

Abstract
Whole-cell recordings from cultured rat hippocampal neurons, from freshly dissociated dorsal root ganglion (DRG) neurons and from human embryonic kidney (HEK) 293 cells expressing the glutamate receptor GluR6 subunit were used to study the modulation of kainate receptor channels by long chain fatty acids.
In all three cell types, application of cis-unsaturated fatty acids caused a dose-dependent reduction in whole-cell currents evoked by kainate. Docosahexaenoic acid (DHA), arachidonic acid (AA), linolenic acid and linoleic acid all produced substantial inhibition at a concentration of 50 μm, whereas inhibition by linolenelaidic acid and linolelaidic acid was significantly weaker. Fully saturated fatty acids were essentially inactive.
With continuous exposure to active fatty acids, the peak current elicited by kainate declined over a time course of several minutes to reach a steady-state level less than 50 % of the initial amplitude. Recovery was slow in control solution, but was speeded up by exposure to bovine serum albumin (0.5 mg ml−1), a protein that binds fatty acids with submicromolar affinity. The inhibition in neurons was half-maximal with 5–15 μm AA or DHA, but potency was at least 10-fold greater at GluR6 in HEK 293 cells.
Inhibition by AA or DHA was unaffected by extracellular nordihydroguaiaretic acid (10 μm), indomethacin (10 μm), 17-octadecynoic acid (30 μm) or 1-(5-isoquinolinylsulphonyl)-2-methylpiperazine dihydrochloride (H-7; 10 μm). Furthermore, inclusion of H-7 (100 μm), BAPTA (10 mm), AA (50 μm), antioxidants, or the protein kinase C inhibitor PKC19-36 (20 μm) in the internal solution had little effect on whole-cell currents and did not prevent inhibition of currents by extracellular application of AA or DHA.
We conclude that the inhibition produced by cis-unsaturated fatty acids does not require conversion to oxidized metabolites or activation of PKC. Instead, active compounds may interact directly with an extracellular, or intramembraneous, site on kainate receptors.
Arachidonic acid (AA) is thought to serve as an intercellular messenger in many parts of the nervous system (Attwell et al. 1993). The release of AA from membrane phospholipids can be stimulated by a number of different neurotransmitters, including glutamate, serotonin, acetylcholine and catecholamines. In the case of glutamate, release of AA is evoked by activation of NMDA receptors (Dumuis et al. 1988) or by the simultaneous activation of AMPA (α-amino-3-hydroxy-5-methylisoxazole-4-propionate) receptors and metabotropic glutamate receptors (Dumuis et al. 1990). Exposure to cis-unsaturated fatty acids, including AA and docosahexaenoic acid (DHA), has been shown to modulate ionic currents in neurons as well as in other cell types (Ordway et al. 1991; Meves, 1994). Potentiation of currents has been reported for NMDA receptors in hippocampal neurons (Miller et al. 1992; Nishikawa et al. 1994), and for K+ channels in Aplysia neurons (Piomelli et al. 1987) and in mammalian cardiac and smooth muscle cells (Kim & Clapham, 1989; Ordway et al. 1989). In contrast, AA application produces inhibition of currents mediated by neuronal AMPA (Kovalchuk et al. 1994) and GABAA (Schwartz & Yu, 1992; Hamano et al. 1996) receptors, as well as inhibition of voltage-gated sodium (Linden & Routtenberg, 1989), calcium (Keyser & Alger, 1990) and M-current (Schmitt & Meves, 1993) channels in neurons or neuroblastoma cells. Inhibition of currents associated with glutamate transport in glial cells has also been reported (Barbour et al. 1989).
There are several different mechanisms by which fatty acids might act on membrane channel proteins (Ordway et al. 1991; Attwell et al. 1993; Meves, 1994). For example, in Aplysia neurons potentiation of K+ channel activity is thought to be mediated by specific oxidized metabolites of AA that are generated via cytoplasmic lipoxygenase pathways (Piomelli et al. 1987). In addition, AA and other cis-unsaturated fatty acids activate protein kinase C (PKC) (Shinomura et al. 1991), which could alter channel properties by phosphorylation (Linden & Routtenberg, 1989; Schmitt & Meves, 1993). Formation of free radicals, which is stimulated by AA, has also been proposed to mediate channel modulation (Keyser & Alger, 1990). These indirect actions mediated by PKC or metabolites of AA would require the passage of exogenously applied AA through the cell membrane into the cytoplasm. In a number of cases, however, it has been suggested that fatty acids may modify channel activity by directly binding to the channel protein or by changing its local environment in the lipid bilayer (Ordway et al. 1991).
Previous work on NMDA (Miller et al. 1992; Nishikawa et al. 1994) and AMPA (Kovalchuk et al. 1994) receptors suggests that their modulation probably does not involve downstream metabolites of fatty acids or activation of PKC. Instead, it has been proposed that cis-unsaturated fatty acids may interact directly with these receptors (Miller et al. 1992; Kovalchuk et al. 1994), possibly by binding to a specific extracellular domain. Although evidence for direct binding of fatty acids to glutamate receptors has not yet been reported, sequence analysis of glutamate receptor subunits has revealed a region of homology between the NR1 subunit of NMDA receptors and a family of small intracellular fatty acid-binding proteins (Petrou et al. 1993). The GluR6 subunit (Egebjerg et al. 1991), which contributes to kainate receptors, also displays significant homology with fatty acid-binding proteins in this extracellular region (Kovalchuk et al. 1994). In contrast, other glutamate receptor subunits, including those which contribute to AMPA receptors, show less homology (Kovalchuk et al. 1994).
In the present study we have tested the actions of AA and DHA on whole-cell currents mediated by kainate-preferring receptors. Native kainate receptors were studied in hippocampal and dorsal root ganglion (DRG) neurons, and recombinant receptors formed by expression of the GluR6 subunit were studied in human embryonic kidney (HEK) 293 cells. In all cases, exposure to AA or DHA produced inhibition of peak currents mediated by these receptors. The properties of this inhibition indicate a direct action on the receptors or on their membrane environment.
METHODS
Cell culture
Primary cultures of hippocampal neurons were prepared as previously described (Wilding & Huettner, 1997), under protocols approved by the Washington University Animal Studies Committee. Long-Evans rats, 2–5 days old, were rapidly decapitated. Hippocampi were isolated and cut into 500 μm slices. Subiculum and entorhinal cortex were removed from each slice, and the slices were incubated at 30–35°C with gentle stirring in Earle's balanced salt solution (EBSS; Life Technologies, Grand Island, NY, USA) containing papain (20 units ml−1; Worthington Biochemical, Freehold, NJ, USA). After 90 min, the tissue was rinsed with EBSS containing bovine serum albumin (BSA) and ovomucoid, both at 1 mg ml−1. Cells were dissociated with a fire-polished Pasteur pipette and plated onto glass coverslips coated with poly-dl-ornithine or matrigel (Becton Dickinson, Bedford, MA, USA). Cultures were maintained at 37°C in Eagle's minimal essential medium (MEM; Life Technologies) with 20 mm glucose, 0.5 mm glutamine, 100 units ml−1 penicillin, 0.1 mg ml−1 streptomycin and 5 % rat serum (serum was prepared from Sprague-Dawley rats killed by inhalation of CO2). Most recordings were obtained from hippocampal cells that had been in culture for 7–21 days.
Freshly dissociated DRG neurons were prepared from rapidly decapitated 1- to 2-week-old rats as described previously (Huettner, 1990; Wilding & Huettner, 1995). Ganglia were cut in half and incubated at 30–35°C for 30 min in EBSS containing protease XXIII (1 mg ml−1; Sigma). Cells were dissociated by trituration in EBSS containing BSA at 1 mg ml−1. The cell suspension was maintained overnight at room temperature (20–23°C) and used the next day.
HEK 293 cells were maintained at 37°C under 5 % CO2 in growth medium composed of MEM, alpha-formulation (Life Technologies), supplemented with 10 % fetal calf serum (JRH Biosciences, Lenexa, KS, USA), 100 units ml−1 penicillin and 0.1 mg ml−1 streptomycin. Cells were cotransfected with cDNAs encoding the fully edited GluR6 (V, C; R) subunit in the pcDNA I/AMP vector (Invitrogen, San Diego, CA, USA) and the mouse L3T4 surface antigen (Tourvieille et al. 1986) in the pRBG4 vector (kindly provided by Dr David Clapham, Harvard Medical School, USA). GluR6-pcDNA (1 μg) and L3T4-pRBG4 (0.5 μg) were incubated for 45 min with SuperFect reagent (15 μg; Qiagen, Santa Clarita, CA, USA) in OptiMEM (100 μl; Life Technologies), then diluted 1 : 5 onto HEK 293 cells at 50–70 % confluence. After 3–4 h, three volumes of growth medium containing 5 mm kynurenic acid were added. On the next day, transfected cells were incubated with protease XXIII (1 mg ml−1; 10–15 min), dissociated into single cells and replated at low density onto nitrocellulose-coated tissue culture plastic. Recordings were obtained 24–48 h after this replating step. The cells were first incubated for 30 min with phycoerytherin-conjugated monoclonal anti-L3T4 (PharMingen, San Diego, CA, USA). Isolated cells that were labelled with the fluorescent antibody were targeted for recording.
Electrophysiology
The recording chamber was perfused at 1–2 ml min−1 with Tyrode solution comprising (mm): 150 NaCl, 4 KCl, 2 MgCl2, 2 CaCl2, 10 glucose and 10 Hepes; pH adjusted to 7.4 with NaOH. Recording pipettes were pulled from boralex glass and filled with an internal solution containing (mm): 140 caesium glucuronate, 10 EGTA, 5 CsCl, 5 MgCl2, 5 ATP, 1 GTP and 10 Hepes; pH adjusted to 7.4 with CsOH. The open tip resistance of whole-cell pipettes was 1–5 MΩ. An agar bridge prepared in 4 M KCl connected the bath to a ground well, filled with internal solution, which contained the reference electrode. Whole-cell currents were recorded with an Axopatch 200A amplifier, filtered at 1 kHz (−3 dB, 4-pole Bessel), and digitized at 5–10 kHz. Membrane potentials have been corrected for a junction potential of −10 mV between the Tyrode solution, in which seals were formed, and the internal solution.
Drug applications
Control or agonist-containing solutions were applied to cells by local perfusion from a multibarrelled pipette as described previously (Wilding & Huettner, 1997). One end of a 250 μl microcap (1.58 mm i.d.; Drummond Scientific, Broomall, PA, USA) was fire polished to an open diameter of approximately 400–500 μm. Eight fused silica tubes (320 μm i.d.; J. & W. Scientific, Folsom, CA, USA) were aligned within the microcap to yield a dead volume of much less than 1 μl. For rapid applications, the drug reservoirs were held under static air pressure (10–15 p.s.i.) and solution flow to the delivery tubes was controlled by computer-gated electronic valves (The Lee Company, Westbrook, CT, USA). The rate of solution exchange was determined by switching from 160 mmN-methyl-D-glucamine chloride to 160 mm NaCl in the continuous presence of 50 μm kainate to activate AMPA receptors. In whole-cell recordings from cultured hippocampal neurons the external solution was exchanged with a time constant of 5 to 15 ms.
Agonists were applied in a solution composed of 160 mm NaCl, 2 mm CaCl2 and 10 mm Hepes; pH adjusted to 7.4 with NaOH. Ascorbic acid was added to this external solution at 0.1 mm in order to impede the spontaneous oxidation of unsaturated compounds. Fatty acids were prepared as 50 mm stock solutions in DMSO and stored at −80°C as 50 or 100 μl aliquots under argon gas. They were purchased from Matreya, Inc. (Pleasant Gap, PA, USA) or from Sigma. GYKI 53655 was generously provided by Eli Lilly and Company. All other compounds were from Research Biochemicals International (Natick, MA, USA) or Sigma.
Data analysis
Except as noted, results are presented as means ±s.e.m. To calculate percentage inhibition, peak currents evoked by kainate were expressed as a fraction of the last current recorded before exposure to fatty acid and plotted as a function of time. Data points obtained from all cells in a data set for the last five kainate applications prior to fatty acid exposure were fitted with the equation for a straight line. Percentage inhibition was calculated relative to this line using currents recorded after 5 or 6 min exposure to fatty acids using the equation: Percentage inhibition = 100 (1 - (mean peak current/value of the fitted line)).
RESULTS
Inhibition by arachidonic acid
We initially tested the action of AA on currents mediated by kainate receptors in cultured postnatal hippocampal neurons (Wilding & Huettner, 1997) and in freshly dissociated DRG cells (Huettner, 1990). For recordings from hippocampal neurons, the non-competitive antagonists GYKI 53655 (100 μm; Paternain et al. 1995; Wilding & Huettner, 1995) and MK-801 (2 μm; Huettner & Bean, 1988) were used to block currents mediated by AMPA and NMDA receptors, respectively. As shown in Fig. 1, exposure to 50 μm AA produced a progressive reduction in currents evoked by 2 s applications of 300 μm kainate. Both the peak and steady-state currents were reduced. Inhibition developed over a time course of 4 to 5 min reaching a maximum of 60–80 % inhibition of peak current at this concentration. In preliminary experiments, we tested whether it was necessary to include AA in the kainate test solution or whether exposure to AA in between test applications was sufficient to inhibit the currents. As shown in Fig. 2A, the time course of inhibition was virtually identical whether or not the test solution contained AA. For convenience, in most of our subsequent experiments fatty acids were only added to the control solution and not to the kainate test solutions.
Figure 1. Time course of inhibition in individual cells.
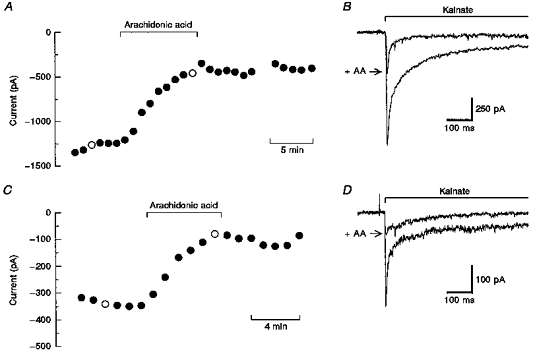
A and C, peak currents evoked by 300 μm kainate in a hippocampal neuron (A) and a DRG neuron (C) plotted as a function of time. The control solution contained 50 μm arachidonic acid (AA) during the period indicated by the horizontal bar. Holding potential, −80 mV. B and D, individual traces corresponding to ○ in A and C, respectively. Kainate was applied by rapid perfusion for the period indicated. Traces recorded before and during exposure to AA are shown superimposed. Fatty acids reduced both the peak and steady-state currents in all cells tested. The relative reduction in peak versus steady-state current showed some variability from one cell to the next (cf. B with D); however, no consistent correlation with cell type or desensitization kinetics was noted.
Figure 2. Arachidonic acid is not required in the test solution (A) and BSA speeds recovery from inhibition (B).
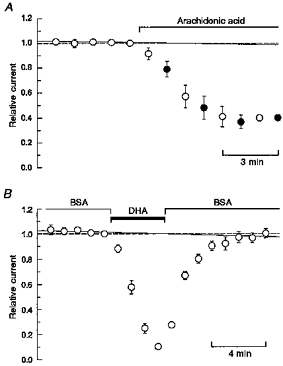
A, peak currents evoked by 300 μm kainate in hippocampal neurons plotted as a function of time. Points show the mean current (±s.e.m.; n= 4 cells) expressed as a fraction of the last current recorded before exposure to AA (50 μm). Once exposure to AA in the control solution began, the cells were tested alternately with 300 μm kainate solutions that lacked AA(○) or that contained AA (•). B, mean peak currents (±s.e.m.; n= 7 cells) evoked by 300 μm kainate in HEK 293 cells expressing the GluR6 subunit plotted as a function of time. Bovine serum albumin (BSA) was included in the control solution at 0.5 mg ml−1 except during the period of exposure to docosahexaenoic acid (DHA; 5 μm). In A and B, a straight (continuous) line was fitted to the 5 peak amplitudes recorded in control solution prior to fatty acid exposure (see text).
In most cells, prolonged exposure to fatty acids eventually produced an irreversible increase in holding current so that it was difficult to monitor recovery after applications that lasted more than a few minutes. In the few cells that remained stable long enough to be returned to control solution, there was relatively little evidence of recovery of the peak current (e.g. Fig. 1). Shorter exposures to AA also produced stable inhibition that was less than maximal. Figure 2B shows an experiment to test whether the lack of recovery in control solution was caused by very slow washout of the hydrophobic fatty acids or by some sort of irreversible change in receptor properties. In these experiments, cells were washed with control solution that contained 0.5 % BSA, a protein that binds fatty acids with submicromolar affinity (Richieri et al. 1993). As shown in Fig. 2B, exposure to BSA speeded up recovery from inhibition, suggesting that the slow recovery observed in control solution was owing to residual fatty acid that remained associated with the cells.
To obtain a quantitative measure of the action of AA and other fatty acids, we determined the percentage inhibition after a 5 or 6 min exposure time. Peak currents recorded at 1 min intervals were normalized to the final peak amplitude obtained in control solution. In this way, we were able to combine results from several cells to visualize the time course of inhibition in the whole population. As illustrated in Fig. 2A and B, the peak current amplitude in control solution was relatively stable over time, although many cells displayed a very gradual decline in current amplitude. To correct for this slow decline and thus provide the most conservative estimate of inhibition, we fitted a straight line to the five peak amplitudes recorded in control solution prior to fatty acid exposure. Percentage inhibition was calculated relative to this line as described in the Methods.
Inhibition by other cis-unsaturated fatty acids
In addition to AA, we tested a number of other fatty acids for their ability to inhibit currents mediated by kainate receptors. The graphs in Fig. 3A and B show the mean percentage inhibition (±s.e.m.) observed at the end of a 5–6 min exposure period for thirteen different compounds. All of the compounds were tested at a concentration of 50 μm. As shown in Fig. 3A, the majority of cis-unsaturated fatty acids produced substantial inhibition. Within this group, six of the compounds produced greater than 50 % inhibition; however, oleic acid (C18, cis-9), docosatrienoic acid (DTA; C22, cis-13,16,19) and the methyl ester of DHA (methyl-DHA) did not cause significant block. Interestingly, we did observe significant, albeit partial, blockade on application of eicosatetraynoic acid (ETYA; C20, cis-5,8,11,14), which is an inhibitor of cyclo-oxygenase, lipoxygenase and cytochrome P450 enzymes for fatty acid metabolism (Meves, 1994). For three of the compounds, we tested both the all-cis and all-trans isomers (Fig. 3B). In all three cases, inhibition by the trans-isomers was significantly weaker than that by the corresponding cis-isomers. In addition, we saw no significant inhibition with five fully saturated fatty acids (myristic, palmitic, stearic, arachidic and behenic). However, the free fatty acid concentration for these saturated compounds was likely to be well below our target of 50 μm, owing to their limited aqueous solubility (Cistola et al. 1988).
Figure 3. Structure-activity (A and B) and concentration-response (C) relationships.
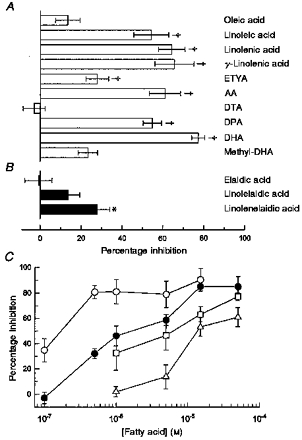
Mean percentage inhibition (±s.e.m.), at 5–6 min exposure time, plotted for 13 different compounds. A, 10 compounds with cis-unsaturated bonds. B, 3 isomers with trans-unsaturated bonds. All compounds were tested at a concentration of 50 μm on cultured hippocampal neurons. The number of cells tested with each compound ranged from 4 to 11; their common names or abbreviations are given to the right of each bar. ETYA, eicosatetraynoic acid; DTA, docosatrienoic acid; DPA, docosapentaenoic acid. Compounds that produced significant inhibition are indicated: *P < 0.05, †P < 0.01. C, mean percentage inhibition (±s.e.m.), at 5–6 min exposure time, plotted versus fatty acid concentration for hippocampal neurons (AA,▵ DHA, □) and HEK 293 cells expressing GluR6 (AA, • DHA, ○). The number of cells tested at each concentration ranged from 3 to 10.
DHA and AA were selected for further analysis at lower concentrations. As shown in Fig. 3C, both compounds produced significant inhibition at a concentration of 15 μm. In hippocampal neurons much weaker block was observed for 5 μm AA, whereas the threshold concentration for significant action by DHA was between 1 and 5 μm. AA and DHA were significantly more potent in HEK 293 cells expressing GluR6. For these recombinant receptors both of the compounds produced significant inhibition at a concentration of 0.5 μm.
Block shows little dependence on agonist concentration or membrane potential
Figure 4A shows experiments designed to test whether or not inhibition by the fatty acids involved a competitive interaction with channel activation by the agonist. Because cells did not survive prolonged exposure to high concentrations of fatty acids, we were not able to evaluate the complete agonist concentration-response relationship in individual cells that were equilibrated with a fixed fatty acid concentration. Instead, we alternated applications of 630 μm kainate with test applications of a lower kainate concentration. As shown in Fig. 4A, the inhibition by AA was not overcome by increasing the concentration of kainate; however, currents evoked by submaximal concentrations of kainate appeared to be more sensitive to inhibition than currents elicited by 160 and 630 μm kainate.
Figure 4. Inhibition shows little dependence on agonist concentration or membrane potential.
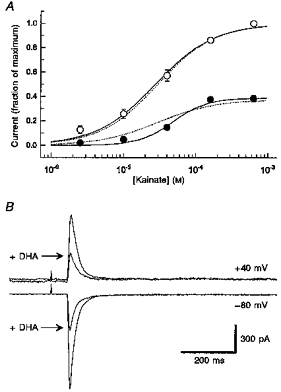
A, current evoked by 2.5, 10, 40, 160 and 630 μm kainate alone (○) or after 5–6 min exposure to 50 μm AA (•). Peak current (±s.e.m.) is plotted as a function of kainate concentration. Continuous curves show the best fits to: I/Imax=b/(1 + (EC50/[kainate])n), where EC50 is the concentration producing half-maximal current, n is the slope factor and Imax is the peak current evoked by 630 μm kainate. For the control curve: EC50= 28 μm, n= 1.0 and b was set to 1. With 50 μm AA: EC50= 49 μm, n= 1.8 and b= 0.39. Dashed curves show the best simultaneous fit in which EC50 and n were constrained to be the same for both data sets (EC50= 29 μm, n= 1.1, b= 0.37). This simultaneous fit was significantly inferior to the two separate fits as judged by the ratio of residual variance (F test). B, whole-cell currents evoked by 300 μm kainate at a holding potential of −80 and +40 mV in a HEK 293 cell expressing GluR6. Traces recorded before and after 5 min exposure to 0.5 μm DHA are shown superimposed.
To test whether inhibition by fatty acids was affected by membrane potential, we examined the inhibition of kainate currents at holding potentials of −80 and +40 mV (Fig. 4B). In five HEK 293 cells expressing GluR6 that were exposed to 0.1–0.5 μm DHA, we observed a mean inhibition of 60.9 ± 5.3 % at +40 mV and 64.7 ± 4.5 %; at −80 mV. In three additional cells tested with 5 μm DHA, we observed 90.6 ± 2.3 %; inhibition at −80 mV and 82.5 ± 4.0 %; blockade at +40 mV. These differences are not significant (Student's paired t test).
Pharmacology of inhibition
In order to determine the mechanism of inhibition, we tested the action of drugs that should interfere with the conversion of AA or DHA to active metabolites (Meves, 1994). As shown in Table 1, continuous superfusion with the cytochrome P450 inhibitor 17-octadecynoic acid (ODYA, 30 μm; Zou et al. 1994) did not prevent the inhibition of kainate receptor-mediated current. Moreover, superfusion with a combination of indomethacin (10 μm) and nordihydroguaiaretic acid (NDGA; 10 μm), which inhibit lipoxygenase and cyclo-oxygenase enzymes, respectively, did not block the inhibition by AA.
Table 1.
Pharmacology of inhibition
| Treatment | Concentration | In/out‡ | Percentage inhibition§ | n |
|---|---|---|---|---|
| Control, hippocampus | (15 μm DHA) | — | 63.2 ± 6.0 | 5 |
| + ODYA | 30 μm | Out | 81.9 ± 8.7† | 4 |
| Control, hippocampus | (50 μm AA) | — | 61.0 ± 7.6 | 9 |
| + NDGA plus indomethacin | 10 μm,10 μm | Out | 62.3 ± 3.7 | 5 |
| + H-7 | 10 μm | Out | 84.1 ± 4.6† | 5 |
| + H-7 | 100 μm | In | 62.9 ± 9.4 | 4 |
| + BAPTA | 10 mm | In | 53.1 ± 4.0* | 4 |
| + AA | 50 μm | In | 56.7 ± 3.7 | 7 |
| + SOD | 100 units ml−1 | Both | 70.7 ± 10.0† | 4 |
| Control, GluR6 | (1 μm DHA) | — | 81.1 ± 9.6 | 5 |
| + PKC19–36 | 20 μm | In | 71.5 ± 6.4† | 6 |
Compounds were added to the internal (in) or external (out) solution.
Mean percentage inhibition (±s.e.m.) at 5–6 min exposure time.
P < 0.05
P < 0.01, significant difference from control. ODYA, 17-octadecynoic acid; NDGA, nordihydroguaiaretic acid; H-7, 1-(5-isoquinolinylsulphonyl)-2-methyl- piperazine dihydrochloride; SOD, superoxide dismutase.
Addition of superoxide dismutase (SOD; 100 units ml−1) to both the internal and external solution (Table 1), or a combination of three antioxidants, catalase (250 units ml−1), tiron (1 mm) and mannitol (200 μm) (data not shown), did not prevent inhibition by AA in either hippocampal neurons or HEK 293 cells. These results indicate that free radical formation is not likely to account for the inhibition of kainate receptor-mediated currents (see Keyser & Alger, 1990).
To test whether the action of AA depended on its ability to activate PKC (Shinomura et al. 1991), we added a number of different reagents to the internal solution, including the kinase inhibitor 1-(5-isoquinolinylsulphonyl)-2-methylpiperazine dihydrochloride (H-7, 100 μm; Hidaka et al. 1984), AA itself (50 μm), the calcium chelator BAPTA (10 mm) and PKC19-36 (20 μm), which is a peptide inhibitor of PKC (House & Kemp, 1987). In control experiments, we determined that none of these agents caused any significant change in the amplitude of peak kainate-activated currents as a function of time after break through to the whole-cell recording mode. Furthermore, inclusion of these compounds in the internal solution did not prevent inhibition of kainate-evoked currents by a subsequent external application of AA or DHA (Table 1). Addition of H-7 (10 μm) to the external superfusion medium was similarly ineffective in blocking the action of AA on the currents evoked by kainate.
DISCUSSION
The present study has demonstrated the reversible inhibition of kainate receptor-mediated whole-cell currents by micromolar concentrations of AA and DHA. We undertook these experiments because of the striking asymmetry in the action of fatty acids on currents mediated by NMDA and AMPA receptors. In previous work, both AA and DHA were shown to produce between 1.5- and 3-fold potentiation of currents evoked by NMDA in cultured cerebellar granule neurons and acutely dissociated cortical neurons (Miller et al. 1992; Nishikawa et al. 1994). This enhancement of NMDA receptor-mediated currents occurred within less than a minute and usually disappeared completely when cells were returned to control solution. At AMPA receptors, by contrast, exposure to AA was found to produce a slow onset inhibition that often showed only partial recovery on washout (Kovalchuk et al. 1994). In the present study, the blockade of kainate receptors took several minutes to develop and recovery from block in control solution was extremely slow. Recovery of kainate receptors from inhibition by fatty acids was greatly speeded up, however, when cells were washed with control solution that contained BSA. Thus, inhibition is not maintained when fatty acids are efficiently removed.
Consistent with previous work on the modulation of NMDA and AMPA receptors, our results suggest that cis-unsaturated fatty acids may exert a direct inhibition of kainate receptor operation. The fact that inhibition persisted during exposure to NDGA plus indomethacin, and ODYA (Table 1) argues that conversion of fatty acid to active metabolites is not required. This conclusion is supported by the partial blockade we observed with ETYA (Fig. 3A), which inhibits the enzymes responsible for AA metabolism (Meves, 1994). Furthermore, the activation of PKC or the formation of free radicals does not appear to be necessary for fatty acid inhibition as evidenced by the weak effects of the kinase inhibitors H-7 and PKC19-36 (Hidaka et al. 1984; House & Kemp, 1987), and the free radical scavenger SOD (Keyser & Alger, 1990). At present, we have insufficient evidence to determine whether active fatty acids interact directly with an extracellular domain of the kainate receptor subunits or whether they partition into the membrane and change its local properties in the region surrounding the channel core. The reversibility of current blockade (Fig. 2B), its concentration dependence (Fig. 3C), and the structure-activity relationship for fatty acid inhibition (Fig. 3A and B) all suggest that extreme membrane perturbation or fatty acid lamella formation (Cistola et al. 1988) are not prerequisites for kainate receptor modulation. Indeed, we are not aware of any effects by fatty acids on ion channel activity that show higher potency than the inhibiton of GluR6 receptors by DHA (IC50 < 0.5 μm; cf. Meves, 1994). The 10-fold difference in potency of AA and DHA at native versus recombinant receptors may reflect the heteromeric subunit composition of neuronal receptors, as opposed to the homomeric channels formed by expression of GluR6 in HEK 293 cells. It seems less likely that there would be a 10-fold difference in fatty acid efficacy or accessibility in neurons versus HEK 293 cells that was based solely on differences in the lipid environment of the two cell types. Hippocampal neurons are thought to express GluR6 together with other kainate receptor subunits, whereas DRG neurons express predominantly GluR5 and KA2 (Hollmann & Heinemann, 1994). Further work will be needed to determine the relative potency of fatty acids for these other subunit combinations.
Petrou et al. (1993) have postulated that a specific binding site for fatty acids exists on the extracellular N-terminal domain of the NMDA NR1 subunit, based on sequence homology with a family of small, soluble fatty acid-binding proteins. Interestingly, among all other ionotropic glutamate receptor subunits, the kainate receptor GluR6 subunit displays the next strongest homology with fatty acid-binding proteins in this region (Kovalchuk et al. 1994). Direct interactions of fatty acids with glutamate receptor subunits have not yet been demonstrated. In future work, however, site-directed mutagenesis of conserved residues, which are known to play a role in fatty acid binding to the small soluble proteins (Sacchettini et al. 1989), should provide a conclusive test for involvement of this receptor domain in channel modulation.
Although the function of neuronal kainate receptors in synaptic transmission is not completely understood, recent studies have shown that they may play both a presynaptic and a postsynaptic role. Thus, the strength of transmission at both excitatory (Chittajallu et al. 1996) and inhibitory (Rodriguez-Moreno et al. 1997; Clarke et al. 1997) synapses is reduced when kainate receptors are selectively activated by exogenous agonists, an effect that is thought to involve presynaptic regulation of transmitter release. Moreover, at specific excitatory synapses, including primary afferent inputs in the spinal cord (P. Li, T. J. Wilding, S. Kim, A. Calejesan, J. E. Huettner & M. Zhuo, unpublished observations) and the mossy fibre pathway from dentate granule cells to CA3 neurons (Vignes & Collingridge, 1997; Castillo et al. 1997), a component of postsynaptic current appears to be mediated by kainate receptors. In this light, it is interesting to consider the consequences that local release of AA or DHA might have on synaptic transmission. Because of the numerous channel types that can be modulated by these fatty acids (Meves, 1994), a complete analysis of their actions will be difficult. However, focusing only on transmitter receptors, it seems likely that transmission through NMDA receptors will be enhanced during acute exposure to fatty acids, whereas transmission mediated by AMPA, kainate and GABAA receptors will be reduced. This combination of effects could help to preserve a strong contribution by NMDA receptors in the face of a net reduction in excitatory drive. In most cases, a reduction in excitatory current through AMPA and kainate receptors would be expected to result in weaker signalling via NMDA receptors, owing to the voltage-dependent blockade of NMDA receptor channels by Mg2+. However, both the direct potentiation of NMDA receptors and the inhibition of GABAA receptors by fatty acids would help to compensate for this effect. In particular, a reduction in GABAA receptor-mediated inhibition would serve to prolong individual EPSPs (Bliss & Collingridge, 1993) and thus take better advantage of the slow kinetics of NMDA receptor channels (Lester et al. 1990).
Acknowledgments
This work was supported by the NIH (NS30888) and by the McDonnell Center for Cellular and Molecular Neurobiology. We are grateful to Dr David Leander of Eli Lilly and Company for LY300168 (GYKI 53655), to Dr Steven Heinemann for providing the clone of GluR6, to Dr David Clapham for providing the vector containing L3T4, to Dorothy Turetsky for HEK 293 cells, to Elizabeth Stack for subcloning GluR6 and to our colleagues in the Department of Cell Biology and Physiology for advice on molecular biology. We also thank Dr David Cistola and Michael Finley of Washington University for many helpful discussions and for critical reading of the manuscript.
References
- Attwell D, Miller B, Sarantis M. Arachidonic acid as a messenger in the central nervous system. Seminars in the Neurosciences. 1993;5:159–169. [Google Scholar]
- Barbour B, Szatkowski M, Ingledew N, Attwell D. Arachidonic acid induces a prolonged inhibition of glutamate uptake into glial cells. Nature. 1989;342:918–920. doi: 10.1038/342918a0. [DOI] [PubMed] [Google Scholar]
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. doi: 10.1038/361031a0. [DOI] [PubMed] [Google Scholar]
- Castillo PE, Malenka RC, Nicoll RA. Kainate receptors mediate a slow postsynaptic current in hippocampal CA3 neurons. Nature. 1997;388:182–186. doi: 10.1038/40645. 10.1038/40645. [DOI] [PubMed] [Google Scholar]
- Chittajallu R, Vignes M, Dev KK, Barnes JM, Collingridge GL, Henley JM. Regulation of glutamate release by presynaptic kainate receptors in the hippocampus. Nature. 1996;379:78–81. doi: 10.1038/379078a0. [DOI] [PubMed] [Google Scholar]
- Cistola DP, Hamilton JA, Jackson D, Small DM. Ionization and phase behavior of fatty acids in water: application of the Gibbs phase rule. Biochemistry. 1988;27:1881–1888. doi: 10.1021/bi00406a013. [DOI] [PubMed] [Google Scholar]
- Clarke VRJ, Ballyk BA, Hoo KH, Mandelezys A, Pellizzari A, Bath CP, Thomas J, Sharpe EF, Davies CH, Ornstein PL, Schoepp DD, Kamboj RK, Collingridge GL, Lodge D, Bleakman D. A hippocampal GluR5 kainate receptor regulating inhibitory synaptic transmission. Nature. 1997;389:599–603. doi: 10.1038/39315. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Pin J-P, Oomagari K, Sebben M, Bockaert J. Arachidonic acid released from striatal neurons by joint stimulation of ionotropic and metabotropic quisqualate receptors. Nature. 1990;347:182–184. doi: 10.1038/347182a0. 10.1038/347182a0. [DOI] [PubMed] [Google Scholar]
- Dumuis A, Sebben M, Haynes L, Pin J-P, Bockaert J. NMDA receptors activate the arachidonic acid cascade in striatal neurons. Nature. 1988;336:68–70. doi: 10.1038/336068a0. 10.1038/336068a0. [DOI] [PubMed] [Google Scholar]
- Egebjerg J, Bettler B, Hermans-Borgmeyer I, Heinemann S. Cloning of a cDNA for a glutamate receptor subunit activated by kainate but not AMPA. Nature. 1991;351:745–748. doi: 10.1038/351745a0. 10.1038/351745a0. [DOI] [PubMed] [Google Scholar]
- Hamano H, Nabekura J, Nishikawa M, Ogawa T. Docosahexaenoic acid reduces GABA response in substantia nigra neuron of rat. Journal of Neurophysiology. 1996;75:1264–1270. doi: 10.1152/jn.1996.75.3.1264. [DOI] [PubMed] [Google Scholar]
- Hidaka H, Inagaki M, Kawamoto S, Sasaki Y. Isoquinolinesulfonamides, novel and potent inhibitors of cyclic nucleotide dependent protein kinase and protein kinase C. Biochemistry. 1984;23:5036–5041. doi: 10.1021/bi00316a032. [DOI] [PubMed] [Google Scholar]
- Hollmann M, Heinemann S. Cloned glutamate receptors. Annual Review of Neuroscience. 1994;17:31–108. doi: 10.1146/annurev.ne.17.030194.000335. 10.1146/annurev.ne.17.030194.000335. [DOI] [PubMed] [Google Scholar]
- House C, Kemp BE. Protein kinase C contains a pseudosubstrate prototope in its regulatory domain. Science. 1987;238:1726–1728. doi: 10.1126/science.3686012. [DOI] [PubMed] [Google Scholar]
- Huettner JE. Glutamate receptor channels in rat DRG neurons: Activation by kainate and quisqualate and blockade of desensitization by Con A. Neuron. 1990;5:255–266. doi: 10.1016/0896-6273(90)90163-a. 10.1016/0896-6273(90)90163-A. [DOI] [PubMed] [Google Scholar]
- Huettner JE, Bean BP. Block of N-methyl-D-aspartate-activated current by the anticonvulsant MK-801: Selective binding to open channels. Proceedings of the National Academy of Sciences of the USA. 1988;85:1307–1311. doi: 10.1073/pnas.85.4.1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyser DO, Alger BE. Arachidonic acid modulates hippocampal calcium current via protein kinase C and oxygen radicals. Neuron. 1990;5:545–553. doi: 10.1016/0896-6273(90)90092-t. 10.1016/0896-6273(90)90092-T. [DOI] [PubMed] [Google Scholar]
- Kim D, Clapham DE. Potassium channels in cardiac cells activated by arachidonic acid and phospholipids. Science. 1989;244:1174–1176. doi: 10.1126/science.2727703. [DOI] [PubMed] [Google Scholar]
- Kovalchuk Y, Miller B, Sarantis M, Attwell D. Arachidonic acid depresses non-NMDA receptor currents. Brain Research. 1994;643:287–295. doi: 10.1016/0006-8993(94)90035-3. [DOI] [PubMed] [Google Scholar]
- Lester RAJ, Clements JD, Westbrook GL, Jahr CE. Channel kinetics determine the time course of NMDA receptor-mediated synaptic currents. Nature. 1990;346:565–567. doi: 10.1038/346565a0. 10.1038/346565a0. [DOI] [PubMed] [Google Scholar]
- Linden DJ, Routtenberg A. cis-Fatty acids, which activate protein kinase C, attenuate Na+ and Ca2+ currents in mouse neuroblastoma cells. The Journal of Physiology. 1989;419:95–119. doi: 10.1113/jphysiol.1989.sp017863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meves H. Modulation of ion channels by arachidonic acid. Progress in Neurobiology. 1994;43:175–186. doi: 10.1016/0301-0082(94)90012-4. 10.1016/0301-0082(94)90012-4. [DOI] [PubMed] [Google Scholar]
- Miller B, Sarantis M, Traynelis SF, Attwell D. Potentiation of NMDA receptor currents by arachidonic acid. Nature. 1992;355:722–725. doi: 10.1038/355722a0. 10.1038/355722a0. [DOI] [PubMed] [Google Scholar]
- Nishikawa M, Kimura S, Akaike N. Facilitatory effect of docosahexaenoic acid on N-methyl-D-aspartate response in pyramidal neurones of rat cerebral cortex. The Journal of Physiology. 1994;475:83–93. doi: 10.1113/jphysiol.1994.sp020051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ordway RW, Singer JJ, Walsh JV., Jr Direct regulation of ion channels by fatty acids. Trends in Neurosciences. 1991;14:96–100. doi: 10.1016/0166-2236(91)90069-7. 10.1016/0166-2236(91)90069-7. [DOI] [PubMed] [Google Scholar]
- Ordway RW, Walsh JV, Jr, Singer JJ. Arachidonic acid and other fatty acids directly activate potassium channels in smooth muscle cells. Science. 1989;244:1176–1179. doi: 10.1126/science.2471269. [DOI] [PubMed] [Google Scholar]
- Paternain AV, Morales M, Lerma J. Selective antagonism of AMPA receptors unmasks kainate receptor-mediated responses in hippocampal neurons. Neuron. 1995;14:185–189. doi: 10.1016/0896-6273(95)90253-8. 10.1016/0896-6273(95)90253-8. [DOI] [PubMed] [Google Scholar]
- Petrou S, Ordway RW, Singer JJ, Walsh JV., Jr A putative fatty acid-binding domain of the NMDA receptor. Trends in Biochemical Sciences. 1993;18:41–42. doi: 10.1016/0968-0004(93)90050-w. 10.1016/0968-0004(93)90050-W. [DOI] [PubMed] [Google Scholar]
- Piomelli D, Volterra A, Dale N, Siegelbaum SA, Kandel ER, Schwartz JH, Belardetti F. Lipoxygenase metabolites of arachidonic acid as second messengers for presynaptic inhibition of Aplysia sensory cells. Nature. 1987;328:38–43. doi: 10.1038/328038a0. 10.1038/328038a0. [DOI] [PubMed] [Google Scholar]
- Richieri GV, Anel A, Kleinfeld AM. Interactions of long-chain fatty acids and albumin: determination of free fatty acid levels using the fluorescent probe ADIFAB. Biochemistry. 1993;32:7574–7580. doi: 10.1021/bi00080a032. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Moreno A, Herreras O, Lerma J. Kainate receptors presynaptically downregulate GABAergic inhibition in the rat hippocampus. Neuron. 1997;19:893–901. doi: 10.1016/s0896-6273(00)80970-8. 10.1016/S0896-6273(00)80970-8. [DOI] [PubMed] [Google Scholar]
- Sacchettini JC, Gordon JI, Banaszak LJ. Crystal structure of rat intestinal fatty-acid-binding protein. Journal of Molecular Biology. 1989;208:327–339. doi: 10.1016/0022-2836(89)90392-6. [DOI] [PubMed] [Google Scholar]
- Schmitt H, Meves H. Protein kinase C as a mediator of arachidonic acid-induced decrease of neuronal M current. Pflügers Archiv. 1993;425:134–139. doi: 10.1007/BF00374513. [DOI] [PubMed] [Google Scholar]
- Schwartz RD, Yu X. Inhibition of GABA-gated chloride channel function by arachidonic acid. Brain Research. 1992;585:405–410. doi: 10.1016/0006-8993(92)91246-b. [DOI] [PubMed] [Google Scholar]
- Shinomura T, Asaoka Y, Oka M, Yoshida K, Nishizuka Y. Synergistic action of diacylglycerol and unsaturated fatty acid for protein kinase C activation: Its possible implications. Proceedings of the National Academy of Sciences of the USA. 1991;88:5149–5153. doi: 10.1073/pnas.88.12.5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tourvieille B, Gorman SD, Field EH, Hunkapiller T, Parnes JR. Isolation and sequence of L3T4 complementary DNA clones: expression in T cells and brain. Science. 1986;234:610–614. doi: 10.1126/science.3094146. [DOI] [PubMed] [Google Scholar]
- Vignes M, Collingridge GL. The synaptic activation of kainate receptors. Nature. 1997;388:179–182. doi: 10.1038/40639. [DOI] [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE. Differential antagonism of AMPA-preferring and kainate-preferring receptors by 2,3-benzodiazepines. Molecular Pharmacology. 1995;47:582–587. [PubMed] [Google Scholar]
- Wilding TJ, Huettner JE. Activation and desensitization of hippocampal kainate receptors. Journal of Neuroscience. 1997;17:2713–2721. doi: 10.1523/JNEUROSCI.17-08-02713.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou A-P, Ma Y-H, Sui Z-H, Ortiz De Montellano PR, Clark JE, Masters BS, Roman RJ. Effects of 17-octadecynoic acid, a suicide-substrate inhibitor of cytochrome P450 fatty acid ω-hydroxylase, on renal function in rats. Journal of Pharmacology and Experimental Therapeutics. 1994;268:474–481. [PubMed] [Google Scholar]