

Abstract
Mitochondrial uncouplers are potent stimulants of the carotid body. We have therefore investigated their effects upon isolated type I cells. Both 2,4-dinitrophenol (DNP) and carbonyl cyanide p-trifluoromethoxyphenyl hydrazone (FCCP) caused an increase in [Ca2+]i which was largely inhibited by removal of extracellular Ca2+ or Na+, or by the addition of 2 mm Ni2+. Methoxyverapamil (D600) also partially inhibited the [Ca2+]i response.
In perforated-patch recordings, the rise in [Ca2+]i coincided with membrane depolarization and was greatly reduced by voltage clamping the cell to −70 mV. Uncouplers also inhibited a background K+ current and induced a small inward current.
Uncouplers reduced pHi by 0.1 unit. Alkaline media diminished this acidification but had no effect on the [Ca2+]i response.
FCCP and DNP also depolarized type I cell mitochondria. The onset of mitochondrial depolarization preceded changes in cell membrane conductance by 3–4 s.
We conclude that uncouplers excite the carotid body by inhibiting a background K+ conductance and inducing a small inward current, both of which lead to membrane depolarization and voltage-gated Ca2+ entry. These effects are unlikely to be caused by cell acidification. The inhibition of background K+ current may be related to the uncoupling of oxidative phosphorylation.
Mitochondrial uncouplers have long been known to be extremely potent stimulants of the carotid body causing a rapid and intense increase in neural discharge in the carotid sinus nerve (Shen & Hauss, 1939; Krylov & Anichkov, 1968; Nye & Torrance, 1980; Mulligan & Lahiri, 1981). More recently it has been shown that 2,4-dinitrophenol (DNP) also causes intense neurosecretion from isolated rabbit type I cells (Obeso et al. 1989; Rocher et al. 1991) suggesting that the excitatory effects of uncouplers are mediated through the type I cell. Although it is largely unknown how these agents act, two disparate theories have emerged. One proposal is that uncouplers excite the carotid body by inhibiting oxidative phosphorylation (Krylov & Anichkov, 1968). Indeed the potent excitatory actions of inhibitors of oxidative phosphorylation are a mainstay of the metabolic hypothesis of O2 chemoreception (Anichkov & Belen'kii, 1963). A more recent proposal, however, is that uncouplers act primarily as acidic stimuli (Rocher et al. 1991; Gonzalez et al. 1992, 1994). It is proposed that, as uncouplers are by nature protonophores, they will induce a large fall in intracellular pH. On the basis of this DNP has been used in studies of acid chemoreception (Rocher et al. 1991). Thus, a dichotomy exists as to whether uncouplers are regarded as surrogate acidic or hypoxic stimuli. Like physiological stimuli, uncouplers provoke a substantial increase in intracellular calcium in type I cells (Biscoe & Duchen, 1990a, b; Duchen & Biscoe, 1992; Buckler & Vaughan-Jones, 1993b), but there is disagreement as to how they accomplish this. Whereas both acidosis and hypoxia evoke membrane depolarization and voltage-gated calcium entry (Buckler & Vaughan-Jones, 1994a, b), uncouplers have been proposed to either provoke calcium release from intracellular stores (Biscoe & Duchen, 1990a, b; Duchen & Biscoe, 1992) or promote reverse mode sodium-calcium exchange (Rocher et al. 1991).
In view of the fact that the actions of uncouplers might provide vital clues as to the mechanisms of either acidic or hypoxic chemoreception we have reinvestigated the effects of these compounds upon isolated rat type I cells.
A preliminary account of some of this work has previously been published (Buckler & Vaughan-Jones, 1993b).
METHODS
Cell isolation
Experiments were performed on type I cells enzymically isolated from the carotid bodies of neonatal rats (10–16 days old). The methods of cell isolation have been described in detail elsewhere (Buckler & Vaughan-Jones, 1993a). Briefly, rat pups were anaesthetized with 4 % fluothane in oxygen and the carotid bodies quickly excised and placed in ice-cold phosphate-buffered saline. The rats were then killed by decapitation. The carotid bodies were incubated in phosphate-buffered saline containing collagenase (0.4 mg ml−1, Type 1, Worthington) and trypsin (0.2 mg ml−1 Sigma) and then mechanically dispersed by the use of forceps followed by trituration through fire-polished glass pipettes. The cell suspension was then centrifuged, resuspended in culture medium (Ham's F-12 supplemented with 10 % (v/v) heat-inactivated fetal calf serum, 100 i.u. ml−1 penicillin, 100 μg ml−1 streptomycin and 84 U l−1 insulin) and plated out onto glass coverslips coated with poly-D-lysine (Sigma). Cells were maintained in culture medium until use (4–36 h).
Measurement of intracellular calcium
Cells were loaded with indo-1 by incubation in a solution of 2.5 μm indo-1 AM in culture medium at room temperature (18–25°C) for 1 h (indo-1 AM was added from a 1 mm stock solution in DMSO). Following this loading protocol > 80 % of the total cell fluorescence was rapidly lost upon treatment of the cells with 10 μm digitonin, indicating that minimal amounts of indo-1 were compartmentalized. Indo-1 fluorescence was excited at 340 ± 5 nm and recorded at 405 ± 16 and 495 ± 10 nm using photomultiplier tubes. The signal for each wavelength was averaged over 500 ms intervals, and the ratio (R) was then calculated and calibrated. In some experiments the data were further filtered by a three point rolling average. The calibration constants Rmin, Rmax and F495, free/bound for indo-1 were obtained from an in situ calibration protocol using ionomycin in a separate group of cells, and Kd was assumed to be 250 nm (see Grynkiewicz et al. 1985; Buckler & Vaughan-Jones, 1993a).
Measurement of intracellular pH
Intracellular pH (pHi) was determined using the pH-sensitive dual emission fluoroprobe carboxy-SNARF-1 (Molecular Probes). Cells were loaded with SNARF-1 by incubation for 8 min in a 5 μm solution of the acetoxy-methyl ester SNARF-1 AM in Hepes-buffered Tyrode solution (see below) at room temperature. SNARF-1-loaded cells were illuminated at 540 ± 12 nm. SNARF-1 fluorescence was measured at 590 ± 5 nm and 640 ± 5 nm. The collected data from each photomultiplier tube were integrated over 500 ms intervals, ratioed, calibrated (see below) and then further filtered with a three to five point rolling average. The 590 nm/640 nm emission ratio was calibrated using nigericin (see Buckler & Vaughan-Jones, 1990). Nigericin calibration solutions contained: KCl, 140 mm; MgCl2, 1.0 mm; nigericin, 10 μm; buffered with one of the following organic buffers (all from Sigma): 20 mm Mes (pH 5.5), 20 mm Hepes (pH 7.5) or 20 mm CAPSO (pH 9.5) and was adjusted to the desired pH at 37°C with NaOH.
Rhodamine-123 fluorescence
Cells were loaded with rhodamine-123 (Rh-123) at 10–15 μg ml−1 in bicarbonate-buffered Tyrode solution at room temperature for 12–15 min. Rh-123 was excited at 485 nm and fluorescence recorded at 535 ± 10 nm.
Electrophysiology
All voltage- and current-clamp recordings were performed using the perforated-patch whole-cell recording technique. Experiments were conducted using an Axopatch-1D patch-clamping system (Axon Instruments). Voltage-clamp commands were generated from a program written by one of the authors (K. J. B.) using an analog-to-digital and digital-to-analog converter (CED1401; Cambridge Electronic Design, Cambridge, UK). It should be noted that all voltage ramps are, therefore, staircases with a step amplitude of 0.78 mV.
Electrodes were fabricated from either Corning 7052 (World Precision Instruments) or Clarke CG150 borosilicate glass capillaries. In some cases electrodes were coated with Sylgard 184 (Dow Corning). Electrodes were fire polished before use. The following filling solutions were used. Perforated-patch filling solution A (mm): potassium gluconate, 140; MgCl2, 5; EGTA, 1; Hepes, 10; adjusted to pH 7.2–7.3 with NaOH (approximately 7 mm). Perforated-patch filling solution B (mm): K2SO4, 70; KCl, 30; MgCl2, 2; EGTA, 1; Hepes, 10; adjusted to pH 7.2–7.3 with NaOH. Perforated-patch filling solution C (mm): K2SO4, 55; KCl, 30; MgCl2, 2; EGTA, 1; Hepes, 10; adjusted to pH 7.2–7.3 with NaOH. Filling solutions also contained 120–240 μg ml−1 amphotericin B (added from a stock solution of 60 mg ml−1 in dimethylsulfoxide). Liquid junction potentials in standard bicarbonate-buffered Tyrode solutions, measured relative to a 3 m KCl reference electrode, were approximately 6 mV for pipette filling solution A and 3 mV for filling solutions B and C. Membrane potentials are presented without correction for liquid junction potentials (corrections can be made by subtracting 6 or 3 mV from the membrane potentials reported).
For electrophysiological recordings we used either a 3 m KCl reference electrode (and amplifier BH-1, Axon Instruments) separate from the bath ground (Ag-AgCl pellet) or the bath was earthed via a 3 m KCl bridge to the Ag-AgCl pellet.
Membrane potential and current were filtered at 1 kHz (3 dB) and digitized at 2–4 kHz via a CED1401 and a program written by K. J. B. Current-voltage relationships were constructed as follows. Cells were subjected to repetitive voltage ramps and the resultant current records from each cell were averaged. Then, since the ramps were essentially staircases (see above), the current was averaged for each step. Finally, mean current-voltage relationships were calculated by averaging the individual current-voltage relationship obtained from each cell in the study.
Solutions and reagents
The standard HCO3−-buffered Tyrode solution contained (mm): NaCl, 117; KCl, 4.5; NaHCO3, 23; MgCl2, 1.0; CaCl2, 2.5; glucose, 11; equilibrated with 5 % CO2-95 % air, pH 7.4–7.45. Ca2+-free solutions lacked CaCl2 and contained 1 mm EGTA. Na+-free solutions lacked both NaCl and NaHCO3 and contained 140 mm NMDG. This solution was made by first adding 117 mm NMDG and adjusting the pH to 7.0 with HCl, then a further 23 mm NMDG was added and the solution continuously bubbled with 5 % CO2 for 1 h until the pH stabilized at around 7.4 (with concomitant generation of HCO3−). Finally, if necessary, any minor adjustments to bring the pH of this solution to 7.4 were made by addition of further HCl or NMDG. Ba2+-containing solutions included 5 mm BaCl2 but contained no CaCl2. DNP was added either as a solid or from a 25 mm stock solution in water. Carbonyl cyanide p-trifluoromethoxyphenyl hydrazone (FCCP), methoxyverapamil (D600) and nifedipine were added from 10 mm stock solutions in ethanol. Solutions were superfused at ∼2–3 ml min−1 through a recording chamber with a volume of ∼80 μl. All experiments were conducted at 33–37°C. Collagenase was obtained from Worthington. Trypsin, culture media, D600, nifedipine, FCCP and DNP were obtained from Sigma/Aldrich. Indo-1 AM was obtained from Calbiochem. SNARF-1 AM and Rh-123 were obtained from Molecular Probes.
Statistics
All data are presented as means ± s.e.m. Significance of differences was assessed using Student's paired t test (unless otherwise stated).
RESULTS
Effects of uncouplers on [Ca2+]i
Application of either DNP or FCCP caused a dose-dependent increase in type I cell [Ca2+]i (see Fig. 1). With DNP, a response was evident in one of three cells tested at 25 μm and four of five cells at 50 μm. DNP-evoked [Ca2+]i responses were maximal at 250 μm (Δ[Ca2+]i: 638 ± 167 nm at 250 μm, 599 ± 184 nm at 500 μm, n = 5 cells, not significant (n.s.)). With FCCP, responses were evident in five of six cells at 0.1 μm and were near-maximal at 0.3 μm (Δ[Ca2+]i: 501 ± 76 nm at 0.3 μm, 614 ± 134 nm at 1.0 μm, n = 6, n.s.). In the following studies DNP and FCCP were used at 250 μm and 0.3 μm, respectively. With both agents, at these concentrations, the rise in [Ca2+]i typically began within 5–10 s after switching to solutions containing uncouplers. Note that part of the observed delay was due to the time taken for solutions to reach and exchange in the recording chamber; a more detailed analysis of the time course of the response to FCCP is given later (see Fig. 10). The rise in [Ca2+]i was also readily reversed upon removal of the uncoupler. These responses are similar to those previously reported in rabbit type I cells when challenged with FCCP (Biscoe & Duchen, 1990a; Duchen & Biscoe, 1992). They are also consistent with reports that uncouplers are rapidly acting and potent chemostimulants (Shen & Hauss, 1939; Krylov & Anichkov, 1968).
Figure 1. Effects of uncouplers on [Ca2+]i in type I cells.
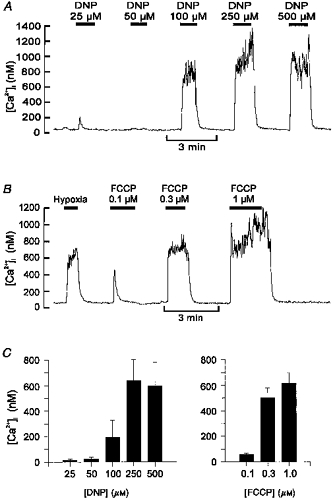
Recordings of [Ca2+]i in individual type I cells and their responses to graded levels of DNP (A) or FCCP (B). Data in B also include a control response to a hypoxic (PO2, ∼5 Torr) stimulus for comparison. C, mean [Ca2+]i responses to graded levels of DNP (n = 3 at 25 μm, n = 5 all other DNP data) and FCCP (n = 6). Error bars indicate s.e.m.
Figure 10. Effects of FCCP on membrane properties and mitochondrial potential.
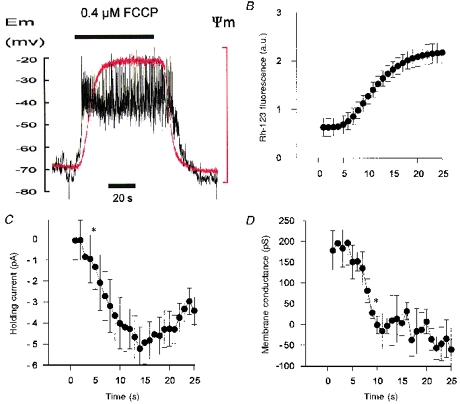
A, simultaneous recording of membrane potential (Vm, black trace) and mitochondrial membrane potential (Ψm, red trace, note that upward deflection equals depolarization) during application of 0.4 μm FCCP. B, C and D, time course of change in mitochondrial potential (Rh-123 fluorescence in arbitrary units, a.u.), holding current and membrane conductance, respectively, following application of 0.4 μm FCCP. Mean ± s.e.m. values from five cells. Asterisks indicate the first data point that is significantly different (P < 0.05) from the control level (control equals data obtained in the first second, i.e. just before FCCP actually reaches the recording chamber). All data were recorded simultaneously in cells voltage clamped at −70 mV and subjected to 500 ms voltage ramps from −100 to −50 mV at 1 Hz. Membrane conductance was calculated by fitting a linear regression over the range −60 to −50 mV). Electrophysiological recordings were performed using the perforated-patch technique with pipette filling solution C.
Cause of uncoupler-induced rise in [Ca2+]i
In order to determine whether the effects of uncouplers were mediated through the release of Ca2+ from intracellular stores or through Ca2+ influx, we studied the effects of both Cao2+ removal and the addition of a non-selective antagonist of calcium entry (Ni2+). The responses to both DNP and FCCP were almost completely ablated in Ca2+-free medium containing 1 mm EGTA (see Fig. 2A and B; Δ[Ca2+]i (control vs. Ca2+-free conditions): for DNP, 336 ± 64 vs. 31 ± 5.2 nm, n = 7, P < 0.005; for FCCP, 1869 ± 320 vs. 69 ± 7 nm, n = 5, P < 0.005). In addition, the response to DNP was greatly reduced by 2 mm Ni2+ (Fig. 2C; Δ[Ca2+]i (control vs. with 2 mm Ni2+): 550 ± 70 vs. 76 ± 4.5 nm, n = 4, P < 0.01). These data indicate that the [Ca2+]i response to uncouplers is mediated mostly through enhanced calcium influx and not by direct release from intracellular stores. One possible route of Ca2+ entry is via reversal of a Na+-Ca2+ exchanger in response to a proton influx causing cellular acidification, activation of Na+-dependent acid extruders (e.g. Na+-H+ exchange), and a rise in [Na+]i. Indeed, it has been shown that the secretory response to DNP is inhibited in the absence of extracellular Na+ (Rocher et al. 1991). We therefore, tested the effects of sodium removal upon the [Ca2+]i response to DNP. Figure 3A shows that replacing with NMDG caused a substantial inhibition of the [Ca2+]i response to DNP (Δ[Ca2+]i (control vs. Na+-free conditions): 447 ± 71 vs. 58 ± 14 nm, n = 4, P < 0.05). These data are consistent with the rise in [Ca2+]i being due to reverse mode Na+-Ca2+ exchange (Rocher et al. 1991). We have, however, previously reported that Na+-free conditions inhibit the [Ca2+]i response to physiological acidic stimuli via an alternative mechanism. The [Ca2+]i response to hypercapnic acidosis is due to membrane depolarization and voltage-gated Ca2+ influx. In Na+-free conditions the membrane hyperpolarizes such that the threshold for voltage-gated Ca2+ entry is not reached and the [Ca2+]i response to hypercapnia is inhibited (Buckler & Vaughan-Jones, 1994b). Thus, the inhibitory effects of sodium removal upon the [Ca2+]i response to DNP are equally consistent with a depolarization-evoked Ca2+ influx through voltage-gated channels. We therefore tested this alternative hypothesis by looking at the effects of Ca2+ channel inhibitors upon the response to DNP and FCCP (Fig. 3B and C). D600 (10 μm) reduced the [Ca2+]i response to DNP by about 55 % (Δ[Ca2+]i (control vs. with D600): 797 ± 271 vs. 326 ± 128 nm, n = 5, P < 0.05), and the response to FCCP by 56 % (Δ[Ca2+]i (control vs. with D600): 1903 ± 533 vs. 831 ± 194 nm, n = 6, P < 0.05). Nifedipine (10 μm) was less effective, reducing the DNP response by only 30 % (Δ[Ca2+]i (control vs. nifedipine): 585 ± 141 vs. 420 ± 68 nm, n = 11, n.s, not shown).
Figure 2. Uncoupler-evoked rise in [Ca2+]i is dependent upon Ca2+ influx.
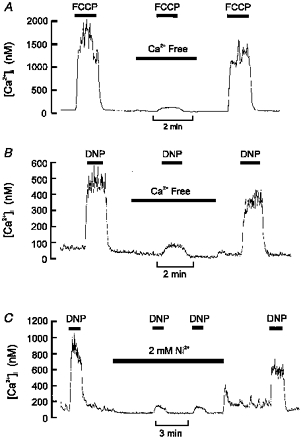
A, effect of removal of extracellular Ca2+ (+ 1 mm EGTA) on [Ca2+]i response to 0.3 μm FCCP. B, effect of removal of extracellular Ca2+ on [Ca2+]i response to 250 μm DNP. C, effect of 2 mm Ni2+ on [Ca2+]i response to 250 μm DNP. Note that both Ca2+ removal and 2 mm Ni2+ substantially inhibited the [Ca2+]i response to uncouplers. A small residual rise in [Ca2+]i remained in most cells.
Figure 3. Effects of Na+ removal and Ca2+ channel antagonists on [Ca2+]i response to uncouplers.
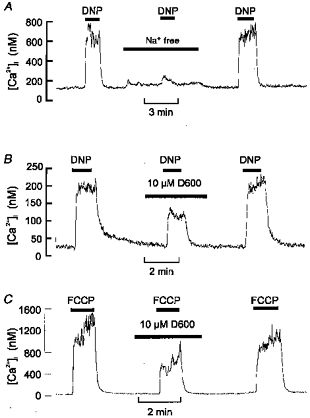
A, effects of removal of extracellular Na+ (replaced with NMDG) on the [Ca2+]i response to 250 μm DNP. B and C, effects of 10 μm D600 on the [Ca2+]i response to 250 μm DNP (B) or 0.3 μm FCCP (C). Note that both removal of extracellular Na+ and addition of D600 inhibited the response to uncoupler (see text for statistics).
The fact that D600 reduces the [Ca2+]i response to DNP and FCCP suggests that uncouplers may evoke some voltage-gated Ca2+ entry. The effect of uncouplers upon membrane potential was therefore determined using the perforated-patch whole-cell recording technique in conjunction with simultaneous measurement of intracellular calcium. Figure 4 shows that under current-clamp conditions (membrane current, Im = 0) both DNP and FCCP evoked a substantial membrane depolarization (membrane potential, Vm (control vs. with uncoupler): for DNP, −51 ± 3.4 vs.−35 ± 2.4 mV, n = 8, P < 0.005; for FCCP, −64 ± 1 vs.−34 ± 3.3 mV, n = 6, P < 0.005). In half of the cells studied uncouplers also evoked spontaneous action potentials, although these were rarely sustained throughout the application of the stimulus. The membrane depolarization rapidly reversed following the removal of uncoupler and, in some cells, led on to a brief hyperpolarization (see Fig. 4). The uncoupler-induced depolarization was accompanied by a rapid rise in [Ca2+]i (Δ[Ca2+]i: for DNP, 537 ± 91 nm; for FCCP, 1448 ± 295 nm). When the same cells were voltage clamped, thus preventing membrane depolarization, the [Ca2+]i response to uncoupler was greatly reduced compared with that under current-clamp conditions (Δ[Ca2+]i: for DNP, 140 ± 35 nm, P < 0.005; for FCCP, 108 ± 16 nm, P < 0.01). These data indicate that the effects of DNP and FCCP upon [Ca2+]i are mediated primarily by membrane depolarization and voltage-gated Ca2+ entry.
Figure 4. Effects of uncouplers on membrane potential and [Ca2+]i.
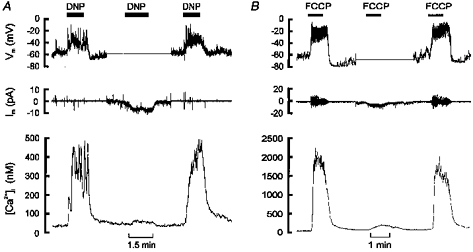
A and B, upper panels show recordings of membrane potential (perforated-patch technique), middle panels membrane current and lower panels [Ca2+]i recorded simultaneously in single type I cells. Each experiment started and finished in current-clamp mode (Im, 0) and shows both a membrane depolarization and a rise in [Ca2+]i in response to uncoupler (250 μm DNP in A, 0.3 μm FCCP in B). In the middle section of each experiment (corresponding to the flat portion of the membrane potential trace) the cells were voltage clamped to −60 (A) or −70 mV (B). Under voltage-clamp conditions DNP and FCCP evoked an inward shift in holding current, consistent with the depolarization seen under current-clamp conditions, and a greatly attenuated rise in [Ca2+]i. Pipette filling solutions A (A) and C (B).
Effects of uncoupler upon resting membrane conductance
Figure 4 also shows that, under voltage-clamp conditions, DNP and FCCP caused a small inward shift in holding current (consistent with the depolarization seen under current-clamp conditions). In order to determine the cause of this inward current, and consequently the cause of membrane depolarization, we measured the effects of DNP and FCCP on the resting membrane conductance of type I cells. Type I cells were voltage clamped, using the perforated-patch technique, and subjected to repetitive voltage ramps from −90 to −30 mV or −100 to −40 mV every 5 s. As can be seen from Fig. 5A, DNP caused both an inward shift in holding current and a substantial decline in the ramp current. Current-voltage (I-V) relationships constructed from these ramps showed that both uncouplers dramatically reduce the conductance of type I cells (control vs. uncoupler: with DNP, 230 ± 29 vs. 21 ± 31 pS, n = 4, P < 0.05; with FCCP, 209 ± 28 vs.−70 ± 28 pS, n = 8, P < 0.005; measured by fitting a linear regression to the I-V curves over the range −50 to −60 mV). It was also notable that whilst the I-V relationships transected the zero current axis under control conditions (zero current potential: DNP control data, −42 ± 6 mV, n = 4; FCCP control data, −63 ± 4.6 mV, n = 8), they did not do so in the presence of uncoupler indicating the absence of a stable resting potential in the range −100 to −30 mV. In two of the cells tested there was also a marked inward rectification of current at potentials positive to −40 mV in the presence of DNP (Fig. 5B). This may represent the activation of voltage-gated Ca2+ current (see Discussion).
Figure 5. Uncouplers reduce membrane conductance.
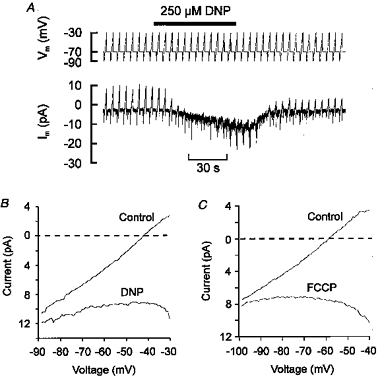
A, a single type I cell voltage clamped at −70 mV and subjected to repetitive voltage ramps (2 s duration) from −90 to −30 mV every 5 s. Pipette filling solution A. Note that DNP caused both an inward shift in holding current and a decline in the amplitude of the ramp current. B, mean current-voltage relationship (n = 4) obtained using the above protocol both under control conditions and in the presence of DNP. C, mean current-voltage relationship (n = 8) obtained using 500 ms voltage ramps from −100 to −40 mV applied every 5 s under control conditions and in the presence of 0.3 μm FCCP. Pipette filling solution C. Note: (a) that membrane conductance is greatly reduced in the presence of either DNP or FCCP; (b) that a zero current (resting) potential does not exist in this voltage range in the presence of DNP or FCCP; and (c) an inwardly rectifying current in the presence of DNP at potentials below −40 mV.
The above data suggest that the uncoupler-induced depolarization is caused by a reduction in membrane conductance resulting from the inhibition of an outward current. One obvious possible cause is the inhibition of a background K+ current. To evaluate the possible role of background K+ current in mediating the response to uncouplers, experiments were performed in which the effects of uncouplers were tested at two different levels of extracellular K+, 4.5 and 20 mm (see Fig. 6A). I-V curves were constructed both under control conditions and in the presence of uncoupler. The uncoupler-sensitive current (i.e. the current that is presumed to be inhibited by the uncoupler) was then determined by subtraction of the I-V curve obtained in the presence of uncoupler from that obtained under control conditions. In normal extracellular [K+] (4.5 mm) most cells showed no evidence of a reversal potential for the uncoupler-sensitive current over the voltage range tested (−100 to −40 mV). In high [K+]o there was a marked rightward and downward shift (towards more positive potentials and negative current) of the uncoupler-sensitive I-V relationship (Fig. 6B and C). Moreover, in 20 mm[K+]o, the uncoupler-sensitive current displayed a clear reversal potential at about −70 mV (for DNP, −71.6 ± 3.1 mV, n = 8; for FCCP, −69.1 ± 2.1 mV, n = 8). This shift in the I-V relationship with high [K+], and the existence of a definite reversal potential at −70 mV, provides clear evidence that at least part of the uncoupler-sensitive current is carried through a background K+ conductance. It was notable, however, that we regularly failed to observe any reversal of the uncoupler-sensitive current in normal [K+]o, and that reversal potentials (Vrev) in 20 mm K+ were negative to the equilibrium potential for K+ (EK) (Vrev, approximately −70 mV; EK, −55 to −50 mV). Although some allowance must be made for voltage-clamp error (due, for example, to diffusion potentials across the perforated patch), the discrepancy between observed and predicted reversal potentials was greater than that found for the oxygen-sensitive background K+ conductance (which was previously studied under the same conditions, Buckler, 1997). This discrepancy suggested that the effects of uncouplers might not be limited to the inhibition of a background K+ current alone.
Figure 6. Current-voltage relationship of uncoupler-sensitive current.
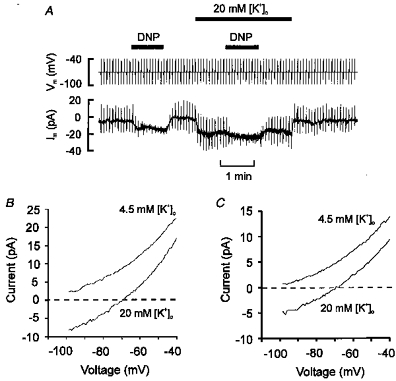
A, type I cell voltage clamped at −70 mV and subjected to 500 ms voltage ramps from −100 to −40 mV at two levels of extracellular K+ in the presence and absence of 250 μm DNP. Pipette filling solution B. B and C, mean current-voltage relationships of DNP (B)- and FCCP (C)-sensitive current obtained by subtracting the I-V relationship obtained in the presence of uncoupler from the control I-V relationship. Note the marked rightward and downward (depolarizing) shift in the uncoupler-sensitive current in high [K+]o. Note also the clear reversal potentials obtained in 20 mm[K+]o with both uncouplers. DNP, n = 8. FCCP, n = 8, pipette filling solution C.
This hypothesis was tested using Ba2+ to reduce the background (or resting) K+ conductance (K. J. Buckler, unpublished data) and thus help to resolve any other current components. Figure 7A shows the I-V relationships obtained using voltage ramps in the presence of 5 mm Ba2+ (no Ca2+). Ba2+ alone reduces the resting membrane conductance at negative potentials (339 ± 78 pS control, 155 ± 65 pS with Ba2+, n = 5, P < 0.01; measured by fitting a linear regression over the range −100 to −70 mV) and also reveals an inwardly rectifying current at potentials positive to −50 mV (presumably due to activation of voltage-gated Ca2+ channels). Under these conditions FCCP induced an inward current at all potentials but had little effect upon membrane conductance (145 ± 62 pS, over the range −100 to −70 mV, n = 5, not significantly different from Ba2+ alone). This FCCP-induced current (Fig. 7B, trace b) showed little sensitivity to changes in voltage over a range of potentials (3.5 pA at −100 mV to 4.5 pA at −70 mV) that would greatly alter the driving force for the diffusion of K+ ions (note that EK≈−90 mV). It is unlikely, therefore, that this current is primarily due to the further inhibition of K+ channels. In contrast, the Ba2+-sensitive current (Fig. 7B, trace a) is markedly dependent upon voltage over the same range with a reversal potential of approximately −90 mV, properties characteristic of a K+ current. Thus FCCP appears to induce an inward current in addition to inhibiting background K+ currents. The nature of this inward current has yet to be determined (see Discussion).
Figure 7. FCCP induces an inward current.
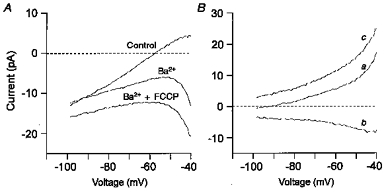
A, mean current-voltage relationship (n = 5) constructed from voltage ramps (−100 to −40 mV) applied every 5 s from a holding potential of −70 mV. Pipette filling solution C. I-V curves for control conditions and those obtained in the presence of 5 mm Ba2+ (no Ca2+) and 5 mm Ba2++ 0.3 μm FCCP are shown. B, difference currents from data in A. Ba2+-sensitive background K+ current (control minus Ba2+) (a). FCCP-induced current (Ba2++ FCCP minus Ba2+) (b). Combined FCCP-induced inward current and Ba2+-sensitive K+ current (control minus Ba2++ FCCP) (c). Note that the FCCP-induced current changed little over the voltage range shown.
Are uncouplers acidic stimuli?
Two mechanisms have been proposed to explain the potent excitatory effects of uncouplers upon the carotid body, the first of which is that they act through the inhibition of oxidative phosphorylation. Other inhibitors of oxidative phosphorylation are also known to be potent chemostimulants (Anichcov & Belen-kii, 1963). The second possibility is that as uncouplers are protonophores they might cause a large fall in intracellular pH thus stimulating the acid chemosensory pathway. The effects of uncouplers upon type I cell pH were directly investigated using SNARF-1 to monitor pHi. In these experiments, uncouplers were indeed found to cause a rapid fall in pHi (see Fig. 8A). Following the initial decrease there was, on average, no secondary pHi recovery (dpH/dt =−0.008 ± 0.008 units min−1 following DNP). The acidification was generally reversible upon removal of uncoupler although pHi did not always return to the control level, but remained slightly (0.03–0.09 units) more acidic. The fall in pHi evoked by uncoupler, however, was not very large (maximum decrease: for DNP, 0.09 ± 0.01, n = 8; for FCCP, 0.1 ± 0.02, n = 6) when compared with that caused by hypercapnic acidosis (10 % CO2) (maximum decrease: 0.23 ± 0.03, n = 7, P < 0.01 Student's unpaired t test against DNP and FCCP data).
Figure 8. Effects of DNP on pHi in comparison with other acidic stimuli.
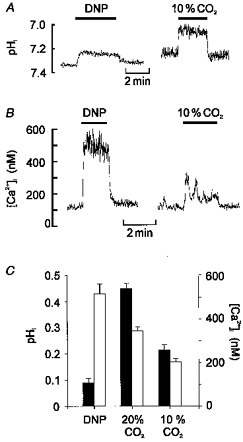
A, effects of 250 μm DNP and hypercapnic acidosis (10 % CO2, pHo 7.15) on pHi. Note that although DNP evokes a rapid fall in pHi the decrease is small in comparison to the effects of hypercapnic acidosis. B, effects of DNP and 10 % CO2 on [Ca2+]i in another cell. C, comparison of the effect of DNP upon both pHi and [Ca2+]i with those of 10 and 20 % CO2. Filled bars represent fall in pHi, open bars rise in [Ca2+]i (CO2 data from Buckler et al. 1991a; Buckler & Vaughan-Jones, 1993a).
Thus uncouplers increase cytosolic levels of both [Ca2+]i and [H+]i. Although this is consistent with the proposal that uncouplers act as acidic stimuli, we were concerned that the fall in pHi seemed rather small compared with the rise in [Ca2+]i. Indeed a comparison of the effects of DNP with data previously obtained in this laboratory using physiological acidic stimuli revealed that DNP causes a disproportionately large rise in [Ca2+]i compared with its effects upon pHi (Fig. 8C). Moreover, in experiments in which both the response to an increase in CO2 from 5 to 10 % and the response to 250 μm DNP were measured in the same cell or cell cluster (Fig. 8B), DNP always caused a substantially larger rise in calcium (peak [Ca2+]i, 492 ± 15 nm, n = 4) than did 10 % CO2 (peak [Ca2+]i, 250 ± 24 nm, P > 0.001; Student's paired t test).
Because the above results indicated that the effects of uncouplers upon [Ca2+]i are poorly correlated to their effects upon pHi we attempted to determine whether cytosolic acidification was necessary at all. Since pHi of type I cells is highly dependent upon the pH of the bathing medium (Buckler et al. 1991a), we attempted to counter the effects of DNP upon pHi by applying a modest elevation of pHo. As is shown in Fig. 9A elevating pHo to 7.7 (by doubling [HCO3−]o) caused an increase in pHi of 0.22 ± 0.03 units (n = 4; yielding a pHi/pHo slope of 0.73 which is similar to the value of 0.68 previously reported by Buckler et al. 1991a). Under these alkalotic conditions addition of DNP failed in three cases to cause any detectable fall in pHi. In a fourth cell there was a fall in pHi of 0.09 units; even in this cell, however, pHi in the presence of DNP (at an extracellular pH of 7.7) remained alkaline compared with control. This protocol therefore prevents DNP from lowering pHi to below the normal resting value. Thus, if DNP were to act primarily through cytosolic acidification, this pre-alkalinizing protocol should block the effects of DNP upon [Ca2+]i. This hypothesis was tested in a separate series of experiments in which [Ca2+]i was measured. As can be seen from Fig. 9B alkalosis had little effect upon the [Ca2+]i response to DNP (Δ[Ca2+]i (control vs. pHo 7.7): 504 ± 276 vs. 449 ± 91 nm, n = 5, n.s.). These experiments suggest that the effects of DNP upon [Ca2+]i are unlikely to be a consequence of cellular acidification.
Figure 9. Effects of extracellular alkalosis on pHi and [Ca2+]i responses to DNP.
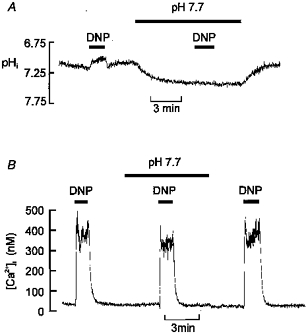
A, effects of increasing extracellular pH upon resting pHi and the response to DNP. pHo was elevated to 7.7 by doubling HCO3− (to 46 mm) with an equimolar reduction in NaCl. Note that the alkaline bathing medium increases pHi and reduces the acidification caused by DNP so that pHi remains alkaline compared with the control level. B, effects of the above alkalinizing protocol on the [Ca2+]i response to DNP. Note that pre-alkalinization does not prevent the [Ca2+]i response to DNP.
Effects of uncouplers on mitochondrial membrane potential
Since uncouplers do not appear to act through cellular acidification, we examined whether they were effective inhibitors of oxidative phosphorylation in type I cells. The effects of DNP and FCCP upon mitochondrial potential were determined by using the Rh-123 fluorescence quenching technique. Because Rh-123 is a cation it is accumulated by the mitochondrion (uptake is driven by the negative potential across the inner mitochondrial membrane). Under appropriate loading conditions, the concentration of Rh-123 within the mitochondrion reaches sufficiently high levels that it quenches its own fluorescence. If the mitochondria depolarize, however, Rh-123 leaks out into the cytoplasm. This redistribution is associated with a reduction in the amount of quenching. Changes in mitochondrial potential are, therefore, revealed as changes in the total fluorescence intensity, with an increase representing mitochondrial depolarization (see e.g. Duchen & Biscoe, 1992). Both 250 μm DNP and 1 μm FCCP caused an abrupt, and reversible, increase in Rh-123 fluorescence (190 ± 16 and 200 ± 17 % of control, respectively, n = 6; not shown). Both agents therefore depolarize type I cell mitochondria, as would be expected for protonophoretic uncouplers.
If the cellular response to uncouplers is due to their effects upon mitochondria, then mitochondrial depolarization should arguably precede, or at least be synchronous with, the electrophysiological response of the cell. We have therefore attempted to define the time course of these two events by simultaneously measuring cell membrane potential (or conductance) and mitochondrial potential. In order to optimize solution exchange time for this series of experiments the level in the recording chamber was minimized and the rate of solution exchange measured by using a fluorescent dye (Rh-123). The initial delay between operating the tap (recorded as an event marker by the data acquisition program) and the first appearance of dye in the bath was 1.4 ± 0.3 s. The half-time of solution exchange, measured from the start of the rise in fluorescence recorded in the bath was 1.0 ± 0.2 s. In the following series of experiments it therefore took approximately 2.5 s to deliver a half-maximal stimulus to the cells.
Measurement of membrane potential revealed that cell depolarization began 3.8 ± 0.6 s (n = 6) after switching to a solution containing 0.4 μm FCCP. The first signs of mitochondrial depolarization, however, were not seen until 4.6 ± 0.3 s (n = 6) after switching solution (see Fig. 10A). Although membrane depolarization appeared to precede mitochondrial depolarization in four of six cells the mean values were not significantly different. One problem with this analysis is that the inherent variability in the resting membrane potential makes determination of the precise point at which cell membrane depolarization begins highly subjective (see for example Fig. 10A). In order to circumvent this problem we also made measurements of membrane conductance and holding current in five of the same cells using 500 ms voltage ramps applied from a holding potential of −70 mV at a frequency of 1 Hz. As can be seen from Fig. 10B the first clear signs of mitochondrial depolarization are discernable in the fifth second following the solution switch. An inward shift in holding current is evident by the fourth second (or possibly even in the third, although this point just failed to reach significance). A significant decline in membrane conductance was not evident until the ninth second (membrane conductance appears to start to decline after 6–7 s). Thus, the development of an inward current would appear to precede the onset of mitochondrial depolarization whereas the decrease in membrane conductance appears to follow it (see Discussion).
DISCUSSION
Effects of uncouplers on [Ca2+]i regulation
This study shows that uncouplers evoke a rapid rise in [Ca2+]i. We propose that, as for acidic and hypoxic stimuli, this [Ca2+]i response is a fundamental part of the chemotransduction process serving as the signal which evokes neurosecretion. This is supported by the observations that both the [Ca2+]i response and DNP-evoked neurosecretion are inhibited by the removal of extracellular calcium (Rocher et al. 1991).
It is apparent that the uncoupler-induced rise in [Ca2+]i is dependent upon cell membrane depolarization (Fig. 4). We have reported previously that Ca2+ entry in response to direct depolarization of rat type I cells occurs mainly through channels with little or no contribution from electrogenic reverse mode Na+-Ca2+ exchange (Buckler & Vaughan-Jones, 1994b). Consequently, the influx seen in the presence of uncoupler is most probably mediated by voltage-gated Ca2+ channels. This is supported by the observation that D600 inhibits the [Ca2+]i response to both DNP and FCCP. The 55 % inhibition of the [Ca2+]i response (to uncoupler) by D600 is consistent with the observation that only 40–70 % of the total Ca2+ current is carried through L-type channels in rat type I cells (e Silva & Lewis, 1995). The reason why nifedipine failed to inhibit significantly the [Ca2+]i response remains unknown.
It was also notable that a small [Ca2+]i response to uncouplers remained both in Ca2+-free conditions, and in the presence of Ni2+. This suggests that some Ca2+ is also released from intracellular stores. Any such release, however, will only be a small component of the overall [Ca2+]i response. We did not attempt to determine the source of this Ca2+, but mitochondrial inhibitors have been reported to cause Ca2+ release from both mitochondrial and non-mitochondrial stores (e.g. Duchen et al. 1990; Jensen & Rehder, 1991). With respect to the role of mitochondrial uncoupler-sensitive stores it has been proposed that they play an important role in the buffering of cytoplasmic [Ca2+] during Ca2+ influx (Friel & Tsien, 1994; Werth & Thayer, 1994; Herrington et al. 1996). If so, in addition to promoting Ca2+ influx, uncouplers may also enhance the resultant [Ca2+]i rise by inhibiting Ca2+ uptake. This possibility remains to be investigated.
Effect of uncouplers upon pHi
Both DNP and FCCP caused a small fall in pHi. Although we have not directly investigated the cause of this, there are a number of possible explanations. As DNP and FCCP are membrane permeant weak acids they might be expected to cause intracellular acidification. This effect would be minimal, however, given the low concentrations used (250 μm DNP and 0.3 μm FCCP). Moreover such an effect would be expected to be transient as acid extruders would subsequently remove the excess H+. Indeed this is what happens during superfusion with other weak acids such as propionate (see Buckler & Vaughan-Jones, 1994c). In contrast, the acidification caused by DNP and FCCP was sustained. The diffusion of DNP and FCCP as weak acids alone cannot therefore account for the observed fall in pHi. Another possibility is that DNP and FCCP act as protonophores at the plasma membrane. This would generate a sustained acid influx that could displace the steady-state pHi (e.g. Boron & De Weer, 1976; Kaila et al. 1989). The final possibility is that metabolic inhibition could impair acid extrusion from the type I cells. There is growing evidence that some forms of the Na+-H+ exchanger are dependent upon intracellular ATP (Demaurex et al. 1997) and are inhibited by metabolic blockade (Wu & Vaughan-Jones, 1994). We have previously reported that Na+-H+ exchange contributes to pH regulation within the type I cell (Buckler et al. 1991b). Its inhibition could therefore contribute to cytosolic acidification.
Uncouplers: acidic or metabolic stimuli?
The hypothesis that DNP acts as a strong acidic stimulus (Rocher et al. 1991; Gonzalez et al. 1994) was not supported by our data. Firstly DNP was found to produce only a weak intracellular acidification, secondly the [Ca2+]i responses observed were disproportionate to the strength of the acidic (pHi) stimulus, and thirdly alkalinizing the cell did not significantly inhibit the [Ca2+]i response to DNP. Nonetheless, we cannot exclude the possibility that the intracellular acidification may contribute, in a small way, to the overall [Ca2+]i response. Equally, there is the possibility that a protonophoretic current induced by uncouplers may contribute to the depolarization (see below).
In contrast to their weak effects upon pHi, DNP and FCCP were very effective at depolarizing mitochondria suggesting that they might act through metabolic inhibition. In this context it should be noted that the concentration dependence of the [Ca2+]i responses to FCCP and DNP are remarkably similar to the concentration dependence for mitochondrial uncoupling, with FCCP being approximately three orders of magnitude more potent than DNP. A wide range of other mitochondrial inhibitors also elevate type I cell [Ca2+]i (Biscoe & Duchen, 1990; Duchen & Biscoe, 1992).. Similar responses have been observed to cyanide, rotenone, myxothiazole and oligomycin. All these agents raise [Ca2+]i primarily through Ca2+ influx (K. J. Buckler, unpublished observations). If the effects of uncouplers are mediated through inhibition of mitochondrial metabolism, then one might expect cell membrane depolarization to lag behind the depolarization of the mitochondrion. In a number of experiments, however, this did not appear to be the case (see Fig. 10). Some caution needs to be exercised here in comparing membrane responses with mitochondrial depolarization. The change in mitochondrial potential has been monitored using a slow, redistribution type, potential-sensitive dye (Rh-123). In experiments performed in isolated mitochondria, the time constant for redistribution of Rh-123 can be many tens of seconds and depends both upon dye concentration and the concentration of mitochondria (Bunting, 1992). It is, therefore, highly probable that the Rh-123 signal lags behind the true time course of mitochondrial depolarization. Consequently, we have used the moment at which the Rh-123 signal begins to change as an index of the time to onset of mitochondrial depolarization and have compared this with the delay in onset of change in the other parameters. From this study, it was apparent that an inward current starts to develop 1–2 s before the first detectable change in Rh-123 fluorescence. The decrease in membrane conductance, on the other hand, did not become apparent until a few seconds after the onset of mitochondrial depolarization. One possible explanation for the time lag between the onset of changes in holding current versus decrease in membrane conductance, is that FCCP may activate the inward current more rapidly than it inhibits the background K+ conductance.
In principle, the FCCP-induced inward current should be accompanied by an increase in membrane conductance. An initial increase in conductance was not seen, however, possibly because it was too small. For example, if the 1 pA inward shift in holding current seen in the first few seconds in Fig. 10C were due to a protonophoretic current, then this would require a protonophoretic conductance of only 20 pS (assuming an equilibrium potential for H+ of −12 mV).
The rapid activation of the FCCP-evoked inward current suggests that it may not be dependent upon mitochondrial depolarization, although there must remain some uncertainty due to the slow response time of the Rh-123 signal. The fact that the decrease in membrane conductance does not appear until after the onset of mitochondrial depolarization, is consistent with the possibility that the background K+ current is dependent upon mitochondrial metabolism. If the inhibition of metabolism is responsible for the reduction in background K+ current, however, then a rapid (i.e. within seconds) means of communication between mitochondrion and cell membrane channels must exist. How this communication is achieved is unknown although one obvious possibility is a change in cytoplasmic ATP or ADP levels. Against this hypothesis, ATP levels measured in the carotid body show little or no decrease in the presence of DNP (Obeso et al. 1989). However, this latter study did not consider the possibility that much of the ATP measured may be sequestered in secretory vesicles rather than in the cytosol. In some neurons uncouplers can cause a substantial reduction in the ATP/ADP ratio within 1 min (Budd & Nicholls, 1996). Thus a role for ATP/ADP in modulating background K+ conductance remains a possibility, although it would require extraordinarily rapid changes in ATP/ADP. Alternatively some other, as yet unidentified, pathway may exist which monitors mitochondrial function and signals changes to extra-mitochondrial targets (in this case ion channels).
Other possible explanations for the effects of uncouplers are: (1) that uncouplers directly inhibit K+ channels or (2) that a protonophore-induced H+ flux across some other membrane delimited organelle triggers a signalling pathway that is capable of regulating K+ channels. There is some precedence for the first possibility in that DNP has been reported to directly modulate (stimulate) ATP-sensitive K+ channel activity (Alekseev et al. 1997). It seems unlikely, however, that two chemically dissimilar compounds such as FCCP and DNP would directly inhibit the same background K+ current at the same concentrations as those which uncouple oxidative phosphorylation. The second possibility cannot, at this stage, be excluded.
Effects of uncouplers upon membrane currents
It has recently been reported that hypoxic depolarization of rat type I cells is mediated through the inhibition of a relatively voltage-insensitive background K+ current (Buckler, 1997). We therefore attempted to determine whether the same was true of the depolarization caused by uncouplers. The current-voltage curves of uncoupler-sensitive current showed a marked positive (depolarizing) shift in 20 mm[K+]o, and a reversal potential (in 20 mm K+) of −70 mV. No other cation conductance could have a reversal potential anywhere near this value, the only other ionic conductance that could would be a chloride current. There is, however, no a priori reason why changing extracellular K+ should cause such a dramatic shift in a chloride current. Another possibility is that the current is carried by a K+-dependent voltage-sensitive electrogenic transporter. The most obvious candidate is the Na+-K+ pump, but an increase in extracellular K+ would have the opposite effect upon pump current, shifting the reversal potential to more negative values. Thus, the only obvious interpretation of this data is that uncouplers inhibit a resting K+-selective channel. The extent to which uncouplers inhibit resting K+ currents can be estimated from the change in total current upon increasing extracellular K+ from 4.5 to 20 mm. For the experiments conducted with DNP, the mean decrease in membrane current at −70 mV upon increasing [K+]o to 20 mm was 16 pA under control conditions but only 7 pA in the presence of DNP. Thus DNP inhibits background K+ currents by approximately 56 % at −70 mV.
Despite the obvious inhibition of background K+ current, we often failed to observe a reversal of the uncoupler-sensitive current in normal [K+]o (at potentials positive to −100 mV), and the reversal potential in 20 mm[K+]o was significantly negative to EK. These inconsistencies prompted a search for other uncoupler-sensitive currents in addition to the background K+ current. In the presence of Ba2+ (to inhibit K+ currents) FCCP was observed to cause an inward shift in current of 3.5–8 pA but had little or no effect upon total membrane conductance over the range −100 to −70 mV. This inward current would cause the I-V relationship of uncoupler-sensitive background K+ current (as determined in Fig. 6) to be displaced upwards to more positive current levels, thus accounting for the failure to observe a reversal potential in normal [K+]o and any reversal potential in 20 mm[K+]o that was negative to EK. Indeed, this effect is recreated in Fig. 7B (trace c) which shows the current- voltage relationship resulting from the combined inhibition of K+ current (with Ba2+) and the generation of the FCCP-induced inward current (note that this generates an outwardly rectifying current which fails to reverse at EK; compare this with total uncoupler-sensitive current in 4.5 mm[K+]o in Fig. 6).
The cause of this inward current is at present unknown although a number of interesting possibilities present themselves. The first is that mitochondrial inhibitors might stimulate a non-selective cation conductance as has recently been reported for CN− in chromaffin cells (Inoue et al. 1998). An alternative possibility is that the FCCP-induced inward current could be due to a directly induced protonophoretic current at the cell membrane. This latter possibility would also account for the cellular acidification caused by uncouplers. Moreover, it could provide an explanation for the intriguing observation that FCCP appears to induce an inward shift in holding current which precedes the onset of mitochondrial depolarization (see above).
Finally, we noted in two cells that there was a marked inward rectification of current in the presence of DNP at potentials positive to −40 mV (this can be seen in the mean I-V relationships in Fig. 5B). Thus, in some cells, DNP may also activate a voltage-dependent inward current at depolarized potentials. Whilst we have not yet established the cause of this current, uncouplers have been reported to cause a hyperpolarizing shift in the voltage activation range of L-type Ca2+ current in arterial smooth muscle (McHugh & Beech, 1996); similar effects have also been reported with cyanide in sensory neurones (Duchen, 1990). A shift in the activation range of voltage-gated Ca2+ current in the type I cell could account for the observed inward rectification. It might also play an important role in the generation of the [Ca2+]i response by enhancing Ca2+ influx in the depolarized cell.
In summary, it is apparent that the inhibition of background K+ current and the generation of an as yet un-characterized inward current play the major role in initiating cell depolarization (possible contributions from voltage-activated Ca2+ currents would only come into play at potentials positive to the resting potential). At the resting potential (−50 to −60 mV) the effective total inward current generated by uncouplers is approximately 10–16 pA (from Fig. 6) of which 6–7 pA is probably due to a true inward current and the remainder (i.e. about half) due to the inhibition of a background outward K+ current.
Conclusions
Uncouplers stimulate the carotid body principally by causing a membrane depolarization, leading to voltage-gated Ca2+ entry and a rapid rise in cytoplasmic [Ca2+]. This [Ca2+]i response is suggested to stimulate neurosecretion leading to excitation of chemo-afferents. The above mechanism is therefore similar to that proposed for the transduction of the physiological stimuli hypoxia (Buckler & Vaughan-Jones, 1994a) and acidosis (Buckler & Vaughan-Jones, 1994b). We did not, however, find any evidence to support the hypothesis that uncouplers act primarily as acidic stimuli. The depolarization was found to be due to the inhibition of a background leak K+ conductance and the generation of an unidentified (possibly protonophoretic) inward current. The effects of uncouplers upon background K+ current are strikingly similar to the effects of hypoxia (Buckler, 1997). The fact that hypoxia and uncouplers cause a substantial inhibition of this current raises the possibility that the same channels are involved in mediating responses to both stimuli. If so, this would add weight to the hypothesis that transduction of hypoxic stimuli is, ultimately, mediated through changes in mitochondrial respiration (Anichcov & Belen-kii, 1963; Biscoe & Duchen, 1990b). Thus the metabolic hypothesis for O2 sensing demands closer scrutiny.
Acknowledgments
This work was supported by the MRC, The Wellcome Trust and the British Heart Foundation.
References
- Alekseev AE, Gomez LA, Aleksandrova LA, Brady PA, Terzic A. Opening of cardiac sarcolemmal KATP channels by dinitrophenol separate from metabolic inhibition. Journal of Membrane Biology. 1997;157:203–214. doi: 10.1007/s002329900229. 10.1007/s002329900229. [DOI] [PubMed] [Google Scholar]
- Anichcov SV, Belen'kii ML. Pharmacology of the Carotid Body Chemoreceptors. New York: Macmillan Publishing; 1963. [Google Scholar]
- Biscoe TJ, Duchen MR. Responses of type I cells dissociated from the rabbit carotid body to hypoxia. The Journal of Physiology. 1990a;428:39–59. doi: 10.1113/jphysiol.1990.sp018199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biscoe TJ, Duchen MR. The cellular basis of transduction in carotid chemoreceptors. American Journal of Physiology. 1990b;258:L271–278. doi: 10.1152/ajplung.1990.258.6.L271. [DOI] [PubMed] [Google Scholar]
- Boron WF, De Weer P. Intracellular pH transients in squid giant axons caused by CO2, NH3, and metabolic inhibitors. Journal of General Physiology. 1976;67:91–112. doi: 10.1085/jgp.67.1.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ. A novel oxygen-sensitive potassium current in rat carotid body type I cells. The Journal of Physiology. 1997;498:649–662. doi: 10.1113/jphysiol.1997.sp021890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Application of a new pH-sensitive fluoroprobe (carboxy-SNARF-1) for intracellular pH measurement in small isolated cells. Pflügers Archiv. 1990;417:234–239. doi: 10.1007/BF00370705. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of acidic stimuli on intracellular calcium in isolated type I cells of the neonatal rat carotid body. Pflügers Archiv. 1993a;425:22–27. doi: 10.1007/BF00374499. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of the metabolic uncoupler DNP on intracellular pH and Ca2+ in type-I cells in enzymically isolated neonatal rat carotid body type I cells. The Journal of Physiology. 1993b;459:345P. [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypoxia on membrane potential and intracellular calcium in rat neonatal carotid body type I cells. The Journal of Physiology. 1994a;476:423–428. doi: 10.1113/jphysiol.1994.sp020143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Effects of hypercapnia on membrane potential and intracellular calcium in rat carotid body type I cells. The Journal of Physiology. 1994b;478:157–171. doi: 10.1113/jphysiol.1994.sp020239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD. Role of intracellular pH and [Ca2+]i in acid chemoreception in type-I cells of the carotid body. In: O'Regan RG, Nolan P, McQueen DS, Paterson DJ, editors. Advances in Experimental Medicine and Biology, Chemoreceptors and Chemoreceptor Reflexes. Vol. 360. New York and London: Plenum Press; 1994c. pp. 155–158. [DOI] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD, Peers C, Lagadic-Gossmann D, Nye PCG. Effects of extracellular pH, PCO2 and HCO3− on intracellular pH in isolated type-I cells of the neonatal rat carotid body. The Journal of Physiology. 1991a;444:703–721. doi: 10.1113/jphysiol.1991.sp018902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckler KJ, Vaughan-Jones RD, Peers C, Nye PCG. Intracellular pH and its regulation in isolated type I carotid body cells of the neonatal rat. The Journal of Physiology. 1991b;436:107–129. doi: 10.1113/jphysiol.1991.sp018542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd SL, Nicholls DG. A reevaluation of the role of mitochondria in neuronal Ca2+ homeostasis. Journal of Neurochemistry. 1996;66:403–411. doi: 10.1046/j.1471-4159.1996.66010403.x. [DOI] [PubMed] [Google Scholar]
- Bunting JR. Influx and efflux kinetics of cationic dye binding to respiring mitochondria. Biophysical Chemistry. 1992;42:163–175. doi: 10.1016/0301-4622(92)85006-p. [DOI] [PubMed] [Google Scholar]
- Demaurex N, Romanek RR, Orlowski J, Grinstein S. ATP dependence of Na+/H+ exchange. Nucleotide specificity and assessment of the role of phospholipids. Journal of General Physiology. 1997;109:117–128. doi: 10.1085/jgp.109.2.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR. Effects of metabolic inhibition on the membrane properties of isolated mouse primary sensory neurones. The Journal of Physiology. 1990;424:387–409. doi: 10.1113/jphysiol.1990.sp018073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Biscoe TJ. Relative mitochondrial membrane potential and [Ca2+]i in type I cells isolated from the rabbit carotid body. The Journal of Physiology. 1992;450:33–61. doi: 10.1113/jphysiol.1992.sp019115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duchen MR, Valdeomillos M, O'Neill SC, Eisner DA. Effects of metabolic blockade on the regulation of intracellular calcium in dissociated mouse sensory neurons. The Journal of Physiology. 1990;424:411–426. doi: 10.1113/jphysiol.1990.sp018074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- e Silva MJ, Lewis DL. L- and N-type Ca2+ channels in adult rat carotid body chemoreceptor type I cells. The Journal of Physiology. 1995;489:689–699. doi: 10.1113/jphysiol.1995.sp021083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel DD, Tsien RW. An FCCP-sensitive store in bullfrog sympathetic neurons and its participation in stimulus-evoked changes in [Ca2+]i. Journal of Neuroscience. 1994;14:4007–4024. doi: 10.1523/JNEUROSCI.14-07-04007.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Oxygen and acid chemoreception in the carotid body chemoreceptors. Trends in Neurosciences. 1992;15:146–153. doi: 10.1016/0166-2236(92)90357-e. [DOI] [PubMed] [Google Scholar]
- Gonzalez C, Almaraz L, Obeso A, Rigual R. Carotid body chemoreceptors: From natural stimuli to sensory discharges. Physiological Reviews. 1994;74:829–898. doi: 10.1152/physrev.1994.74.4.829. [DOI] [PubMed] [Google Scholar]
- Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. Journal of Biological Chemistry. 1985;260:3440–3450. [PubMed] [Google Scholar]
- Herrington J, Park YB, Babcock DF, Hille B. Dominant role of mitochondria in clearance of large Ca2+ loads from rat adrenal chromaffin cells. Neuron. 1996;16:219–228. doi: 10.1016/s0896-6273(00)80038-0. [DOI] [PubMed] [Google Scholar]
- Inoue M, Fujishiro N, Imanaga I. Hypoxia and cyanide induce depolarisation and catecholamine release in dispersed guinea-pig chromaffin cells. The Journal of Physiology. 1998;507:807–818. doi: 10.1111/j.1469-7793.1998.807bs.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JR, Rehder V. FCCP releases Ca2+ from a non-mitochondrial store in an identified Helisoma neuron. Brain Research. 1991;551:311–314. doi: 10.1016/0006-8993(91)90947-t. [DOI] [PubMed] [Google Scholar]
- Kaila K, Mattesson K, Vopio J. Fall in intracellular pH and increase in resting tension induced by a mitochondrial uncoupling agent in crayfish muscle. The Journal of Physiology. 1989;408:271–293. doi: 10.1113/jphysiol.1989.sp017459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krylov SS, Anichkov SV. The effect of metabolic inhibitors on carotid chemoreceptors. In: Torrance RW, editor. Arterial Chemoreceptors. Blackwell; 1968. pp. 103–109. [Google Scholar]
- McHugh D, Beech DJ. Modulation of the Ca2+ channel activity by ATP metabolism and internal Mg2+ in guinea-pig basilar artery smooth muscle cells. The Journal of Physiology. 1996;492:359–376. doi: 10.1113/jphysiol.1996.sp021314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulligan E, Lahiri S. Dependence of carotid chemoreceptor stimulation by metabolic agents on PaO2 and PaCO2. Journal of Applied Physiology. 1981;50:884–891. doi: 10.1152/jappl.1981.50.4.884. [DOI] [PubMed] [Google Scholar]
- Nye PCG, Torrance RW. A breath of carbon monoxide reduces chemoreceptor discharge in the cat. The Journal of Physiology. 1980;307:46–47P. [Google Scholar]
- Obeso A, Almaraz L, Gonzalez C. Effects of cyanide and uncouplers on chemoreceptor activity and ATP content of the cat carotid body. Brain Research. 1989;481:250–257. doi: 10.1016/0006-8993(89)90801-9. [DOI] [PubMed] [Google Scholar]
- Rocher A, Obeso A, Gonzalez C, Herreros B. Ionic mechanisms for the transduction of acidic stimuli in rabbit carotid body glomus cells. The Journal of Physiology. 1991;433:533–548. doi: 10.1113/jphysiol.1991.sp018442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen TCR, Hauss WH. Influence of dinitrophenol, dinitroartocreasol and paranitrophenol upon the carotid sinus chemoreceptors of the dog. Archives Internationales de Pharmacodynamie et de Thérapie. 1939;63:251–258. [Google Scholar]
- Wu M-L, Vaughan-Jones RD. Effect of metabolic inhibitors and second messengers upon Na+-H+ exchange in the sheep cardiac Purkinje fibre. The Journal of Physiology. 1994;478:301–313. doi: 10.1113/jphysiol.1994.sp020251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werth JL, Thayer SA. Mitochondria buffer physiological calcium loads in cultured rat dorsal root ganglion neurones. Journal of Neuroscience. 1994;14:348–356. doi: 10.1523/JNEUROSCI.14-01-00348.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]