal to fura-2 signal as a qualitative and relative index of [Ca2+]i homogeneity. Unfortunately, we cannot convert calculated ratios from experiments into estimates of both [Ca2+] and the relative volume of these two regions. Calculation of both would require another measure of homogeneity. Furthermore, real cells will have varying levels of [Ca2+]i in multiple, not just two, regions. See our prior publication for further discussion of these calculations and predictions (Rembold et al. 1995).
The aequorin/fura-2 ratio formula was empirically derived (Rembold et al. 1995). We made the assumption that [Ca2+]i was the most homogeneous when the tissue was either (1) bathed in PSS with no extracellular Ca2+ or (2) maximally stimulated with histamine in the presence of extracellular Ca2+. In the absence of extracellular Ca2+ there would be no Ca2+ influx, and therefore possibilities for focal increases in Ca2+ would be minimized at a low mean [Ca2+]i. During sustained histamine stimulation, activation (i.e. myosin phosphorylation and force) is maximal despite modest increases in [Ca2+]i. In typical experiments, we find the fura-2 signal in zero Ca2+ is ∼1.0 and it increases to ∼2.2 with 10 min of maximal histamine stimulation. Under the same conditions, the aequorin signal in zero Ca2+ is ∼-0.2 and increases to a sustained value of ∼0.4. Given these values, we calculated the aequorin/fura-2 ratio from mean aequorin and fura-2 data in the top two panels with the following formula:
The resulting corrected aequorin/fura-2 ratio was 1.0 both in zero Ca2+ and with maximal histamine stimulation. Increases in this ratio represent a relatively less homogeneous Ca2+ distribution. Supporting our original assumptions was the finding that observed aequorin/fura-2 ratios never significantly decreased below 1.0. In the presence of homogeneous [Ca2+]i, the calculated aequorin/fura-2 ratio remains relatively independent of [Ca2+]i at [Ca2+]i above 50 nM. Below 50 nM [Ca2+]i the aequorin/fura-2 ratio depended on [Ca2+]i (specifically it decreased with lower [Ca2+]). Prior studies revealed that histamine or 20–40 mM [K+]o did not alter the aequorin/fura-2 ratio, suggesting that stimuli that primarily increase global [Ca2+]i do not alter the aequorin/fura-2 ratio (Rembold et al. 1995). Prior studies also revealed that loading swine carotid artery with both aequorin and fura-2 did not alter the aequorin or fura-2 signal in response to histamine or high [K+]o (Gilbert et al. 1991).
Fura-2-estimated Mn2+ influx
Ca2+ influx was estimated in a third set of intact swine carotid medial tissues loaded with fura-2 as above. The 360 nm fluorescence was collected and normalized such that fluorescence before addition of Mn2+ to the bathing solution was 1.0 and the minimal fluorescence after lysis with 5 mM MnCl2 was 0.0. Mn2+ influx rates were calculated from the slope of the linear regression line of the 360 nm signal measured at 1 s intervals from 10 to 130 s after addition of 0.5 mM Mn2+ (Chen & Rembold, 1992).
Chemicals. Aequorin was obtained from John Blinks (Friday Harbor Laboratories, Friday Harbor, WA, USA). Fura-2 AM was obtained from Molecular Probes. Histamine and other chemicals were obtained from Sigma.
RESULTS
Mechanisms for refilling of the sarcoplasmic reticulum with Ca2+
Our first goal was to evaluate the sources of Ca2+ responsible for Ca2+ refilling of the sarcoplasmic reticulum after Ca2+ depletion. We hypothesized that increases in [Ca2+]pm were necessary for refilling of the sarcoplasmic reticulum with Ca2+. Figure 1 shows the basic protocol. At 10 min, extracellular Ca2+ was nominally removed by changing the bathing solution to a solution with no added Ca2+. This solution has an extracellular [Ca2+] of approximately 10 μM caused by Ca2+ contamination from the glassware and the other salts in the physiological saline solution. Nominal removal of extracellular Ca2+ decreased both the aequorin and fura-2 signals and the aequorin/fura-2 ratio. This decrease in the aequorin/fura-2 ratio is consistent with development of a more homogeneous Ca2+ distribution. At 20 min, the tissues were stimulated with 100 μM histamine for 5 min. This induced a large contraction and large increases in the aequorin and fura-2 signals indicating a Ca2+ release from the intracellular store. There was an increase in the aequorin/fura-2 ratio consistent with our prior report that histamine stimulation in the absence of extracellular Ca2+ induced an inhomogeneous increase in [Ca2+]i (Rembold et al. 1995). At 30 min, the tissues were restimulated with 100 μM histamine for 15 min. This induced a modest contraction and a smaller increase in the aequorin and fura-2 signal consistent with the prior partial release of Ca2+ from the sarcoplasmic reticulum. At 45 min, histamine was washed out repeatedly. At 60 min, extracellular Ca2+ was restored to 1.6 mM for 10 min. Restoration of extracellular Ca2+ was associated with a large increase in the aequorin signal and the aequorin/fura-2 ratio. There was a smaller increase in the fura-2 signal and force. These data suggest that the increase in [Ca2+]i occurred only in a focal region of the cell, probably the subplasmalemmal space. After removal of extracellular Ca2+, the tissues were for a third time stimulated with 100 μM histamine at 80 min, inducing a contraction and an increase in the aequorin and fura-2 signals. This indicates that the 10 min treatment with extracellular Ca2+ partially refilled the sarcoplasmic reticulum with Ca2+ allowing subsequent histamine-induced Ca2+ release.
Figure 1. Demonstration of the protocol for studying the mechanism for refilling of the sarcoplasmic reticulum with Ca2+.
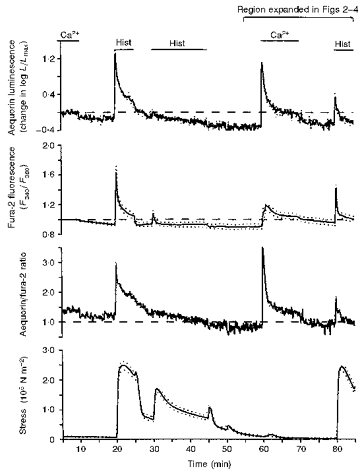
Time courses are shown for aequorin-estimated [Ca2+]i (change in logL/Lmax, top panel), fura-2-estimated [Ca2+]i (background corrected 340/380 fluorescence ratio, second panel), the aequorin/fura-2 ratio (third panel), and active stress (× 105 N m−2, bottom panel). Data are from for swine carotid medial tissues (n= 4–6). Extracellular Ca2+ was nominally removed at 10 min by changing the bathing solution to a solution with no added Ca2+. At 20 min, some of the tissues were treated with 10 μM CPA (e.g. Fig. 4, not shown in this experiment), which was then present in all subsequent solutions and other tissues were not treated with CPA. At 20 min, the tissues were stimulated with 100 μM histamine (Hist) for 5 min and the histamine was then washed out. At 30 min, the tissues were stimulated with 100 μM histamine for 15 min and the histamine was then washed out. At 60 min, extracellular Ca2+ was restored to the tissues for 10 min. In some experiments this solution contained other activating agents. At 70 min, extracellular Ca2+ was removed. At 80 min, the tissues were stimulated with 100 μM histamine. The data collected from 55 to 85 min are expanded in the subsequent Figs 2–4. Data were collected at 1 s intervals and averaged over 10 s for plotting. Mean data values are shown as a continuous line and the dotted lines represent ± 1 s.e.m. Only stress from the fura-2 experiments are shown for clarity (the aequorin stress data were similar).
To evaluate the sources of Ca2+ responsible for Ca2+ refilling of the sarcoplasmic reticulum, we altered the conditions present during restoration of extracellular Ca2+. The data in Figs 2–4 show only the end of the protocol described in Fig. 1 (specifically the data from 55 to 85 min in Fig. 1). All tissues were treated identically with the exception that the Ca2+ restoration solution, present from 60 to 70 min in the protocol, was varied.
Figure 2. Demonstration that histamine inhibited and high [K+]o enhanced refilling of the sarcoplasmic reticulum with Ca2+.
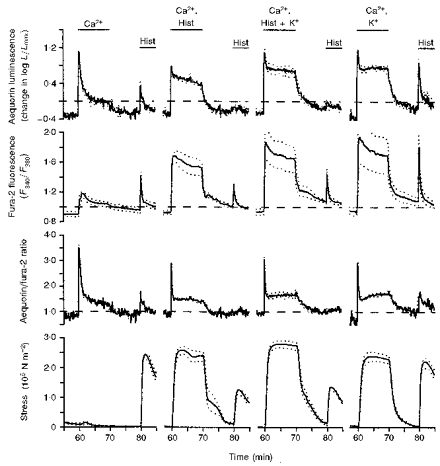
Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are from four sets of swine carotid medial tissues (n= 4−6 for each). All tissues had been treated with low extracellular Ca2+ and two histamine stimuli as shown in Fig. 1. Each tracing starts at the 57 min time point on Fig. 1, and shows the response to adding back extracellular Ca2+ to 1.6 mM at 60 min, then removal of extracellular Ca2+ at 70 min, and stimulation with 100 μM histamine (Hist) at 80 min. The first set had Ca2+ restored in a normal physiological saline solution (left panel, same data as in Fig. 1). The second set had Ca2+ restored in the presence of 100 μM histamine (second from left panel). The third set had Ca2+ restored in the presence of 100 μM histamine and 109 mM [K+]o (second from right panel). The fourth set had Ca2+ restored in the presence of 109 mM [K+]o (right panel). Data are plotted as in Fig. 1.
Figure 4. Demonstration that the SERCA inhibitor cyclopiazonic acid (CPA) inhibited choline+-induced refilling of the sarcoplasmic reticulum with Ca2+ and that CPA increased the fura-2 signal and contractile force during Ca2+ restoration.
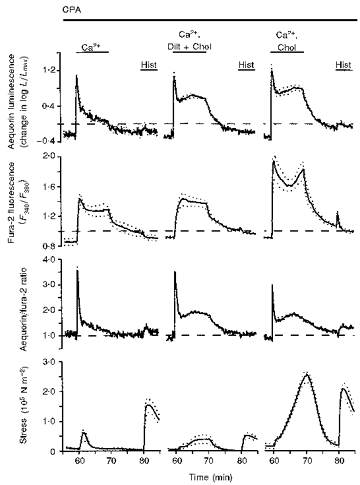
Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are from three sets of swine carotid medial tissues (n= 4–6 for each). All tissues had been treated with low extracellular Ca2+ and two histamine stimuli as described in Fig. 2. Additionally, 10 μM cyclopiazonic acid (CPA) was added prior to the first histamine stimulation (at 20 min in Fig. 1). This corresponds to 37 min prior to each recording in the current figure. CPA was present throughout the remaining protocol (from 20 to 85 min). The first set had Ca2+ restored in a normal physiological saline solution (left panel). The second set had Ca2+ restored in the presence of 10 μM diltiazem and 140 mM choline (substituted isosmotically for Na+, centre panel). The third set had Ca2+ restored in the presence of 140 mM choline (substituted isosmotically for Na+, right panel). Data are plotted as in Fig. 1.
First, we compared tissues that had Ca2+ restored with (1) no other treatment (i.e. the control shown in Fig. 1), (2) 100 μM histamine, (3) 100 μM histamine plus 109 mM [K+]o, and (4) 109 mM [K+]o without histamine. The presence of histamine during Ca2+ restoration inhibited Ca2+ refilling of the sarcoplasmic reticulum (Fig. 2, second from the left panel). During Ca2+ restoration, 100 μM histamine induced large increases in force and the aequorin and fura-2 signals. After removal of extracellular Ca2+, repeat histamine stimulation induced smaller increases in force and the aequorin signal than those observed when Ca2+ restoration occurred in the absence of histamine (the control is the left panel in Fig. 2). The fura-2 signal was modestly decreased. Ca2+ restoration in the presence of both 100 μM histamine and 109 mM [K+]o induced force, aequorin, and fura-2 signals similar to those observed with Ca2+ restoration in the presence of histamine alone (Fig. 2, second from the right panel). This suggests that histamine, rather than the high [Ca2+] during refilling, was responsible for the decreased refilling of the sarcoplasmic reticulum.
High [K+]o alone during Ca2+ restoration enhanced Ca2+ refilling of the sarcoplasmic reticulum. During Ca2+ restoration, 109 mM [K+]o alone induced large increases in force and the aequorin and fura-2 signals similar to those observed with histamine or histamine plus 109 mM [K+]o (Fig. 2, right panel). After removal of extracellular Ca2+, repeat histamine stimulation induced larger increases in the aequorin and fura-2 signals and in force than those observed when Ca2+ restoration occurred in the presence of histamine (either with or without high [K+]o). The aequorin and fura-2 signals observed with histamine stimulation after high [K+]o Ca2+ restoration were both larger than the control response. These data suggest that the form of stimulation, rather than high [Ca2+]per se, determined sarcoplasmic reticulum Ca2+ refilling. Histamine, in spite of producing higher [Ca2+], inhibited sarcoplasmic reticulum Ca2+ refilling. One potential explanation is that histamine-dependent increases in [1,4,5-IP3] could prevent refilling by continued Ca2+ release into the cytosol through open IP3 receptor channels. Another possibility is that the histamine receptor was desensitized by prior histamine stimulation.
We recently found a protocol that increases the aequorin signal to high levels with only modest increases in the fura-2 signal or force. This is consistent with the protocol inducing a focal increase in subplasmalemmal [Ca2+] ([Ca2+]pm) without a large increase in central [Ca2+] ([Ca2+]c). The experimental protocol involves blocking L-type Ca2+ channels with 10 μM diltiazem and substituting Na+ with choline+ to increase [Ca2+]i primarily via inhibition of Na+-Ca2+ exchange (Rembold et al. 1992). We compared tissues that had Ca2+ restored with (1) no other treatment (the control from Fig. 1), (2) diltiazem plus choline+ substitution for Na+, and (3) choline+ substitution for Na+ without diltiazem. As previously reported (Van Riper et al. 1996), diltiazem plus choline+ induced a large increase in the aequorin signal with only modest increases in force and the fura-2 signals (Fig. 3, centre panel). This produced a large increase in the aequorin/fura-2 ratio indicating a primarily focal increase in [Ca2+]i. After removal of extracellular Ca2+, histamine stimulation induced a smaller contraction than, but similar increases in the aequorin and fura-2 signals to, those observed in the control Ca2+ restoration. These data suggest that the focal increase in [Ca2+]i in the subplasmalemmal region did not enhance sarcoplasmic reticulum Ca2+ refilling. Ca2+ restoration in the presence of choline+ alone induced large increases in both the aequorin and fura-2 signals (Fig. 3, right panel). After removal of extracellular Ca2+, repeat histamine stimulation induced a larger contraction and larger increases in the aequorin and fura-2 signals than those observed in either the control or diltiazem-choline+ Ca2+ restoration. These data suggest that higher [Ca2+] in the central cytoplasm (i.e. induced by either choline+ or high [K+]o) was required to enhance Ca2+ refilling. It is probable that increases in [Ca2+]pm are an important mechanism for partially refilling the sarcoplasmic reticulum as stated in the first hypothesis. However, complete filling of the sarcoplasmic reticulum required increases in [Ca2+]c.
Figure 3. Demonstration that reversal of Na+-Ca2+ exchange with choline+ increased the fura-2 signal and enhanced refilling of the sarcoplasmic reticulum with Ca2+; both were inhibited by the L-type Ca2+ channel blocker diltiazem (Dilt).
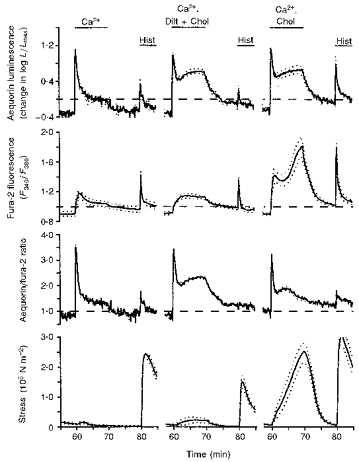
Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are from three sets of swine carotid medial tissues (n= 4–6 for each). All tissues had been treated with low extracellular Ca2+ and two histamine stimuli as described in Fig. 2. The first set had Ca2+ restored in a normal physiological saline solution (left panel, same data as in Fig. 2, left panel). The second set had Ca2+ restored in the presence of 10 μM diltiazem and 140 mM choline (substituted isosmotically for Na+, centre panel). The third set had Ca2+ restored in the presence of 140 mM choline (Chol; substituted isosmotically for Na+, right panel). Data are plotted as in Fig. 1.
Next, we evaluated the role of SERCA, the Ca2+ pump located in the sarcoplasmic reticulum membrane, in refilling of the sarcoplasmic reticulum with Ca2+. Tissues were treated as in Fig. 3 with the exception that cyclopiazonic acid (CPA), a water soluble specific inhibitor of SERCA, was added with the first addition of histamine and was present throughout the remaining protocol (i.e. from 20 to 85 min in Fig. 1). Restoration of extracellular Ca2+ alone was associated with large increases in the aequorin signal and the aequorin/ fura-2 ratio (Fig. 4, left panel). There was a larger increase in the fura-2 signal and force than that observed in the absence of CPA (cf. left panels in Figs 3 and 4). After removal of extracellular Ca2+, stimulation with 100 μM histamine induced a small contraction associated with no change in the aequorin or fura-2 signals. This indicates that 10 μM CPA prevented sarcoplasmic reticulum Ca2+ refilling during the 10 min exposure to 1.6 mM extracellular Ca2+. The contraction observed in the absence of measurable changes in [Ca2+] may result from histamine-induced increases in the [Ca2+]i sensitivity of myosin light chain phosphorylation at basal [Ca2+]i levels (Rembold & Murphy, 1988). These data suggest that the modest increases in [Ca2+]pm observed with Ca2+ restoration alone (Fig. 3, left panel) were sufficient to partially reload the sarcoplasmic reticulum with Ca2+. This process was inhibited by blocking SERCA with 10 μM CPA (Fig. 4, left panel).
Restoration of extracellular Ca2+ in the presence of CPA, diltiazem and choline+ induced a large increase in the aequorin signal with only modest increases in force and the fura-2 signals similar to those observed with diltiazem and choline+ alone. After removal of extracellular Ca2+, histamine stimulation induced a smaller contraction and no change in the aequorin or fura-2 signals, indicating that CPA inhibited Ca2+ refilling even with high [Ca2+]pm (the small contraction may represent increases in Ca2+ sensitivity). Ca2+ restoration in the presence of CPA and choline+ induced biphasic increases in both the aequorin and fura-2 signals. The initial increase in the fura-2 signal was larger than that observed with choline+ alone. Intriguingly, only the second phase of the increase in fura-2 signal was associated with a contraction. Since the aequorin signal was also high, the initial increase in the fura-2 signal may represent a very high increase in [Ca2+]pm that is detected by both fura-2 and aequorin.
After removal of extracellular Ca2+, repeat histamine stimulation induced a large contraction and small increases in the aequorin and fura-2 signals. These were smaller than observed in the absence of CPA, but suggest that either the CPA did not totally inhibit SERCA or that there are other, CPA insensitive mechanisms responsible for Ca2+ uptake into the sarcoplasmic reticulum at high [Ca2+].
Role of the sarcoplasmic reticulum in regulating [Ca2+]i homogeneity
Our second hypothesis was that sarcoplasmic reticulum Ca2+ handling maintains a [Ca2+] gradient within unstimulated smooth muscle cells. We evaluated this hypothesis with CPA. In Fig. 4, restoration of extracellular Ca2+ in the presence of CPA was associated with a larger increase in force and the fura-2 signal than that observed in the absence of CPA (Fig. 3). These data suggest that Ca2+ uptake by the sarcoplasmic reticulum was partially responsible for preventing Ca2+ influx from reaching the central cytoplasm where it could induce a contraction (this is the ‘buffer barrier’ hypothesis). These data suggest a role for the sarcoplasmic reticulum in maintaining a [Ca2+] gradient. We tested this in unstimulated tissues. In the presence of 1.6 mM extracellular Ca2+, addition of 10 μM CPA increased the fura-2 signal (Fig. 5), consistent with prior reports (Abe et al. 1996). However, CPA did not induce a significant change in the aequorin signal or contractile stress. As a result the aequorin/fura-2 ratio decreased, suggesting that CPA induced a more homogeneous [Ca2+]i distribution. These data are consistent with the hypothesis that activity of SERCA is at least partially responsible for maintenance of inhomogeneous [Ca2+]i distribution in unstimulated smooth muscle.
Figure 5. Demonstration that inhibition of SERCA increased the fura-2 signal without altering the aequorin signal; this suggests that SERCA function was responsible for maintaining an inhomogeneous [Ca2+]i distribution in resting smooth muscle (i.e. maintaining [Ca2+]pm > [Ca2+]c).
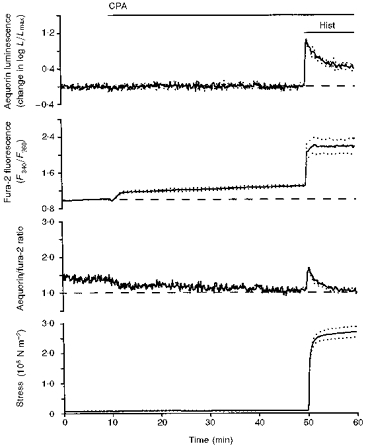
Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are from swine carotid medial tissues (n= 4–6). In the presence of 1.6 mM extracellular Ca2+, tissues were first exposed to 10 μM cyclopiazonic acid (CPA) at 10 min and then 100 μM histamine was added at 50 min. Data are plotted as in Fig. 1. The maintenance of maximal contractile stress at peak values despite decreasing aequorin-estimated [Ca2+] (cf. 51 vs. 60 min) is a manifestation of the latch phenomenon (Rembold & Murphy, 1988).
Subsequent addition of 100 μM histamine induced large increases in force and the aequorin and fura-2 signals. The aequorin/fura-2 ratio transiently increased and then decreased back to prior values.
A part of the second hypothesis was that basal levels of Ca2+ influx are responsible for elevated [Ca2+]pm. We further evaluated the second hypothesis by studying the response of carotid artery to removal of extracellular Ca2+ with or without CPA to inhibit SERCA. The effect of extracellular Ca2+ removal with or without cyclopiazonic acid is shown in Fig. 6. Nominal removal of extracellular Ca2+ reduced the aequorin signal relatively more than the fura-2 signal (Fig. 6, filled circles; also see Rembold et al. 1995). This resulted in a decrease in the aequorin/fura-2 ratio suggesting that removal of extracellular Ca2+ induced a more homogeneous [Ca2+]i distribution. These data are consistent with the second hypothesis that Ca2+ influx is responsible for maintaining the Ca2+ inhomogeneity in unstimulated smooth muscle. Addition of 10 μM CPA 1 min after nominal removal of extracellular Ca2+ induced a significant increase in the fura-2 signal with little change in the aequorin signal. The aequorin signal was similar to the aequorin signal observed with removal of extracellular Ca2+ without CPA. Addition of CPA transiently decreased the aequorin/fura-2 ratio to values lower than that observed with only removal of extracellular Ca2+. These data are consistent with the hypothesis that the sarcoplasmic reticulum continually ‘leaks’ Ca2+ into the cytosol. Inhibition of SERCA can ‘uncover’ the Ca2+ leak by preventing reuptake of the leaked Ca2+ back into the sarcoplasmic reticulum.
Figure 6. Demonstration that removal of extracellular [Ca2+] reduced the aequorin signal and reduces [Ca2+]i inhomogeneity; this suggests that Ca2+ influx was responsible for [Ca2+]i inhomogeneity in resting smooth muscle.
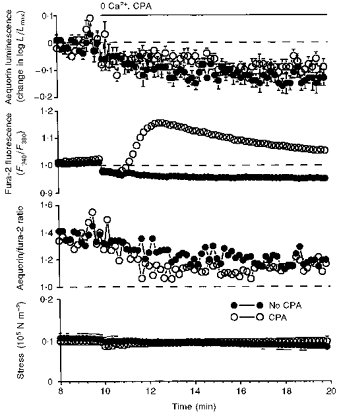
The data show the response to removal of extracellular Ca2+ with and without CPA. Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are means ±s.e.m. from two sets of swine carotid medial tissues (n= 4–6 for each). Extracellular Ca2+ was nominally removed at 10 min by changing the bathing solution to a solution with no added Ca2+. At 10.5 min, one set of the tissues was treated with 10 μM CPA (○), and the other set of tissues was not treated with CPA (•). Data were collected at 1 s intervals and averaged over 10 s for plotting. Symbols without error bars reflect errors less than the size of the symbol. Only stress from the fura-2 experiments is shown for clarity (the aequorin stress data were similar).
Our third hypothesis was that sarcoplasmic reticulum Ca2+ handling was important in removal of Ca2+ from the centre of the cell upon removal of stimulation (i.e. vectorial Ca2+ efflux). The response to histamine stimulation with or without CPA in the absence of extracellular Ca2+ is shown in Fig. 7. In both the absence and presence of CPA, 100 μM histamine increased both the aequorin and fura-2 signals and induced a contraction. There was a transient increase in the aequorin/fura-2 ratio, suggesting the Ca2+ release was relatively inhomogeneous. The responses were similar in both the presence and absence of CPA, suggesting that (1) the 10 min CPA treatment had not substantially depleted the sarcoplasmic reticulum of Ca2+ and (2) the focal Ca2+ release was not significantly modified by CPA-inhibitable SERCA function. Upon washout of histamine, the fura-2 signal from the CPA-treated tissues decreased more slowly than the fura-2 signal from the tissues not treated with CPA. The aequorin signal and contractile stress decreased with a similar time course regardless of the presence of CPA. This result suggests that sarcoplasmic reticulum Ca2+ handling is important in reducing [Ca2+] in the central cytoplasm (which is best measured with fura-2). Subplasmalemmal Ca2+, as measured with aequorin, decreased at a similar rate regardless of the presence of CPA. This is consistent with the hypothesis that Ca2+ can be removed from the subplasmalemmal region by the action of plasma membrane Ca2+ pumps and Na+-Ca2+ exchange which are not inhibited by CPA.
Figure 7. Demonstration that SERCA function was important in vectorial Ca2+ efflux; after washout of histamine in the absence of extracellular Ca2+ (25 min), cyclopiazonic acid slowed the decline in fura-2-estimated [Ca2+]c.
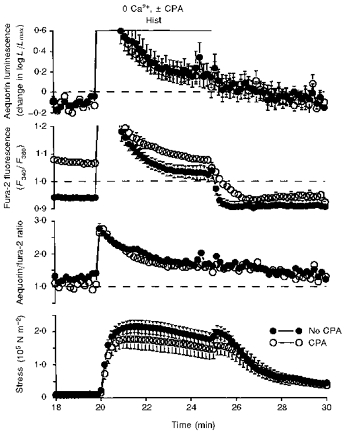
The data show the response to histamine stimulation with and without CPA in the absence of extracellular Ca2+. Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are from two sets of swine carotid medial tissues (n= 4–6 for each). One set of tissues was treated with 10 μM CPA (○), and the other set of tissues was not treated with CPA (○). The data are a continuation of the experiment shown in Fig. 6. At 10 min, extracellular Ca2+ was nominally removed and, at 10.5 min, the first set of tissues was incubated in 10 μM CPA, which was then present for the remaining protocol. At 20 min, the tissues were stimulated with 100 μM histamine, and the histamine washed out at 25 min. Data were collected and plotted as in Fig. 6.
Our fourth hypothesis was that Ca2+ release from the sarcoplasmic reticulum is crucial for the initial phase of agonist-induced contractions. Our goal was to evaluate this hypothesis in the presence of extracellular Ca2+. Therefore, we developed a protocol in which the tissues had varying amounts of Ca2+ in the sarcoplasmic reticulum. We accomplished this by removing extracellular Ca2+ and then stimulating with varying [histamine] to variably release Ca2+ in the presence of CPA to prevent Ca2+ reuptake into the sarcoplasmic reticulum. Controls without CPA were also included. We performed an abbreviated protocol similar to Fig. 1. At 10 min, extracellular Ca2+ was nominally removed. At this point, some of the tissues were treated with 10 μM CPA, which was then present in all subsequent solutions. At 20 min, the tissues were either (1) stimulated with 100 μM histamine to induce a large release of Ca2+ from the intracellular store, (2) stimulated with 3 μM histamine to induce a more modest Ca2+ release, or (3) not stimulated to minimize Ca2+ release. At 25 min, the histamine was washed out. At 40 min, extracellular Ca2+ was restored to all tissues (see Rembold et al. 1995) for a demonstration of this type of experiment). Finally, at 50 min, all tissues were stimulated with a maximally effective concentration of histamine (100 μM). Figure 8 shows the response to the final histamine stimulation depending on prior treatment. There was no significant difference in force or in the aequorin or fura-2 signal during the sustained phase of the contraction (5–10 min after histamine stimulation, data not shown). The only differences were in the first minute after stimulation. The only group showing a delay in force generation was the tissues previously treated with 100 μM histamine in the presence of CPA. These data show that depletion of Ca2+ from the sarcoplasmic reticulum can transiently attenuate subsequent contraction; however, the effect was relatively small. There were larger effects on the Ca2+ signals. The presence of CPA attenuated the histamine-induced aequorin and fura-2 transients regardless of prior level of histamine stimulation. This is the expected result from CPA-induced inhibition of Ca2+ refilling of the sarcoplasmic reticulum. Prior stimulation of CPA-treated tissues with 100 μM histamine attenuated the subsequent histamine-induced aequorin and fura-2 transient when compared with the untreated and 3 μM histamine-treated tissues. These data suggest that release of Ca2+ from the sarcoplasmic reticulum has at least a small role in the initial phase of histamine-induced swine carotid arterial contraction. It appears that other contractile mechanisms can compensate for the loss of sarcoplasmic reticulum Ca2+ release with only modest changes in contraction kinetics.
Figure 8. Demonstration that prior depletion of the sarcoplasmic reticulum (with histamine and CPA) modestly attenuated the subsequent histamine-induced Ca2+ transient and contraction.
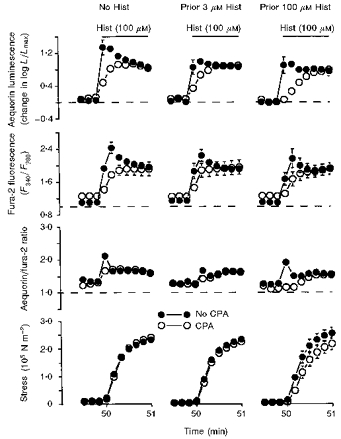
Time courses are shown for aequorin-estimated [Ca2+]i, fura-2-estimated [Ca2+]i, the aequorin/fura-2 ratio and active stress. Data are from six sets of swine carotid medial tissues (n= 4–6 for each). At 10 min, extracellular Ca2+ was nominally removed and, at 10.5 min, the three of the six sets of tissues were incubated in 10 μM CPA (○) which was then present for the remaining protocol. The other three sets of tissues were not treated with CPA (○). At 20 min, the tissues were then either (1) not stimulated (left panel), (2) stimulated with 3 μM histamine (centre panel), or (3) stimulated with 100 μM histamine (right panel). The experiments labelled ‘prior H100′ are a continuation of the experiments shown in Figs 6 and 7. At 25 min, all histamine was washed out. At 40 min, 1.6 mM extracellular [Ca2+] was restored to all tissues. At 50 min, all tissues were stimulated with 100 μM histamine in the presence of 1.6 mM extracellular [Ca2+]; the plot shows 30 s prior to and 60 s after this addition of histamine. Data were collected and plotted as in Fig. 6.
These data also show that prior concerns of histamine-induced aequorin consumption (Rembold & Murphy, 1988; Abe et al. 1995) are not a major concern in these experiments. The tissues previously treated with 100 μM histamine 25 min before restimulation had aequorin transients (Fig. 8, right panel, filled symbols) only modestly decreased compared with tissues not exposed to histamine stimulation (Fig. 8, left panel, filled symbols).
Finally, we investigated whether inhibition of SERCA altered Ca2+ influx. We measured Mn2+-induced quenching of fura-2 fluorescence as a method to estimate Ca2+ influx in intact smooth muscle tissues (Chen & Rembold, 1992). Mn2+ can traverse L-type Ca2+ channels and the Mn2+ influx rate through Ca2+ channels is proportional to the Ca2+ influx rate. Mn2+ binding to fura-2 rapidly quenches fura-2 fluorescence and therefore Mn2+ binding to fura-2 is not rate limiting. If extracellular Mn2+ is added to cells loaded with fura-2, the rate of decline in fura-2 fluorescence (measured at the isosbestic wavelength of 360 nm) is proportional to the rate of Mn2+ influx and is an index of the rate of Ca2+ influx. We previously found that histamine stimulation or high [K+]o depolarization increased Mn2+ influx rates and that diltiazem, forskolin and nitroglycerine attenuated histamine-stimulated Mn2+ influx (Chen & Rembold, 1992). The protocol was similar to that shown in Fig. 8. At 10 min, extracellular Ca2+ was nominally removed. At this point, some of the tissues were treated with 10 μM CPA, which was then present in all subsequent solutions. At 20 min, the tissues were either unstimulated or stimulated with 100 μM histamine. At 25 min, all histamine was washed out. At 40 min, 0.5 mM MnCl2 was added to the tissues and the fluorescence at 360 nm measured. In the absence of extracellular Ca2+, we found that treatment with CPA and/or prior histamine treatment (to deplete the sarcoplasmic reticulum) had no significant effect on Mn2+ influx rates (Fig. 9). As expected, Mn2+ influx in the absence of extracellular Ca2+ was significantly higher than the Mn2+ influx observed in unstimulated controls in the presence of extracellular Ca2+.
Figure 9. Demonstration that Mn2+ influx rates were not changed by depletion of the sarcoplasmic reticulum or the presence of CPA.
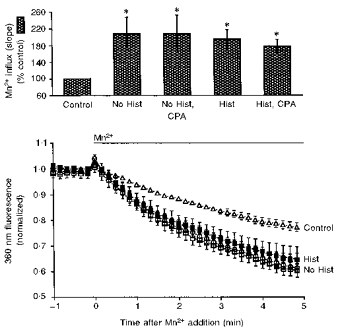
These data suggest that the Ca2+ influx induced by sarcoplasmic reticulum depletion did not enter via Mn2+-permeable channels. In one set of tissues (control), MnCl2 (0.5 mM) was added to tissues without prior manipulation. In the other four sets of tissues, extracellular Ca2+ was nominally removed for 10 min. At 10.5 min, the two of the sets were incubated in 10 μM CPA which was then present for the remaining protocol. At 20 min, two sets of tissues were stimulated with 100 μM histamine for 5 min. At 40 min, MnCl2 (0.5 mM) was added to all four sets of tissues (n= 4–6 for each set). The lower panel is the mean (continuous lines) ± 1 s.e.m. (dotted lines) change in 360 nm fluorescence observed after MnCl2 addition to each set of tissues. The time course of change in 360 nm fluorescence was similar in all four restoration studies and all were significantly faster than the control. Lysis refers to the addition of hypotonic 5 mM MnCl2 to determine background fluorescence. Data were collected at 1 s and plotted at 10 s intervals. *P < 0.05 vs. control as compared with the Newman-Keuls test.
DISCUSSION
In 1977, Van Breemen observed a dissociation between the rate of 45Ca2+ entry and contraction in Ca2+ depleted smooth muscle tissues that were activated by restoration of extracellular Ca2+ (Van Breemen, 1977). He proposed that addition of extracellular Ca2+ increased [Ca2+]i focally, i.e. [Ca2+]pm increased more than [Ca2+]c. Van Breemen's hypothesis (termed the ‘buffer barrier’ hypothesis) requires Ca2+ removal by SERCA, the Ca2+ pump of the sarcoplasmic reticulum. According to this hypothesis, basal Ca2+ influx rates are moderately high in unstimulated smooth muscle cells. Ca2+ pumps in the subsarcolemmal sarcoplasmic reticulum and plasma membrane and Na2+-Ca2+ exchanger in the plasma membrane continually remove Ca2+ from the subsarcolemmal space. Ca2+ pump activity prevents the increased [Ca2+]pm from diffusing to the central cytoplasm where it can increase [Ca2+]c, activate myosin light chain kinase, and induce a contraction. Ca2+ pumps near the plasma membrane are a functional ‘barrier’ to Ca2+ diffusion between the subsarcolemmal space and the bulk cytoplasm. If the sarcoplasmic reticulum is Ca2+ depleted, then Ca2+ influx into the subsarcolemmal space can refill the sarcoplasmic reticulum without increasing [Ca2+]c or inducing a contraction (Van Breemen, 1977; Van Breemen et al. 1986). This hypothesis is supported by recent structural studies: electron microscopy reveals that some sarcoplasmic reticulum is near the plasma membrane (Devine et al. 1972) and this region contains proteins necessary for Ca2+ release and uptake (Villa et al. 1993; Moore et al. 1993). Such a barrier was predicted by computer models of Ca2+ dynamics in smooth muscle cells. These models predicted that intermittently opening Ca2+ channel would maintain higher [Ca2+]pm and that Ca2+ pump function and passive intracellular Ca2+ buffers maintain [Ca2+]c at lower levels (Bers & Peskoff, 1991; Kargacin & Fay, 1991).
The goal of this study was to evaluate some of the characteristics of buffer barrier function. The buffer barrier hypothesis predicts several interrelationships between focal changes in [Ca2+]i and sarcoplasmic reticulum in intact arterial smooth muscle. We found that sarcoplasmic reticulum function regulates Ca2+ homogeneity and that specific types of Ca2+ inhomogeneity can determine the degree of Ca2+ uptake by the sarcoplasmic reticulum. We evaluated four specific hypotheses. These hypotheses are summarized in Fig. 10.
Figure 10. Proposed schema for Ca2+ distribution in smooth muscle cells.
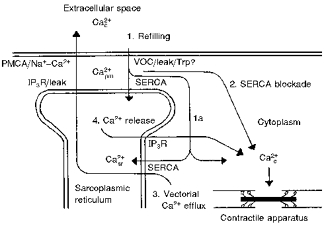
Shown is a section of a smooth muscle cell showing at the left the extracellular space, at the top the subplasmalemmal space (labelled [Ca2+]pm), and the sarcoplasmic reticulum. At the bottom right is the cytosol with the contractile apparatus. The proposed movements of Ca2+ are shown as arrows numbered 1–4. Abbreviations: PMCA, plasma membrane calcium ATPase (calcium pump); Na+-Ca2+, sodium-calcium exchanger; IP3R, the inositol trisphosphate receptor calcium release channel; Leak, the leak channel; VOC, voltage-operated L-type calcium channel; Trp, the trp channel; SERCA the sarco(endo)plasmic reticulum calcium ATPase (calcium pump); other abbreviations as in text.
(1) We hypothesized that increases in [Ca2+]pm were primarily responsible for refilling of the sarcoplasmic reticulum with Ca2+. This hypothesis was partially correct. We found that restoration of extracellular Ca2+ induced a large increase in the aequorin signal and a smaller increase in the fura-2 signal, suggesting an increase in [Ca2+]pm without a large increase in [Ca2+]c (Fig. 10, mechanism 1). This increase in [Ca2+]pm was associated with partial reloading of the sarcoplasmic reticulum with Ca2+ as evidenced by subsequent histamine-induced aequorin and fura-2 transients (Fig. 2, left panel). CPA inhibited reloading, indicating that SERCA function was important for reloading (Fig. 4, left panel). Restoration of extracellular Ca2+ in the presence of high [K+]o or choline+ further increased [Ca2+]c. This increase in [Ca2+]c was associated with enhanced reloading of the sarcoplasmic reticulum (right panels in Figs 2, 3 and 10, mechanism 1a). The latter was partly inhibited by CPA (Fig. 4, right panel), suggesting that reloading can also occur via SERCA in the central sarcoplasmic reticulum when [Ca2+]c is high. Large increases in [Ca2+]pm without large increases in [Ca2+]c, produced by choline+ with diltiazem (Fig. 3, centre panel, Fig. 10, mechanism 1), did not enhance reloading of the sarcoplasmic reticulum. These data suggest that sarcoplasmic reticulum Ca2+ reloading is bimodal. Transient increases in [Ca2+]pm partially filled the sarcoplasmic reticulum. Larger increases in [Ca2+]c were required to fully fill the sarcoplasmic reticulum. It is possible that a ‘full’ sarcoplasmic reticulum is actually ‘overloaded.’ A full sarcoplasmic reticulum may only be present transiently when Ca2+ is being transported vectorially from the central cytoplasm.
(2) We hypothesized that sarcoplasmic reticulum Ca2+ handling maintains a [Ca2+] gradient in unstimulated smooth muscle cells. Specifically, we hypothesized that basal levels of Ca2+ influx increase subplasmalemmal [Ca2+] ([Ca2+]pm) to higher levels than central myoplasmic [Ca2+] ([Ca2+]c) in unstimulated smooth muscle. Blocking SERCA with CPA increased the fura-2 signal without altering the aequorin signal (Figs 5 and 10, mechanism 2). This decreased the aequorin/fura-2 ratio, suggesting a more homogeneous [Ca2+]i distribution. These data are consistent with a [Ca2+]pm being higher than [Ca2+]c in resting smooth muscle tissues. Removal of extracellular Ca2+ without CPA also reduced the aequorin/fura-2 ratio, suggesting that basal levels of Ca2+ influx were responsible for maintaining a higher levels of basal [Ca2+]pm (Fig. 6). These data suggest that one function of the unstimulated sarcoplasmic reticulum is to maintain low [Ca2+]c even if [Ca2+]pm is higher from high Ca2+ influx. These data are consistent with the ‘buffer barrier’ hypothesis of Van Breemen (Van Breemen, 1977; Van Breemen et al. 1986). Similar results were observed when tissues were treated with choline+ plus diltiazem to reverse Na+-Ca2+ exchange. In the absence of CPA, there was a large increase in the aequorin signal without a large increase in the fura-2 signal indicating an increase in [Ca2+]pm (Fig. 3). When SERCA was blocked with CPA, the fura-2 signal increased more, indicating that sarcoplasmic reticulum function was necessary to prevent Ca2+ in the subplasmalemmal region from diffusing to the central cytoplasm (Figs 4 and 10, mechanism 2). The increased [Ca2+]pm is probably due to reversal of the Na+-Ca2+ exchange as well as to unspecified leak sources (Ashida & Blaustein, 1990). The increased Ca2+ in the subsarcolemmal region did not appear to cause increased Ca2+ in the bulk cytoplasm, demonstrating that the sarcoplasmic reticulum exerted considerable buffering of incoming Ca2+ (Sturek et al. 1992).
(3) We hypothesized that the sarcoplasmic reticulum is important in removal of Ca2+ from the centre of the cell upon removal of stimulation (Fig. 10, mechanism 3). This has been termed vectorial Ca2+ efflux (Stehno-Bittel & Sturek, 1992; Chen & Van Breemen, 1993). Upon washout of histamine, the fura-2 signal from the CPA-treated tissues decreased more slowly than the fura-2 signal from the tissues not treated with CPA (Fig. 7). This result suggests that sarcoplasmic reticulum Ca2+ handling is important in reducing [Ca2+] in the central cytoplasm, which is best measured with fura-2. Vectorial Ca2+ efflux may be important in reducing [Ca2+]c after high levels of stimulation such as high [K+]o (Fig. 2). Subplasmalemmal Ca2+, as measured with aequorin, decreased at a similar rate regardless of the presence of CPA. This is consistent with the hypothesis that Ca2+ is removed from the subplasmalemmal region by the action of plasma membrane Ca2+ pumps and Na+-Ca2+ exchange, which are not inhibited by CPA.
(4) We hypothesized that Ca2+ release from the sarcoplasmic reticulum is crucial for the initial phase of agonist-induced contractions (Fig. 10, mechanism 4). Sarcoplasmic reticulum Ca2+ depletion by prior treatment with 100 μM histamine and CPA transiently attenuated the response to subsequent histamine stimulation (Fig. 8). Histamine-induced aequorin and fura-2 transients were substantially attenuated. The effect on contraction was smaller: a delay in contraction of approximately 10 s. These data suggest that release of Ca2+ from the sarcoplasmic reticulum has at least a small role in the initial phase of histamine-induced swine carotid arterial contraction. It appears that other contractile mechanisms can compensate for the loss of sarcoplasmic reticulum Ca2+ release with only modest changes in contraction kinetics.
These data suggest that the sarcoplasmic reticulum has an important role in regulating [Ca2+] in at least two cytoplasmic compartments of smooth muscle cells. In unstimulated smooth muscle, Ca2+ pumping by SERCA maintains lower [Ca2+]c despite basal levels of Ca2+ influx which increase [Ca2+]pm. After stimulation, Ca2+ pumping by SERCA is important for reductions in [Ca2+]c. Agonist-induced Ca2+ release can induce contraction; however, if this mechanism is inhibited, there are other contractile mechanisms that can initiate and maintain contraction without a large effect on contractile kinetics. These data are consistent with Van Breemen's ‘buffer barrier’ hypothesis (Van Breemen, 1977; Van Breemen et al. 1986).
Acknowledgments
The authors would like to thank Barbara Weaver for technical assistance. Smithfield Co. (Smithfield, VA, USA) donated the arteries. C. M. R. is a Lucille P. Markey Scholar and grants from the Lucille P. Markey Charitable Trust, the PHS (HL38918), and the Virginia Affiliate of the American Heart Association supported this research.
References
- Abe F, Karaki H, Endoh M. Effects of cyclopiazonic acid and ryanodine on cytosolic calcium and contraction in vascular smooth muscle. British Journal of Pharmacology. 1996;118:1711–1716. doi: 10.1111/j.1476-5381.1996.tb15596.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abe F, Mitsui M, Karaki H, Endoh M. Calcium compartments in vascular smooth muscle cells as detected by aequorin signal. British Journal of Pharmacology. 1995;116:3000–3004. doi: 10.1111/j.1476-5381.1995.tb15955.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashida T, Blaustein MP. Regulation of cell calcium and contractility in mammalian smooth muscle: the role of sodium-calcium exchange. The Journal of Physiology. 1990;392:617–635. doi: 10.1113/jphysiol.1987.sp016800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Augustine GJ, Neher E. Calcium requirements for secretion in bovine chromaffin cells. The Journal of Physiology. 1992;450:247–271. doi: 10.1113/jphysiol.1992.sp019126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bers DM, Peskoff A. Diffusion around a cardiac calcium channel and the role of surface bound calcium. Biophysical Journal. 1991;59:703–721. doi: 10.1016/S0006-3495(91)82284-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blinks JR, Wier WG, Hess P, Prendergast FG. Measurement of Ca2+ concentrations in living cells. Progress in Biophysics and Molecular Biolology. 1982;4026:1–114. doi: 10.1016/0079-6107(82)90011-6. [DOI] [PubMed] [Google Scholar]
- Brose N, Petrenko AG, Sudhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256:1021–1025. doi: 10.1126/science.1589771. [DOI] [PubMed] [Google Scholar]
- Chen Q, Van Breemen C. The superficial buffer barrier in venous smooth muscle: Sarcoplasmic reticulum refilling and unloading. British Journal of Pharmacology. 1993;109:336–343. doi: 10.1111/j.1476-5381.1993.tb13575.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X-L, Rembold CM. Cyclic nucleotide dependent regulation of Mn2+ influx, [Ca2+]i, and arterial smooth muscle relaxation. American Journal of Physiology. 1992;263:C468–473. doi: 10.1152/ajpcell.1992.263.2.C468. [DOI] [PubMed] [Google Scholar]
- Devine CE, Somlyo AV, Somlyo AP. Sarcoplasmic reticulum and excitation-contraction coupling in mammalian smooth muscles. Journal of Cell Biology. 1972;52:690–718. doi: 10.1083/jcb.52.3.690. 10.1083/jcb.52.3.690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert EK, Weaver BA, Rembold CM. Depolarization decreases the [Ca2+]i-sensitivity of myosin light chain kinase in arterial smooth muscle: a comparison of aequorin and Fura 2 [Ca2+] estimates. FASEB Journal. 1991;5:2593–2599. doi: 10.1096/fasebj.5.11.1868983. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz AM, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Kargacin G, Fay FS. Ca2+ movement in smooth muscle cells studied with one- and two-dimensional diffusion models. Biophysical Journal. 1991;60:1088–1100. doi: 10.1016/S0006-3495(91)82145-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lederer WJ, Niggli E, Hadley RW. Sodium-calcium exchange in excitable cells: Fuzzy space. Science. 1990;248:283. doi: 10.1126/science.2326638. [DOI] [PubMed] [Google Scholar]
- Moore EDW, Etter EF, Phillipson KD, Carrington WA, Fogarty KE, Lifshitz LM, Fay FS. Coupling of the Na+/Ca2+ exchanger, Na+/K+ pump and sarcoplasmic reticulum in smooth muscle. Nature. 1993;365:657–660. doi: 10.1038/365657a0. 10.1038/365657a0. [DOI] [PubMed] [Google Scholar]
- Nagai R, Kuro-o M, Babij P, Periasamy M. Identification of two types of smooth muscle myosin heavy chain isoforms by cDNA cloning and immunoblot analysis. Journal of Biological Chemistry. 1989;264:9734–9737. [PubMed] [Google Scholar]
- Neher E, Augustine GJ. Calcium gradients and buffers in bovine chromaffin cells. The Journal of Physiology. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osipchuk YV, Wakui M, Yule DI, Gallacher DV, Petersen OH. Cytoplasmic Ca2+ oscillations evoked by receptor stimulation, G-protein activation, internal application of inositol trisphosphate or Ca2+: simultaneous microfluorimetry and Ca2+ dependent Cl− current recording in single pancreatic acinar cells. EMBO Journal. 1990;9:697–704. doi: 10.1002/j.1460-2075.1990.tb08162.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM. Desensitization of swine arterial smooth muscle to transplasmalemmal Ca2+ influx. The Journal of Physiology. 1989;416:273–290. doi: 10.1113/jphysiol.1989.sp017760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rembold CM. Electromechanical and pharmacomechanical coupling. In: Barany M, editor. Biochemistry of Smooth Muscle Contraction. Chicago: Academic Press; 1996. pp. 227–239. [Google Scholar]
- Rembold CM, Murphy RA. Myoplasmic [Ca2+] determines myosin phosphorylation in agonist-stimulated swine arterial smooth muscle. Circulation Research. 1988;63:593–603. doi: 10.1161/01.res.63.3.593. [DOI] [PubMed] [Google Scholar]
- Rembold CM, Richard HL, Chen X-L. Na+/Ca2+ exchange, myoplasmic [Ca2+], and contraction of arterial smooth muscle. Hypertension. 1992;19:308–313. doi: 10.1161/01.hyp.19.4.308. [DOI] [PubMed] [Google Scholar]
- Rembold CM, VanRiper DA, Chen X-L. Focal [Ca2+]i increases detected by aequorin but not fura-2 in histamine and caffeine stimulated swine carotid artery. The Journal of Physiology. 1995;488:549–564. doi: 10.1113/jphysiol.1995.sp020989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuttleworth TJ, Thompson JL. Effect of temperature on receptor-activated changes in [Ca2+]i and their determination using fluorescent probes. Journal of Biological Chemistry. 1991;266:1410–1414. [PubMed] [Google Scholar]
- Smith SJ, Augustine GJ. Calcium ions, active zones and synaptic transmitter release. Trends in Neurosciences. 1988;11:458–464. doi: 10.1016/0166-2236(88)90199-3. 10.1016/0166-2236(88)90199-3. [DOI] [PubMed] [Google Scholar]
- Stehno-Bittel L, Sturek M. Spontaneous sarcoplasmic reticulum calcium release and extrusion from bovine, not porcine, coronary artery smooth muscle. The Journal of Physiology. 1992;451:49–78. doi: 10.1113/jphysiol.1992.sp019153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sturek M, Kunda K, Hu Q. Sarcoplasmic reticulum buffering of myoplasmic calcium in bovine coronary artery smooth muscle. The Journal of Physiology. 1992;451:25–48. doi: 10.1113/jphysiol.1992.sp019152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takemura H, Putney JW., Jr Capacitative calcium entry in parotid acinar cells. Biochemical Journal. 1989;258:409–412. doi: 10.1042/bj2580409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uto A, Arai H, Ogawa Y. Reassessment of Fura-2 and the ratio method for determination of intracellular Ca2+ concentrations. Cell Calcium. 1991;12:29–37. doi: 10.1016/0143-4160(91)90082-p. 10.1016/0143-4160(91)90082-P. [DOI] [PubMed] [Google Scholar]
- Van Breemen C. Calcium requirement for activation of intact aortic smooth muscle. The Journal of Physiology. 1977;272:317–329. doi: 10.1113/jphysiol.1977.sp012046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Breemen C, Lukeman S, Leijten P, Yamamoto H, Loutzenhiser R. The role of superficial SR in modulating force development induced by Ca entry into arterial smooth muscle. Journal of Cardiovascular Pharmacology. 1986;8:S111–116. doi: 10.1097/00005344-198600088-00023. [DOI] [PubMed] [Google Scholar]
- Van Riper DA, Chen XL, Gould EM, Rembold CM. Focal increases in [Ca2+]i may account for apparent low Ca2+ sensitivity in swine carotid artery. Cell Calcium. 1996;19:501–508. doi: 10.1016/s0143-4160(96)90059-7. 10.1016/S0143-4160(96)90059-7. [DOI] [PubMed] [Google Scholar]
- Villa A, Podini P, Panzeri MC, Söling HD, Volpe P, Meldolesi J. The endoplasmic-sarcoplasmic reticulum of smooth muscle: Immunocytochemistry of vas deferens fibers reveals specialized subcompartments differently equipped for the control of Ca2+ homeostasis. Journal of Cell Biology. 1993;121:1041–1051. doi: 10.1083/jcb.121.5.1041. 10.1083/jcb.121.5.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshikawa A, Van Breemen C, Isenberg G. Buffering of plasmalemmal Ca2+ current by sarcoplasmic reticulum of guinea pig urinary bladder myocytes. American Journal of Physiology. 1996;271:C833–841. doi: 10.1152/ajpcell.1996.271.3.C833. [DOI] [PubMed] [Google Scholar]
- Yue DT, Wier WG. Estimation of intracellular [Ca 2+] by nonlinear indicators. A quantitative analysis. Biophysical Journal. 1998;48:533–537. doi: 10.1016/S0006-3495(85)83810-8. [DOI] [PMC free article] [PubMed] [Google Scholar]