

Abstract
We constructed rat homologues (S252F and +L264) of two human α4 nicotinic mutations - α4(S248F) and α4(777ins3) - that have been linked to autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) and co-expressed them with wild-type rat β2 subunits in Xenopus oocytes.
The S252F and +L264 mutations had three common effects on the ACh response. First, they caused use-dependent potentiation of the response during a train of brief 100 nm ACh pulses. Second, they delayed the rise times of the 5–15 nm (+L264) and 30 nm (S252F) ACh responses. Third, they reduced extracellular Ca2+-induced increases in the 30 μm ACh response.
Beside these shared effects, the S252F mutation also reduced the channel burst duration measured from voltage-jump relaxations, enhanced steady-state desensitization and reduced the single-channel conductance. In contrast, the +L264 mutation prolonged the channel burst duration, did not affect desensitization and slightly increased single-channel conductance. Neither mutation affected the number of surface receptors measured by antibody binding but the S252F mutation reduced the maximum ACh response.
The ACh concentration dependence of use-dependent potentiation and the delay in the rising phase of the mutant ACh response suggest that these effects are caused by a slow unblocking of the closed mutant receptors. Use-dependent potentiation of the mutant response during a series of high-frequency cholinergic inputs to the presynaptic terminal could trigger ADNFLE seizures by suddenly increasing nicotinic-mediated transmitter release.
Two mutations in the M2 region of the human α4 neuronal nicotinic subunit - α4(S248F) and α4(776ins3) - have been linked to autosomal dominant nocturnal frontal lobe epilepsy (ADNFLE) (Steinlein et al. 1995, 1997). The α4(S248F) mutation is a serine to phenylalanine substitution at position 248 in the human α4 nicotinic subunit. The α4(776ins3) mutation is a 3-base pair insertion that adds a leucine at position 259 in the amino acid sequence of the human α4 subunit. Photo-affinity labelling and structure- function experiments show that the M2 region of the nicotinic subunits forms the conducting pore of the receptor (reviewed in Karlin & Akabas, 1995). Thus, both ADNFLE mutations lie in the conducting pore of the nicotinic receptor.
ADNFLE patients suffer from brief and occasionally violent nocturnal seizures (Scheffer et al. 1995). However, the physiological mechanism responsible for these seizures has not been established. Previous studies show that the predominant brain nicotinic receptor subtype is α4β2 (Whiting & Lindstrom, 1987; Flores et al. 1991; Whiting et al. 1991). Receptors formed by co-expressing α4(S248F) or α4(776ins3) subunits with wild-type (WT) β2 subunits in Xenopus oocytes (Weiland et al. 1996; Steinlein et al. 1997; Kuryatov et al. 1997) differ from the WT receptor in several ways but no common effects of the two mutations on the acetylcholine (ACh) response have been reported previously.
To determine whether the ADNFLE mutations have any common effects on the ACh response, we constructed two rat homologues (S252F and +L264) of the human ADNFLE mutations α4(S248F) and α4(777ins3), co-expressed them with rat β2 subunits in Xenopus oocytes, and studied the properties of the expressed receptors. We also constructed the rat double mutation V247I:S252F, which combined the S252F mutation with a second V247I mutation that converted the only rat/human residue substitution in the α4 M2 region to the corresponding human residue. All three mutant receptors displayed a novel use-dependent potentiation of the ACh response during a train of brief 100 nm ACh applications. We suggest that use-dependent increases in the amplitude of the mutant synaptic currents could trigger seizures by suddenly increasing nicotinic-mediated transmitter release. Some of the results have appeared previously in abstract form (Figl et al. 1997).
METHODS
Molecular biology
We used the Stratagene QuikChange kit (La Jolla, CA, USA) to make the mutations and verified them by DNA sequencing. To increase receptor expression, the rat α4 and β2 subunit cDNAs were subcloned into a modified pBluescript vector between a 5′ untranslated region from the alfalfa mosaic virus which enhances protein synthesis (Jobling & Gehrke, 1987) and a long 3′ polyA tail. Xenopus oocytes were isolated as previously described (Quick & Lester, 1994) in accordance with the guidelines of the University of California Animal Use Committee. In brief, female Xenopus were anaesthetized by immersion in cold (∼4°C) water containing 0.2% ethylmetaaminobenzoate (MS-222, Acros Corp., Pittsburgh, PA, USA) for 30–60 min. The animals were allowed to recover in individual tanks for 24 h before being returned to their home tank. The oocytes were incubated for 24–72 h in a modified Barth's solution (96 mm NaCl, 5 mm Hepes, 2.5 mm sodium pyruvate, 2 mm KCl, 1.8 mm CaCl2, 1 mm MgCl2, 2.5 μg ml−1 gentamicin, 5% horse serum, pH 7.4) at 18°C before the experiments began.
Electrophysiology
Whole oocytes were voltage clamped with a GeneClamp amplifier (Axon Instruments) using two microelectrodes filled with 3 m KCl. During the experiments, the oocytes were continually superfused with a nominal Ca2+-free saline (ND98; 98 mm NaCl, 5 mm Hepes, 1 mm MgCl2, pH 7.5) at ambient temperature (18–23°C). To prevent activation of the endogenous Ca2+-activated Cl− current in the experiments with 2.5 mm extracellular Ca2+, we soaked the oocytes for 5–7 h in 70 μm BAPTA AM (Sands et al. 1993). We digitally recorded the voltage-clamp current using a personal computer equipped with a DigiData 1200 A/D interface and pCLAMP version 6 software (Axon Systems). To prevent aliasing, the data were filtered at 1/3 to 1/5 of the sampling frequency with an 8-pole, low-pass Bessel filter. To measure the ACh-induced voltage-jump relaxation current, we jumped the membrane potential from −50 mV to +50 mV and then to a final voltage between −60 and −150 mV, in the presence and absence of ACh. The difference between the relaxation current with and without ACh was taken as the ACh-induced relaxation current. The voltage-jump relaxation currents were filtered at ≤ 800 Hz. ACh was applied by bath perfusion or rapid U-tube microperfusion (Cohen et al. 1995). The ACh concentration-response relations were analysed as described previously (Cohen et al. 1995).
Excised patches were bathed in symmetrical 100 mm KCl, 10 mm EGTA, 10 mm Hepes, 5 mm KOH and 2 mm MgCl2 (pH 7.4). The data were filtered at 1–3 kHz, recorded on videotape using a Neuro-corder DR-290 pulse code modulator (Neuro Data Instruments, New York, NY, USA) and sampled at 5 times the filter frequency. We used pCLAMP version 6.0 to construct and fit the single-channel amplitude histograms. We measured the single-channel conductance from all-points histograms of the patch current.
125I-antibody and [3H]epibatidine binding
To compare the number of mutant and WT surface receptors, we used an 125I-sheep anti-rat F(ab′)2 antibody fragment (Amersham Life-Science, Arlington Heights, IL, USA) to detect the primary antibodies mAb 270 and mAb 299 binding to the expressed surface receptors. To reduce non-specific 125I-secondary antibody binding, we pre-incubated the oocytes with a panel of unlabelled mouse IgG proteins (Southern Biotech, Birgmingham, AL, USA) before exposing them to the primary antibody (mAb 299 and mAb 270). After this pre-incubation, we incubated the oocytes with the unlabelled primary antibody for 15 min, rinsed them 3 times with ND98 (above), and blocked them with 3% bovine serum albumin (BSA) in ND98 for 15 min. After blocking with BSA, we rinsed 3 times with ND98 and incubated the oocytes with the 125I-secondary antibody (3 μg antibody (ml ND98)−1) for 15 min. To measure background 125I labelling, a group of matched control oocytes were treated identically but were not exposed to the primary antibody. 125I labelling was measured with a γ-counter. Background 125I labelling from the secondary antibody fragment was ≤ 50% of the total labelling.
We followed previously published protocols (Peng et al. 1994; Gerzanich et al. 1995) to measure ±[3H]epibatidine (New England Nuclear, Boston, MA, USA) binding to mAb 299-immunoprecipitated receptors. The wells of enhanced immunoassay/radioimmunoassay (EIA/RIA) strip plates (Costar Corning Corp., Cambridge, MA, USA) were coated with mAb 299 by adding 0.5–2.0 μg of the antibody to each well in 100 μl of 10 mm sodium bicarbonate (pH 8.8) for an overnight incubation at 4°C. After the overnight incubation, the coated wells were blocked with 3% BSA in 200 μl of PBS-Tween buffer (100 mm NaCl, 10 mm sodium phosphate, 0.05% Tween 20, pH 7.5) for 2 h at room temperature (20–24°C) and rinsed 3 times with PBS-Tween buffer. Next, we solubilized the WT and mutant receptors by vortexing the injected oocytes for several minutes in 5–7 volumes of a lysis buffer (2% Triton X-100, 50 mm NaCl, 50 mm sodium phosphate buffer, pH 7.5; 1 tablet of CompleteTM protease inhibitor (Boehringer Manheim) per 40 ml of lysis buffer) in a 1.5 ml microcentrifuge tube. The lysed oocytes were gently rotated in the tube for 20 min at 4°C and then microcentrifuged for 20 min at 14 000 r.p.m. (16 000 g). We removed the clear supernatant and added 100 μl aliquots of the supernatant to the antibody-coated wells for an overnight incubation at 4°C. We rinsed the wells 3 times with PBS- Tween after the overnight incubation and then added the appropriate [3H]epibatidine concentration in PBS-Tween for 40 min at 4°C. To avoid agonist depletion at low [3H]epibatidine concentrations (0.001–0.3 nm), we incubated each well in 2 ml of the [3H]epibatidine solution. We used a volume of 200 μl per well for the higher [3H]epibatidine concentrations (≥ 1 nm). The amount of receptor added to the wells was adjusted to ensure that depletion of the free agonist was ≤ 10%. After the 40 min incubation, the wells were rinsed 3 times with PBS-Tween buffer, placed whole into 1 ml of scintillation fluid and counted. Non-specific binding was measured using several methods. First, we added 100 μm cold (-)nicotine to all the [3H]epibatidine solutions and measured [3H]epibatidine binding to the WT receptors. Second, we measured [3H]epibatidine binding to antibody-treated wells incubated with solubilized un-injected oocytes. Third, and finally, we measured [3H]epibatidine binding to blocked wells that were not pre-treated with antibody or homogenized receptor. There were no significant differences between the amount of non-specific binding measured by these three methods. Non-specific [3H]epibatidine binding was 5–10% of the maximum total [3H]epibatidine binding.
RESULTS
Use-dependent potentiation of the +L264 and S252F ACh response
Previous experiments suggest that the α4(S248F) mutation induces some kind of use-dependent potentiation of the ACh response (Kuryatov et al. 1997). To study this phenomenon in more detail and to determine if both the S252F and +L264 mutations had this effect, we measured the WT, +L264, S252F and V247I:S252F responses to ten 100 nm ACh pulses (150 ms) spaced 5 s apart. The WT response remained constant during the pulse train (Fig. 1A). However, all three mutant responses displayed cumulative, use-dependent potentiation (Fig. 1B–D). The peak +L264 response increased ∼2-fold by the end of the pulse train (Fig. 1B and D). The peak S252F (Fig. 1C) and V247I:S252F responses increased ≥ 3-fold (Fig. 1D).
Figure 1. Use-dependent potentiation of the +L264, S252F and V247I:S252F ACh responses.
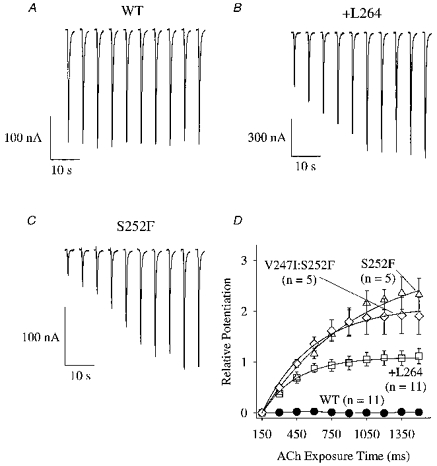
A-C, voltage-clamped WT, +L264 and S252F responses (downward deflections) to a series of 100 nm ACh pulses (ten 150 ms pulses, 5 s apart) at −50 mV. D, relative potentiation of the responses as a function of the total 100 nm ACh exposure time. Lines are least squares fits to eqn (1). See text for fit parameters. Symbols and error bars are mean ±s.e.m.; n is the number of oocytes.
We defined the relative potentiation (RP) of the responses within the pulse train as:
Thus, for a constant response, the RP was zero. When the RP values of the +L264, S252F and V247I:S252F responses were plotted against the total time that the receptors were exposed to ACh (tACh), they approached a maximum (RPmax) as a negative exponential function of tACh:
| (1) |
where 150 ms was the duration of the first pulse and τp was the time constant of potentiation. Fits of the relative potentiation data to eqn (1) yielded values of RPmax = 1 and τp= 300 ms for the +L264 receptor, RPmax = 3 and τp= 830 ms for the S252F receptor, and RP = 2.1 and τp= 440 ms for the V247I:S252F receptor (Fig. 1D). Thus, all three mutants displayed significant use-dependent potentiation of the 100 nm ACh response with time constants in the 0.3–0.8 s range, and the rat/human residue substitution in the α4 M2 region had little effect on use-dependent potentiation of the 100 nm ACh response. The ACh pulses in these experiments were too short to reach a uniform ACh concentration around the entire oocyte. Thus, the peak ACh concentration at the oocyte surface may have been less than the concentration applied from the U-tube.
Several factors affected use-dependent potentiation of the mutant ACh response (Fig. 2A and B). Increasing the inter-pulse interval from 5 s to 40 s abolished potentiation of the +L264 response (Fig. 2A). Additionally, potentiation occurred only when the probability of channel opening was low (Fig. 2B). When we raised the ACh concentration to a nearly saturating level (500 μm), the WT and +L264 responses to a train of ten 500 μm ACh pulses (10 ms) spaced 5 s apart remained constant and the S252F response decreased by > 50% (Fig. 2B). The decrease in the S252F response was probably due to cumulative desensitization of the receptors. The ACh concentration dependence of potentiation suggests that it represents use-dependent unblocking of the closed mutant receptors.
Figure 2. Several factors affected use-dependent potentiation of the +L264 response.
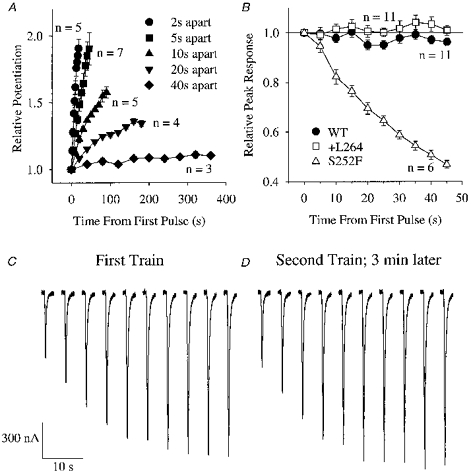
A, relative potentiation of the +L264 response during a train of ten 150 ms pulses of 100 nm ACh spaced 2, 5, 10, 20 or 40 s apart. Error bars are ±s.e.m.B, changes in the peak WT, +L264 and S252F responses during a train of ten 500 μm ACh pulses (10 ms) spaced 5 s apart. Holding potential, Vh =−50 mV. C and D, potentiation of the +L264 100 nm ACh response was reversible. C, potentiation of the +L264 response during a series of ten 300 ms pulses of 100 nm ACh spaced 5 s apart. Downward deflections of traces show the individual ACh responses. D, potentiation of the +L264 response in the same oocyte 3 min after the first pulse train. Note that the initial ACh response in D has returned to nearly the same level as the initial response in C and that both pulse trains induce the same amount of potentiation.
Potentiation of the mutant ACh response was reversible (Fig. 2C and D). Figure 2C shows potentiation of the +L264 100 nm ACh response during a train of 300 ms ACh pulses spaced 5 s apart. Three minutes later, we re-applied the same pulse train to the same oocyte (Fig. 2D). The amplitude of the first +L264 response in both pulse trains was nearly identical. Likewise, the amplitudes of the final responses in both pulse trains were nearly the same. Thus, the +L264 ACh response returned to its original size within 3 min after the first pulse train and displayed the same level of potentiation during a second pulse train.
Mutations slow the rise of the 5–30 nm ACh response
The +L264 and S252F mutations had an unusual effect on the rising phase of the response to very low ACh concentrations (Fig. 3A–D). The WT response to a 15–30 nm ACh concentration jump approached steady state sigmoidally with a half-rise time (t½) of 0.7 ± 0.1 s (mean ±s.e.m., n = 12). The rate of ACh concentration change at the oocyte surface is not spatially uniform when ACh is applied from the U-tube microperfusion system. Since the diameter of the oocyte is so large (∼0.5 mm), the ACh concentration on the side facing the U-tube aperture rises faster than the ACh concentration on the side facing away from the U-tube. Therefore, the half-times of the rising and falling phases of the macroscopic ACh response reflect the change in the mean ACh concentration at the oocyte surface. These times should not be taken as an indication of the rate of solution exchange on the side of the oocyte facing the U-tube, which probably occurs much faster. As the Hill coefficient (nH) of the WT ACh response is close to unity (nH = 0.9, see below), at ACh concentrations << EC50, the WT ACh response should be nearly directly proportional to the mean ACh concentration surrounding the oocyte. Therefore, if we make the reasonable assumption that ACh binding to and unbinding from the WT receptors is in rapid equilibrium during the rising and falling phase of our ACh applications, then the time course of the WT response at low ACh concentrations should reflect the time course of the mean ACh concentration change around the oocyte (Fig. 3A). In contrast to the WT response, the +L264 15 nm ACh response took more than 4 times longer to reach its t½ (3.3 ± 0.4 s, n = 7) and this delay increased to 5.2 ± 0.4 s (n = 8) with 10 nm ACh and 12.4 ± 1.8 s (n = 7) with 5 nm ACh (Fig. 3B). Similarly, the t½ for the S252F 30 nm ACh response (2.5 ± 0.4 s, n = 10) was > 3 times longer than that for the WT (Fig. 3C). Inspection of the 5–15 nm+L264 and 30 nm S252F responses showed that they increased just as rapidly as the WT response at the onset of the ACh concentration jump but slowed down shortly thereafter (Fig. 3B and C). The rapid initial phase was particularly noticeable in the 5 nm+L264 response (Fig. 3B) but was also visible in the other mutant responses. The rapid initial component of the +L264 5 nm ACh response represented 14 ± 2% (n = 7) of the final steady-state response. The end point of the initial phase of the S252F 30 nm response was too indistinct to accurately measure its amplitude.
Figure 3. The +L264 and S252F mutations delayed the rising phase of the 5–15 nm (+L264) and 30 nm (S252F) ACh response.
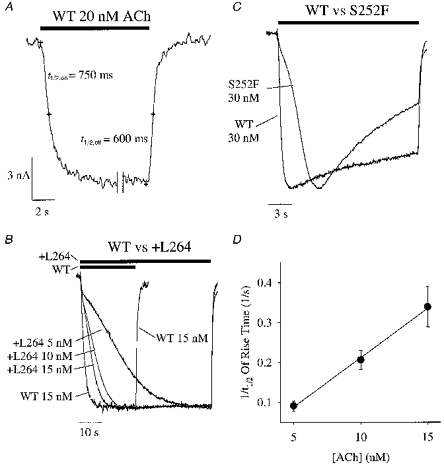
A, the rising and falling phases of the WT response to 20 nm ACh at an expanded time scale. We deleted the steady-state portion of the response between the double vertical lines. The small crosses superimposed on the traces denote from left to right the start of the ACh application, the half-rise time of the ACh response (t½,on), the end of the ACh application, and the half-fall time of the ACh response (t½,off). Note that the t½,on and t½,off values were approximately equal for the WT response. The bar above the traces indicates the timing of ACh application. B, WT response to a 15 nm ACh concentration jump superimposed on +L264 responses to 5, 10 and 15 nm ACh concentration jumps (+L264 traces from a single oocyte) after normalizing the responses to the same peak value. Bars above the traces show the timing of the ACh application. C, superimposed and normalized S252F and WT 30 nm ACh responses. D, the reciprocal of the half-rise time (1/t½) of the +L264 ACh response increased linearly with the ACh concentration ([ACh]). Error bars are ±s.e.m. (n = 7–8). Vh =−50 mV.
The mutant ACh responses in Fig. 3B and C also show that the mutations prolong the rising, but not the falling, phases of the ACh responses. The similarity of the time course of the falling phases of the WT and mutant ACh responses in Fig. 3B and C shows that the time course of solution exchange around the WT and mutant oocytes is the same even though the mutant ACh response rises more slowly. The delay in the mutant response to low ACh concentrations could be caused by a slow unblocking of the closed mutant channels. The WT response to < 15 nm ACh and the S252F response to < 30 nm ACh were too small for accurate measurements of t½. The rise times of the +L264, S252F and WT responses to ≥ 50 nm ACh were not significantly different.
Mutations reduce Ca2+ potentiation of the ACh response
Previous experiments show that adding 2.5 mm extracellular Ca2+ to nominal Ca2+-free saline increases the peak amplitude of the 30 μm human α4β2 ACh response roughly 3-fold at −100 mV (Steinlein et al. 1997). The α4(776ins3) mutation reduces Ca2+-induced increases in the ACh response by 45% (Steinlein et al. 1997). To determine whether the +L264 and S252F mutations similarly affected Ca2+-induced increases in the ACh responses, we measured the amplitude of the WT, +L264 and S252F 30 μm ACh responses in nominal Ca2+-free (ND98) and 2.5 mm extracellular Ca2+ saline (ND98 + 2.5 mm Ca2+), at voltages of −50 and −100 mV, and before and after a 5–7 h incubation in the membrane-permeable Ca2+ chelator BAPTA AM (70 μm). The WT response increased ∼3-fold after raising the extracellular [Ca2+] to 2.5 mm (Table 1). The +L264 and S252F responses increased only ∼2-fold (Table 1). The effect of Ca2+ on the amplitude of the ACh response was not voltage dependent between −50 and −100 mV (Table 1). Pre-loading the oocytes with 70 μm BAPTA AM likewise failed to affect Ca2+-induced increases in the ACh response (Table 1). Therefore, activation of the endogenous oocyte Ca2+-activated Cl− current by Ca2+ influx through the nicotinic receptors was not responsible for the Ca2+-induced increases in the ACh response.
Table 1.
Relative increases in the 30 μm ACh response after adding 2.5 mm extracellular Ca2+
| Receptor | Voltage (mV) | BAPTA AM treatment | Relative increase in the ACh response |
|---|---|---|---|
| WT (n = 4) | −50 | No | 2.9 ± 0.1 |
| S252F (n = 5) | −50 | No | 1.5 ± 0.1 * |
| +L264 (n = 6) | −50 | No | 1.7 ± 0.1 * |
| WT (n = 3) | −100 | No | 3.4 ± 0.1 |
| S252F (n = 5) | −100 | No | 1.7 ± 0.1 * |
| +L264 (n = 3) | −100 | No | 2.1 ± 0.2 * |
| WT (n = 3) | −100 | Yes | 3.2 ± 0.2 |
| S252F (n = 3) | −100 | Yes | 1.8 ± 0.1 * |
| +L264 (n = 3) | −100 | Yes | 2.2 ± 0.2 * |
Significantly different from the WT value (P < 0.05).
Mutations have little effect on the ACh concentration-response relation
The S252F or +L264 mutations did not significantly change the EC50 or apparent Hill coefficient (nH) of the ACh concentration-response relation at −50 mV (Fig. 4A–D). The WT EC50 (2.6 ± 0.2 μm) and nH (0.90 ± 0.03, n = 6) were within the range (EC50 = 3–5 μm, nH = 0.7–1.5) previously reported for recombinantly expressed rat (Vibat et al. 1995) and human α4β2 nicotinic receptors (Weiland et al. 1996; Buisson et al. 1996; Kuryatov et al. 1997; Steinlein et al. 1997). The S252F EC50 (3.2 ± 0.2 μm) and nH (1.20 ± 0.05, n = 3) were similar to the WT values. The S252F EC50 was within the range (EC50 = 2–12 μm) previously reported for human α4(S248F)β2 receptors (Weiland et al. 1996; Kuryatov et al. 1997) but the nH was slightly larger than the previous values (nH = 0.9–1). The +L264 EC50 (1.8 ± 0.2 μm) and nH (1.10 ± 0.07, n = 9) were also similar to the WT values. The +L264 EC50 at −50 mV was considerably larger than the EC50 (0.3 μm) for the α4(776ins3)β2 receptor at −100 mV (Steinlein et al. 1997) and nH was slightly larger than the previously reported value (0.8). However, the +L264 channel burst duration increased as the membrane potential became more negative (below). Therefore, the apparent difference between the +L264 and α4(776ins3)β2 EC50 values may be partly due to membrane potential.
Figure 4. WT, S252F and +L264 ACh concentration-response relations at −50 mV.
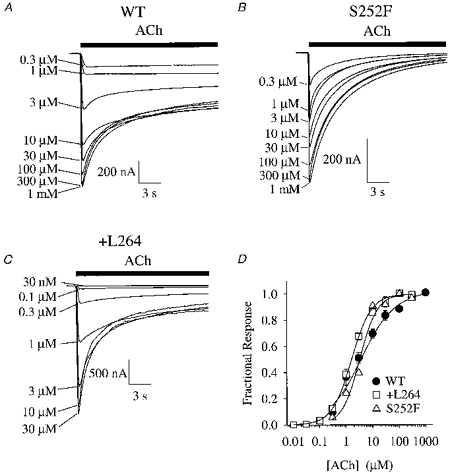
A-C, WT, S252F and +L264 responses to a series of increasing ACh concentrations. Bars above the traces indicate the duration of the step application of ACh from the microperfusion system. Note the rapid desensitization of the S252F response. D, normalized WT, S252F and +L264 ACh concentration-response relations. Lines are fits to the Hill equation (see Methods). See text for fit parameters. Error bars are ± se.m. Symbols obscure most error bars.
Mutations have little effect on [3H]epibatidine binding
[3H]epibatidine is a high-affinity neuronal nicotinic agonist (Badio & Daly, 1994). To determine whether the +L264 and S252F mutations affected [3H]epibatidine affinity, we measured [3H]epibatidine binding to mAb 299-immunopurified +L264, S252 and WT receptors (Fig. 5). The S252F and +L264 mutations had little effect on [3H]epibatidine affinity. The apparent KD for [3H]epibatidine binding to the WT receptor was 0.66 ± 0.06 nm (mean ± standard error of the estimate, n = 4 repeats). The KD for binding to the +L264 receptor (0.24 ± 0.02 nm, n = 2) was slightly smaller than the WT value. However, the KD for the S252F receptor (1.20 ± 0.07 nm, n = 2) was slightly larger (Fig. 5). The mutant and WT KD values for [3H]epibatidine binding were not significantly different. They fell within the range (KD = 0.4–1.6 nm) previously reported for [3H]epibatidine binding to low-affinity [3H]epibatidine receptors in rat forebrain homogenates (Houghtling et al. 1995) and human α4β2 nicotinic receptors expressed in HEK 293 cells (Eilers et al. 1997). The values of nH for the +L264 (1.1 ± 0.1, n = 2), S252F (1.2 ± 0.1, n = 2) and WT receptors (1.1–1.2) were not significantly different.
Figure 5. WT, +L264 and S252F [3H]epibatidine concentration- binding relations.
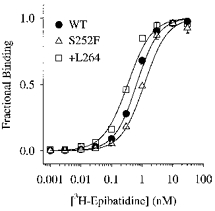
WT, S252F and +L264 receptors were immunoprecipitated onto EIA/RIA strip plate wells using the anti-α4 antibody mAb 299. Curves are fits to the Hill equation. See text for fit parameters.
+L264 prolongs voltage-jump relaxations
The channel burst duration determines the rate of nicotinic synaptic current decay at the neuromuscular junction (reviewed in Edmonds et al. 1995). We used voltage-jump relaxations to study the effects of the +L264 and S252F mutations on the channel burst duration as channel activity in the +L264, S252F and WT outside-out patches washed out too rapidly to construct reliable burst histograms. Besides providing a direct and simple method of estimating the synaptic current decay rate, relaxation analysis has the added advantage of avoiding arbitrary definitions of the critical shut time between bursts (Colquhoun & Sigworth, 1995) and length-biased sampling. The time constant of the voltage-jump relaxation current approaches the time constant of the slow burst duration and the synaptic current decay at low fractional receptor occupancies (Edmonds et al. 1995). The ACh concentrations we used to measure the WT (50 nm), +L264 (30 nm) and S252F (100 nm) relaxations represented fractional receptor occupancies of 1, 2 and 3%. We calculated the fractional occupancies of the receptors at these ACh concentrations using the Hill equation, and the WT and mutant EC50 and nH values measured above.
The relaxation currents were well fitted by the sum of two negative exponentials and a constant term (Fig. 6A–D). We measured the fast time constant (τf), the slow time constant (τs) and the percentage amplitude of the fast component (Af/Atot) of the relaxation currents (Fig. 6E–G). The WT τf, τs and Af/Atot were not significantly voltage dependent between −60 and −150 mV (Fig. 6E–G). The WT τf, τs and Af/Atot were 5.0 ± 0.3 ms, 50 ± 1 ms and 55 ± 1% (n = 5) for a voltage jump from +50 to −150 mV and 4.8 ± 0.6 ms, 47 ± 5 ms and 63 ± 2% (n = 5) for a jump from +50 to −70 mV. Previous studies of α3β2 and α3β4 nicotinic receptor relaxation currents (Figl et al. 1996) suggest that these two exponential relaxation components represent two receptor subpopulations with different channel burst durations. Interestingly, human WT α4β2 channels also display long and short openings (Kuryatov et al. 1997). Our WT τf and τs were similar to the mean open times (3.7 and 23 ms) reported previously for the smaller of the two human α4β2 channels (Kuryatov et al. 1997). If the open times of the rat and human WT α4β2 channels are similar, then the rat WT bursts probably contain few channel openings.
Figure 6. WT, S252F and +L264 voltage-jump relaxation currents.
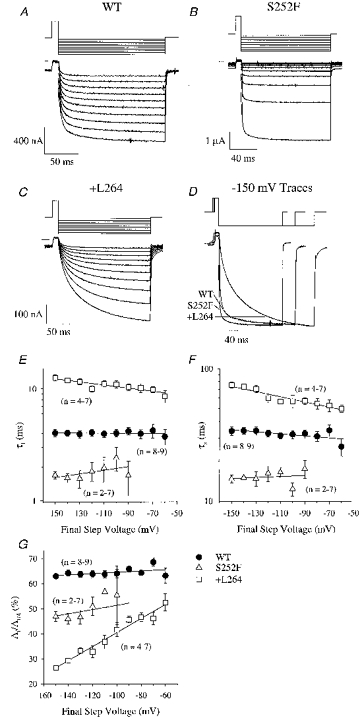
A-C, series of WT, S252F and +L264 relaxation currents produced by jumps from an initial voltage of +50 mV to a final voltage of −150 to −60 mV (in 10 mV increments). The WT, S252F and +L264 ACh concentrations were 50, 100 and 30 nm, respectively. Voltage-jump protocols are shown above traces. The lines superimposed on the relaxations are fits to the sum of two negative exponentials and a constant. The apparent increase in the rectification of the S252F response was probably due to desensitization of the response during the voltage-jump protocol (the first jump was to −150 mV). D, normalized WT, S252F and +L264 relaxation currents for a jump from +50 to −150 mV. The WT and S252F voltage-jump protocols were shorter than the +L264 protocol. E-G, voltage dependence of τf, τs and Af/Atot for the WT, S252F and +L264 relaxations. Error bars are ±s.e.m. Only the +L264 τf, τs and Af/Atot displayed significant voltage dependence. E, slope of the +L264 τf regression line was −0.0014 mV−1 (r2 = 0.8). The τf at 0 mV was 7.6 ms. F, slope of the +L264 τs regression line was −0.002 mV−1 (r2 = 0.9). The τs at 0 mV was 37 ms. G, slope of the +L264 Af/Atot regression line was 0.3% per millivolt (r2 = 0.97). The Af/Atot at 0 mV was 70%.
The S252F mutation reduced the relaxation time constants (Fig. 6D). For a jump from +50 to −150 mV, the S252F τf (1.7 ± 0.1 ms, n = 5) was < one-half that of the WT (above). The S252F τs (15 ± 1 ms, n = 5) was < one-third. The S252F Af/Atot (47 ± 2%, n = 5) was slightly less than the WT value. Similar to the WT relaxations, the S252F τf, τs and Af/Atot displayed no significant voltage dependence between −90 and −150 mV (Fig. 6E–G). The S252F relaxations were too small to accurately resolve at voltages more positive than −90 mV. Previous results show that, similar to the human WT channels, the α4(S248F)β2 channels display short and long channel openings (Kuryatov et al. 1997). The S252F τf was nearly identical to the short mean open time (1.9 ms) of the α4(S248F)β2 channel (Kuryatov et al. 1997). The S252F τs was ∼3 times longer than the long mean open time (4 ms). Thus, the short S252F bursts (τf) may represent a single channel opening.
The +L264 mutation prolonged the relaxation currents in a voltage-dependent manner. The +L264 τf and τs displayed weak voltage dependence between −60 and −150 mV (Fig. 6E and F). The +L264 Af/Atot displayed strong voltage dependence (Fig. 6G). The +L264 τf increased e-fold per −311 mV (Fig. 6E). The τs increased e-fold per −217 mV (Fig. 6F). The Af/Atot decreased linearly from 46 ± 1% (n = 4) at −70 mV to 26 ± 1% (n = 7) at −150 mV (Fig. 6G). For a jump from +50 to −150 mV, the +L264 τf (13 ± 1 ms, n = 7) was > 2-fold larger than the WT value (above) and the +L264 τs (75 ± 4 ms, n = 7) was 50% larger. The +L264 Af/Atot (26 ± 1%, n = 7) was nearly 2-fold larger than the WT value. The +L264 mutation had less of an effect on the relaxation currents as the final jump voltage approached −70 mV. However, even at −70 mV, the +L264 τf (10.0 ± 0.6 ms, n = 4) was still 2-fold larger than the WT value. The +L264 τs (53 ± 6 ms, n = 4) and Af/Atot (46 ± 2%, n = 4) were similar to the WT values. If synapses involving α4β2 neuronal nicotinic AChRs are functional in the CNS, then +L264 should prolong the synaptic current decay and the S252F mutation should shorten it. Ultrastructural analysis combined with mAb 299 labelling shows that the α4 nicotinic subunit is present at synapses in the supraoptic nucleus (Shioda et al. 1997).
Mutations do not affect the number of surface receptors
To determine whether the +L264 and S252F mutations affected the number of surface receptors, we measured surface expression of the +L264, S252F and WT receptors using the anti-α4 antibody mAb 299 (Whiting & Lindstrom, 1988), the anti-β2 antibody mAb 270 (Whiting & Lindstrom, 1987) and an 125I-sheep anti-rat secondary antibody fragment (see Methods). The +L264 and S252F mutations did not affect 125I labelling of the oocytes pretreated with mAb 299 (Fig. 7A and C) or mAb 270 (Fig. 7B and D). They also had no effect on background 125I labelling of the oocytes treated with the 125I-secondary antibody fragment only (Fig. 7). Therefore, injecting equal amounts of WT, S252F and +L264 RNA produced equal numbers of surface receptors. Previous results show that the α4(S248F) mutation also has no effect on the oocyte surface receptor expression measured by 125I-mAb 290 binding (Kuryatov et al. 1997).
Figure 7. The +L264 and S252F mutations did not affect surface mAb 299 and mAb 270 antibody binding.
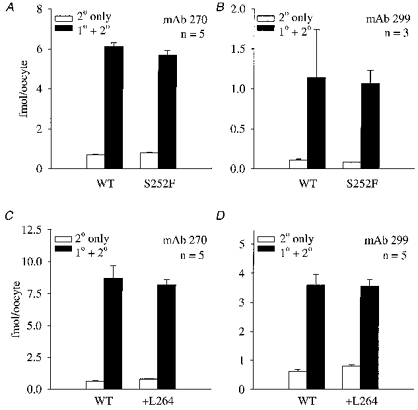
A-D, [125I]secondary antibody labelling of the S252F, +L264 and WT receptors. Each mutant was paired with a matched WT control. We used both anti-α4 (mAb 299) and anti-β2 (mAb 270) primary antibodies to measure surface receptor expression. Filled bars show 125I labelling of the receptors treated with both the primary (1°) and secondary (2°) antibodies. Open bars show 125I labelling of the receptors treated with the secondary (2°) antibody only. Error bars are +s.e.m.
S252F reduces the maximum ACh response
Central presynaptic terminals probably release a nearly saturating concentration of neurotransmitter during synaptic transmission (reviewed in Clements, 1996). If synapses involving α4β2 nicotinic receptors are functional in the CNS, then the maximum ACh response should determine the peak amplitude of the synaptic current. To determine whether the mutations affected the maximum ACh response, we measured the peak response of the +L264, S252F and WT receptors to 500 μm ACh at −50 mV at the same time as antibody binding experiments (above). The WT response (1680 ± 270 nA, n = 14) was not significantly different from the +L264 response (2100 ± 600 nA, n = 13), similar to previous results for the α4(776ins3) mutation (Steinlein et al. 1997). However, the S252F response (572 ± 96 nA, n = 13) was significantly less (Dunn's test, P < 0.01, n = 13–14) than the WT response. This reduction could be due to a decrease in the S252F single-channel conductance (below). However, the faster desensitization of the S252F receptor could have also contributed to the reduction in peak response. Previous results show that the α4(S248F) mutation does not significantly affect the response to 1 μm ACh (Weiland et al. 1996).
S252F enhances slow receptor desensitization
To study the effects of the +L264 and S252F mutations on the conformational changes underlying desensitization of the open state, we measured the time course of desensitization during a step application of a nearly saturating ACh concentration (Fig. 8). Nicotinic receptor desensitization is a complex cyclic process (Katz & Thesleff, 1957). However, at a saturating ACh concentration, the kinetics of desensitization can be simplified by making three reasonable assumptions. First, we assumed that ACh is a full agonist. Second, we assumed that desensitization is minimal in the absence of ACh (Franke et al. 1993). Third, we assumed that the receptor has two serial desensitized states. The kinetic model for the desensitization produced by a saturating ACh concentration jump then becomes: where O, Df and Ds are the open, fast-desensitized and slow-desensitized states, and kf, k−f, ks and k−s are rate constants. Model 1 predicts that the response IACh(t) to a saturating ACh concentration jump should decay bi-exponentially to a steady-state value (I∞),
| (2) |
where Af and As are the amplitudes of the fast and slow exponential components, τf and τs are the fast and slow desensitization time constants and t is the time after the peak response. Subject to the initial condition that all the receptors are open at time zero (assumption 1 above), we solved Model 1 to obtain the rate constants kf, k−f, ks and k−s in terms of τf, τs, Af, As and I∞. Defining the current at time zero (I0) as:
we obtained kf from τf, τs, Af, As, I0 and the following equation:
Figure 8. S252F mutation enhanced steady-state desensitization.
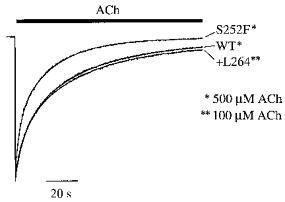
Normalized WT, S252F and +L264 responses to nearly saturating ACh concentration jumps at −50 mV. Bar above the traces shows the application of ACh. Fits of the falling phase of the responses to eqn (2) superimposed on the traces. See text for mean fit parameters.
Model 1.
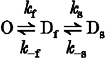
Using the calculated value of kf, we then obtained k−s as follows:
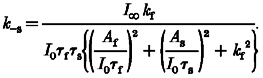 |
If τs >> τf, then the time constants of desensitization (τs and τf) are related to the underlying rate constants in Model 1 (kf, k−f, ks, k−s) by the following equations:
and
(Adams & Feltz, 1977). To obtain k−f and ks, we solved these two equations for k−f and ks in terms of τs, τf, kf and k−s:
and
Desensitization of the S252F, +L264 and WT responses to 500, 100 and 500 μm ACh was fitted well by eqn (2) (Fig. 8). Based on the S252F, +L264 and WT ACh concentration- response relations (Fig. 4D), these ACh concentrations represented fractional receptor occupancies of > 99%. Similar to the α4(776ins3) mutation (Steinlein et al. 1997), the +L264 mutation did not affect desensitization. However, the S252F mutation increased the level of steady-state desensitization (1 - I∞/(I∞+Af+As)) by 4–5 times. At steady state, only 2% of the peak S252F response remained, compared to 8–10% for the WT and +L264 responses (Fig. 8). The S252F τs (23 ± 1 s, n = 10) was also significantly less than the WT (36 ± 1 s, n = 11) and +L264 (42 ± 2 s, n = 9) values. The S252F τf (4.0 ± 0.2 s, n = 10) was slightly less than the WT (5.1 ± 0.3 s, n = 9–11) and +L264 values (6.0 ± 0.4 s, n = 9). The relative amplitude of the fast S252F component (37 ± 3% of the peak response, n = 10) was slightly larger than the WT (30 ± 2%, n = 11) and +L264 values (33 ± 1%, n = 9). The values for the desensitization rate constants (kf, k−f, ks, k−s in Table 2) show that there are two reasons why the S252F mutation enhanced steady-state desensitization. First, it increased the rate constant kf for entering the fast desensitized state from the open state. Second, it enhanced the stability (k−s/ks) of the slow desensitized state. The S252F rate constant for leaving the slow desensitized state k−s was nearly 3 times smaller than the WT and +L264 values (Table 2). Thus, S252F receptors entered the fast desensitized state more rapidly than the WT or +L264 receptors and left the slow desensitized state more slowly.
Table 2.
Desensitization rate constants for the WT, S252F and +L264 receptors
| Receptor | kf (10−3 s−1) | k−f (10−3 s−1) | ks (10−3 s−1) | k−s (10−3 s−1) |
|---|---|---|---|---|
| WT (n = 11) | 75 ± 5 | 81 ± 8 | 74 ± 6 | 1.9 ± 0.2 |
| S252F (n = 10) | 118 ± 8 | 84 ± 8 | 93 ± 12 | 0.6 ± 0.1 |
| +L264 (n = 9) | 80 ± 10 | 63 ± 5 | 55 ± 4 | 2.0 ± 0.2 |
Mutations affect single-channel conductance
Lastly, we measured the chord conductance of single +L264, S252F and WT channels in symmetrical 100 mm KCl solutions at −100 mV. Amplitude histograms of the WT single-channel currents (Fig. 9) revealed two main conductance states at 34 ± 2 pS (n = 6 patches) and 49 ± 1 pS (n = 5). The 34 pS state was present in all the WT patches (6/6 patches) and the 49 pS state was present in 5/6 of the patches. The largest WT conductance state was similar to the 46 pS value for recombinantly expressed human α4β2 nicotinic receptors (Buisson et al. 1996). The smaller WT conductance state (34 pS) was similar to a previous value (34 pS) for rat α4β2 nicotinic receptors expressed in oocytes (Charnet et al. 1992). To calculate the proportion of the open time spent in each WT open state, we divided the area under each component of the amplitude histogram that represented an open state by the total area under all the components representing open states. In the single patch without 49 pS openings, we assigned a percentage area of zero to the 49 pS component and 100% to the 34 pS component. The WT channels spent 60 ± 11% (mean ±s.e.m., n = 6 patches) of the total open time in the 34 pS state and 40 ± 11% (n = 6) of the total open time in the 49 pS state. The S252F amplitude histograms showed a single 32.2 ± 0.1 pS conductance state in all the patches (4/4 patches). However, there may have been an additional smaller state that was too brief or rare to resolve (Fig. 9). The +L264 amplitude histograms revealed two main conductance states at 49.4 ± 0.1 pS (4/4 patches) and 14.7 ± 0.1 pS (2/4). A single +L264 patch contained an additional 40 pS state (Fig. 9). Based on the relative areas of the open-state components of the amplitude histogram, the +L264 channels spent 14 ± 9% of the total open time in the 14.7 pS state, 6 ± 6% in the 40 pS state and 79 ± 12% in the 49.4 pS state (n = 4). The S252F mutation should reduce the mean single-channel conductance because it eliminated the 49 pS WT state. In contrast, the +L264 mutation should have relatively little effect on the mean single-channel conductance because the decreased conductance of the lesser main state (14.7 pS) should be offset by the increased frequency of the greater main states (49 pS). Similar to our results for the S252F mutation, previous results show that the α4(S248F) mutation eliminates the larger of the two WT conductance states (Kuryatov et al. 1997).
Figure 9. Conductance states of the WT, S252F and +L264 channels.
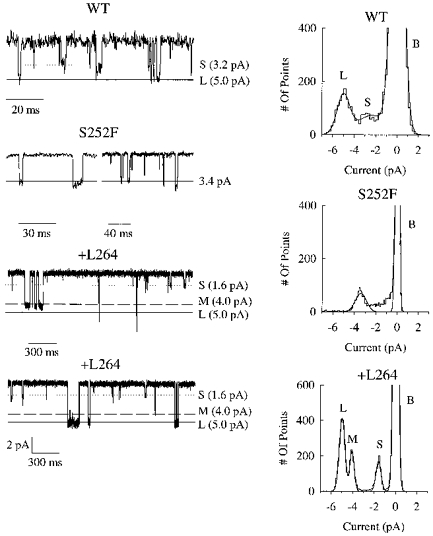
Left panels, sample traces of WT, S252F and +L264 channel openings (downward) in outside-out patches. Recordings were filtered at 1 kHz and sampled at 5 kHz. Vertical scale (under the +L264 traces) is identical for all three receptors. Horizontal lines show the mean current levels determined from the all-points histograms on the right. Right panels, all-points histograms of single-channel currents from patches shown on left (includes data not shown in traces). B, S, m and L denote the baseline current and the small, medium and large channel currents. Smooth curves are fits to the sum of multiple Gaussians. Vh =−100 mV.
DISCUSSION
The S252F and +L264 mutations have three common effects on the ACh response. First, they potentiate the response to a train of short ACh pulses in a use- and ACh concentration-dependent manner. Second, they delay the response to low (nm) ACh concentrations. Third, they reduce Ca2+-induced increases in the 30 μm ACh response. Besides these three common effects, the +L264 mutation increases the voltage-jump relaxation time constants and the percentage amplitude of the slow relaxation in a voltage-dependent manner, and eliminates the smaller (34 pS) of the two main WT conductance states. In contrast, the S252F mutation reduces the voltage-jump relaxation time constants in a non-voltage-dependent manner, reduces the maximum ACh response, increases the level of steady-state desensitization and reduces the mean single-channel conductance. Neither mutation affects the number of surface receptors or significantly shifts the EC50 for ACh or the KD for [3H]epibatidine binding. Except for the difference between the S252F and WT maximum response and the absence of a significant leftward shift in the +L264 EC50 for ACh, our results are consistent with the previous results for the α4(S248F) (Weiland et al. 1996; Kuryatov et al. 1997) and α4(776ins3) mutations (Steinlein et al. 1997). The contrasting effects of the +L264 and S252F mutations on the relaxation kinetics and single-channel conductance suggest that these effects do not cause ADNFLE seizures even though they may alter the synaptic response.
Previous results suggest that some biophysical and pharmacological properties of neuronal nicotinic receptors depend on the host cell type (Lewis et al. 1997). Mouse fibroblasts expressing rat α3β4 channels display two kinds of α3β4 channels. One channel type has a conductance of 30–40 pS and a brief lifetime similar to native nicotinic channels in rat superior cervical ganglion neurons. The other channel type has a conductance of 20–26 pS and opens in long bursts, similar to rat α3β4 channels expressed in oocytes (Lewis et al. 1997). The absence of the high-conductance, short-lived channels in the oocytes may mean that oocytes cannot assemble rat α3β4 channels in the same way as mammalian cells. However, other studies show that despite these apparent differences, the ACh concentration- response relations for rat α3β4 receptors expressed in oocytes (EC50 = 219 ± 12 μm, nH = 2.2 ± 0.1, Cohen et al. 1995) and rat α3β4 receptors expressed in HEK 293 (human embryonic kidney) cells (EC50 = 202 ± 32 μm, nH = 1.9 ± 0.4, Wong et al. 1995) are not significantly different. In contrast to α3β4 receptors, the single-channel conductance of the larger rat α4β2 channel expressed in oocytes (49 pS) matches that (46 pS) of human α4β2 channels expressed in HEK 293 cells (Buisson et al. 1996). Moreover, the EC50 (2.6 ± 0.2 μm) and nH (0.90 ± 0.03) of the rat α4β2 ACh concentration-response relation in oocytes is similar to the EC50 (3 μm) and nH (1.2) of the human α4β2 ACh concentration-response relation in HEK 293 cells (Buisson et al. 1996). Thus, we have as yet no conclusive data showing that the properties of α4β2 nicotinic receptors expressed in oocytes differ significantly from those expressed in cell lines or the native tissue.
Two mechanisms could produce apparent use-dependent potentiation of the ACh response. First, the mutations could prolong the lifetime of the closed mono-liganded receptor state. However, the mono-liganded mutant state would have to persist long enough after the first ACh pulse to increase the response to the subsequent pulses, and the 5 s inter-pulse interval in the pulse train makes this mechanism unlikely. Moreover, one would expect a significant decrease in the EC50 for ACh for such a large increase in the lifetime of the mono-liganded state. Second, the mutations could create a site in the channel pore that binds a blocking molecule more tightly when the channel is closed than when it is open (closed-channel block). Once the mutant channels open, the blocker would dissociate and permit ions to permeate the channel. The relief of block during a train of ACh pulses could produce apparent use-dependent potentiation of the response.
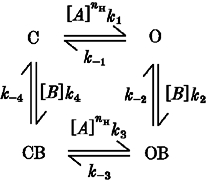
Closed-channel block is a novel but plausible mechanism. Most models of channel block assume that the blocking site is not accessible in the closed state. However, hydrophilic sulfhydryl-specific reagents reach cysteine-substituted residues in M2 in the closed state and block ion permeation (reviewed in Karlin & Akabas, 1995). Therefore, a blocker can reach a binding site in M2 even when the channel is closed. We can illustrate closed-channel block with the following allosteric model: where B is the blocking molecule, C is the closed state, CB is the closed blocked state, O is the open state, OB is the open blocked state, [B] is the blocker concentration, [A] is the ACh concentration and the ki values are rate constants. For simplicity, all the steps involved in agonist activation of the channel have been combined into single steps in this model (O ⇌ C, CB ⇌ OB). If channel opening is much faster than channel unblocking, then the macroscopic rate constant of this reaction is [CB]k−4+[C][B]k4+[OB]k−2+[O][B]k2 (square brackets denote the fractional occupancies of states C, CB, OB and O). ACh binding relieves closed-channel block because a closed-channel blocker has a higher affinity for the closed state than the open state. Reducing the probability of channel opening will slow down the apparent rate of channel unblocking which explains why potentiation is easier to observe at low ACh concentrations. Slow unblocking of the closed channels at low ACh concentrations could also explain why the mutations delay the rise of the ACh response at very low fractional receptor occupancies.
Reblocking of the closed channels during the inter-pulse interval must be slow, otherwise equilibrium is restored and use-potentiation will not occur. Closed unblocked channels therefore represent a sink and closed blocked channels (CB) represent a pool of receptors that contribute to use-dependent potentiation. This phenomenon is illustrated by the slow rise times of the +L264 and S252F responses to 5–15 nm and 30 nm ACh, respectively (Fig. 3B and C). The initial rapidly rising phase of the response represents the opening of unblocked channels. This rise is as fast as the ACh concentration rises on the oocyte surface. The more slowly rising phase represents the opening of closed blocked channels (CB) which have to unblock before they can pass current. Since this rate is slower than the rise in the surface ACh concentration, the apparent rate by which closed unblocked channels reach the open state is faster than the rate through which closed blocked channels (CB) reach the open state. This slower rate is ACh concentration dependent because as we increase the ACh concentration, we increase the fractional occupancy of the OB state. Figure 3D shows that, as the ACh concentration increases, so does the rate of rise (proportional to 1/t½) of the slowly rising phase of the +L264 response. The relative amplitude of the rapid initial phase of the +L264 5 nm ACh response suggests that 86% of the closed +L264 receptors are blocked in the absence of ACh. Could potentiation occur during synaptic transmission? The answer depends on the relative values of k−2 and k−3 in Model 2. If k−3 is comparable to or > k−2, then the mutant channels would not unblock completely during a brief, saturating ACh impulse and use-dependent potentiation of the synaptic current could occur. We have not determined the identity of the blocking molecule, but previous studies of ion-channel block suggest that it could be Mg2+ (Ifune & Steinbach, 1992), a polyamine (Lopatin et al. 1994), or even part of the protein itself as for the Shaker channel (Zagotta et al. 1990).
Model 2.
Both mutations should reduce the peak synaptic current under physiological conditions because of their effects on Ca2+-induced increases in the ACh response. The +L264 and S252F mutations reduce Ca2+-induced increases in the ACh response by the same amount (40–50%) as the α4(776ins3) mutation does (Steinlein et al. 1997). Our results suggest that the maximum WT ACh response should be ≥ 30% larger than the +L264 and S252F ACh responses in 2.5 mm extracellular Ca2+. We used 2.5 mm Ca2+ for our experiments to facilitate comparison with the previous α4(776ins3) results (Steinlein et al. 1997). However, the free extracellular Ca2+ concentration in the interstitial space is actually ∼1.2 mm (Guyton & Hall, 1996). Therefore, the difference between the maximum WT and mutant ACh response may be less under physiological conditions.
The mechanism responsible for Ca2+-induced increases in the neuronal nicotinic response is not completely understood. Experiments on central nicotinic receptors suggest that extracellular Ca2+ increases the channel opening rate (Mulle et al 1992). If the +L264 and S252F mutations do not affect Ca2+-induced increases in the channel opening rate, then the decrease in Ca2+-induced potentiation of the mutant receptors may mean that Ca2+ ions block ion permeation through the mutant channels. The α4(S248F) mutation reduces the Ca2+ permeability of the receptor but measurements of the residual current in Na+-free saline suggest that Ca2+ ions carry only 6% of the current through the WT channels (Kuryatov et al. 1997). The +L264 and S252F mutations reduce Ca2+-induced increases in the ACh response by 40–50% (Table 2). Therefore, a reduction in the mutant Ca2+ permeability would not be sufficient to account for the effects of the mutants on Ca2+-induced increases in the ACh response. Ca2+ ions may therefore block ion permeation through the mutant channels.
We can envisage two kinds of mechanisms that could generate ADNFLE seizures. The first mechanism is that the ADNFLE mutations reduce cortical nicotinic-mediated inhibitory neurotransmitter release by reducing the Ca2+ permeability of presynaptic nicotinic receptors. A reduction in inhibitory transmitter levels could lead to an increase in cortical neuronal firing. Presynaptic α4β2 nicotinic receptors can increase Ca2+ influx (and therefore neurotransmitter release) by two mechanisms. First, activation of the receptors will depolarize the synaptic terminal and cause voltage-dependent Ca2+ channels to open. Second, the receptors transport Ca2+ directly into the cell. The α4(S248F) mutation reduces Ca2+ permeability (Kuryatov et al. 1997) and, therefore, it should reduce direct Ca2+ transport into the cell. However, previous studies suggest that, with the possible exception of α7 nicotinic receptors, the activation of voltage-dependent Ca2+ channels probably accounts for most of the nicotinic receptor-mediated presynaptic Ca2+ influx (reviewed in Wonnacott, 1997). Therefore, a reduction in the Ca2+ permeability of the mutant receptors may have a relatively minor effect on α4β2-induced neurotransmitter release. It is also unclear why a reduction in presynaptic Ca2+ influx should selectively reduce inhibitory transmitter release. The second mechanism that could generate ADFNLE seizures is that use-dependent potentiation of the mutant response produces a sudden increase in nicotinic-mediated neurotransmitter release. Use-dependent potentiation could enhance both inhibitory and excitatory neurotransmitter release, and both effects may contribute to synchronous neuronal firing in the cortex. Use-dependent potentiation of the mutant response will affect nicotinic receptor-mediated transmitter release in a frequency-dependent manner. High-frequency presynaptic cholinergic stimulation should relieve closed-channel block of the mutant receptors and increase nicotinic-mediated neurotransmitter release. Therefore, a sudden increase in the firing rate of an ascending cholinergic cortical projection could trigger seizures by suddenly increasing the amount of neurotransmitter released by cortical presynaptic nicotinic receptors. Ordinarily, ADNFLE patients experience an average of eight brief (< 60 s) nocturnal seizures per night (Scheffer et al. 1995). Except for these seizures, ADNFLE patients exhibit no chronic neurological deficits. The absence of chronic neurological deficits and the brevity of the seizures suggests that the physiological conditions that trigger ADNFLE seizures occur relatively rarely. A mechanism such as use-dependent potentiation, which should occur only under special conditions, therefore seems to be more compatible with the transitory nature of the ADNFLE seizures than a mechanism such as reduced receptor Ca2+ permeability, which should produce only a minor tonic decrease in nicotinic receptor-mediated inhibitory transmitter release in the brain.
Acknowledgments
We thank Dr Henry Lester and Dr Cesar Labarca (California Institute of Technology) for providing the rat α4 clone, the rat β2 clone and the modified pBluescript expression vector, Dr Kenneth Dorshkind (UCLA) for the mAb 270 antibody, and Hyosuk Leathers (UCLA) for technical assistance. This work was supported by grants from the American Heart Association (nos 96–254 and 96–112), and the UC Tobacco-Related Disease Research Program (6KT-0208).
References
- Adams PR, Feltz A. Interaction of a fluorescent probe with acetylcholine-activated synaptic membrane. Nature. 1977;269:609–611. doi: 10.1038/269609a0. [DOI] [PubMed] [Google Scholar]
- Badio B, Daly JW. Epibatidine, a potent analgetic and nicotinic agonist. Molecular Pharmacology. 1994;45:563–569. [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human α4β2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch clamp study. Journal of Neuroscience. 1996;16:7880–7891. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charnet P, Labarca C, Cohen BN, Davidson N, Lester HA, Pilar G. Pharmacological and kinetic properties of α4β2 neuronal nicotinic acetylcholine receptors expressed in Xenopus oocytes. The Journal of Physiology. 1992;450:375–394. doi: 10.1113/jphysiol.1992.sp019132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements JD. Transmitter time course in the synaptic cleft: its role in central synaptic function. Trends in Neurosciences. 1996;19:163–171. doi: 10.1016/s0166-2236(96)10024-2. [DOI] [PubMed] [Google Scholar]
- Cohen BN, Figl A, Quick MW, Labarca C, Davidson N, Lester HA. Regions of β2 and β4 responsible for differences between the steady state dose-response relationships of the α3β2 and α3β4 neuronal nicotinic receptors. Journal of General Physiology. 1995;105:745–764. doi: 10.1085/jgp.105.6.745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D, Sigworth F. Fitting and statistical analysis of single-channel records. In: Sakmann B, Neher E, editors. Single-Channel Recording. New York: Plenum Press; 1995. pp. 483–587. [Google Scholar]
- Edmonds B, Gibb AJ, Colquhoun D. Mechanisms of activation of muscle nicotinic acetylcholine receptors and the time course of endplate currents. Annual Review of Physiology. 1995;57:469–493. doi: 10.1146/annurev.ph.57.030195.002345. 10.1146/annurev.ph.57.030195.002345. [DOI] [PubMed] [Google Scholar]
- Eilers H, Schaeffer E, Bickler PE, Forsayeth JR. Functional deactivation of the major neuronal nicotinic receptor caused by nicotine and a protein kinase C-dependent mechanism. Molecular Pharmacology. 1997;52:1105–1112. [PubMed] [Google Scholar]
- Figl A, Labarca C, Davidson N, Lester HA, Cohen BN. Voltage-jump relaxation kinetics for wild-type and chimeric β subunits of neuronal nicotinic receptors. Journal of General Physiology. 1996;107:369–379. doi: 10.1085/jgp.107.3.369. 10.1085/jgp.107.3.369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figl A, Viseshakul N, Forsayeth J, Cohen BN. A mutation associated with epilepsy enhances desensitization of the α4β2 neuronal nicotinic receptor. Biophysical Journal. 1997;72:A150. [Google Scholar]
- Flores CM, Rogers SW, Pabreza LA, Wolfe BB, Kellar KJ. A subtype of nicotinic cholinergic receptor in rat brain is composed of α4 and β2 subunits and is up-regulated by chronic nicotine treatment. Molecular Pharmacology. 1991;41:31–37. [PubMed] [Google Scholar]
- Franke C, Parnas H, Hovav G, Dudel J. A molecular scheme for the reaction between acetylcholine and nicotinic channels. Biophysical Journal. 1993;64:339–356. doi: 10.1016/S0006-3495(93)81374-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerzanich V, Peng X, Wang F, Wells G, Anand R, Fletcher S, Lindstrom J. Comparative pharmacology of epibatidine: a potent agonist for neuronal nicotinic acetylcholine receptors. Molecular Pharmacology. 1995;48:774–782. [PubMed] [Google Scholar]
- Guyton AC, Hall JE. Textbook of Medical Physiology. Philadelphia, PA, USA: W. B. Saunders; 1996. p. 986. [Google Scholar]
- Houghtling RA, Davila-Garcia MI, Kellar KJ. Characterization of (±)-[3H]epibatidine binding to nicotinic cholinergic receptors in rat and human brain. Molecular Pharmacology. 1995;48:280–287. [PubMed] [Google Scholar]
- Ifune CK, Steinbach JH. Inward rectification of acetylcholine-elicited currents in rat phaeochromocytoma cells. Journal of Physiology. 1992;457:143–165. doi: 10.1113/jphysiol.1992.sp019369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jobling SA, Gehrke L. Enhanced translation of chimaeric messenger RNAs containing a plant viral untranslated leader sequence. Nature. 1987;325:622–625. doi: 10.1038/325622a0. 10.1038/325622a0. [DOI] [PubMed] [Google Scholar]
- Karlin A, Akabas MH. Toward a structural basis for the function of nicotinic acetylcholine receptors and their cousins. Neuron. 1995;15:1231–1244. doi: 10.1016/0896-6273(95)90004-7. 10.1016/0896-6273(95)90004-7. [DOI] [PubMed] [Google Scholar]
- Katz B, Thesleff S. A study of the ‘desensitization’ produced by acetylcholine at the motor end-plate. Journal of Physiology. 1957;138:63–80. doi: 10.1113/jphysiol.1957.sp005838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuryatov A, Gerzanich V, Nelson M, Olale F, Lindstrom J. Mutation causing autosomal dominant nocturnal frontal lobe epilepsy alters Ca2+ permeability, conductance, and gating of human α4β2 nicotinic acetylcholine receptors. Journal of Neuroscience. 1997;17:9035–9047. doi: 10.1523/JNEUROSCI.17-23-09035.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TM, Harkness PC, Sivilotti LG, Colquhoun D, Millar NS. The ion channel properties of a rat recombinant neuronal nicotinic receptor are dependent on the host cell type. Journal of Physiology. 1997;505:299–306. doi: 10.1111/j.1469-7793.1997.299bb.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature. 1994;24:366–369. doi: 10.1038/372366a0. 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Mulle C, Lena C, Changeux J-P. Potentiation of nicotinic receptor response by external calcium in rat central neurons. Neuron. 1992;8:937–945. doi: 10.1016/0896-6273(92)90208-u. 10.1016/0896-6273(92)90208-U. [DOI] [PubMed] [Google Scholar]
- Peng X, Gerzanich B, Anand R, Whiting PJ, Lindstrom J. Nicotine-induced increase in neuronal nicotinic receptors results from a decrease in the rate of receptor turnover. Molecular Pharmacology. 1994;46:523–530. [PubMed] [Google Scholar]
- Quick MW, Lester HA. Methods for expression of excitability proteins in Xenopus oocytes. In: Conn PM, editor. Methods in Neurosciences. Vol. 19. San Diego: Academic Press; 1994. pp. 261–279. [Google Scholar]
- Sands SB, Costa ACS, Patrick J. Barium permeability of neuronal nicotinic receptor α7 expressed in Xenopus oocytes. Biophysical Journal. 1993;65:2614–2621. doi: 10.1016/S0006-3495(93)81296-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheffer IE, Bhatia KP, Lopez-Cendes I, Fish DR, Marsden D, Andermann E, Andermann R, Desbiens R, Keene D, Cendes F, Manson JI, Constantinou JEC, McIntosh A, Berkovic SF. Autosomal dominant nocturnal frontal lobe epilepsy: a distinctive clinical disorder. Brain. 1995;118:61–73. doi: 10.1093/brain/118.1.61. [DOI] [PubMed] [Google Scholar]
- Shioda S, Yada T, Muroya S, Takigawa M, Nakai Y. Nicotine increases cytosolic Ca2+ in vasopressin neurons. Neuroscience Research. 1997;29:311–318. doi: 10.1016/s0168-0102(97)00103-x. 10.1016/S0168-0102(97)00103-X. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Magnusson A, Stoodt J, Bertrand S, Weiland S, Berkovic SF, Nakken KO, Propping P, Bertrand D. An insertion mutation of the CHRNA4 gene in a family with autosomal dominant nocturnal frontal lobe epilepsy. Human Molecular Genetics. 1997;6:943–947. doi: 10.1093/hmg/6.6.943. 10.1093/hmg/6.6.943. [DOI] [PubMed] [Google Scholar]
- Steinlein OK, Mulley JC, Propping P, Wallace RH, Phillips HA, Sutherland GR, Scheffer IE, Berkovic SF. A missense mutation in the neuronal nicotinic acetylcholine receptor α4 subunit is associated with autosomal dominant nocturnal frontal lobe epilepsy. Nature Genetics. 1995;11:201–203. doi: 10.1038/ng1095-201. 10.1038/ng1095-201. [DOI] [PubMed] [Google Scholar]
- Vibat CRT, Lasalde JA, McNamee MG, Ochoa ELM. Differential desensitization properties of rat neuronal nicotinic acetylcholine receptor subunit combinations expressed in Xenopus oocytes. Cellular and Molecular Neurobiology. 1995;15:411–425. doi: 10.1007/BF02071877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weiland S, Witzemann V, Villaroel A, Propping P, Steinlein O. An amino acid exchange in the second transmembrane segment of a neuronal nicotinic receptor causes partial epilepsy by altering its desensitization kinetics. FEBS Letters. 1996;398:91–96. doi: 10.1016/s0014-5793(96)01215-x. 10.1016/S0014-5793(96)01215-X. [DOI] [PubMed] [Google Scholar]
- Whiting P, Lindstrom J. Purification and characterization of a nicotinic acetylcholine receptor from rat brain. Proceedings of the National Academy of Sciences of the USA. 1987;84:595–599. doi: 10.1073/pnas.84.2.595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting PJ, Lindstrom J. Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. Journal of Neuroscience. 1988;8:3395–3404. doi: 10.1523/JNEUROSCI.08-09-03395.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Whiting P, Schoepfer R, Lindstrom J, Priestley T. Structural and pharmacological characterization of the major brain nicotinic acetylcholine receptor subtype stably expressed in mouse fibroblasts. Molecular Pharmacology. 1991;40:463–472. [PubMed] [Google Scholar]
- Wong ET, Holstad SG, Mennerick SJ, Hong SE, Zorumski CF, Isenberg KE. Pharmacological and physiological properties of a putative ganglionic nicotinic receptor, α3β4, expressed in transfected eucaryotic cells. Molecular Brain Research. 1995;28:101–109. doi: 10.1016/0169-328x(94)00189-l. 10.1016/0169-328X(94)00189-L. [DOI] [PubMed] [Google Scholar]
- Wonnacott S. Presynaptic nicotinic ACh receptors. Trends in Neurosciences. 1997;20:92–98. doi: 10.1016/s0166-2236(96)10073-4. 10.1016/S0166-2236(96)10073-4. [DOI] [PubMed] [Google Scholar]
- Zagotta WN, Hoshi T, Aldrich RW. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 1990;250:568–571. doi: 10.1126/science.2122520. [DOI] [PubMed] [Google Scholar]