

Abstract
An important mechanism by which the tumor suppressor p53 maintains genomic stability is to induce cell cycle arrest through activation of the cyclin-dependent kinase inhibitor p21WAF1/Cip1 gene. We show that the gene encoding the gut-enriched Krüppel-like factor (GKLF, KLF4) is concurrently induced with p21WAF1/Cip1 during serum deprivation and DNA damage elicited by methyl methanesulfonate. The increases in expression of both Gklf and p21WAF1/Cip1 due to DNA damage are dependent on p53. Moreover, during the first 30 min of methyl methanesulfonate treatment, the rise in Gklf mRNA level precedes that in p21WAF1/Cip1, suggesting that GKLF may be involved in the induction of p21WAF1/Cip1. Indeed, GKLF activates p21WAF1/Cip1 through a specific Sp1-like cis-element in the p21WAF1/Cip1 proximal promoter. The same element is also required by p53 to activate the p21WAF1/Cip1 promoter, although p53 does not bind to it. Potential mechanisms by which p53 activates the p21WAF1/Cip1 promoter include a physical interaction between p53 and GKLF and the transcriptional induction of Gklf by p53. Consequently, the two transactivators cause a synergistic induction of the p21WAF1/Cip1 promoter activity. The physiological relevance of GKLF in mediating p53-dependent induction of p21WAF1/Cip1 is demonstrated by the ability of antisense Gklf oligonucleotides to block the production of p21WAF1/Cip1 in response to p53 activation. These findings suggest that GKLF is an essential mediator of p53 in the transcriptional induction of p21WAF1/Cip1 and may be part of a novel pathway by which cellular responses to stress are modulated.
A principal function of the tumor suppressor p53 is to maintain genomic stability. It does so by eliciting cellular changes in response to various forms of stress such as DNA damage, hypoxia, and nucleotide deprivation (1–3). The amount of p53 protein increases in response to these so-called genotoxic stresses. In addition, covalent modifications such as phosphorylation are involved in its activation (4, 5). Once activated, p53 exerts potent regulatory effects on diverse aspects of cellular events that cumulate in cell cycle arrest or apoptosis (3). Many of these “downstream” events are dependent upon the ability of p53 to function as a transcription factor in activating the expression of “target” genes (2, 6). Notably, an important consequence of p53 activation is the transcriptional induction of the gene encoding the cyclin-dependent kinase (Cdk)1 inhibitor p21 (also called WAF1 or Cip1) (7, 8). p21WAF1/Cip1 inhibits the activity of several cyclin-Cdk complexes such as cyclin D1-Cdk4, cyclin E1-Cdk2, and cyclin A-Cdk2, which results in cell cycle arrest at the G1-S transition checkpoint (9, 10).
The gut-enriched Krüppel-like factor (GKLF, KLF4) (11) is a recently identified and developmentally regulated transcription factor, the expression of which is enriched in the epithelial cells of the gastrointestinal tract (12–14), skin (14, 15), and thymus (16) and in vascular endothelial cells (17). Both the in vivo (12–16) and in vitro (12) patterns of expression of Gklf are indicative of a growth arrest-associated nature. Upon stimulation of quiescent cultured cells by fresh serum, levels of Gklf mRNA are decreased significantly during the G1-S transition phase of the cell cycle (12). Conversely, constitutive expression of GKLF inhibits DNA synthesis (12). In vivo, Gklf transcripts are highly enriched in the population of terminally differentiated, post-mitotic epithelial cells of the intestinal tract and skin (12–15). Moreover, the intestinal expression of Gklf is down-regulated in two independent mouse models of intestinal tumorigenesis or hyperproliferation (18, 19). Taken together, these studies suggest that GKLF is potentially a negative regulator of proliferation; however, the mechanism by which it accomplishes this task is not well defined.
The established binding site for GKLF is rich in GC content (20) and overlaps with that for the transcription factor Sp1 (21, 22). By coincidence, the proximal promoter of the p21WAF1/Cip1 gene contains a number of GC-rich elements (7), some of which have been shown to bind Sp1 (23–29). These Sp1-binding sites have been shown to be important in controlling expression of p21WAF1/Cip1 in several physiologically diverse processes, including the gene’s responsiveness to phorbol ester (23), transforming growth factor-β (24–26), and sodium butyrate (27), and in keratinocyte differentiation (28). As both Gklf and p21WAF1/Cip1 are growth arrest-associated genes, we sought to determine whether GKLF is involved in regulating p21WAF1/Cip1 expression. We demonstrate that GKLF not only transactivates the p21WAF1/Cip1 proximal promoter, but also mediates the activating effects of p53 in response to DNA damage on the same promoter. This study suggests that GKLF may be an important component of the p53 tumor suppressor network of regulatory proteins.
EXPERIMENTAL PROCEDURES
Plasmid Constructs, Reagents, and Cell Lines
The eukaryotic expression vector PMT3 and its derivatives containing various forms of GKLF were previously described (12, 20, 30, 31). They include full-length GKLF (PMT3-GKLF-(1–483)), truncated GKLF lacking the three zinc fingers (PMT3-GKLF-(1–401)), and truncated GKLF containing the zinc fingers only (PMT3-GKLF-(350–483)). pC53-SN3 and pC53-SX3, two cytomegalovirus-based expression constructs containing wild-type p53 and mutant p53 with a missense mutation at codon 143 in the DNA-binding domain (DBD) of p53, respectively, were kindly provided by B. Vogelstein and K. Kinzler (32). The reporter constructs linking various regions of the p21WAF1/Cip1 promoter to chloramphenicol acetyltransferase (CAT) have previously been described (23). They include the CAT reporter linked to either a 2320-nt 5′-flanking sequence of the p21WAF1/Cip1 gene containing an upstream p53-binding site at nt −2301 (33) or the same 2320-nt 5′-flanking sequence with a small internal deletion of the sequence between nt −122 and −61 of the p21WAF1/Cip1 promoter that removed the first four of the six Sp1 sites from the proximal promoter (33). Reporter constructs containing the proximal promoter region of the p21WAF1/Cip1 gene with various 5′-end points as well as internal deletions or point mutations affecting the various Sp1 sites in the proximal promoters have all been described (23). The WWP-Luc and DM-Luc constructs are two luciferase reporters that contain 2.4 and 2.2 kb, respectively, of the 5′-flanking sequence of the p21WAF1/Cip1 gene and were kindly provided by B. Vogelstein and K. Kinzler (7). The DM-Luc construct lacks the upstream p53-binding site at nt −2301 in the p21WAF1/Cip1 promoter (7).
The polyclonal rabbit anti-GKLF serum was described (12). Anti-p53 serum was purchased from Santa Cruz Biotechnology (sc-6243), and the monoclonal antibody against p21WAF1/Cip1 was purchased from Pharmingen (SXM30). The p53−/− and p53+/+ mouse embryo fibroblasts (MEFs) were generously provided by L. Donehower (34). The 10(1)-p53 val135 cell line was provided by A. Levine (35). This cell line, which was derived from the parental 10(1) cell line (36), is an immortalized murine embryo fibroblast line that lacks endogenous p53 expression, but contains a stably transfected temperature-sensitive p53 protein, val135 (37). At the nonpermissive temperature of 38.5 °C, p53 val135 is transcriptionally inactive, whereas at the permissive temperature of 31.5 °C, it is transcriptionally competent (35). The sense and antisense oligonucleotides to GKLF contain nucleotide sequences corresponding to amino acid codons 7–13 of GKLF in the sense and antisense orientations, respectively. At the center of this sequence (amino acid 10) is the second of two initiation methionine codons of GKLF, which was felt to be in a translationally more favorable context than the first (14). The nucleotide sequence of the antisense oligonucleotide is 5′-GCT GAC AGC CAT GTC AGA CTC-3′, and that of the sense oligonucleotide is 5′-GAG TCT GAC ATG GCT GTC AGC-3′. Note that the underlined sequence represents the initiation methionine codon at amino acid 10 (12).
Conditions of Cell Treatments and Northern and Western Blot Analyses
For the serum deprivation experiments, the content of fetal calf serum in the cell medium was reduced from 10 to 0.5% to induce a growth-arrested state (12). To cause DNA damage, methyl methanesulfonate (MMS) was added to cells at a concentration of 100 μg/ml, which has previously been shown to result in cell cycle arrest (38). After various treatment periods, total RNA was isolated from cells using Triazol (Life Technologies, Inc.). Twenty μg of RNA from each sample were studied by Northern blot analyses using conditions previously described (12). Blots were probed with a full-length cDNA encoding GKLF (12), p21WAF1/Cip1 (7), or glyceraldehyde-3-phosphate dehydrogenase (CLONTECH). The conditions for Western blot analysis were also previously described, using a 1:1000 dilution of an affinity-purified polyclonal anti-GKLF serum (12).
Transfection and Luciferase and CAT Assays
All transfections were performed by lipofection as described (20, 21, 30, 31). Unless otherwise specified, all reactions contained 5 μg each of the reporter and effector constructs/10-cm dish. Luciferase and CAT assays were performed as described (20, 21).
Reverse Transcription-Polymerase Chain Reactions
RNA was extracted from human embryonic kidney (HEK) 293 cells and human colonic carcinoma HT29 cells (39). The content of Gklf transcript from each cell line was determined using reverse transcription-PCR. The content of the β-actin transcript was similarly determined as a control. One μg of RNA was reverse-transcribed in an 80-μl volume containing 50 mm Tris-HCl, pH 8.3, 75 mm KCl, 10 mm dithiothreitol, 3 mm MgCl2, 0.5 mm dGTP, 0.5 mm dATP, 0.5 mm dTTP, 0.5 mm dCTP, 80 units of RNase inhibitor, 100 pmol of random primer, pd(N)6, and 200 units of Moloney murine leukemia virus reverse transcriptase (Life Technologies, Inc.) at 42 °C for 1 h. The cDNA was then amplified in a 50-μl reaction that contained 10 mm Tris-HCl, pH 8.3, 50 mm KCl, 10 mm MgCl2, 0.1% gelatin, 2.5 units of REDTaq DNA polymerase (Sigma), 0.2 mm dGTP, 0.2 mm dATP, 0.2 mm dTTP, 0.2 mm dCTP, and 40 pM each of the forward and reverse primers (see below) at the following settings: 94 °C for 45 s, 45 °C for 1 min, and 72 °C for 1.5 min for a total of 40 cycles. The PCR products were then visualized on a 1.5% agarose gel stained with ethidium bromide.
The primers used in the PCR were synthesized according to the published cDNA sequences encoding human GKLF and β-actin (Gen-Bank™ Data Bank accession numbers AF105036 and X00351, respectively). The forward primer sequence for GKLF is 5′-AGGTCGGAC-CACCTCGCCTTACACATG-3′, and the reverse primer sequence is 5′-AAGGTAAAGAGAATACAAGGTGATCTTTTATGC-3′. The length of the expected PCR product was 345 bp. The forward and reverse primer sequences for β-actin are 5′-TACGCCAACACAGTGCTGTCTGG-3′ and 5′-TACTCCTGCTTGCTGATCCACAT-3′, respectively, with the expected PCR product measuring 206 bp.
Electrophoretic Mobility Shift Assays
EMSAs were performed as described (20). Preparation of nuclear extracts from COS-1 cells transfected with PMT3 expression constructs containing full-length GKLF, truncated GKLF containing only the zinc fingers or lacking the zinc fingers, or PMT3 vector alone was as described previously (20, 21). Purified p53 containing the DBD was kindly provided by N. Pavletich (40). This domain contains the core portion of p53 between amino acids 102 and 292, which binds with high affinity to a p53 recognition site (40, 41). The purification of recombinant p53 DBD expressed from the pET3d bacterial expression vector (Novagen) in transformed Escherichia coli BL21(D3) cells was as described previously (40). The protein was supplied at a concentration of 14 mg/ml in a solution of 50 mm BisTris propane HCl, pH 6.8, 200 mm sodium phosphate, and 5 mm dithiothreitol and had a >98% purity of the core domain.
The wild-type p21 oligonucleotide used in EMSA contains the sequence between nt −129 and −99 of the p21WAF1/Cip1 promoter, which includes both Sp1-1 and Sp1-2 sites (27). The mutant p21 oligonucleotide contains a 3-bp substitution in the Sp1-1 site. The sequences in the sense orientation for the two oligonucleotides are shown below.

The oligonucleotide probe containing the binding site for p53 was derived from the p53-response sequence in the promoter of the human GADD45 gene (42) and has the sequence 5′-TACAGAACATGTCTAAGCATGCTGGGG-3′ in the sense orientation. When indicated, unlabeled competitor oligonucleotides were added in 10-, 20-, or 50-fold molar excess of the probe to the reaction.
In Vitro Synthesis of p53 and Immunoprecipitation
[35S]Methionine-labeled p53 was synthesized by the TNT Coupled Reticulocyte Lysate system (Promega) using a full-length cDNA encoding p53 cloned in pBluescript (provided by B. Vogelstein). Ten μl of the translation product were mixed with 50 μg of nuclear extracts prepared from transfected COS-1 cells in a final volume of 100 μl containing 20 mm HEPES, pH 7.5, 40 mm KCl, 3 mm MgCl2, 1 mm dithiothreitol, and 5% glycerol at 4 °C for 2 h. At the completion of the incubation, 15 μg of affinity-purified anti-GKLF serum or preimmune serum were added to the reaction, which was gently rotated overnight at 4 °C. Fifty μl of packed protein A-Sepharose beads (Amersham Pharmacia Biotech) were then added to each reaction, and the incubation was continued for 1 h at 4 °C. The beads were subsequently collected by centrifugation, washed three times with the incubation buffer, and resuspended in sample buffer before electrophoresis.
RESULTS
Both Gklf and p21WAF1/Cip1 Are Induced during Growth Arrest—
Previously, we showed that the levels of the Gklf transcript were low in actively proliferating cells, but were increased in cells that had been deprived of serum (12). Results of the Northern blot analysis in Fig. 1A recapitulate this event. Fig. 1A also shows that upon serum deprivation, the levels of the p21WAF1/Cip1 transcript rose concomitantly with those of Gklf. To determine whether Gklf is induced during growth arrest under a different condition, we treated NIH 3T3 cells with MMS, which causes DNA damage and subsequently cell cycle arrest (38). As shown in Fig. 1B, the levels of Gklf mRNA were increased 2 h after the addition of MMS, as were those of p21WAF1/Cip1 mRNA. When normalized to the expression of the control glyceraldehyde-3-phosphate dehydrogenase gene, which was not affected by the treatment, the degree of induction of p21WAF1/Cip1 was higher than that of Gklf between 2 and 8 h of MMS treatment (Fig. 1B, bar graph). This contrasts with the changes in mRNA levels of the two genes during the initial 30 min of treatment, in which the rise in Gklf preceded that in p21WAF1/Cip1 (Fig. 1C). These results suggest that both Gklf and p21WAF1/Cip1 respond similarly to signals elicited during growth arrest due to DNA damage. However, the induction of Gklf begins slightly earlier than that of p21WAF1/Cip1 during the initial phase of DNA damage.
Fig. 1. Northern blot analysis of Gklf and p21WAF1/Cip1 in NIH 3T3 cells during growth arrest.
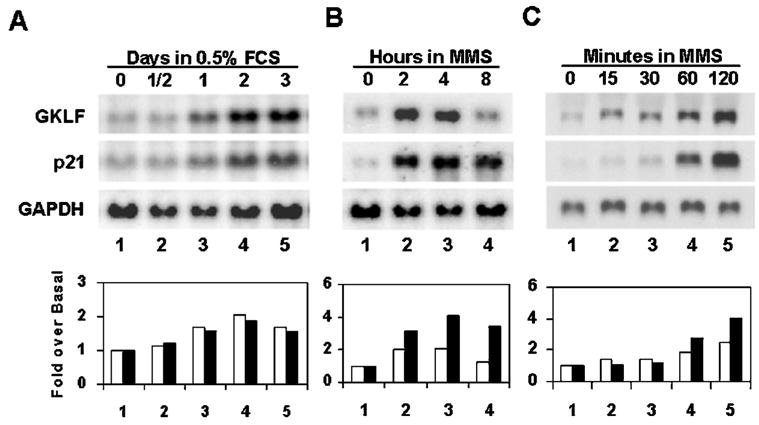
Growth arrest was induced in actively proliferating NIH 3T3 cells maintained in a medium containing 10% fetal calf serum (FCS) by the reduction of serum content to 0.5% (A) or by the addition of 100 μg/ml MMS to the medium (B and C). RNA was isolated at the indicated time points, and 20 μg were loaded in each lane and analyzed for the message content of Gklf, p21WAF1/Cip1, or glyceraldehyde-3-phosphate dehydrogenase (GAPDH). The bar graphs show the quantitative information of -fold induction of Gklf (open bars) and p21WAF1/Cip1 (closed bars) at each treatment time point over the untreated (Basal) value for each experiment. The calculation was performed first by normalizing the band intensity of the Gklf or p21WAF1/Cip1 transcript to that of glyceraldehyde-3-phosphate dehydrogenase at each time point and then comparing the normalized value of Gklf or p21WAF1/Cip1 at each treatment time point with that of untreated cells (time 0).
Induction of GKLF and p21WAF1/Cip1 by MMS Is Dependent on p53
To determine whether the inductive responses of Gklf and p21WAF1/Cip1 to MMS treatment are dependent on p53, we compared the expression of the two genes in MEFs isolated from mice that contained (p53+/+) or lacked (p53−/−) p53 created by homologous recombination (34). As shown in Fig. 2 (lanes 1 and 2), neither fibroblasts contained appreciable amounts of GKLF and p21WAF1/Cip1 in the untreated state, despite a relative abundance of p53 in the p53+/+ cells. Upon the addition of MMS, there was a dramatic increase in the levels of GKLF and p21WAF1/Cip1 beginning at 2 h but, only in p53+/+ MEFs (lanes 4, 6, and 8). In contrast, although there was a p53-independent response in GKLF and p21WAF1/Cip1 production to MMS in p53−/− cells (lanes 3, 5, and 7), this component appeared minor compared with the p53-proficient cells. We conclude that the increase in expression of Gklf in response to MMS-induced DNA damage, like that of p21WAF1/Cip1, is dependent on the presence of p53.
Fig. 2. Western blot analysis of GKLF and p21WAF1/Cip1 in MEFs proficient or deficient in p53.
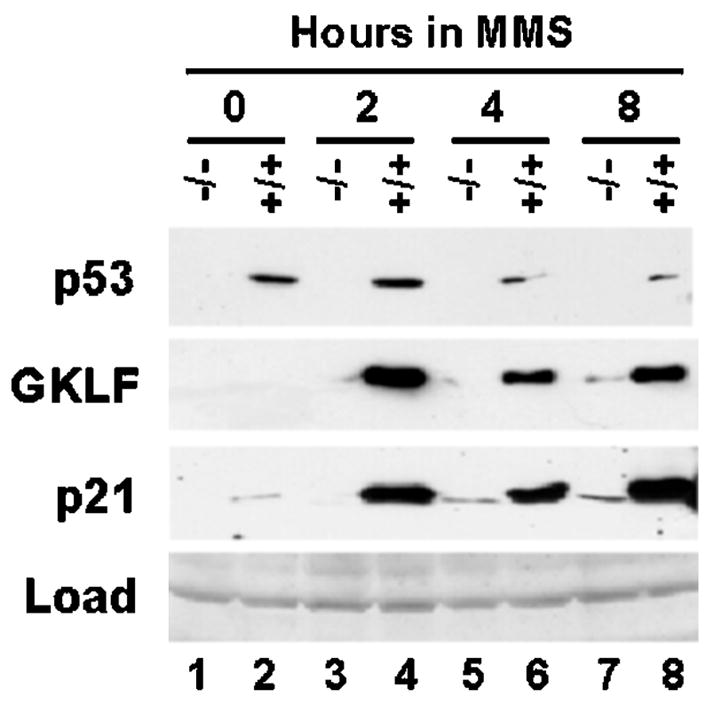
MEFs were prepared from p53-deficient (−/−) mouse embryos (34) or their wild-type littermate control (+/+) and treated with 100 μg/ml MMS for the time periods indicated. Proteins were isolated and analyzed for the content of p53, GKLF, or p21WAF1/Cip1 by Western blot analysis. Load represents a portion of the gel stained with Coomassie Blue before electrophoretic transfer.
Both GKLF and p53 Transactivate the p21WAF1/Cip1 Proximal Promoter through an Identical cis-Element
The sequential pattern of expression in Gklf followed by p21WAF1/Cip1 immediately after the addition of MMS raised the intriguing question of whether GKLF might be responsible in part for the induction of p21WAF1/Cip1. We considered this plausible, as the promoter of the p21WAF1/Cip1 gene contains a number of GC-rich cis-elements that resemble Sp1-binding sites (7, 23–29), and GKLF has been shown to bind to a GC-rich DNA sequence with which Sp1 also interacts (20, 21).
To determine whether GKLF regulates the p21WAF1/Cip1 promoter, we performed cotransfection experiments in HEK 293 cells using a series of p21WAF1/Cip1 promoter-reporter constructs (23) and an expression construct containing either wild-type GKLF or mutant GKLF with its zinc fingers deleted (Fig. 3A, effectors 2 and 3, respectively) (12, 20, 30, 31). Since p53 has been shown to transcriptionally activate a reporter gene when linked to the 5′-flanking sequence of p21WAF1/Cip1 (7, 23), expression constructs containing wild-type p53 or mutant p53 that lost its ability to bind DNA (Fig. 3A, effectors 4 and 5, respectively) (32) were also included in the analysis. Consistent with a previous report (13), HEK 293 cells contained a negligible amount of Gklf transcript at the base-line level as determined by reverse transcription-PCR relative to a human colon cancer carcinoma cell line, HT29 (Fig. 3L). This low level of base-line Gklf expression in HEK 293 cells allowed a better delineation of the effects of GKLF on the p21WAF1/Cip1 promoter activity.
Fig. 3. GKLF and p53 transactivate the p21WAF1/Cip1 promoter.
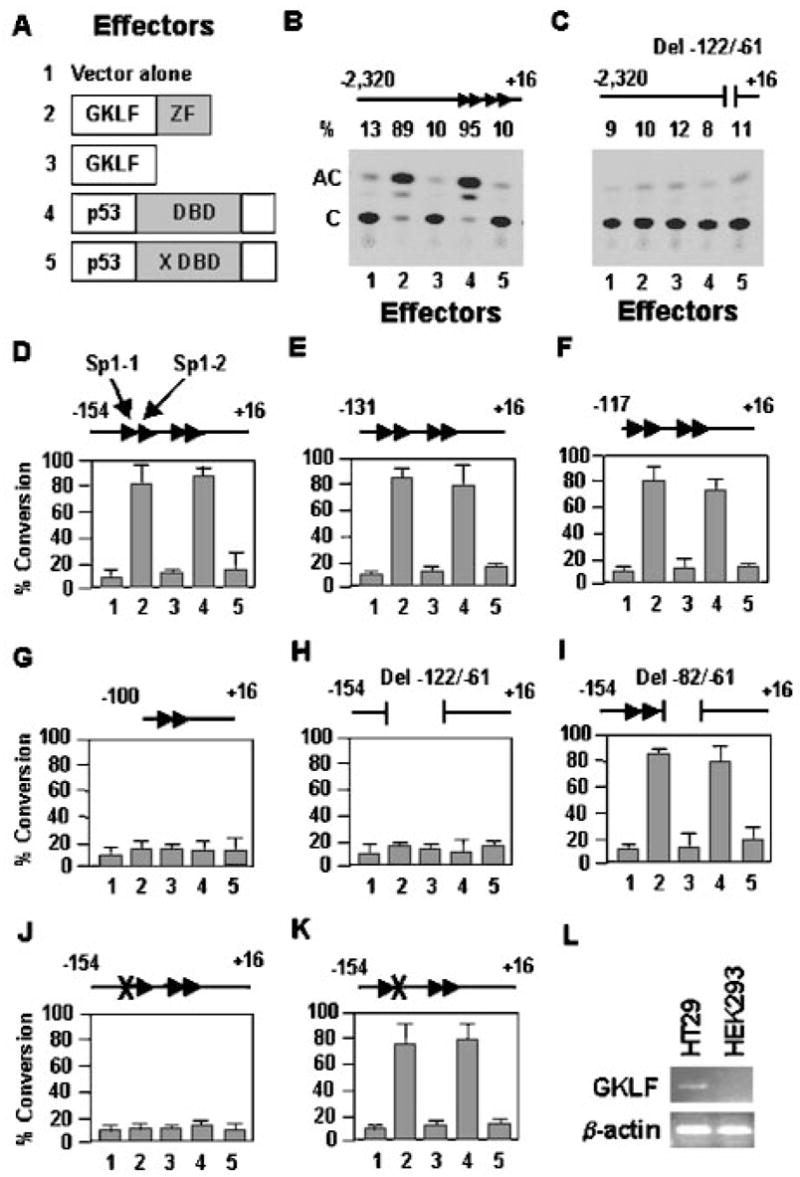
A depicts the five effectors used throughout the cotransfection studies. Effector 1 is the PMT3 expression vector alone. Effectors 2 and 4 are expression constructs of wild-type GKLF and p53, respectively. Effector 3 is a mutant GKLF that does not have its zinc fingers (ZF) (30). Effector 5 is a mutant p53 with a missense mutation at codon 143 (X) in the DBD of p53 (32). Various regions of the p21WAF1/Cip1 promoter were linked to the CAT reporter (B–K) and cotransfected with an equivalent quantity of the various effectors into HEK 293 cells. The four Sp1-binding sites (27) between nt −122 and −61 of the promoter are represented by the four arrowheads. The locations for Sp1-1 and Sp1-2 are identified in D. The × in J and K represents a 3-bp mutation in the first and second Sp1-binding sites, respectively. The numbers on the x axis in D–K correspond to the five effectors shown in A. C is the substrate chloramphenicol, and AC represents the acetylated product. % Conversion = (AC/(AC + C)) × 100. Shown in D–K are the means of three independent experiments. Bars represent S.D. L shows the results of reverse transcription-PCR of the mRNA levels of Gklf and β-actin in HT29 and HEK 293 cells.
Fig. 3B shows that both wild-type GKLF and p53 (effectors 2 and 4, respectively), but not mutant GKLF and p53 (effectors 3 and 5, respectively), transactivated the CAT reporter gene linked to nt −2320 to +16 of the p21WAF1/Cip1 promoter sequence. However, neither wild-type GKLF nor wild-type p53 was able to transactivate the same promoter that had a small internal deletion in the sequence between nt −122 and −61 (Fig. 3C). These results indicate that GKLF, like p53, is capable of activating the p21WAF1/Cip1 promoter and that in order for both proteins to act on the promoter, the sequence between nt −122 and −61 is essential. The dependence of p53 on this proximal region of the p21WAF1/Cip1 promoter was unexpected since the binding sites for p53 in 2320 nt of the promoter were previously localized to sequences much farther upstream from the immediate flanking region of the p21WAF1/Cip1 gene (43).
The sequence between nt −122 and −61 of the p21WAF1/Cip1 promoter contains four GC-rich elements that resemble Sp1-binding sites, which have previously been designated Sp1-1 to Sp1-4 sites in the 5′ to 3′ direction (27). To precisely define the cis-element(s) within this sequence that mediates the activating effect of GKLF and p53 on the p21WAF1/Cip1 promoter, we performed additional cotransfection experiments in which the CAT reporter gene was linked to either −154 to +16 bp of the promoter (Fig. 3D) or to one that contained various 5′- and internal deletions or point mutations (Fig. 3, E–K). It is clear from the results of these experiments that the transactivating effects of GKLF and p53 were co-localized to an identical cis-element, which was the first Sp1-binding site (Sp1-1 site) beginning at nt −116 in the p21WAF1/Cip1 promoter.
GKLF, but Not p53, Binds to the Sp1-1 Element in the p21WAF1/Cip1 Promoter
To determine whether GKLF or p53 binds to the Sp1-1 sequence identified above, we performed EMSAs between GKLF and a labeled oligonucleotide containing the sequence between nt −129 and −99 of the p21WAF1/Cip1 promoter (Fig. 4A, p21 (wt) Probe) or between the DBD of p53 (40, 41) and an established p53-binding sequence (Fig. 4B, p53 Probe). Nuclear extracts prepared from COS-1 cells transfected with the PMT3 expression vector containing either full-length (FL) GKLF (Fig. 4A, lane 1) or the zinc finger (ZF) portion of GKLF (lane 9) bound to the wild-type p21 probe. The resulting DNA-protein complexes (C1 and C2) were competed away by an unlabeled wild-type p21WAF1/Cip1 sequence (Fig. 4A, lanes 2–4 and 10–12, respectively), but not by a mutated competitor in which the Sp1-1 site was destroyed due to a 3-bp substitution (lanes 5–7 and 13–15, respectively). As controls, nuclear extracts prepared from either vector alone-transfected cells (C, lane 16) or cells transfected with a mutant GKLF construct lacking the zinc fingers (ΔZF, lane 17) did not exhibit any appreciable binding to the wild-type p21 probe. The results in Fig. 4A therefore provide strong evidence that GKLF interacts directly with the Sp1-1 site of the p21WAF1/Cip1 promoter. In contrast, the DNA-binding domain of p53, although clearly capable of binding to an established p53-binding sequence (Fig. 4B, lane 2), failed to interact with the p21WAF1/Cip1 sequence since an unlabeled wild-type p21 probe did not compete at all (lanes 6–8). Moreover, p53 DBD failed to form a complex with a labeled wild-type p21 oligonucleotide (data not shown). These results suggest that although the transactivating effect of p53 on the p21WAF1/Cip1 proximal promoter depends on the Sp1-1 site, as does GKLF, it is mediated by a mechanism that does not involve the direct binding of p53 to the DNA.
Fig. 4. Relationship among p53, GKLF, and the Sp1-1 site of the p21WAF1/Cip1 promoter.
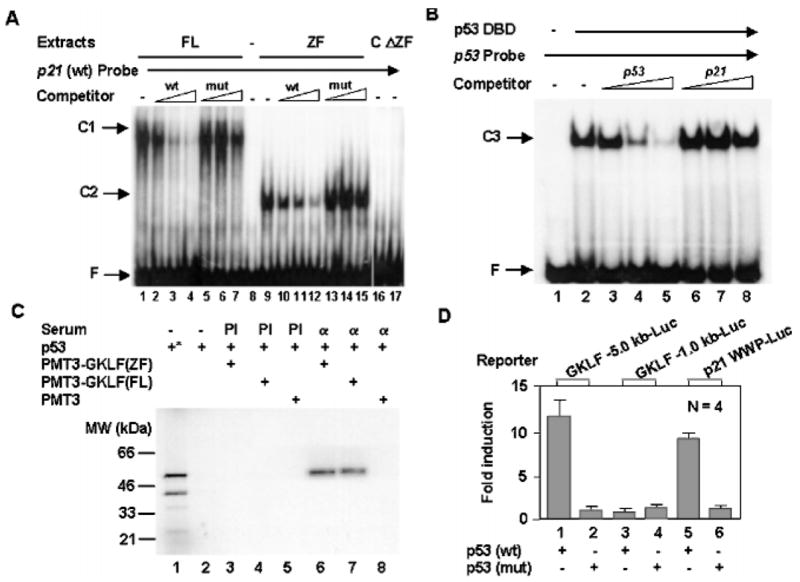
A, GKLF binds to the Sp1-1 site. EMSAs were performed using nuclear extracts prepared from COS-1 cells transfected with an expression construct containing the full-length (FL) GKLF (lanes 1–7) or the zinc finger (ZF) region of GKLF (lanes 9–15) and a radiolabeled oligonucleotide probe containing the sequence between nt −129 and −99 of the p21WAF1/Cip1 promoter, which includes both Sp1-1 and Sp1-2 sites (27). Where indicated, increasing amounts of unlabeled oligonucleotides representing either the wild-type (wt) sequence or a mutated (mut) sequence that contains a 3-bp substitution in the Sp1-1 site were included. Lane 8 contains the probe alone without added proteins. Lanes 16 and 17 contain nuclear extracts obtained from COS-1 cells transfected with the PMT3 vector alone (C) and the GKLF construct that lacks the zinc fingers (ΔZF) as in Fig. 3A, respectively. C1 is the complex formed between full-length GKLF and the probe, and C2 is formed between the zinc fingers and the probe. F is free probe. B, p53 does not interact with the sequence between nt −129 and −99 of the p21WAF1/Cip1 promoter. EMSAs were performed with the purified DBD of p53 (40) and a labeled probe representing an established p53-binding site. Competitors include unlabeled p53-binding sequence (lanes 3–5) and unlabeled wild-type p21WAF1/Cip1 sequence between nt −129 and −99 (lanes 6–8). C3 indicates the complex between p53 DBD and the probe. C, GKLF interacts with p53. 35S-Labeled p53 synthesized by in vitro transcription and translation was mixed with nuclear extracts from COS-1 cells transfected with PMT3-GKLF(ZF), PMT3-GKLF(FL), or PMT3 vector alone and precipitated with either preimmune (PI) serum or anti-GKLF serum (α). Lane 1 (*) contains the input p53, and lane 2 is p53 mixed with protein A-Sepharose beads without added serum. The precipitated materials were resolved by denaturing polyacrylamide gel electrophoresis and visualized by autoradiography. D, p53 transactivates the Gklf promoter. Either 5.0 or 1.0 kb of the 5′-flanking sequence of the mouse Gklf gene was linked to a luciferase reporter and cotransfected into Chinese hamster ovary cells with an expression construct containing either wild-type p53 or mutant p53 that no longer binds DNA (see Fig. 3A). Included was a p21 WWP-Luc construct containing 2.4 kb of the p21WAF1/Cip1 promoter sequence linked to the luciferase reporter as a control (7). Shown are the means of four experiments. Bars are S.D.
p53 Physically Interacts with GKLF and Transcriptionally Activates the GKLF Promoter, Resulting in a Synergistic Activation of the p21WAF1/Cip1 Promoter by p53 and GKLF
One potential method by which p53 may accomplish its indirect effect on the p21WAF1/Cip1 proximal promoter is by forming a physical complex with GKLF, thus gaining access to the promoter. To test this hypothesis, we performed co-immunoprecipitation experiments using in vitro synthesized p53 and GKLF produced in transfected cells. As shown in Fig. 4C, anti-GKLF serum (α) specifically coprecipitated p53 when p53 was combined with nuclear extracts from cells transfected with either the zinc finger region of GKLF (lane 6) or full-length GKLF (lane 7), but not with those transfected with the PMT3 vector alone (lane 8). No p53 was detected in any of the reactions incubated with preimmune (PI) serum (Fig. 4C, lanes 3–5). These findings provide strong evidence for a physical interaction between p53 and GKLF in a region that includes the zinc fingers of GKLF. It is of interest to note that only full-length p53, but not an internally initiated p53 with an estimated molecular mass of 40 kDa (Fig. 4C, lane 1), was recovered in the immunoprecipitates (lanes 6 and 7). This suggests that the N-terminal portion of p53 may be necessary for the interaction with GKLF.
The dependence of Gklf induction on p53 as shown in Fig. 2 suggests that Gklf, like p21WAF1/Cip1, is regulated by p53 during DNA damage. Indeed, the results in Fig. 4D show that p53 transcriptionally activated Gklf since a luciferase reporter linked to 5.0 kb (bar 1), but not 1.0 kb (bar 3), of the mouse Gklf promoter was transactivated by wild-type p53. In contrast, a mutant p53 failed to activate either reporter (Fig. 4D, bars 2 and 4). The degree of induction of the −5.0-kb Gklf promoter activity by p53 was comparable to that seen for 2.4 kb of the p21WAF1/Cip1 promoter (p21 WWP-Luc) (Fig. 4D, compare bars 1 and 5). These results therefore suggest that there is a p53-response element(s) between −5.0 and −1.0 kb of the Gklf promoter. The exact location of this element(s) has not been determined since only the first kb of the Gklf promoter has been sequenced so far (22) and does not contain a p53-binding site.
To determine whether the physical interaction between GKLF and p53 and the transcriptional induction of Gklf by p53 are physiologically relevant to the regulation of the p21WAF1/Cip1 promoter, we performed cotransfection experiments using subsaturating concentrations of expression vectors containing GKLF, p53, or both and a luciferase reporter gene containing 2.4 kb (WWP-Luc) or 2.2 kb (DM-Luc) of the p21WAF1/Cip1 promoter sequence (7). The two reporters differed from each other in that WWP-Luc included an upstream p53-binding site located at nt −2301 (33). As shown in Fig. 5, the combination of GKLF and p53 resulted in a synergistic induction of the p21WAF1/Cip1 promoter either in the presence (bar 3) or absence (bar 6) of the upstream p53-binding sequence. These results suggest that GKLF and p53 act in a cooperative manner to activate p21WAF1/Cip1 gene expression by a mechanism that does not require the upstream p53-binding site of the p21WAF1/Cip1 promoter.
Fig. 5. Synergistic activation of the p21WAF1/Cip1 promoter by GKLF and p53.

Cotransfection experiments were performed in HEK 293 cells with a luciferase reporter linked to 2.4 or 2.2 kb of the p21WAF1/Cip1 promoter sequence (WWP-Luc, which contains an upstream p53-binding site at nt −2301, or DM-Luc, which does not, respectively (7 and 33)) and subsaturating amounts of expression constructs containing GKLF, p53, or both. Shown are the means of four independent experiments. Bars represent S.D.
GKLF Is Necessary for the Inductive Effect of p53 on the p21WAF1/Cip1 Promoter
The sequential induction of Gklf and p21WAF1/Cip1 during the early phase of DNA damage and the physical dependence of p53 on GKLF in activating the p21WAF1/Cip1 proximal promoter raised the possibility that GKLF may be important in mediating the effect of p53 on stimulating p21WAF1/Cip1 gene expression. To address this possibility, we examined a system in which activation of p53 is inducible due to a temperature-sensitive mutation. As shown in Fig. 6A, induction of wild-type p53 activity by shifting 10(1) cells stably transfected with the temperature-sensitive p53 val135 (35, 37) from the nonpermissive (38.5 °C) to the permissive (31.5 °C) temperature resulted in a considerable accumulation of GKLF as well as p21WAF1/Cip1 to a degree similar to that observed in MMS-treated p53+/+ MEFs (Fig. 2). Next, to assess the role of GKLF in mediating the inductive effect of p53 on p21WAF1/Cip1 expression, we treated cells with a 21-nt anti-sense oligonucleotide containing the sequence that surrounds the initiation methionine codon of GKLF in an attempt to block the translation of Gklf mRNA. We then determined the effects of such treatments on the production of p21WAF1/Cip1 at the permissive temperature. A sense oligonucleotide with the complementary sequence to the antisense oligonucleotide was used as a control. As shown in Fig. 6B (lanes 3, 5, and 7), cells treated with increasing concentrations of the antisense oligonucleotide contained progressively lower levels of GKLF. Importantly, the same cells produced correspondingly lower levels of p21WAF1/Cip1 as well. Although there was some decrease in the levels of GKLF (and consequently, p21WAF1/Cip1) in cells treated with the highest concentration (3.0 μm) of the control sense oligonucleotide (Fig. 6B, lane 6), this was probably due to a nonspecific, perhaps toxic side effect of the high oligonucleotide concentration. Be that as it may, the results in Fig. 6B indicate that a decreased production of GKLF leads to an inhibition of p21WAF1/Cip1 synthesis. We therefore conclude that GKLF is an important mediator of the action of p53 in inducing p21WAF1/Cip1 gene expression.
Fig. 6. GKLF mediates the inductive effect of p53 on p21WAF1/Cip1.
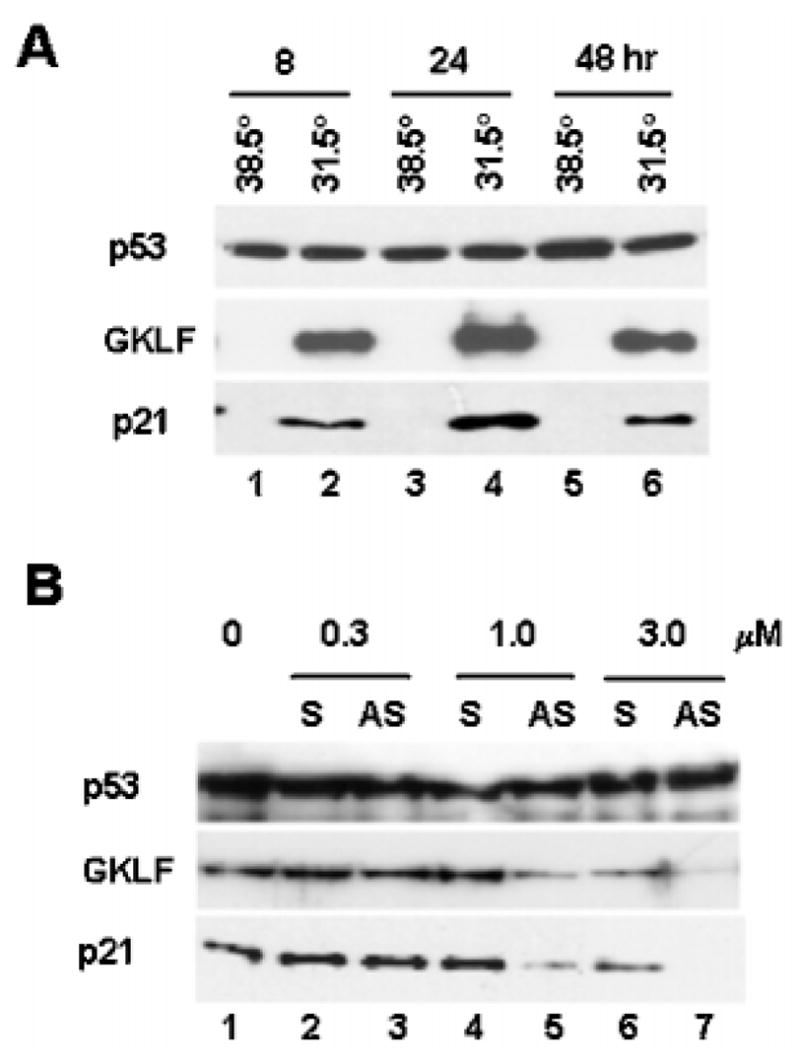
A, induction of Gklf and p21WAF1/Cip1 in 10(1) cells (36) containing a temperature-sensitive mutant p53 protein. 10(1) cells stably expressing the temperature-sensitive p53 val135 mutant (35, 37) were propagated at either the nonpermissive temperature of 38.5 °C or the permissive temperature of 31.5 °C for the time periods indicated. Proteins were harvested and analyzed for p53, GKLF, or p21WAF1/Cip1 by Western blot analysis. Both GKLF and p21WAF1/Cip1 were absent at time 0 when cells were maintained at 38.5 °C (data not shown). B, inhibition of p21WAF1/Cip1 formation in the 10(1)-p53 val135 cell line by antisense oligonucleotides to GKLF. Cells were transfected by lipofection with increasing amounts of sense (S) or antisense (AS) oligonucleotides to GKLF at 38.5 °C for 5 h and shifted to 31.5 °C for an additional 24 h before being harvested for quantification of p53, GKLF, or p21WAF1/Cip1 by Western blot analysis.
DISCUSSION
This study reveals a novel mechanism by which expression of the p21WAF1/Cip1 gene is modulated during cellular stress induced by DNA damage. At lease five lines of evidence in the study suggest that GKLF plays a physiologically relevant and possibly crucial role in mediating the activating effect of p53 on p21WAF1/Cip1 expression: 1) the sequential manner in which Gklf and p21WAF1/Cip1 are expressed in the immediate period following DNA damage (Fig. 1C); 2) the requirement of the GKLF-response element in the p21WAF1/Cip1 promoter (i.e. the Sp1-1 site) for the transactivating effect of p53, despite the presence of other bona fide p53-response elements in the same promoter (Fig. 3, B and C); 3) the physical interaction between p53 and GKLF (Fig. 4C) and the transcriptional induction of Gklf by p53 (Fig. 4D); 4) the cooperative manner in which GKLF and p53 activate the p21WAF1/Cip1 promoter (Fig. 5); and 5) the ability of antisense GKLF oligonucleotides to inhibit p21WAF1/Cip1 synthesis upon p53 activation (Fig. 6). These findings demonstrate that p53 may depend on GKLF to activate the p21WAF1/Cip1 promoter, thus implicating GKLF as an important component of the p53 network of cell cycle regulators.
Based on the observations of this study, we propose a model that portrays the regulation of the p21WAF1/Cip1 proximal promoter by GKLF and p53 during cellular stress elicited by DNA damage. In this model, activation of p53 represents an immediate response to DNA damage as depicted by numerous previous studies (reviewed in Refs. 1–3). The activated p53 causes an increase in the quantity of GKLF, which is mediated, at least in part, at the level of transcription (Fig. 4D). In addition, p53 physically interacts with GKLF and consequently allows it to gain access to the p21WAF1/Cip1 proximal promoter through the Sp1-1 site to which GKLF alone binds. A result of this complex relationship is the synergistic induction of the activity of the p21WAF1/Cip1 promoter (Fig. 5). This model would therefore predict an immediate and possibly maximal induction of expression of the gene encoding p21WAF1/Cip1 following DNA damage. This would assure the immediate cessation of cell cycle progression due to the potent inhibitory effect of p21WAF1/Cip1 on cyclin-dependent kinases (48). It is of note that despite the involvement of the various Sp1 elements in the p21WAF1/Cip1 promoter in mediating the responses of the promoter to numerous other physiological stimuli (Fig. 7), the lone utilization of the Sp1-1 site by GKLF and consequently by p53 has not previously been documented.
Fig. 7. Model for the regulation of the p21WAF1/Cip1 proximal promoter by p53 and GKLF.
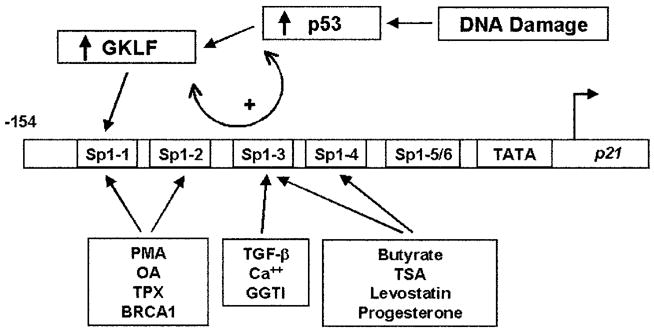
The locations of the six Sp1-like elements within −154 bp of the p21WAF1/Cip1 promoter are designated according to a previous report (27). The model illustrates that the activation of p53 by DNA damage leads to both an increase in GKLF synthesis and an interaction between p53 and GKLF (double arrow), which cumulates in the binding of GKLF to the Sp1-1 element of the p21WAF1/Cip1 promoter. The various Sp1 cis-elements that mediate the functions of other physiological stimuli are also indicated. They include the phorbol ester phorbol 12-myristate 13-acetate (PMA) and okadaic acid (OA) (23); trapoxin (TPX), a histone deacetylase inhibitor (44); BRCA1, the breast cancer tumor suppressor gene (45); transforming growth factor-β (TGF-β) (24); Ca2+, which is important in keratinocyte differentiation (28); a geranylgeranyltransferase I (GGTI) inhibitor (46); butyrate (27) and trichostatin A (TSA) (29), both also histone deacetylase inhibitors; levostatin, a 3-hydroxy-3-methylglutaryl-coenzyme A reductase inhibitor (47); and progesterone (43).
The mechanism by which GKLF participates in the regulation of the p21WAF1/Cip1 promoter by p53 is reminiscent of that for another growth arrest-associated gene, GADD45 (49). Like Gklf and p21WAF1/Cip1, expression of GADD45 is induced by genotoxic stresses such as DNA damage (50). In addition to a strong p53-binding element in an intronic sequence of GADD45 (42), p53 was shown to contribute to the stress response of the GADD45 promoter (50). Much of this stress responsiveness was localized to a GC-rich motif of the proximal promoter to which the tumor suppressor WT1 (Wilms’ tumor 1) (52) binds, but p53 does not. The mechanism by which p53 activates the promoter is thought to be mediated by its ability to physically interact with WT1 (50). This resulted in a strong and cooperative induction of the GADD45 promoter when p53 and WT1 were concurrently introduced (50). Finally, abrogation of WT1 function by an antisense vector markedly reduced the induction of the GADD45 promoter (50). Similar to the conclusion of the present study, it was concluded that p53 contributes to the positive regulation of the GADD45 promoter primarily by protein-protein interactions.
Recent literature provides another example in which the p21WAF1/Cip1 promoter can be cooperatively regulated by multiple proteins with important functions in cell cycle control. In a previous study (45), BRCA1 was shown to transactivate the p21WAF1/Cip1 proximal promoter through the region between nt −117 and −93, which contains both Sp1-1 and Sp1-2 sites. This resulted in an inhibition of progression into the S phase in cells that overexpressed BRCA1 (45). Importantly, p53 potentiated the BRCA1-dependent activation of the p21WAF1/Cip1 promoter by physically interacting with BRCA1 (53). Thus, it appears that p53 activates expression of its target genes such as p21WAF1/Cip1 and GADD45 by multiple but perhaps interrelated mechanisms. These mechanisms include direct binding of p53 to the classical p53-response elements and indirect interaction with non-consensus binding sites through physical contacts with other regulatory proteins, including GKLF, WT1, and BRCA1.
Another potential mechanism responsible for the synergistic induction of the p21WAF1/Cip1 promoter by p53 and GKLF may involve the participation of other regulatory proteins. In this regard, both p53 (54, 55) and GKLF (31) have been shown to interact with a group of transcriptional coactivators, including p300 and CBP (56–59). In fact, the ability of GKLF to activate transcription is dependent on its interaction with p300/CBP (31). Thus, it is possible to modify the model proposed in Fig. 7 to include p300/CBP, which can serve as a bridge between p53/GKLF and the basal transcriptional machinery such as the TATA-binding factor and RNA polymerase II (60, 61). It is of interest to note that p300 and CBP are enzymes that display histone acetylase activity (62, 63) and that the activity of the p21WAF1/Cip1 promoter is subject to regulation by compounds that alter chromatin structure due to acetylation such as butyrate, trichostatin A, and trapoxin (Fig. 7). Moreover, the Sp1-like cis-elements responsible for the action of these compounds appear to differ among one another (Fig. 7). It is formally possible that the targets of regulation by these compounds may be unique transcription factors that recognize the different Sp1-like elements in the p21WAF1/Cip1 promoter.
The results in Fig. 1 indicate that both Gklf and p21WAF1/Cip1 are induced by serum deprivation and by DNA damage. However, the kinetics in which the two genes are induced by the two conditions are distinctly different from each other. During the course of serum deprivation, the extent of induction for both Gklf and p21WAF1/Cip1 is quite similar (Fig. 1A). However, the time course of induction for Gklf and p21WAF1/Cip1 in cells treated with MMS is different from that of serum deprivation. Thus, with the exception of an earlier induction of Gklf during the initial 30 min of MMS treatment (Fig. 1C), the level of induction of p21WAF1/Cip1 after 1 h of MMS treatment is significantly higher than that of Gklf (Fig. 1, B and C). These results suggest that factors in addition to GKLF may be involved in the rise in p21WAF1/Cip1 transcript level after the immediate phase of DNA damage. However, the parallel rise in the levels of both Gklf and p21WAF1/Cip1 transcripts during serum deprivation suggests a potentially more uniform mechanism of induction of the two genes, perhaps including a mechanism that is independent of p53, as has been demonstrated in other systems (64). Experiments are in progress to address this potentially p53-independent component of Gklf and p21WAF1/Cip1 activation.
In the intestinal tract, the Gklf transcript is detected primarily in terminally differentiated, post-mitotic epithelial cells (12–14). Interestingly, the p21WAF1/Cip1 transcript is also distributed in the same cell population (65). Moreover, the intestinal expression of p21WAF1/Cip1 both during development and in the adult mouse has been shown to be independent of p53 under basal conditions (51). Whether the in vivo expression of Gklf is also independent of p53 is unclear at this point. However, it is clear that the induction of both Gklf and p21WAF1/Cip1 in response to genotoxic stress is highly dependent on p53 (Fig. 2). Moreover, this inductive response is not limited to the intestinal cell lineage and includes fibroblasts such as NIH 3T3 and MEFs. Thus, the in vitro behavior of Gklf as modulated by stress is more ubiquitous than its in vivo tissue distribution. This may be viewed as additional evidence for the potentially broader significance of GKLF in mediating the “guardian” function of p53.
Acknowledgments
We thank B. Vogelstein, K. Kinzler, A. Levine, L. Donehower, and N. Palvetich for providing plasmids, reagents, and cell lines.
Footnotes
This work was in part supported by grants from the National Institutes of Health (to V. W. Y., K. H. K., and A. S. K.).
The abbreviations used are: Cdk, cyclin-dependent kinase; GKLF, gut-enriched Krüppel-like factor; KLF4, Krüppel-like factor 4; DBD, DNA-binding domain; CAT, chloramphenicol acetyltransferase; nt, nucleotide(s); kb, kilobase(s); bp, base pair(s); MEFs, mouse embryo fibroblasts; MMS, methyl methanesulfonate; PCR, polymerase chain reaction; HEK, human embryonic kidney; EMSA, electrophoretic mobility shift assay; BisTris propane, 1,3-bis]tris(hydroxymethyl)methylamino[propane; CBP, cAMP-response element-binding protein-binding protein.
References
- 1.Agarwal ML, Taylor WR, Chernov MV, Chernova OB, Stark GR. J Biol Chem. 1998;273:1–4. doi: 10.1074/jbc.273.1.1. [DOI] [PubMed] [Google Scholar]
- 2.Ko LJ, Prives C. Genes Dev. 1996;10:1054–1072. doi: 10.1101/gad.10.9.1054. [DOI] [PubMed] [Google Scholar]
- 3.Levine AJ. Cell. 1997;88:323–331. doi: 10.1016/s0092-8674(00)81871-1. [DOI] [PubMed] [Google Scholar]
- 4.Giaccia AJ, Kastan MB. Genes Dev. 1998;12:2973–2983. doi: 10.1101/gad.12.19.2973. [DOI] [PubMed] [Google Scholar]
- 5.Prives C. Cell. 1998;95:5–8. doi: 10.1016/s0092-8674(00)81774-2. [DOI] [PubMed] [Google Scholar]
- 6.El-Deiry WS. Semin Cancer Biol. 1998;8:345–357. doi: 10.1006/scbi.1998.0097. [DOI] [PubMed] [Google Scholar]
- 7.El-Deiry WS, Tokino T, Velculescu VE, Levy DB, Parsons R, Trent JM, Lin D, Mercer WE, Kinzler KW, Vogelstein B. Cell. 1993;75:817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 8.Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. Cell. 1993;75:805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 9.Hartwell LH, Kastan MB. Science. 1994;266:1821–1828. doi: 10.1126/science.7997877. [DOI] [PubMed] [Google Scholar]
- 10.Sherr CJ, Roberts JM. Genes Dev. 1995;9:1149–1163. doi: 10.1101/gad.9.10.1149. [DOI] [PubMed] [Google Scholar]
- 11.Turner J, Crossley M. Trends Biochem Sci. 1999;24:236–240. doi: 10.1016/s0968-0004(99)01406-1. [DOI] [PubMed] [Google Scholar]
- 12.Shields JM, Christy RJ, Yang VW. J Biol Chem. 1996;271:20009–20017. doi: 10.1074/jbc.271.33.20009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jenkins TD, Opitz OG, Okano J, Rustgi AK. J Biol Chem. 1998;273:10747–10754. doi: 10.1074/jbc.273.17.10747. [DOI] [PubMed] [Google Scholar]
- 14.Garrett-Sinha LA, Eberspaecher H, Seldin MF, de Crombrugghe B. J Biol Chem. 1996;271:31384–31390. doi: 10.1074/jbc.271.49.31384. [DOI] [PubMed] [Google Scholar]
- 15.Segre JA, Bauer C, Fuchs E. Nat Genet. 1999;22:356–360. doi: 10.1038/11926. [DOI] [PubMed] [Google Scholar]
- 16.Panigada M, Porcellini M, Sutti F, Doneda L, Pozzoli O, Consalez GG, Guttinger M, Grassi F. Mech Dev. 1999;81:103–113. doi: 10.1016/s0925-4773(98)00237-8. [DOI] [PubMed] [Google Scholar]
- 17.Yet SF, McA’Nulty MM, Folta SC, Yen HW, Yoshizumi M, Hsieh CM, Layne MD, Chin MT, Wang H, Perrella MA, Jain MK, Lee ME. J Biol Chem. 1998;273:1026–1031. doi: 10.1074/jbc.273.2.1026. [DOI] [PubMed] [Google Scholar]
- 18.Ton-That H, Kaestner KH, Shields JM, Mahatanankoon CS, Yang VW. FEBS Lett. 1997;419:239–243. doi: 10.1016/s0014-5793(97)01465-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kaestner KH, Silberg DG, Traber PG, Schutz G. Genes Dev. 1997;11:1583–1595. doi: 10.1101/gad.11.12.1583. [DOI] [PubMed] [Google Scholar]
- 20.Shields JM, Yang VW. Nucleic Acids Res. 1998;26:796–802. doi: 10.1093/nar/26.3.796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang W, Shields JM, Sogawa K, Fujii-Kuriyama Y, Yang VW. J Biol Chem. 1998;273:17917–17925. doi: 10.1074/jbc.273.28.17917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mahatan CS, Kaestner KH, Geiman DE, Yang VW. Nucleic Acids Res. 1999;27:4562–4569. doi: 10.1093/nar/27.23.4562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Biggs JR, Kudlow JE, Kraft AS. J Biol Chem. 1996;271:901–906. doi: 10.1074/jbc.271.2.901. [DOI] [PubMed] [Google Scholar]
- 24.Datto MB, Yu Y, Wang XF. J Biol Chem. 1995;270:28623–28628. doi: 10.1074/jbc.270.48.28623. [DOI] [PubMed] [Google Scholar]
- 25.Li JM, Datto MB, Shen X, Hu PP, Yu Y, Wang XF. Nucleic Acids Res. 1998;26:2449–2456. doi: 10.1093/nar/26.10.2449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moustakas A, Kardassis D. Proc Natl Acad Sci U S A. 1998;95:6733–6738. doi: 10.1073/pnas.95.12.6733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nakano K, Mizuno T, Sowa Y, Orita T, Yoshino T, Okuyama Y, Fujita T, Ohtani-Fujita N, Matsukawa Y, Tokino T, Yamagishi H, Oka T, Nomura H, Sakai T. J Biol Chem. 1997;272:22199–22206. doi: 10.1074/jbc.272.35.22199. [DOI] [PubMed] [Google Scholar]
- 28.Prowse DM, Bolgan L, Molnar A, Dotto GP. J Biol Chem. 1997;272:1308–1314. doi: 10.1074/jbc.272.2.1308. [DOI] [PubMed] [Google Scholar]
- 29.Sowa Y, Orita T, Minamikawa S, Nakano K, Mizuno T, Nomura H, Sakai T. Biochem Biophys Res Commun. 1997;241:142–150. doi: 10.1006/bbrc.1997.7786. [DOI] [PubMed] [Google Scholar]
- 30.Shields JM, Yang VW. J Biol Chem. 1997;272:18504–18507. doi: 10.1074/jbc.272.29.18504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Geiman DE, Ton-That H, Johnson JM, Yang VW. Nucleic Acids Res. 2000;28:1106–1113. doi: 10.1093/nar/28.5.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Baker SJ, Markowitz S, Fearon ER, Willson JK, Vogelstein B. Science. 1990;249:912–915. doi: 10.1126/science.2144057. [DOI] [PubMed] [Google Scholar]
- 33.Gartel AL, Tyner AL. Exp Cell Res. 1999;246:280–289. doi: 10.1006/excr.1998.4319. [DOI] [PubMed] [Google Scholar]
- 34.Jones SN, Sands AT, Hancock AR, Vogel H, Donehower LA, Linke SP, Wahl GM, Bradley A. Proc Natl Acad Sci U S A. 1996;93:14106–14111. doi: 10.1073/pnas.93.24.14106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sabbatini P, Lin J, Levine AJ, White E. Genes Dev. 1995;9:2184–2192. doi: 10.1101/gad.9.17.2184. [DOI] [PubMed] [Google Scholar]
- 36.Harvey DM, Levine AJ. Genes Dev. 1991;5:2375–2385. doi: 10.1101/gad.5.12b.2375. [DOI] [PubMed] [Google Scholar]
- 37.Wu X, Bayle JH, Olson D, Levine AJ. Genes Dev. 1993;7:1126–1132. doi: 10.1101/gad.7.7a.1126. [DOI] [PubMed] [Google Scholar]
- 38.Fornace AJ, Alamo I, Hollander MC. Proc Natl Acad Sci U S A. 1988;85:8800–8804. doi: 10.1073/pnas.85.23.8800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.von Kleist S, Chany E, Burtin P, King M, Fogh J. J Natl Cancer Inst. 1975;55:555–560. doi: 10.1093/jnci/55.3.555. [DOI] [PubMed] [Google Scholar]
- 40.Pavletich NP, Chambers KA, Pabo CO. Genes Dev. 1993;7:2556–2564. doi: 10.1101/gad.7.12b.2556. [DOI] [PubMed] [Google Scholar]
- 41.Cho Y, Gorina S, Jeffrey PD, Pavletich NP. Science. 1994;265:346–355. doi: 10.1126/science.8023157. [DOI] [PubMed] [Google Scholar]
- 42.Hollander MC, Alamo I, Jackman J, Wang MG, McBride OW, Fornace AJ. J Biol Chem. 1993;268:24385–24393. [PubMed] [Google Scholar]
- 43.Owen GI, Richer JK, Tung L, Takimoto G, Horwitz KB. J Biol Chem. 1998;273:10696–10701. doi: 10.1074/jbc.273.17.10696. [DOI] [PubMed] [Google Scholar]
- 44.Sambucetti LC, Fischer DD, Zabludoff S, Kwon PO, Chamberlin H, Trogani N, Xu H, Cohen D. J Biol Chem. 1999;274:34940–34947. doi: 10.1074/jbc.274.49.34940. [DOI] [PubMed] [Google Scholar]
- 45.Somasundaram K, Zhang H, Zeng YX, Houvras Y, Peng Y, Zhang H, Wu GS, Licht JD, Weber BL, El-Deiry WS. Nature. 1997;389:187–190. doi: 10.1038/38291. [DOI] [PubMed] [Google Scholar]
- 46.Adnane J, Bizouarn FA, Qian Y, Hamilton AD, Sebti SM. Mol Cell Biol. 1998;18:6962–6970. doi: 10.1128/mcb.18.12.6962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lee SJ, Ha MJ, Lee J, Nguyen P, Choi YH, Pirnia F, Kang WK, Wang XF, Kim SJ, Trepel JB. J Biol Chem. 1998;273:10618–10623. doi: 10.1074/jbc.273.17.10618. [DOI] [PubMed] [Google Scholar]
- 48.Xiong Y, Hannon GJ, Zhang H, Casso D, Kobayashi R, Beach D. Nature. 1993;366:701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 49.Zhan Q, Chen IT, Antinore MJ, Fornace AJ., Jr Mol Cell Biol. 1998;18:2768–2778. doi: 10.1128/mcb.18.5.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fornace AJ, Jr, Nebert DW, Hollander MC, Luethy JD, Papathanasion M, Fargnoli J, Holbrook NJ. Mol Cell Biol. 1989;9:4196–4203. doi: 10.1128/mcb.9.10.4196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Macleod KF, Sherry N, Hannon G, Beach D, Tokino T, Kinzler K, Vogelstein B, Jacks T. Genes Dev. 1995;9:935–944. doi: 10.1101/gad.9.8.935. [DOI] [PubMed] [Google Scholar]
- 52.Rauscher FJ, III, Morris JF, Tournay OE, Cook DM, Curran T. Science. 1990;250:1259–1262. doi: 10.1126/science.2244209. [DOI] [PubMed] [Google Scholar]
- 53.Zhang H, Somasundaram K, Peng Y, Tian H, Zhang H, Bi D, Weber BL, El-Deiry WS. Oncogene. 1998;16:1713–1721. doi: 10.1038/sj.onc.1201932. [DOI] [PubMed] [Google Scholar]
- 54.Gu W, Shi XL, Roeder RG. Nature. 1997;387:819–823. doi: 10.1038/42972. [DOI] [PubMed] [Google Scholar]
- 55.Lill NL, Grossman SR, Ginsberg D, DeCaprio J, Livingston DM. Nature. 1997;387:823–827. doi: 10.1038/42981. [DOI] [PubMed] [Google Scholar]
- 56.Eckner R. Biol Chem. 1996;377:685–688. [PubMed] [Google Scholar]
- 57.Ludlow JW, Skuse GR. Virus Res. 1995;35:113–121. doi: 10.1016/0168-1702(94)00094-s. [DOI] [PubMed] [Google Scholar]
- 58.Chrivia JC, Kwok RP, Lamb N, Hagiwara M, Montminy MR, Goodman RH. Nature. 1993;365:855–859. doi: 10.1038/365855a0. [DOI] [PubMed] [Google Scholar]
- 59.Kwok RP, Lundblad JR, Chrivia JC, Richards JP, Bachinger HP, Brennan RG, Roberts SG, Green MR, Goodman RH. Nature. 1994;370:223–226. doi: 10.1038/370223a0. [DOI] [PubMed] [Google Scholar]
- 60.Abraham SE, Lobo S, Yaciuk P, Wang HG, Moran E. Oncogene. 1993;8:1639–1647. [PubMed] [Google Scholar]
- 61.Neish AS, Anderson SF, Schlegel BP, Wei W, Parvin JD. Nucleic Acids Res. 1998;26:847–853. doi: 10.1093/nar/26.3.847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Wu C. J Biol Chem. 1997;272:28171–28174. doi: 10.1074/jbc.272.45.28171. [DOI] [PubMed] [Google Scholar]
- 63.Howe L, Brown CE, Lechner T, Workman JL. Crit Rev Eukaryotic Gene Expression. 1999;9:231–243. doi: 10.1615/critreveukargeneexpr.v9.i3-4.80. [DOI] [PubMed] [Google Scholar]
- 64.Modiano JF, Ritt MG, Wojcieszyn J, Smith R., III DNA Cell Biol. 1999;8:357–367. doi: 10.1089/104454999315259. [DOI] [PubMed] [Google Scholar]
- 65.Gartel AL, Serfas MS, Gartel M, Goufman E, Wu GS, El-Deiry WS, Tyner AL. Exp Cell Res. 1996;227:171–181. doi: 10.1006/excr.1996.0264. [DOI] [PubMed] [Google Scholar]