

Abstract
Sphingosylphosphorylcholine (SPC) is a powerful vasoconstrictor, but in vitro its EC50 is ∼100-fold more than plasma concentrations. We examined whether sub-contractile concentrations of SPC (≤1 μmol/l) modulated vasoreactivity of rat intrapulmonary arteries (IPA), using myography and measurement of intracellular [Ca2+] ([Ca2+]i). SPC (1 μmol/l) had no effect on force or [Ca2+]i on its own, but dramatically potentiated constrictions induced by ∼25 mmol/l [K+], such that at 40 min force and [Ca2+]i (Fura PE3 340/380 ratio) were increased by 429 ± 96% and 134 ± 26% respectively. The potentiation was stereospecific, apparent at concentrations >100 nmol/l SPC, and independent of the endothelium, 2-APB-sensitive Ca2+ entry, and Rho kinase. It was abolished by the PLC inhibitor U73122, the broad spectrum PKC inhibitor Ro 31-8220 and the PKCδ inhibitor rottlerin, but not by Gö6976, which is ineffective against PKCδ. The potentiation could be attributed to enhancement of Ca2+ entry. SPC also potentiated the responses to PGF2α and U436619, which activate a 2-APB sensitive non-selective cation channel (NSCC) in IPA. In this case potentiation was partially inhibited by diltiazem, but abolished by 2-APB, Ro31-8220 and rottlerin. 1 μmol/l SPC caused translocation of PKCδ to the perinuclear region and cytoskeleton of cultured IPA smooth muscle cells. We present the novel finding that low, sub-contractile concentrations of SPC potentiate Ca2+ entry in IPA through both voltage-dependent and independent pathways, via a receptor-dependent mechanism involving PKCδ. This has implications for the physiological role of SPC, especially in cardiovascular disease where SPC is reported to be elevated.
Keywords: Pulmonary artery, Lysosphingolipids, Sphingosylphosphorylcholine, L-type Ca2+ channels, Protein kinase Cδ
Introduction
The lysosphingolipid sphingosylphosphorylcholine (SPC) is an important cardiovascular mediator derived from sphingomyelin and carried by plasma lipoproteins1,2. It has been associated with multifarious processes including proliferation, angiogenesis, inflammation and vasoconstriction1-7, and elevated SPC concentrations may contribute to cardiovascular disease2, including hypercholesterolemia and atherosclerosis3,8, cerebral and coronary vasospasm6,7 and hypertension9. In systemic arteries SPC-induced vasoconstriction is reported to be mediated via pertussis toxin (PTX) sensitive Gi-proteins, IP3-induced Ca2+ release, Rho kinase (RhoK)-mediated Ca2+ sensitization, and Ca2+ entry via voltage-dependent L-type channels1,2,10, although in some arteries Ca2+ sensitization may be the major mechanism6,7. In contrast, we recently reported that in rat small intrapulmonary arteries (IPA) the SPC-induced elevation of [Ca2+]i is primarily due to activation of a voltage-independent Ca2+ entry pathway, that is insensitive to PTX and Ca2+ release from stores4.
A conceptual problem regarding any physiological role for SPC is that the EC50 for most of its effects in vitro is in the range of 7-18 μmol/l1,2,4-6, whereas plasma concentrations may be as low as 50 nmol/l, though in serum this rises to 130 nmol/l suggesting release from activated platelets11. It has been argued that lysophospholipids act in a paracrine or autocrine fashion, with higher local concentrations than in plasma, especially at sites of thrombus formation, atheroscelerosis and inflammation1,2,6,7; tissue concentrations of ∼10 μmol/l SPC are reported for certain types of inflammation12. Studies on SPC are hindered by lack of specific antagonists, and its receptors remain unidentified. SPC is a low affinity ligand for sphingosine-1-phosphate (S1P) receptors, but this cannot account for the majority of its actions. Although GPR4, OGR1 and G2A have been proposed as SPC receptors, recent evidence suggests this family responds to protons and not SPC13,14.
We considered whether concentrations of SPC insufficient to exert direct vasoactive effects might potentiate vasoconstriction induced by other means, as demonstrated for some other agonists in IPA15, and examined the effects of sub-contractile concentrations of SPC (≤ 1 μmol/l) on depolarization- and agonist-induced vasoconstriction of rat IPA. We report the novel and potentially important finding that these low concentrations of SPC substantially potentiate IPA vasoreactivity via a PKCδ-dependent enhancement of both voltage-dependent and independent Ca2+ entry, and that this mechanism differs from that underlying vasoconstriction induced by higher concentrations of SPC.
Materials and Methods
Male Wistar rats (200-300g) were killed by cervical dislocation; the investigation conforms with the Guide for the Care and Use of Laboratory Animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996). Small IPA (3rd-4th branch; 150-450 μm i.d.) were mounted in a myograph (Danish MyoTechnology, Denmark) containing physiological salt solution (PSS) gassed with 95% air / 5% CO2 (pH 7.4) at 37°C, as previously described16. Endothelial denudation was achieved by rubbing the lumen, and confirmed by loss of relaxation to acetylcholine. Pulmonary artery smooth muscle cells (PASMCs) were dispersed from similar sized IPA using collagenase (type XI, 2mg/ml) and papain (1mg/ml)16, and used immediately for electrophysiology or cultured in DMEM containing 10% FCS. PASMCs from passages 3-4 were grown on 13mm coverslips and growth-arrested in serum-free medium for 24 hrs before use; each cell line was verified as smooth muscle by immunostaining for smooth muscle α-actin, calponin and desmin (Santa Cruz Biotechnology, CA).
Estimation of [Ca2+]i
IPA were incubated for 1 hour at 37°C in PSS with 4 μmol/l Fura PE-3/AM followed by washing with PSS. The myograph was mounted on an inverted microscope and microfluorimeter (Cairn Ltd., Faversham, U.K.). Force was recorded simultaneously with the ratio of emission intensities at >510 nm from excitation wavelengths of 340 and 380 nm (F340/380).
Electrophysiology
Freshly dispersed PASMCs were studied at ∼20°C using whole cell patch clamp (Axopatch-200c, Axon Instruments Inc., CA). The bath was continuously perfused with HEPES-buffered PSS containing 2 mmol/l tetraethylammonium (TEA) to block BKCa current. The pipette solution contained (mmol/l): KCl 140; MgCl2 2; EGTA 5; HEPES 10; MgATP 2.0, Li2GTP 0.2; pH adjusted to 7.2 with KOH. Current-voltage relationships were obtained using a voltage ramp protocol, with a holding potential of -60 mV and a 0.5 s ramp from -90 to +100mV every 5 s.
α-toxin permeabilization of IPA
Isometric force was recorded in α-toxin-permeabilized arteries, as described previously4. IPA were mounted as above, but incubated at 26°C and permeabilized with 60 μg/ml α-toxin at pCa 6.5. IPA were relaxed with solution containing 10 mmol/l EGTA before sub-maximal vasoconstriction was induced by increasing [Ca2+] to 200 nmol/l (pCa 6.7) by adjusting the K2EGTA/CaEGTA ratio.
PKCδ translocation
Cultured PASMCs were exposed to 1μM SPC for 10 min, before fixation with 4% paraformaldehyde and 4% PEG6000, and permeabilization with 0.1% Triton. Cells were stained with anti-PKCδ antibody (Santa Cruz Biotechnology) overnight at 4C, followed by Alexa 488 labelled secondary antibody (goat anti-rabbit IG, Invitrogen) for 2 hrs at room temperature. Coverslips were viewed using a CARVII confocal imager (BD Biosciences) and Zeiss Axovert microscope. Specificity of anti-PKCδ was confirmed by Western blot; staining was negative with secondary antibody alone.
Solutions and drugs
Physiological salt solution (PSS) contained (mmol/l): NaCl 118; NaHCO3; 24; KCl 4; CaCl2 1.8; MgSO4 1; NaH2PO4 0.434; glucose 5.56. In experiments where [K+] was altered, osmolarity was maintained by equimolar substitution of K+ for Na+. HEPES-buffered PSS contained (mmol/l) NaCl 130; KCl 4: CaCl2 2.0, MgCl2 2; glucose 5; HEPES 10, buffered to pH 7.4 with NaOH. Chemicals were obtained from Calbiochem (Notts., UK), except 2-APB (2-aminoethoxydiphenylborane; Tocris Cookson Ltd., Bristol, UK), and D-erythro-SPC and L-threo-SPC (Avanti Lipids Inc., AL). Unless otherwise stated, diastereomeric SPC was used in experiments. Lysosphingolipids were dissolved in 2:1 chloroform:methanol for storage at -80°C. Before use, solvent was evaporated and the residue dissolved in PSS.
Calculations and statistical analysis
Changes in force and [Ca2+]i (F340/380) were normalized to the response to a 2 min exposure to KPSS4,17, and expressed as %KPSS. Values for EC50 (IC50), and maximum response were derived from fitting to the Hill equation (Sigmaplot, Systat Software Inc., CA). Results are shown as mean ± S.E.M., and compared using paired or unpaired Student’s t test, or ANOVA with a Holm-Sidak post hoc as appropriate (SigmaStat, Systat Software Inc., CA).
Results
As we previously reported for diastereomeric SPC4, D-erythro-SPC induced vasoconstriction of IPA only at concentrations >1 μmol/l, with a similar Hill coefficient (2.4 ± 0.9), maximum vasoconstriction (88 ± 6% KPSS), and EC50 (7.7 ± 0.2 μmol/l; n=3). L-threo-SPC only elicited constriction at 30 μmol/l (11 ± 4% KPSS; n=3).
Sub-contractile concentrations of SPC and depolarization-induced IPA vasoconstriction
IPA were challenged with sequential 5 min applications of PSS containing 21-25 mmol/l [K+] to give a rise in tension ∼15% of that induced by KPSS. SPC (1 μmol/l) was added to the bath, and the procedure repeated at 20 min intervals in the continued presence of SPC. Alone, 1 μmol/l SPC had no effect on tension and caused only a very small, transient (<3 min) elevation in [Ca2+]i (1.9 ± 0.7% KPSS, n=13). However, it subsequently caused a dramatic potentiation of the response to depolarization that gradually increased to a plateau at ∼40-60 min (Fig. 1), such that at 20 min force was increased to 294 ± 47% and [Ca2+]i (F340/380) to 198 ± 14% (n=7, p<0.01), and at 40 min to 529 ± 96% and 234 ± 26% respectively (n=6, p<0.001). In the absence of SPC the response to repeated depolarization was unchanged over the entire experimental period (95 ± 10% of control at 80 min, n=4). The SPC-induced potentiation of force was apparent at concentrations above 100 nmol/l, concentration dependent and stereospecific (Fig. 2A).
Figure 1.

A typical experimental trace of simultaneous force and [Ca2+]i (expressed as F340/380) recordings in a small IPA, illustrating the sequential challenge protocol with PSS containing 25 mmol/l [K+]. Note that 1 μmol/l SPC had no effect on force or [Ca2+]i on its own, but caused a substantial, slowly developing potentiation of the constriction elicited by 25 mmol/l [K+]. The response to depolarization with KPSS is shown for comparison.
Figure 2.

A. Concentration-dependence of SPC-induced potentiation at 40 min of constriction elicited by ∼25mmol/l [K+], showing stereospecificity. Diastereomeric SPC (open bars); D-erythro-SPC (black bars); L-threo-SPC (grey bars). Bars are mean ± S.E.M, n=4-5. * = p<0.05, ** = p<0.01 versus control (100%); ## = p<0.01 D-erythro-SPC versus L-threo-SPC. B. Cumulative concentration-response relationship for K+ (equimolar substitution for Na+) in IPA (filled circles; EC50: 36 ± 2 mmol/l; maximum: 109 ± 3% KPSS; n=9), and following preincubation with 1 μmol/l SPC (open circles; EC50: 31 ± 1 mmol/l, p<0.001; maximum: 120 ± 4% KPSS, p<0.001; n=9). Symbols are mean ± S.E.M.
We examined the effects of SPC on the concentration-response relationship to [K+] (Fig. 2B). Pre-incubation with 1 μmol/l SPC for 40 min shifted this relationship to the left (EC50: control: 36 ± 2 mmol/l [K+]; SPC: 31 ± 1 mmol/l [K+]; n=9, p<0.001). The maximum response was increased (control: 109 ± 3% KPSS; SPC: 120 ± 4% KPSS; p<0.001), indicating enhancement of voltage-gated Ca2+ entry and/or increased Ca2+ sensitivity rather than depolarization. 1μmol/l SPC had no effect on KV current amplitude in myocytes from IPA, with an unchanged zero current potential (control: -24 ± 1mV; SPC: -22 ± 1mV; n=5). Direct examination of voltage-activated Ca2+ current (ICav) proved impossible, as this current is small and highly variable in IPA and ran down over the period required (>30min).
Mechanism of SPC-induced potentiation of vasoconstriction
Using the sequential K+ challenge protocol shown in Fig. 1, we examined the effects of endothelial denudation, 2-APB (75 μmol/l), cyclopiazonic acid (CPA, SERCA inhibitor, 10 μmol/l), Y27632 (RhoK inhibitor, 3 μmol/l), U73122 (PLC inhibitor, 10μmol/l), and three PKC inhibitors: Ro31-8220 (broad spectrum, 3 μmol/l), Gö6976 (conventional and some novel isozymes, but not PKCδ, 3 μmol/l), and rottlerin (PKCδ, 1 μmol/l). IPA were pre-incubated with inhibitor for 20 min, then challenged four times at 20 min intervals with PSS containing ∼24 mmol/l [K+], with SPC (1 μmol/l) applied during the final two challenges. Tension developed during these last two challenges was compared with the paired controls, i.e. following preincubation with inhibitor but before addition of SPC.
SPC-induced potentiation of tension was unaffected by removal of the endothelium, 2-APB, CPA, Y27632 or Gö6976, but was substantially reduced or abolished by U73122, Ro31-8220, and rottlerin (Fig. 3). This suggests the mechanism of potentiation involves PLC and PKCδ, but not endothelium-derived mediators, store release or 2-APB-sensitive Ca2+ entry, RhoK-mediated Ca2+ sensitization, or other PKC isozymes.
Figure 3.

Potentiation of depolarization (∼25mmol/l [K+])-induced vasoconstriction of IPA by 1 μmol/l SPC measured at 40 min, and following removal of the endothelium or preincubation with inhibitors. Data were compared with their respective paired controls (100%), i.e. following preincubation with inhibitor but before addition of SPC. Bars are mean ± S.E.M., n = 4-31. * = p<0.05, ** = p<0.01 versus control; † = p<0.05, †† = p<0.01 versus SPC.
Quantification of changes in [Ca2+]i in the presence of PKC inhibitors was complicated by their fluorescence, but in 3 experiments we were able to show that Ro 31-8220 abolished both the SPC-induced potentiation of tension and the rise in [Ca2+]i (tension: 99 ± 15% control; [Ca2+]i (F340/380): 104 ± 20% control) (Fig. 4A & B). PdBu (100 nmol/l), an activator of conventional and novel PKC isozymes, also potentiated the depolarization-induced rise in [Ca2+]i (147 ± 11% control, n=3, p<0.05; Fig. 4C).
Figure 4.

Representative experimental traces of simultaneous force and intracellular [Ca2+] (expressed as F340/380) recordings, showing the control response superimposed on the response in the same IPA following 40 min incubation with 1 μmol/l SPC (panels A, B & D) or 100 nmol/l PdBu (panel C). In panels A, B & C constriction was elicited by PSS containing 23 mmol/l [K+], and in panel D by the TP agonist U-46619 (20 nmol/l) in the presence of diltiazem (10 μmol/l).
1μmol/l SPC had no detectable effect on tension (n=4) in α-toxin permeabilized IPA constricted to ∼30% of maximum response with a pCa of 6.7, obviating any contribution from PKC-mediated Ca2+ sensitization.
Effect of 1 μmol/l SPC on agonist-induced IPA vasoconstriction
SPC (1 μmol/l) caused a significant left-ward shift of the PGF2α concentration-response relationship (Fig. 5A). PGF2α-induced vasoconstriction of IPA is mediated via TP receptors and associated with Ca2+ entry via a 2-APB-sensitive voltage-independent pathway and diltiazem-sensitive L-type channels17. As expected, diltiazem (10 μmol/l) partially suppressed the response to PGF2α, but only reduced and did not abolish the SPC-induced left-ward shift of the PGF2α concentration-response relationship (Fig. 5B). Consistent with this, and using the sequential challenge protocol shown in Fig. 1 but with PGF2α, SPC (1 μmol/l) in the presence of diltiazem potentiated 3μmol/l PGF2α-induced constriction to 341 ± 125% (n=3) at 40 min, and U436619 (20 nmol/l)-induced tension and elevation in [Ca2+]i (tension: 563 ± 159%; [Ca2+] (F340/380): 212 ± 57%; n=3; Fig. 4D), indicating that SPC also potentiates voltage-independent Ca2+ entry.
Figure 5.

Cumulative concentration-response relationships for PGF2α in IPA. The left hand panel shows the effect of preincubation with 1 μmol/l SPC (Control, filled circles; SPC, open circles); the right hand panel shows the response in the presence of 10 μmol/l diltiazem. In both cases control and experimental curves were significantly different (-diltiazem: p<0.01, n=4; +diltiazem: p<0.05, n=5; 2 way ANOVA); symbols are mean ± S.E.M., * = p<0.01 (Holm-Sidak post hoc).
Repeated challenges with PGF2α showed less consistency than for K+, so in order to characterise in more detail the SPC-mediated potentiation of the response to PGF2α, we applied SPC during a stable established PGF2α-induced constriction equivalent to ∼25% KPSS, with potentiation measured at 10 min (Fig. 6A). Due to the slowly developing nature of the SPC-induced potentiation, its magnitude using this shortened protocol was smaller than for the repeated challenge protocol, but was still highly significant (140 ± 6.2% control, n=12, p<0.001). For comparison, SPC-induced potentiation of K+-induced constriction was 165 ± 26% (n=3) at 10 min using this protocol.
Figure 6.

A: Representative trace illustrating the second, shorter protocol where SPC is added on top of an established stable vasoconstriction induced by PGF2α. SPC was added once developed force reached a plateau, and caused a further increase in force; in the absence of SPC tension was stable for at least 10 min, illustrated by the dashed line. B: Increase in PGF2α-induced force caused by 1 μmol/l SPC measured after 10 min using the protocol shown in A., and following preincubation with inhibitors; 100% represents tension in the presence of inhibitors but before addition of SPC. Bars are mean ± S.E.M., n = 4-12. * = p<0.05, ** = p<0.01 versus control; † = p<0.05, †† = p<0.01 versus SPC.
Using the above protocol we investigated the effects of diltiazem (10 μmol/l), 2-APB (75 μmol/l), Y27632 (3 μmol/l), Ro31-8220 (3 μmol/l), rottlerin (1 μmol/l) and CPA (10 μmol/l) on the potentiation induced by 1 mol/l SPC. IPA were preincubated with inhibitors for 20 min; PGF2α was then applied to induce a constriction of ∼25% KPSS (3-10 μmol/l PGF2α). Fig. 6B shows the effects of these inhibitors on SPC-induced potentiation. Consistent with the data shown in Fig. 3 for depolarization-induced constriction, the potentiation was unaffected by Y27632 but substantially reduced or abolished by Ro31-8220 and rottlerin, and consistent with figs. 4D and 5B diltiazem significantly reduced the potentiation by ∼50%, whereas 2-APB abolished it. Following constriction with U-46619 (20 nmol/l) in the presence of diltiazem, SPC (1 μmol/l) caused a further rise in both tension (148 ± 10% control, n=6, p<0.01) and [Ca2+]i (184 ± 16%, p<0.01), again confirming that SPC potentiates voltage-independent Ca2+ entry.
Effect of 1 μmol/l SPC on PKCδ translocation in cultured PASMCs
The data obtained in the presence of the three PKC inhibitors strongly suggested a role for PKCδ; we therefore examined the effect of 1 μmol/l SPC on PKCδ sub-cellular localization in PASMCs cultured from IPA. In untreated cells PKCδ staining was strongest within the nucleus, with diffuse staining throughout the cytoplasm (Fig. 7). In SPC treated cells staining in the nucleus was reduced, with translocation of PKCδ to the perinuclear region, cytoskeleton and possibly membrane.
Figure 7.
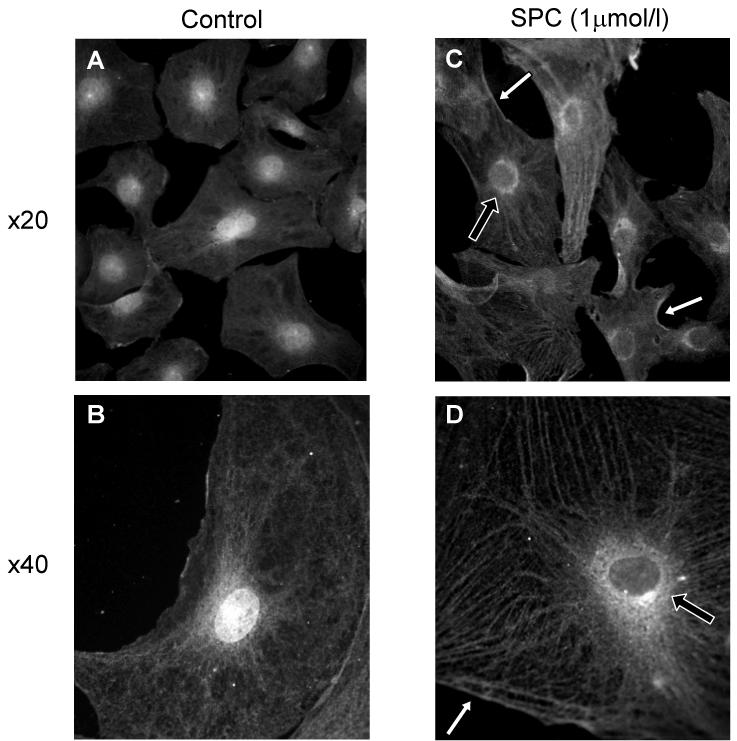
SPC induced translocation of PKCδ immunoreactivity in PASMCs from the nucleus to the perinuclear regions (e.g. black arrows) and cytoskeleton, with some evidence for translocation to the membrane (e.g. white arrows). A & B: control; C & D: treatment with 1 μmol/l SPC for 10 min. B & D are at higher magnification (oil immersion objective) to show detail. Figures typical for 6-10 coverslips from each of 6 cell lines derived from different animals. >90% cells demonstrated translocation.
Discussion
Our key finding is that concentrations of SPC insufficient to cause vasoconstriction or elevation of [Ca2+]i in IPA can nevertheless substantially enhance vasoconstriction elicited by other means. Such concentrations (300-1000 nmol/l) are >10 fold smaller than the EC50 reported for SPC-induced vasoconstriction per se, or indeed most other actions of SPC (∼12 μmol/l)1,2,4-6, and are consequently much closer to those reported for plasma11. Notably, SPC was stereospecific for the effects elicited by both high and low concentrations, implying that both are receptor mediated (Fig. 2A).
The signalling pathways underlying potentiation of IPA vasoreactivity by ≤1 μmol/l SPC differ from those underlying vasoconstriction to higher concentrations. We have shown that the latter is mediated by increased RhoK-mediated Ca2+ sensitivity and activation of 2-APB-sensitive, voltage-independent Ca2+ entry pathway, but is independent of Ca2+ release from stores and PLC4. In contrast, the potentiation of depolarization-induced constriction by 1 μmol/l SPC was unaffected by 2-APB or inhibition of RhoK, but was strongly suppressed by inhibition of PLC and PKC (Fig. 3).
The related sphingolipid S1P inhibits KV channels and depolarises cerebral arteries by via a PKC-dependent mechanism18. However, although the [K+]/force relationship was shifted to the left by 1 μmol/l SPC, the maximum response was also enhanced (Fig. 2B), and there was no effect on KV currents in IPA smooth muscle cells. This suggests that neither KV channel inhibition nor depolarization can account for the potentiation of vasoreactivity. Both RhoK and PKC modulate Ca2+ sensitivity in pulmonary artery19, but as inhibition of RhoK was without effect and 1 μmol/l SPC did not alter Ca2+ sensitivity of α-toxin permeabilized IPA, this suggests the potentiation is mediated entirely via the observed enhancement of the elevation of [Ca2+]i (Figs. 1 and 4).
PKC enhances Ca2+ entry via L-type channels in smooth muscle20 by increasing channel availability and prolonging open time21,22, and there is some evidence for differential effects of conventional and novel PKC isozymes20. Ro31-8220 has equivalent potencies against conventional (α, βI, βII, γ), novel (δ, ε, η, θ, μ) and atypical (ι, λ) PKC isozymes (IC50 <1 μmol/l), whereas Gö6976 has selectivity for conventional isozymes (IC50 <1 μmol/l), but is relatively ineffective against PKCδ (IC50 ≥30 μmol/l). Rottlerin has a 10-fold greater potency for PKCδ (IC50 ∼1-6 μmol/l) than for PKC α, β and γ, and ∼50-fold for ε, θ and ζ19,23. Ro31-8220 and rottlerin abolished the SPC-induced potentiation of vasoreactivity whereas Gö6976 had no effect, suggesting that PKCδ is a critical component of the signal transduction pathway. Accordingly, 1 μmol/l SPC induced PKCδ translocation in PASMCs from the nucleus to the perinuclear region, cytoskeleton and possibly membrane (Fig. 7); activation of PKCδ has previously been associated with a similar sub-cellular translocation in vascular smooth muscle and cardiomyocytes24,25.
The potentiation of PGF2α-induced vasoconstriction was inhibited by diltiazem by only 50%, and unlike that for depolarization-induced vasoconstriction was abolished by 2-APB (Fig. 6B). This implies that SPC also potentiates voltage-independent Ca2+ entry, and this was found to be the case (see Fig. 4D). This is also PKCδ-dependent as the potentiation was abolished by Ro31-8220 and rottlerin (Fig. 6B). The complete block by 2-APB and partial block by diltiazem is consistent with a model whereby TP receptor agonists activate NSCC, resulting in both Ca2+ and Na+ entry, with the latter causing depolarization and additional Ca2+ entry via L-type channels (see Fig. 5 and 17).
Consistent with our results, other studies have shown that SPC activates PKCδ and PLC in pancreatic islet endothelial cells26, and PKCδ has been reported to mediate the hypoxia-induced enhancement of Ca2+ entry via L-type channels in carotid body glomus cells27. Moreover, SPC has also been reported to potentiate 2-APB-sensitive Ca2+ entry in thyroid FRO cells via a PKC- and PLC-dependent mechanism, independent of intracellular Ca2+ release or any increase in inositol phosphates28. SPC-induced vasoconstriction in systemic arteries is in part mediated by PLC/IP3-induced Ca2+ release1,10, though this is not the case for IPA4; this reflects our previous finding that activation of PLC/IP3-mediated Ca2+ release does not couple to vasoconstriction in IPA17. Considering the above, we speculate that the potentiation of IPA vasoreactivity elicited by SPC is mediated via a GPCR linked to PLC, with subsequent activation of PKCδ by DAG and PKCδ-dependent phosphorylation of L-type channels and a 2-APB-sensitive NSCC.
In summary, we report the novel and potentially important finding that sub-contractile concentrations (≤1 μmol/l) of SPC cause significant potentiation of IPA vasoreactivity via a PKCδ-mediated enhancement of Ca2+ entry via both L-type channels and a voltage-independent, 2-APB-sensitive NSCC. This pathway involves different mechanisms from those previously shown to underlie vasoconstriction induced by higher concentrations of SPC in IPA4 and other vascular beds see1,2,10, possibly implying the existence of more than one GPCR for SPC.
Perspectives
Our results have considerable implications for the pathophysiological role of SPC. Whilst lysosphingolipids may act in a paracrine or autocrine fashion, with localised concentrations considerably higher than in plasma especially at sites of thrombus formation, atherosclerosis and inflammation1,2,6,7, the discrepancy between plasma SPC concentrations11 and those required for most of its reported effects is still in the order of 100 fold. The phenomenon we describe here however was apparent at 300 nmol/l, and could conceivably be active in healthy tissues, and certainly if SPC increases in cardiovascular disease as suggested2,9. This could be of particular importance in the lung, as pulmonary vascular disease is commonly associated with inflammation and microemboli, factors that would be expected to increase SPC. However the phenomenon does not seem to be limited to IPA, as in preliminary experiments we have found that it also occurs in a range of small systemic arteries. As SPC potentiates the response to TP agonists and is released from activated platelets11, an intriguing speculation is that this phenomenon could also contribute to the initial vasoconstrictor response associated with haemostasis and platelet-derived thromboxane A2.
Acknowledgments
Source of funding
The Wellcome Trust (grants 078075, 068160) and British Heart Foundation (FS/06/003/20402).
Footnotes
Conflict of Interest/Disclosure Statement None
References
- 1.Meyer zu Heringdorf D, Himmel HM, Jakobs KH. Sphingosylphosphorylcholine-biological functions and mechanisms of action. Biochim Biophys Acta. 2002;1582:178–189. doi: 10.1016/s1388-1981(02)00154-3. [DOI] [PubMed] [Google Scholar]
- 2.Alewijnse AE, Peters SL, Michel MC. Cardiovascular effects of sphingosine-1-phosphate and other sphingomyelin metabolites. British journal of pharmacology. 2004;143:666–684. doi: 10.1038/sj.bjp.0705934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Duong CQ, Bared SM, Abu-Khader A, Buechler C, Schmitz A, Schmitz G. Expression of the lysophospholipid receptor family and investigation of lysophospholipid-mediated responses in human macrophages. Biochim Biophys Acta. 2004;1682:112–119. doi: 10.1016/j.bbalip.2004.03.002. [DOI] [PubMed] [Google Scholar]
- 4.Thomas GD, Snetkov VA, Patel R, Leach RM, Aaronson PI, Ward JP. Sphingosylphosphorylcholine-induced vasoconstriction of pulmonary artery: activation of non-store-operated Ca2+ entry. Cardiovascular research. 2005;68:56–64. doi: 10.1016/j.cardiores.2005.05.013. [DOI] [PubMed] [Google Scholar]
- 5.Bischoff A, Czyborra P, Fetscher C, Meyer Zu Heringdorf D, Jakobs KH, Michel MC. Sphingosine-1-phosphate and sphingosylphosphorylcholine constrict renal and mesenteric microvessels in vitro. British journal of pharmacology. 2000;130:1871–1877. doi: 10.1038/sj.bjp.0703515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakao F, Kobayashi S, Mogami K, Mizukami Y, Shirao S, Miwa S, Todoroki-Ikeda N, Ito M, Matsuzaki M. Involvement of Src family protein tyrosine kinases in Ca(2+) sensitization of coronary artery contraction mediated by a sphingosylphosphorylcholine-Rho-kinase pathway. Circulation research. 2002;91:953–960. doi: 10.1161/01.res.0000042702.04920.bf. [DOI] [PubMed] [Google Scholar]
- 7.Shirao S, Kashiwagi S, Sato M, Miwa S, Nakao F, Kurokawa T, Todoroki-Ikeda N, Mogami K, Mizukami Y, Kuriyama S, Haze K, Suzuki M, Kobayashi S. Sphingosylphosphorylcholine is a novel messenger for Rho-kinase-mediated Ca2+ sensitization in the bovine cerebral artery: unimportant role for protein kinase C. Circulation research. 2002;91:112–119. doi: 10.1161/01.res.0000026057.13161.42. [DOI] [PubMed] [Google Scholar]
- 8.Morikage N, Kishi H, Sato M, Guo F, Shirao S, Yano T, Soma M, Hamano K, Esato K, Kobayashi S. Cholesterol primes vascular smooth muscle to induce Ca2 sensitization mediated by a sphingosylphosphorylcholine-Rho-kinase pathway: possible role for membrane raft. Circulation research. 2006;99:299–306. doi: 10.1161/01.RES.0000235877.33682.e9. [DOI] [PubMed] [Google Scholar]
- 9.Hemmings DG. Signal transduction underlying the vascular effects of sphingosine 1-phosphate and sphingosylphosphorylcholine. Naunyn-Schmiedeberg’s archives of pharmacology. 2006;373:18–29. doi: 10.1007/s00210-006-0046-5. [DOI] [PubMed] [Google Scholar]
- 10.Meyer Zu Heringdorf D. Lysophospholipid receptor-dependent and -independent calcium signaling. J Cell Biochem. 2004;92:937–948. doi: 10.1002/jcb.20107. [DOI] [PubMed] [Google Scholar]
- 11.Liliom K, Sun G, Bunemann M, Virag T, Nusser N, Baker DL, Wang DA, Fabian MJ, Brandts B, Bender K, Eickel A, Malik KU, Miller DD, Desiderio DM, Tigyi G, Pott L. Sphingosylphosphocholine is a naturally occurring lipid mediator in blood plasma: a possible role in regulating cardiac function via sphingolipid receptors. The Biochemical journal. 2001;355:189–197. doi: 10.1042/0264-6021:3550189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murata Y, Ogata J, Higaki Y, Kawashima M, Yada Y, Higuchi K, Tsuchiya T, Kawainami S, Imokawa G. Abnormal expression of sphingomyelin acylase in atopic dermatitis: an etiologic factor for ceramide deficiency? The Journal of investigative dermatology. 1996;106:1242–1249. doi: 10.1111/1523-1747.ep12348937. [DOI] [PubMed] [Google Scholar]
- 13.Meyer Zu Heringdorf D, Jakobs KH. Lysophospholipid receptors: Signalling, pharmacology and regulation by lysophospholipid metabolism. Biochim Biophys Acta. 2007;1768:923–940. doi: 10.1016/j.bbamem.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 14.Seuwen K, Ludwig MG, Wolf RM. Receptors for protons or lipid messengers or both? Journal of receptor and signal transduction research. 2006;26:599–610. doi: 10.1080/10799890600932220. [DOI] [PubMed] [Google Scholar]
- 15.MacLean MR, Morecroft I. Increased contractile response to 5-hydroxytryptamine1-receptor stimulation in pulmonary arteries from chronic hypoxic rats: role of pharmacological synergy. British journal of pharmacology. 2001;134:614–620. doi: 10.1038/sj.bjp.0704273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Snetkov VA, Aaronson PI, Ward JP, Knock GA, Robertson TP. Capacitative calcium entry as a pulmonary specific vasoconstrictor mechanism in small muscular arteries of the rat. British journal of pharmacology. 2003;140:97–106. doi: 10.1038/sj.bjp.0705408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Snetkov VA, Knock GA, Baxter L, Thomas GD, Ward JP, Aaronson PI. Mechanisms of the prostaglandin F2{alpha}-induced rise in [Ca2+]i in rat intrapulmonary arteries. J Physiol. 2006;571:147–163. doi: 10.1113/jphysiol.2005.101394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Coussin F, Scott RH, Nixon GF. Sphingosine 1-phosphate induces CREB activation in rat cerebral artery via a protein kinase C-mediated inhibition of voltage-gated K+ channels. Biochem.Pharmacol. 2003;66:1861–1870. doi: 10.1016/s0006-2952(03)00546-x. [DOI] [PubMed] [Google Scholar]
- 19.Ward JP, Knock GA, Snetkov VA, Aaronson PI. Protein kinases in vascular smooth muscle tone--role in the pulmonary vasculature and hypoxic pulmonary vasoconstriction. Pharmacol.Ther. 2004;104:207–231. doi: 10.1016/j.pharmthera.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 20.Keef KD, Hume JR, Zhong J. Regulation of cardiac and smooth muscle Ca(2+) channels (Ca(V)1.2a,b) by protein kinases. American journal of physiology. 2001;281:C1743–1756. doi: 10.1152/ajpcell.2001.281.6.C1743. [DOI] [PubMed] [Google Scholar]
- 21.Obejero-Paz CA, Auslender M, Scarpa A. PKC activity modulates availability and long openings of L-type Ca2+ channels in A7r5 cells. The American journal of physiology. 1998;275:C535–543. doi: 10.1152/ajpcell.1998.275.2.C535. [DOI] [PubMed] [Google Scholar]
- 22.Schuhmann K, Groschner K. Protein kinase-C mediates dual modulation of L-type Ca2+ channels in human vascular smooth muscle. FEBS Lett. 1994;341:208–212. doi: 10.1016/0014-5793(94)80458-3. [DOI] [PubMed] [Google Scholar]
- 23.Gschwendt M, Muller HJ, Kielbassa K, Zang R, Kittstein W, Rincke G, Marks F. Rottlerin, a novel protein kinase inhibitor. Biochemical and biophysical research communications. 1994;199:93–98. doi: 10.1006/bbrc.1994.1199. [DOI] [PubMed] [Google Scholar]
- 24.Disatnik MH, Buraggi G, Mochly-Rosen D. Localization of protein kinase C isozymes in cardiac myocytes. Experimental cell research. 1994;210:287–297. doi: 10.1006/excr.1994.1041. [DOI] [PubMed] [Google Scholar]
- 25.Li C, Wernig F, Leitges M, Hu Y, Xu Q. Mechanical stress-activated PKCdelta regulates smooth muscle cell migration. Faseb J. 2003;17:2106–2108. doi: 10.1096/fj.03-0150fje. [DOI] [PubMed] [Google Scholar]
- 26.Jeon ES, Kang YJ, Song HY, Im DS, Kim HS, Ryu SH, Kim YK, Kim JH. Sphingosylphosphorylcholine generates reactive oxygen species through calcium-, protein kinase Cdelta- and phospholipase D-dependent pathways. Cellular signalling. 2005;17:777–787. doi: 10.1016/j.cellsig.2004.11.004. [DOI] [PubMed] [Google Scholar]
- 27.Summers BA, Overholt JL, Prabhakar NR. Augmentation of L-type calcium current by hypoxia in rabbit carotid body glomus cells: evidence for a PKC-sensitive pathway. Journal of neurophysiology. 2000;84:1636–1644. doi: 10.1152/jn.2000.84.3.1636. [DOI] [PubMed] [Google Scholar]
- 28.Afrasiabi E, Blom T, Ekokoski E, Tuominen RK, Tornquist K. Sphingosylphosphorylcholine enhances calcium entry in thyroid FRO cells by a mechanism dependent on protein kinase C. Cellular signalling. 2006;18:1671–1678. doi: 10.1016/j.cellsig.2006.01.005. [DOI] [PubMed] [Google Scholar]