

Abstract
Previous studies that investigated the role of inflammation in the neurotoxicity of manganese (Mn) found that Mn enhanced the production of inflammogen (lipopolysaccharide; LPS)-induced proinflammatory cytokines such as IL-6 and TNF-α. Although we have shown that the enhanced cytokine production occurs via a NF-κB-dependent mechanism, the role of upstream kinases in this Mn-induced enhancement has not been explored. As other studies have demonstrated that p38 mitogen activated protein kinase (p38) is necessary for LPS-induced, NF-κB-dependent expression of proinflammatory cytokines, we hypothesized that Mn enhancement of LPS-induced production of IL-6 and TNF-α may be associated with p38 activation and conducted a series of experiments to address our hypothesis. We found that pre-treatment of microglial cells with a p38-inhibitor (SB203580) prevented Mn+LPS- induced production of IL-6 and TNF-α. Moreover, potentiation of IL-6 and TNF-α production, which occurred in both concurrent and sequential (3 h apart) exposures to Mn and LPS, was inhibited by inhibition of p38. Additionally, Mn exposure enhanced the phosphorylation and activity of p38 and this effect was persistent. Although p38 activity declined over time in vehicle and LPS-exposed cells, it persisted in cells exposed to Mn or Mn+LPS. Thus, the increased production of proinflammatory cytokines by LPS-activated microglia exposed to Mn is associated with increased and persistent activation of p38.
Keywords: manganese, microglia, cytokines, inflammation, p38 MAPK
INTRODUCTION
Manganese (Mn), while an essential metal, is also a common environmental contaminant. The presence of Mn in alloys, fertilizers, batteries, and fungicides, as well as the re-introduction of the fuel additive methylcyclopentdienyl manganese tricarbonyl (MMT), is of environmental and occupational concern (Aschner, 2000; Frumkin et al., 1997). Occupational exposure to Mn has been linked to a specific neuropathology, manganism, that is characterized by clinical signs and lesions similar to Parkinson’s Disease (PD; Meco et al., 1994). Manganese is thought to exert its effects, at least partially, by disrupting mitochondrial respiration leading to increased oxidative stress (Aschner and Aschner, 1991; Gavin et al., 1999). This is supported by studies demonstrating that Mn-containing compounds, such as the fungicide Maneb and the fuel additive MMT, can inhibit mitochondrial respiration (Auttissier et al., 1977; Zhang et al., 2003).
While Mn is directly toxic to neuronal cells, neurons are not the only CNS cells that are associated with and contribute to Mn neurotoxicity. Astrocytes, for example, accumulate Mn and may produce reactive oxygen species (ROS) and other substances that may be damaging to neurons (Aschner et al., 2000). Importantly, it has been demonstrated that the other CNS resident cells, the microglia, and/or the astrocytes may produce inflammatory mediators that could be involved in the mechanisms of Mn neurotoxicity, especially in cases where an additional inflammatory stimulus is present (Chang and Liu, 1999; Filipov et al., 2005; Spranger et al., 1998).
Microglia have been implicated in PD (humans and animal models) and research utilizing the model PD toxicant MPTP (1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine) has shown that activated microglia persist long after exposure to MPTP has ended (McGeer et al., 1988, 2003). Additionally, it has been demonstrated that prior exposure to Mn before challenge with MPTP will result in greater basal ganglia pathology than exposure to Mn or MPTP alone (Takahashi et al., 1989). Therefore, this study suggests that Mn exposure has the potential of interacting with other basal ganglia toxicants, thus causing greater neurotoxicity. However, the likelihood of such exposure (i.e. Mn + MPTP) is remote. On the other hand, a more relevant model, may involve Mn and lipopolysaccharide (LPS). LPS is a common environmental contaminant (Niehaus and Lange, 2003) and model inflammogen due to its ability to stimulate microglia to produce cytokines, nitric oxide (NO), and ROS (Chao et al., 1992; Jeohn et al., 2002; Liu et al., 2002). Importantly, LPS exposure is also associated with basal ganglia toxicity (Niehaus and Lange, 2003) and it has been used as a model for PD in in vitro and in vivo studies (Liu et al., 2002; Castano et al., 1998)
Binding of LPS to CD14 and TLR4 cell surface receptors leads to the activation of intracellular kinases, including the mitogen activated protein kinases (MAPK; Bhat et al., 1998; Jeohn et al., 2002). The MAPK family of proteins is comprised of the extracellular signal-regulated kinases (ERK), stress-activated or c-Jun N-terminal kinases (SAPK/JNK), big MAPK 1 (BMK1), and the p38 MAPK (Koistinaho and Koistinaho, 2002). Of these MAPK, p38 MAPK (p38) and ERK appear to be primarily involved in the production of inflammatory mediators by microglia. In primary microglia and microglial cell lines, LPS has been shown to dose- and time-dependently increase the phosphorylation of ERK and p38, as well as increase the expression of iNOS and TNF-α (Bhat et al., 1998; Lee et al., 1994; Lee et al., 1993). Of note, the p38-dependent increases in NO production require not only the phosphorylation of p38 but increased kinase activity as well (Jeohn et al., 2002). Additionally, by exposing microglia to ERK-and p38-inhibitors prior to exposure to LPS, the LPS-induced increases in NO and TNF-α were inhibited (Bhat et al., 1998). Furthermore, LPS-induced, p38-dependent, increases in NO and TNF-α by microglia have been shown to decrease neuronal survivability in neuronal-glial co-culture (Jeohn et al., 2002). The fact that this effect can be inhibited by pretreatment with inhibitors of p38 suggests that p38 appears to play a dominant role in the process.
Although inflammatory responses are essential for the maintenance and defense of tissues, uncontrolled or chronic inflammation can be detrimental to tissue homeostasis, especially in sensitive tissues like the nervous system. In fact, abnormally high levels of inflammatory cytokines, such as TNF-α, have been implicated in the etiology of PD (Nagatsu et al., 2000). Within the context of Mn neurotoxicity, Mn enhances the production of inflammatory mediators by microglia. Indeed, exposure to Mn potentiates LPS-induced production of inflammatory cytokines (TNF-α & IL-6) and NO in vitro (Filipov et al., 2005). Additionally, this effect is NF-kB-dependent as inhibitors of NF-kB were able to prevent the potentiation observed in Mn+LPS exposed cells (Filipov et al., 2005). At present, it is not known whether the potentiation of inflammatory cytokine production by Mn occurs at the level of NF-kB or further upstream in the intracellular signaling cascade. Since potential upstream targets include p38 and ERK and because a p38 inhibitor alone or in combination with an ERK inhibitor prevents the LPS-induced production of inflammatory mediators (Bhat et al., 1998), we conducted preliminary studies examining the effect of MAPK inhibition on cytokine production in Mn-exposed microglial cells activated with LPS (Crittenden and Filipov, 2004). From these studies we determined that inhibition of p38, but not of ERK, eliminated the potentiation of LPS-induced cytokine production in N9 microglial cells.
After we established the role of p38 in the enhancement of LPS-induced proinflammatory cytokine production by Mn, our objectives in this study were to (i) examine in detail the functional activation of p38 by exposure to Mn by itself or in combination with LPS and (ii) evaluate the time-window during the proinflammatory cytokine production process which is dependent upon p38 in Mn+LPS-exposed microglia.
MATERIALS AND METHODS
Chemicals
Unless specified, all chemicals and reagents were purchased from Sigma-Aldrich (Sigma; St. Louis, MO) and MnCl2 with purity >99% was used.
Cell Culture
The N9 murine microglial cell line used in the experiments was a gift kindly provided by Dr. P. Ricciardi-Castagnoli (University of Milan, Italy). These cells, derived by a retroviral immortalization of day-13 embryonic mouse brain cultures, are similar to primary microglia in that, upon activation, they produce proinflammatory cytokines, such as IL-1β, IL-6, and TNF-α as well as NO (Righi et al., 1989). For example, side by side comparisons of the N13 microglial cell line, which is almost identical with the N9 cell line and it was developed by the same group of researchers (Righi et al., 1989), with primary mouse microglia cells demonstrated that both produced comparable amounts of IL-6 following stimulation with a concentration-range of LPS (Heyen et al., 2000). The cultures were maintained (5% CO2, 95% air, at 37°C) in RPMI-1640 supplemented with 10% FBS, 0.075% sodium bicarbonate, 1 mM sodium pyruvate, 1 mM non-essential amino acids, 2 mM L-glutamine, 50 μM 2-mercaptoethanol, 25 μg/ml gentamycin, 100 U/ml penicillin G, and 100 mg/ml streptomycin (all from Invitrogen, Carlsbad, CA). For pharmacological inhibition and cytokine studies, cells were seeded at 0.25 × 106 cells/well (0.5 ml volume) in 48-well plates (Costar; Fisher Scientific, Pittsburgh, PA). For western blot protein analysis, cells were seeded at 2.5 × 106 cells/well (5 ml volume) in 6-well plates (Costar), whereas for the flow cytometry analysis, the density was 2.0 × 106 cells/well (4 ml volume). Cells were incubated for up to 24 h in the presence of Mn (250 μM) and/or LPS (Escherichia coli 0111: B4; 100 ng/ml).
In our preliminary studies (Crittenden and Filipov, 2004), and in Filipov et al. (2005), Mn concentrations of 50–1000 μM and LPS of 10–1000 ng/ml were used, and the potentiation of cytokine production was both Mn- and LPS-dependent. Hence, the concentrations of Mn and LPS used in this study were selected based on previous experiments that indicated no significant N9 cell death following exposure to Mn and/or LPS at these concentrations (Filipov et al., 2005) and are in line with numerous other in vitro studies where levels of Mn (Mn2+) range from 10 μM to 4 mM with the most typical exposure range being 100 to 500 μM (i.e., Li et al., 2005; Malthankar et al., 2004). Moreover, the above concentrations and the concentrations used in our study are representative of Mn levels found in brains of non-human primates following exposure to manganese dioxide for 3 months (ranging from 35 to 350 μM; Suzuki et al., 1975). Eelevated levels of Mn in the basal ganglia are not associated only with occupational exposure, but have been also observed in the brains of PD patients (Yase 1972), as well as in autopsied brains of patients with cirrhosis (Pomier-Layrargues et al., 1995). Of note, the concentration of LPS we have used in the present study (100 ng/ml) is relatively low and we have already reported that when greater amounts of LPS are present in the culture medium, less Mn is required for a potentiating effect on cytokine production to be observed (Filipov et al., 2005).
Cytokine Analysis
The amounts of interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) in the supernatants were determined using DuoSet ELISA kits (R&D; Systems, Inc., Minneapolis, MN) as per manufacturer’s instructions. Briefly, 96-well microplates (Costar) were coated with 1:180 dilution of respective capture antibody in PBS and incubated overnight at room temperature (RT). The plates were then washed with PBS-Tween buffer (3x) and blocked (1% BSA, 5% sucrose in PBS) for 1 h at RT. Next, plates were washed again (3x) and either samples or standards were dispensed into the wells. After incubating for 2 h at RT the plates were washed (3x), incubated with detection antibody for 2 h, and washed again (3x). Strepavidin-HRP conjugate was then added for 20 min, plates were washed (3x), and TMB-based substrate was added to each well and allowed to develop up to 30 min. The reaction was stopped by addition of 50 μl of 2N H2SO4 per well and the absorbance was read using a Spectramax Plate Reader (Molecular Devices, Sunnyvale, CA) at 450 nm. All samples for each cytokine were assayed in duplicate, and the mean was used in the subsequent statistical analysis. A standard curve was generated for each plate.
Pharmacological manipulations
The p38 inhibitor, SB203580 (Calbiochem, San Diego, CA), was added 3 h prior to, or after, the addition of Mn, LPS, or Mn and LPS to the cell culture. The inhibitor was dissolved in DMSO (26.5 mM) and stored at −80°C until diluted to a working concentration (50 μM) prior to use. The final DMSO concentration was < 0.2% for vehicle and p38-inhibitor exposed cells.
Immunoblot analysis of p38 and phospho-p38
After incubation for 15 min, 1 and 4 h, cells were removed from the culture well via scraping and the cell suspension was centrifuged (300g; 10 min; 4°C). Following centrifugation, the supernatants were discarded, cells were resuspended in 100μl of RIPA (modified radioimmuno-precipitation) lysis buffer (1x PBS, 1% Igepal, 0.5% sodium deoxycholate, 0.1% SDS) containing PMSF (Sigma), protease and phosphatase inhibitors (Protease Inhibitor Cocktail, Sigma, and Halt Phosphatase Inhibitor Cocktail, Pierce [Rockford, IL], respectively), and held on ice for 30 min with occasional pipetting to disrupt cell membranes. Protein concentration in the cell lysates was determined using the Bradford method with reagents obtained from Bio-Rad (Hercules, CA) and with BSA as a standard. Aliquots of each sample were diluted in reducing sample buffer and heat denatured for 5 min at 95 °C. Twenty micrograms of total protein were loaded and separated on a 10% SDS-PAGE gel, transferred to a PVDF membrane, and the membranes were blocked in 5% milk for 1 h at RT. The membranes were then incubated overnight with rabbit antibodies specific for the non-phosphorylated or phosphorylated p38 (Cell Signaling Technology, Beverly, MA) at 1:1000 and 1:500 dilutions, respectively, washed 3x, and then probed with goat anti-rabbit secondary antibody conjugated with horseradish peroxidase (Bio-Rad) at a 1:10,000 dilution. The blots were exposed to SuperSignal West Pico chemiluminescent substrate (Pierce) for up to 5 min and then enclosed in transparent covers prior to exposure to x-ray film. Band density was analyzed using the UN-SCAN-IT software package (Silk Scientific Inc., Orem, UH).
Phospho-p38 analysis by Flow Cytometry
Four ml of 0.5 × 106 cells/ml N9 cell suspension (2 ×106 cells/well) were plated overnight in 6-well plates as described above. Cells were incubated with Mn/LPS for 15 min, 1, and 4 h. Following incubation, cells were lifted from the culture wells with a cell lifter, spun down (10 min; 300 xg; 4 °C), and resuspended in 0.5 ml of PBS. Cells were then fixed by adding of 0.5 ml of 4% paraformaldehyde (2% final concentration) to the tubes and incubating them for 10 min at 37°C. Next, the cells were chilled for 1 min on ice, centrifuged (10 min; 300 ×g; 4 °C), and permeabilized by resuspending them in 1 ml of 90% methanol and an incubation for 30 min on ice, followed by an overnight incubation at −20 °C. The next day, cells were centrifuged to remove the methanol and washed twice with a binding buffer (1% BSA in PBS, 2 ml for each wash). Next, 1 ml of the cell suspension (approximately 0.6 × 106 cells) was transferred to flow tubes, spun down, and resuspended in 100 μl of binding buffer. Following 10 min incubation at RT, 10 μl/tube of the anti-phospho-p38 (T180/Y182)-PE pre-titrated antibody, or an appropriate PE-conjugated isotype control (mouse IgG1; both from BD Biosciences, San Diego, CA) were added and the tubes incubated for 60 min at RT in the dark. At the end of this incubation, the cells were washed twice with a binding buffer, resuspended in 0.5 ml of PBS and analyzed by FACS Calibur flow cytometer (Becton Dickinson, San Jose, CA).
p38 activity analysis
The enzymatic activity of the phosphorylated p38 was determined by a western blot analysis using the p38 MAP Kinase Assay Kit (Cell Signaling Technology). Briefly, following exposure to Mn and/or LPS, the cells were rinsed once with ice-cold PBS followed by incubation with 0.5 ml of ice-cold 1x lysis buffer containing 1mM PMSF for 5 min. Cells were scraped and transferred to 1.5 ml tubes, sonicated 4x for 5 sec, and centrifuged (14,000 × g) for 10 min at 4 °C. Next, 20 μl of bead-immobilized antibody slurry was added to 200 μl of sample supernatant and incubated overnight at 4 °C. After centrifugation (14,000 × g) for 10 min at 4 °C), the samples were washed (2x) in 1x lysis buffer followed by 2 washes in 1x kinase buffer. Finally, each sample was resuspended in 50 μl of 1x kinase buffer supplemented with 200 μM ATP and 2 μg of kinase substrate (ATF-2 fusion protein). All samples were incubated for 30 min at 30 °C. The kinase reaction was terminated by denaturing at 95 °C for 5 min in 3x SDS sample buffer containing 150 mM DTT. The samples were electrophoretically separated (10% SDS-PAGE gel) and transferred to PVDF membranes. The membranes were blocked in 5% w/v nonfat milk and probed with a polyclonal, rabbit IgG anti-Phospho-ATF-2 (Thr71) antibody at a 1:2000 dilution. Following washing (3X), the membranes were incubated with HRP-conjugated anti-rabbit IgG and HRP-conjugated anti-biotin antibody (for biotinylated protein molecular weight markers). The membranes were incubated for 1 min in LumiGLO substrate and wrapped in sheet protectors. The membranes were exposed to X-ray film for 10 s, the films developed and band density analyzed using the UN-SCAN-IT software package.
Statistical Analysis
Data were analyzed using analysis of variance (ANOVA). When statistical differences were detected (P < 0.05), treatment means were separated by the Student Newman Keuls (SNK) post hoc test. All data are presented as means ± S.E.M.
RESULTS
Pharmacological manipulation
To determine the time-dependency in the ability of Mn to enhance LPS-induced pro-inflammatory cytokine production and its association with p38, in addition to exposing cells to Mn+LPS as before (Filipov et al., 2005), we exposed cells to 250 μM Mn 3 h before or after activation by 100 ng/ml LPS. As previously observed, exposure to Mn+LPS potentiated the microglial production of TNF-α (Figure 1A) and IL-6 (Figure 1B). Cells activated with LPS 3 h before or after exposure to 250 μM Mn produced more TNF-α (Figure 1A) and IL-6 (Figure 2B) in comparison to LPS-alone. In terms of the timing of exposure to Mn or LPS, the observed potentiation effect was the lowest when Mn was added 3 h after LPS, but, nevertheless, the potentiation was still present.
Figure 1.
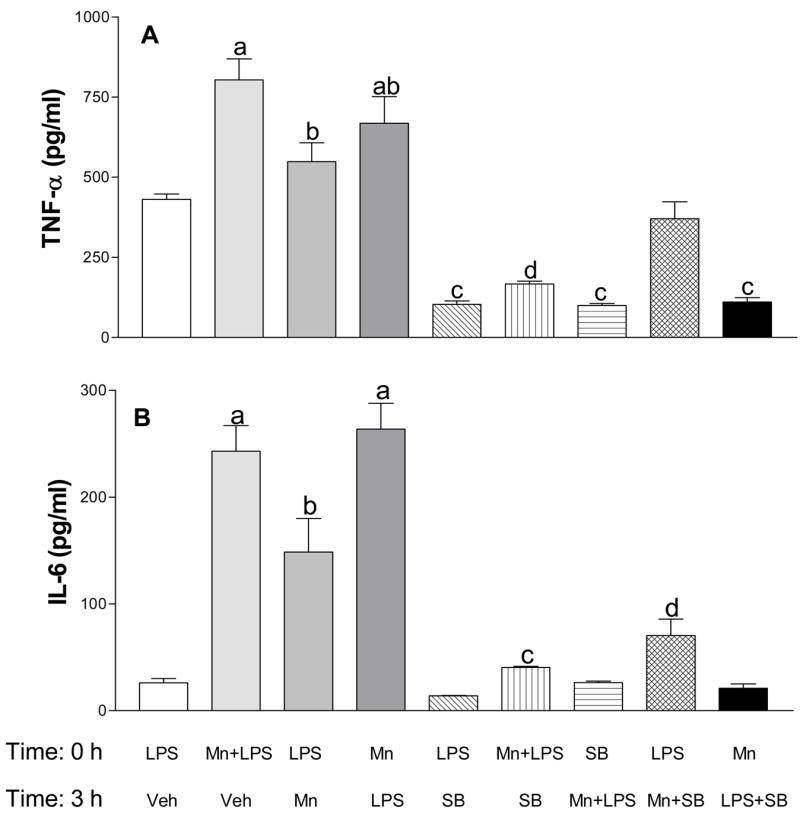
Effect of delayed exposure to manganese (Mn) and/or SB203580 (SB, a p38 inhibitor) on lipopolysaccharide (LPS) activated microglial cytokine production. TNF-α (1A) and IL-6 (1B) production was analyzed in supernatants collected 24 h after exposure of N9 microglial cells to LPS (100 ng/ml, time 0) then vehicle (Veh, 3 h later), Mn (250 μM) + LPS (time 0) then Veh (3 h later), LPS (time 0) then Mn (3 h later), Mn (time 0) then LPS (3 h later), LPS (time 0) then SB (50 μM, 3 h later), LPS (time 0) then Mn + SB (3 h later), Mn (time 0) then LPS + SB (3 h later), Mn + LPS (time 0) then SB (3 h later), and SB (time 0) then Mn + LPS (3 h later). Media levels of TNF-α and IL-6 were analyzed by respective DuoSet ELISA as described in the Materials and Methods. Data shown in each bar represent the mean ± S.E.M. of 4–8 independent replicates. a, b, c, d Presence of letters on top of bars indicate treatment differences in the TNF-α (1A) and IL-6 (1B) levels with bars with different letters being different from each other and from bars without a letter (P < 0.05).
Figure 2.

Densitometric analysis (left panels) and representative western blots (right panels) of phosphorylated p38 kinase in N9 microglia exposed to Mn, LPS, or Mn+LPS. Phosphorylated p38 protein from N9 cells exposed to vehicle, 250 μM Mn, 100 ng/ml LPS, or 250 μM Mn+100 ng/ml LPS for 15 min (A), 1 h (B) and 4 h (C) was determined by western blot analysis. Western blots and densitometric analyses were performed as described in the Materials and Methods. Data shown in each bar represent the mean ± S.E.M. of the adjusted pixel density for phosphorylated p38 normalized to non-phosphorylated p38. For abbreviations please refer to the legend on Figure 1. Each bar represents a minimum of 3 independent replicates. a Presence of letters on top of bars indicate treatment differences in phosphorylated p38 levels within a time point with bars with different letters being different from each other and from bars without a letter (P < 0.05).
Exposure to 250 μM Mn and 50 μM SB203580 (SB, a p38 inhibitor) 3 h after LPS activation prevented Mn-induced potentiation of TNF-α and IL-6 (Figures 1A and 1B). Similarly, exposure to Mn followed by SB and LPS 3 h later also prevented Mn-induced potentiation of TNF-α and IL-6 (Figures 1A and 1B, respectively). Additionally, SB inhibited cytokine production in cells exposed simultaneously to Mn+LPS, regardless of whether Mn+LPS exposure occurred before, or after, SB203580 (Figures 1A and 1B).
Immunoblot analysis of p38 and phospho-p38 protein levels
To examine p38 activation, we studied the induction of p38 phosphorylation following exposure to Mn (250 μM) with and without LPS (100 ng/ml) for up to four hours after exposure. The levels of non-phosphorylated p38 (all exposures) did not change in response to Mn and/or LPS exposure (data not shown). However, induction of p38 phosphorylation (normalized to non-phosphorylated p38) was observed as early as 15 min following Mn and/or LPS exposure (Figure 2A). Increased phosphorylation was observed at 1 h for Mn+LPS and this effect persisted through the 4 h time point (Figures 2B and 2C, respectively). On the other hand, the LPS-induced phosphorylation had almost returned to control levels, while Mn by itself increased p38 phosphorylation at the 4 h time point (Figure 2C).
Phospho-p38 analysis by Flow Cytometry
Similar to the western blot data (Figure 2), flow cytometry results indicated a time-dependent effect of LPS, Mn, and Mn+LPS on phospho-p38 (Figure 3). Thus, in the absence of Mn, 100 ng/ml LPS-induced increase was rapid (15 min), but it plateaued at 1 h and begun to subside at the 4 h time point. On the other hand, 250 μM Mn increased the phospho-p38 levels by 15 min numerically with the effect being significant at the 1 and 4 h time points. While at 15 min the Mn+LPS combination increased phospho-p38 to an extent similar to LPS, at the 1 (significant) and 4 (non-significant) h time points, the Mn+LPS effects were greater than the effects of either Mn or LPS alone. Importantly, similar to the western blot data, by 4 h the LPS-induced phosphorylation had returned to near control levels.
Figure 3.
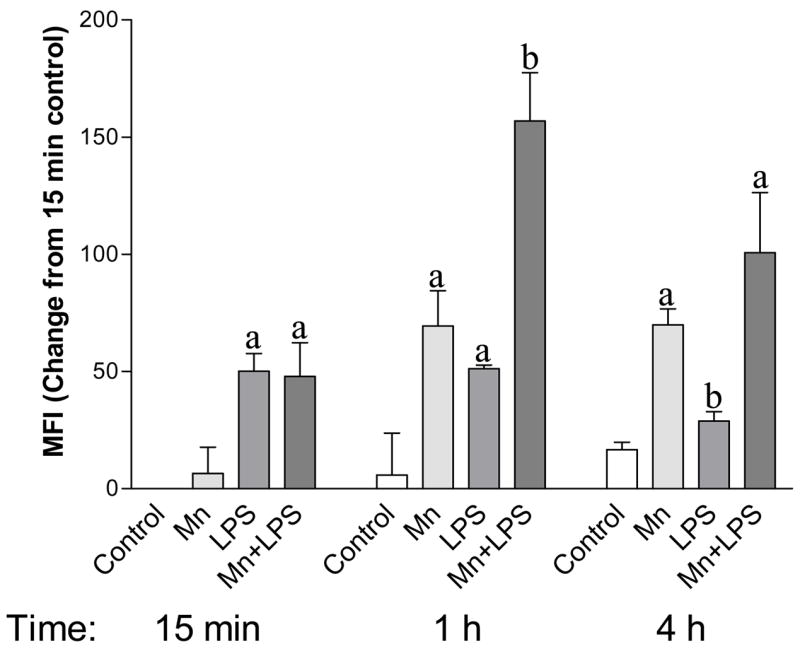
Phosphorylated p38 protein levels in whole cell lysate of N9 microglia exposed to 250 μM with or without 100 ng/ml LPS. Whole cell lysates collected after 15 min, 1 h, or 4 h were analyzed by flow cytometry as described in the Materials and Methods. Data shown represent the mean fluorescence intensity (MFI) change from the 15 min control ± S.E.M. for two independent experiments where each experimental condition was independently replicated (n = 4). For abbreviations please refer to the legend on Figure 1. a, b Presence of letters on top of bars indicate treatment differences in phosphorylated p38 levels within a time point with bars with different letters being different from each other and from bars without a letter (P < 0.05).
p38 activity
The kinase activity of p38 was measured using western blot detection of a p38-phosphorylated substrate (ATF-2 Fusion Protein). Exposure to 250 μM Mn, 100 ng/ml LPS, or Mn+LPS increased p38-kinase activity at 15 min and 4 h compared to control (Figures 4A, and 4B). This effect persisted through the 4 h time point, but only for the Mn and Mn+LPS exposed cells (Figure 4C).
Figure 4.
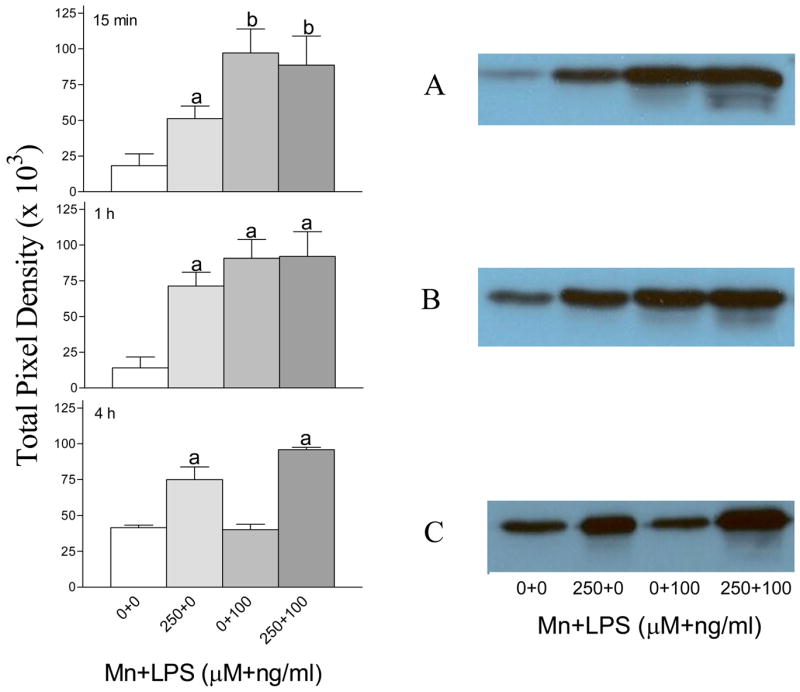
p38 activity following exposure to vehicle, 250 μM Mn and/or 100 ng/ml LPS for 15 min (A), 1 h (B), or 4 h (C) in N9 microglial cells. The p38 activity in whole cell lysates was analyzed by western blot for the detection of a phosphorylated p38 substrate (ATF-2 Fusion Protein) followed by densitometric analyses (left hand panel) as described in the Materials and Methods. Blots representative of three independent experiments are shown in the right hand panel. Data shown in each bar represent the mean ± S.E.M. of the total pixel density for 3 independent replicates. For abbreviations please refer to the legend on Figure 1. a,b Presence of letters on top of bars indicate treatment differences in p38 activity within a time point with bars with different letters being different from each other and from bars without a letter (P < 0.05).
DISCUSSION
Although it has been demonstrated that microglia play a role in basal ganglia neurotoxicity (Castano et al., 1998; Olanow and Tatton, 1999), how environmental toxicants like Mn contribute to microglial-mediated inflammation has only recently been explored. Brain regions that possess an abundance of microglial cells are especially sensitive to inflammatory stimuli, particularly the bacterial endotoxin LPS (Kim et al., 2000). Activation of macrophages and microglia by LPS is thought to occur (primarily) by binding of LPS to TLR4, leading to activation of intracellular kinases (i.e. MAPK) and transcription factors like NF-κB (Akira and Takeda, 2004; Barton and Medzhitov, 2003). Moreover, activation of the MAPK and NF-κB by LPS results in the production of inflammatory mediators such as NO and TNF-α (Bhat el al., 1998; Lee et al., 1993; Lee et al., 1994).
Previous research has demonstrated that Mn exposure, in combination with LPS, can increase the production of iNOS and NO by microglial cells (Chang and Liu, 1999; Chen et al., 2006; Filipov et al., 2005), as well as the production of the proinflammatory cytokines TNF-α (Chen et al., 2006; Filipov et al., 2005) and IL-6 (Filipov et al., 2005). On its own, Mn exposure increases the cytokine production only slightly and at higher concentrations, suggesting that microglial activation must be present in order for the Mn effects on TNF-α and IL-6 to be observed. In this study, similar to Filipov et al. (2005), we observed a strong potentiation of TNF-α and IL-6 production by N9 microglial cells following exposure to Mn+LPS compared to exposure to LPS alone. Interestingly, Mn-potentiation of IL-6 in activated microglia was greater than for TNF-α, with IL-6 concentrations increasing 5-fold or more, in comparison to 2-fold changes in TNF-α. On the other hand, exposure to the p38 inhibitor SB203580 inhibited TNF-α in Mn+LPS-exposed cells to a greater extent than the inhibition observed for IL-6, suggesting that although both cytokines depend on p38 for their synthesis, the degree of dependency may be different for the two cytokines
Importantly, we were also able to demonstrate that Mn can potentiate LPS-induced cytokine production if the Mn-exposure occurs 3 h after the exposure to the inflammatory stimulus, or, alternatively, that prior (3 h) exposure to Mn can potentiate the LPS-induced proinflammatory cytokine production. To our knowledge, this is the first report of this observation, providing additional data to support the hypothesis that Mn exposure can potentiate the production of proinflammatory cytokines in vitro and, possibly, inflammation in vivo not only in cases of co-exposure to an inflammogen and Mn, but also in cases where the exposure is, within a (possibly) narrow window, sequential.
LPS has been shown to activate the MAPK family of kinases, leading to the production of inflammatory mediators such as NO and TNF-α (Bhat et al., 1998; Lee et al., 1993; Lee et al., 1994). While for microglia, both p38 and ERK MAPK have been shown to contribute to the production of inflammatory mediators (Bhat et al., 1998; Jeohn et al., 2002: Koistinaho and Koistinaho, 2002; Zarubin and Han, 2005), p38 appears to play a dominant role. For example, LPS-stimulated production of NO and cytokines by microglia can be inhibited by prior exposure to a p38 inhibitor (Bhat et al., 1998; Jeohn et al., 2002). This is also the case with Mn potentiation of LPS-induced cytokine expression in the present study, as exposure to SB203580 before or after exposure to Mn+LPS inhibited the production of IL-6 and TNF-α by N9 microglial cells. In support, our western blot and flow cytometry p38 activity data indicated that the increased p38 activity caused by Mn persists for at least 4 h.
In contrast, the ERK inhibitor (PD98059) has a very small effect on IL-6 and TNF-α production by the Mn+LPS exposed microglia (Crittenden and Filipov, 2004; data not shown), suggesting that activation of other MAPK which are known to contribute to the production of inflammatory mediators in microglia (Bhat et al., 1998; Koistinaho and Koistinaho, 2002; Zarubin and Han, 2005), play only a minor role in the Mn+LPS-induced production of TNF-α and IL-6.
Two other recent studies have investigated the role of intracellular kinases in the Mn effects on NO/iNOS and/or TNF-α production using the microglial cell line BV-2 (Bae et al., 2006) or primary mixed glial cells (Chen et al., 2006). Bae et al. (2006) examined the effects of Mn (100 μM to 1 mM) by itself only on iNOS and suggested that multiple MAP kinases were phosphorylated by Mn and that, based on inhibitor data, the p38 kinase appeared not to play a major role in the Mn-induced iNOS. Nevertheless, even though not quantified, these authors did report that p38 phosphorylation was increased by Mn at 1, 3 and 6 h post exposure, thus agreeing with our data. In their studies with primary glial cells, which contain both astrocytes and microglia (85 and 15%, respectively), Chen et al. (2006) observed that (i) the phosphorylation of multiple (p38, ERK, JNK) kinases was induced by 30 min exposure to Mn, either by itself or in combination with LPS/IFN-γ, (ii) similar to our present data and to Filipov et al. (2005), while having no effect on its own, Mn potentiated TNF-α production by glial cells exposed to LPS/IFN-γ, and (iii) pharmacological inhibition of p38 was one of the ways of inhibiting the potentiating effect of Mn on TNF-α production. Interestingly, these authors also suggested that iNOS/NO production may be regulated in a manner different from that of TNF-α. Importantly, similar to our present studies, Chen et al. (2006) indicated that an activation of MAPKs by Mn is not sufficient to trigger the glial cell production of TNF-α, but that it is critical for the potentiating effects of Mn on LPS/IFN-γ-stimulated TNF-α production to be observed.
Having observed that p38 was required for the Mn-potentiation of cytokine production, it was necessary to examine p38 phosphorylation directly as enhanced phosphorylation is associated with increased activity and thus could increase the production of IL-6 and TNF-α. Indeed, this observation was supported by western blot analysis and flow cytometry. Although LPS did increase the phosphorylation of p38 through the 1 h time point, by 4 h this effect had diminished. In contrast, prolonged phosphorylation of p38 was observed in Mn-exposed cells, persisting through the 4 h time point. Similarly, increased p38 activation was observed in Mn-exposed cells, an effect that persisted through the 4 h time point. LPS alone increased p38 activity, however, by 4 h, the LPS-induced increase in p38 activity had largely subsided. Moreover, we have observed enhanced p38 activation 24 h following exposure to Mn or Mn+LPS (data not shown).
While p38 is clearly associated with the Mn-potentiation of TNF-α and IL-6 by activated microglia, how Mn enhances the phosphorylation and activity of p38 is not apparent at this time. One possibility is that Mn acts at a site other than p38, perhaps upstream at the level of the MAPK kinase (MKK). The MKKs are responsible for phosphorylating MAPK to its active form. Two MKK that are known to activate p38 are MKK3 and MKK6 (Koistinaho and Koistinaho, 2002; Zarubin and Han, 2005). Increased activity by MKK3 or MKK6 could lead to enhanced p38 MAPK activation (Enslen et al., 1998). Alternatively, inhibition of the enzymes responsible for inactivating p38, the dual specificity phosphatases (DSP; Groom et al., 1996), may be responsible for the prolonged phosphorylation following exposure to Mn/LPS that was observed in the current study. In this regard, another metal, arsenic, has been shown to inhibit a different DSP responsible for maintaining low basal levels of JNK (Cavigelli et al., 1996). Additionally, it has been demonstrated that ERK can negatively regulate p38 via the MAPK phosphatase MKP-1 (Carter and Hunninghake, 2000). It is possible that Mn inhibits either ERK-mediated activation of MKP-1 or, MKP-1 activity.
Our earlier study indicated that the Mn-induced potentiation of IL-6 and TNF-α is NF-κB dependent (Filipov et al., 2005). With our present study we demonstrate that p38 is critical for the Mn-induced potentiation of cytokine production to occur. Exactly how NF-κB and p38 are interacting in Mn+LPS exposed microglia is the subject of future studies. A possible mechanism involves NF-κB and regulation of gene transcription. Active p38 phosphorylates the TATA-binding protein (TBP), resulting in the association of TBP with the TATA-promoter as it has already been demonstrated that p38 phosphorylation of TBP is required for NF-κB-dependent gene expression, including TNF-α and IL-6 (Carter et al., 1999). As Mn-potentiation of LPS-induced inflammatory cytokine production appears to require both p38 and NF-κB, it is possible that the observed increases in p38 activity may increase phosphorylation of TBP leading to enhanced NF-κB-dependent gene expression of IL-6, TNF-α and, possibly, other inflammatory mediators. This is further supported by the observation in the present study that increased phosphorylation of p38 was observed in cells exposed to Mn in the absence of LPS. However, Mn exposure alone resulted in negligible amounts of IL-6 and TNF-α produced by the N9 microglia as demonstrated in our previous study (Filipov et al., 2005). As synthesis of IL-6 and TNF-α is NF-κB-dependent, and exposure to LPS leads to activation of NF-κB, it is likely that Mn-potentiation of inflammatory cytokine production occurs via a p38-dependent mechanism, but requires the subsequent/concurrent activation of NF-κB. Considering the interactive nature of intracellular signaling, it is possible that Mn may be acting at multiple sites leading to increased p38 phosphorylation and activity and subsequent IL-6 and TNF-α production as proposed in Figure 5.
Figure 5.
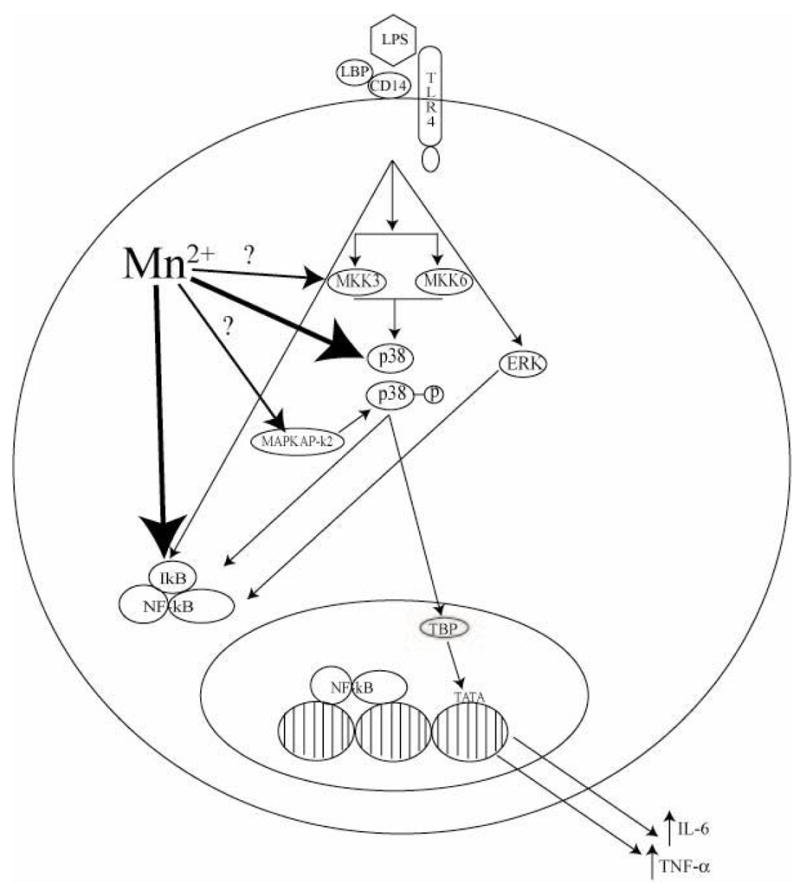
Diagram of potential/known Mn sites of action in microglial intracellular signaling following activation with LPS. Small arrows indicate known intracellular signaling pathways while larger arrows indicate possible sites of interaction between signaling molecules and Mn. Large arrows with a ‘?’ denote potential sites of action for Mn-enhanced p38 acitivity. Abbreviations: LPS (Lipopolysaccharide), LBP (LPS Binding Protein), TLR4 (Toll Like Receptor 4), MKK3/6 (Mitogen Activated Protein Kinase Kinase) p38 (p38 Mitogen Activated Protein Kinase), p38-p (phosphorylated p38), ERK (Extracellular Signal-Regulated Kinase), MAPKAP-k2 (MAPK Phosphatase), IkB (Inhibitor of NF-kB), NF-kB (Nuclear Factor kappa B), TBP (TATA-Binding Protein), TATA (TATA Box).
In summary, enhanced production of inflammatory cytokines by microglia exposed to Mn+LPS is associated with persistent activation of p38 and inhibition of p38 prevents this enhancement. Importantly, the increase cytokine production by Mn+LPS occurs regardless whether the exposure to Mn and LPS is concurrent or sequential (3 h apart); in all cases, the potentiation is prevented by inhibition of p38. Overall, our data suggest that exposure to environmental toxicants, such as Mn, may prolong microglial activation by utilizing the p38 pathway and contribute to chronic inflammation and neurotoxicity.
Acknowledgments
This project was supported by research grant from the National Institute of Environmental Health Sciences (NIH), ES011654. Portions of this research were presented at the annual meeting of the Society of Toxicology in San Diego, CA (March 2006).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Aschner M. Manganese: Brain transport and emerging research needs. Environmental Health Prespectives. 2000;108(Suppl 3):429–432. doi: 10.1289/ehp.00108s3429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aschner M, Aschner JL. Manganese neurotoxicity: Cellular effects and blood-brain barrier transport. Neuroscience Biobehavior Review. 1991;15:334–340. doi: 10.1016/s0149-7634(05)80026-0. [DOI] [PubMed] [Google Scholar]
- Bae JH, Jang BC, Suh SI, Ha E, Baik HH, Kim SS, Lee MY, Shin DH. Manganese induces inducible nitric oxide synthase (iNOS) expression via activation of both MAP kinase and PI3K/Akt pathways in BV2 microglial cells. Neuroscience Letters. 2006;398:151–154. doi: 10.1016/j.neulet.2005.12.067. [DOI] [PubMed] [Google Scholar]
- Bhat NR, Zhang P, Lee JC, Hogan EL. Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-a gene expression in endotoxin-stimulated primary glial cultures. Journal of Neuroscience. 1998;18:1633–1641. doi: 10.1523/JNEUROSCI.18-05-01633.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter AB, Knudtson KL, Monick MM, Hunninghake GW. The p38 mitogen-activated protein kinase is required for NF-κB-dependent gene expression. Journal of Biological Chemistry. 1999;274:30858–30863. doi: 10.1074/jbc.274.43.30858. [DOI] [PubMed] [Google Scholar]
- Carter AB, Hunninghake GW. A constitutive active MEK→ERK pathway negatively regulates NF-κB-dependent gene expression by modulating TATA-binding protein phosphorylation. Journal of Biological Chemistry. 2000;275:27858–27864. doi: 10.1074/jbc.M003599200. [DOI] [PubMed] [Google Scholar]
- Castano A, Herrera AJ, Cano J, Machado A. Lipopolysaccharide intranigral injection induces inflammatory reaction and damage in nigrostriatal dopaminergic system. Journal of Neurochemistry. 1998;70:1584–1592. doi: 10.1046/j.1471-4159.1998.70041584.x. [DOI] [PubMed] [Google Scholar]
- Cavigelli M, Wilfred WL, Anning L, Bing S, Katsuji Y, Karin M. The tumor promoter arsenite stimulates AP-1 activity by inhibiting a JNK phosphatase. European Molecular Biology Organization Journal. 1996;15:6269–6279. [PMC free article] [PubMed] [Google Scholar]
- Chao CC, Hu S, Close K, Choi CS, Molitor TW, Novick WJ, Peterson PK. Cytokine release from microglia: Differential inhibition by pentoxifylline and dexamethasone. Journal of Infectous Disease. 1992;166:847–853. doi: 10.1093/infdis/166.4.847. [DOI] [PubMed] [Google Scholar]
- Chang JY, Liu LZ. Manganese potentiates nitric oxide production by microglia. Brain Research Molecular Brain Research. 1999;68:22–28. doi: 10.1016/s0169-328x(99)00082-0. [DOI] [PubMed] [Google Scholar]
- Chen CJ, Ou YC, Lin SY, Liao SL, Chen SY, Chen JH. Manganese modulates pro-inflammatory gene expression in activated glia. Neurochemistry International. 2006;49:62–71. doi: 10.1016/j.neuint.2005.12.020. [DOI] [PubMed] [Google Scholar]
- Crittenden PL, Filipov NM. Manganese/LPS induced proinflammatory cytokine production is p-38 kinase dependent. Abstracts, Annual Meeting of the South Central Chapter of the Society of Toxicology; 2004. [Google Scholar]
- Enslen H, Raingeaud J, Davis RJ. Selective activation of p38 mitogen-activated protein (MAP) kinase isoforms by the MAP kinase kinases MKK3 and MKK6. Journal of Biological Chemistry. 1998;273:1741–1748. doi: 10.1074/jbc.273.3.1741. [DOI] [PubMed] [Google Scholar]
- Filipov NM, Seegal RF, Lawrence DA. Manganese potentiates in vitro production of proinflammatory cytokines and nitric oxide by microglia through a nuclear factor kappa B-dependent mechanism. Toxicological Science. 2005;84:139–148. doi: 10.1093/toxsci/kfi055. [DOI] [PubMed] [Google Scholar]
- Frumkin H, Solomon G. Manganese in the U.S. gasoline supply. American Journal of Industrial Medicine. 1997;31:107–115. doi: 10.1002/(sici)1097-0274(199701)31:1<107::aid-ajim16>3.0.co;2-6. [DOI] [PubMed] [Google Scholar]
- Gavin CE, Gunter KK, Gunter TE. Manganese and calcium transport in mitochondria: Implications for manganese toxicity. Neurotoxicology. 1999;20:445–453. [PubMed] [Google Scholar]
- Groom LA, Sneddon AA, Alessi DR, Dowd S, Keyse SM. Differential regulation of the MAP, SAP and RK/p38 kinases by Pyst1, a novel cytosolic dual-specificity phosphatase. European Molecular Biological Organization Journal. 1996;15:3621–3632. [PMC free article] [PubMed] [Google Scholar]
- Heyen JRR, Ye S, Finck BN, Johnson RW. Interleukin (IL)-10 inhibits IL-6 production in microglia by preventing activation of NF-kB. Molecular Brain Research. 2000;77:138–147. doi: 10.1016/s0169-328x(00)00042-5. [DOI] [PubMed] [Google Scholar]
- Jeohn GH, Cooper CL, Wilson B, Chang RCC, Jang KJ, Kim HC, Liu B, Hong JS. p38 MAP kinase is involved in lipopolysaccharide-induced dopaminergic neuronal cell death in rat mesencephalic neuron-glia cultures. Annals of the New York Academy of Sciences. 2002;962:332–346. doi: 10.1111/j.1749-6632.2002.tb04078.x. [DOI] [PubMed] [Google Scholar]
- Jeohn GH, Cooper CL, Wilson B, Chang RCC, Jang KJ, Kim HC, Hong JS. Go6976 protects mesencephalic neurons from lipopolysaccharide-elicited death by inhibiting p38 MAP kinase phosphorylation. Annals of the New York Academy of Sciences. 2002;962:347–359. doi: 10.1111/j.1749-6632.2002.tb04079.x. [DOI] [PubMed] [Google Scholar]
- Koistinaho M, Koistinaho J. Role of p38 and p44/42 mitogen-activated protein kinases in microglia. Glia. 2002;40:175–183. doi: 10.1002/glia.10151. [DOI] [PubMed] [Google Scholar]
- Lee JC, Laydon JT, McDonnel PC, Gallagher TF, Kumar S, Green D, McNulty D, Blumenthal MJ, Heys JR, Landvatter SW, Strickler JE, McLaughlin MM, Siemens IR, Fisher SM, Livi GP, White JR, Adams JL, Young PR. A protein kinase involved in the regulation of inflammatory cytokine biosynthesis. Nature. 1994;372:739–746. doi: 10.1038/372739a0. [DOI] [PubMed] [Google Scholar]
- Lee JC, Liu W, Dickson DW, Brosnan CF, Berman JW. Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1b. Journal of Immunology. 1993;150:2659–2667. [PubMed] [Google Scholar]
- Li GJ, Zhao Q, Zhang W. Alteration at translational but not transcriptional level of transferrin receptor expression following manganese exposure at the blood-CSF barrier in vitro. Toxicology Applied Pharmacology. 2005;205(2):188–200. doi: 10.1016/j.taap.2004.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B, Gao HM, Wang JY, Jeohn GH, Cooper CL, Hong JS. Role of nitric oxide in inflammation-mediated neurodegeneration. Annals of the New York Academy of Sciences. 2002;962:318–331. doi: 10.1111/j.1749-6632.2002.tb04077.x. [DOI] [PubMed] [Google Scholar]
- Malthankar GV, White BK, Bhishan A, Daniels CK, Rodnick KJ, Lai JC. Differential lowering by manganese treatment of activities of glycolytic and tricarboxylic acid (TCA) cycle enzymes investigated in neuroblastoma and astrocytoma cells is associated with manganese-induced cell death. Neurochemical Research. 2004;29(4):709–717. doi: 10.1023/b:nere.0000018841.98399.ce. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Itagaki S, Boyes BE, McGeer EG. Reactive microglia are positive for HLA-DR in the substantia nigra of Parkinson’s and Alzheimer’s disease brains. Neurology. 1988;38:1285–1291. doi: 10.1212/wnl.38.8.1285. [DOI] [PubMed] [Google Scholar]
- McGeer PL, Schwab C, Parent A, Doudet D. Presence of reactive microglia in monkey substantia nigra years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine administration. Annals of Neurology. 2003;54:599–604. doi: 10.1002/ana.10728. [DOI] [PubMed] [Google Scholar]
- Meco G, Bonifati V, Vanacore N, Fabrizio E. Parkisonism after chronic exposure to the fungicide maneb (manganese ethylene-bis-dithiocarbamate) Scandanavian Journal of Work, Environment, and Health. 1994;20:301–305. doi: 10.5271/sjweh.1394. [DOI] [PubMed] [Google Scholar]
- Nagatsu T, Mogi M, Ichinose H, Togari A. Cytokines in Parkinson’s disease. Journal of Neural Transmission Suppl. 2000;58:143–151. [PubMed] [Google Scholar]
- Niehaus I, Lange JH. Endotoxin: is it an environmental factor in the cause of Parkinson’s disease? Occupational and Environmental Medicine. 2003;60:378. doi: 10.1136/oem.60.5.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olanow CW, Tatton WG. Etiology and pathogenesis of Parkinson’s disease. Annual Review of Neuroscience. 1999;22:123–144. doi: 10.1146/annurev.neuro.22.1.123. [DOI] [PubMed] [Google Scholar]
- Pomier-Layrargues G, Spahr L, Butterworth RF. Increased manganese concentrations in pallidum of cirrhotic patients. Lancet. 1995;345:735. doi: 10.1016/s0140-6736(95)90909-5. [DOI] [PubMed] [Google Scholar]
- Spranger M, Schwab S, Desiderato S, Bonmann E, Krieger D, Fandrey J. Manganese augments nitric oxide synthesis in murine astrocytes: A new pathogenetic mechanism in manganism? Experimental Neurology. 1998;149:277–283. doi: 10.1006/exnr.1997.6666. [DOI] [PubMed] [Google Scholar]
- Suzuki Y, Mouri T, Nishiyama K, Fujii N. Study of subacute toxicity of manganese dioxide in monkeys. Tokushima Journal of Experimental Medicine. 1975;22:5–10. [PubMed] [Google Scholar]
- Takahashi RN, Rogerio R, Zanin M. Maneb enhances MPTP neurotoxicity in mice. Research Communications in Chemical Pathology and Pharmacology. 1989;66:167–170. [PubMed] [Google Scholar]
- Yase Y. Lancet. Vol. 2. 1972. The pathogenesis of amyotrophic lateral sclerosis; p. 292. [DOI] [PubMed] [Google Scholar]
- Zarubin T, Han J. Activation and signaling of the p38 MAP kinase pathway. Cell Research. 2005;15:11–18. doi: 10.1038/sj.cr.7290257. [DOI] [PubMed] [Google Scholar]
- Zhang J, Fitsanakis VA, Gu G, Jing D, Ao M, Amarnath V, Montine TJ. Manganese ethylene-bis-dithiocarbamate and selective dopaminergic neurodegeneration in rat: A link through mitochondrial dysfunction. Journal of Neurochemistry. 2003;84:336–346. doi: 10.1046/j.1471-4159.2003.01525.x. [DOI] [PubMed] [Google Scholar]