

Abstract
Human immunodeficiency virus-1 (HIV-1) encephalitis is characterized by brain infiltration of virus-infected monocytes and macrophages. Cellular products and viral proteins secreted by infected cells likely play an important role in blood–brain barrier (BBB) impairment and the development of HIV-1-associated dementia (HAD). We previously demonstrated that HIV-1 envelope glycoprotein gp120 induces toxicity and alters expression of tight junction proteins in human brain microvascular endothelial cells (HBMECs). Here, we delineate the mechanisms of gp120-induced BBB dysfunction. Human brain microvascular endothelial cells expressed HIV-1 co-receptors (CCR5 and CXCR4). Exposure of HBMECs to gp120 derived from macrophage (CCR5) or lymphocyte (CXCR4)-tropic viruses decreased BBB tightness, increased permeability, and enhanced monocyte migration across in vitro BBB models. Blood–brain barrier integrity was restored after gp120 removal. CCR5 antibodies and inhibitors of myosin light chain kinase or protein kinase C (PKC) blocked gp120-enhanced monocyte migration and permeability of BBB in vitro. Exposure of HBMECs to gp120 induced release of intracellular calcium ([Ca2 +]i) that was prevented by CCR5 antibody and partially blocked by CXCR4 antagonist. Human immunodeficiency virus-1 gp120 activated three PKC isoforms in HBMECs [PKC-α/βII, PKC(pan)-βII and PKC-ζ/λ]. Furthermore, specific PKC inhibitors (acting at the ATP-binding and calcium release site) blocked gp120-induced PKC activation and prevented increase in BBB permeability, supporting the biologic significance of these results. Thus, gp120 can cause dysfunction of BBB via PKC pathways and receptor mediated [Ca2 +]i release leading to cytoskeletal alterations and increased monocyte migration.
Keywords: brain endothelial cells, chemokine receptors, HIV-1gp120, PKC
Introduction
Blood–brain barrier (BBB) compromise is common in HIV-1-infected individuals and is implicated in the pathogenesis of HIV-1-associated dementia (HAD) (Burger et al, 1997; Nottet et al, 1996; Dallasta et al, 1999; Avison et al, 2004). Under steady-state conditions, the BBB functions as an interface between the blood and the brain parenchyma, strictly regulating influx of ions, molecules and leukocytes into the central nervous system (CNS). During progressive HIV-1 infection this function breaks down. Human immunodeficiency virus-1 invades the brain in the early stage of infection and causes various clinical and pathologic abnormalities, ranging from subclinical and mild cognitive motor impairment to overt dementia. Despite the beneficial effects of antiretroviral therapy (ART), 76% to 83% of AIDS autopsy cases show brain involvement (Masliah et al, 2000; Jellinger et al, 2000; Vago et al, 2002). Even with ART, approximately 10% of HIV-1-infected individuals develop HAD, and its prevalence is rising due to the longer lifespan of HIV-1-infected patients (Dore et al, 1999; McArthur, 2004; Kaul et al, 2005).
The BBB plays a central role in the neuropathogenesis of HIV-1 infection. This is substantiated by a number of laboratory, animal model and human studies demonstrating BBB breakdown as a consequence of progressive viral infection and immune compromise (Burger et al, 1997; Dallasta et al, 1999; Persidsky et al, 2000; Kanmogne et al, 2002, 2005a). Breech of the BBB allows progeny virus and activated, HIV-1-infected monocytes or monocyte-derived macrophages (MDM) to infiltrate the brain and spread virus to resident glia including perivascular macrophages, microglia and astrocytes. Brain microvascular endothelial cells are a major component of the BBB and are connected by tight junctions (TJ) limiting paracellular flux and restricting permeability (Hawkins and Davis, 2005).
Members of the seven-transmembrane domain family of chemokine receptors can function as co-receptors for HIV-1 (Deng et al, 1996; Dragic et al, 1996). In addition to the CD4 receptor, the CCR5 co-receptor is utilized by macrophage-tropic strains, while CXCR4 is used by most T-lymphocyte-tropic strains for infection. Other members of the chemokine receptor family, CCR3 and CCR2b, have also been shown to act as co-receptors for a limited number of primary HIV-1 isolates (Connor et al, 1997; Bjorndal et al, 1997). Human immunodeficiency virus-1 gp120 binds directly to CCR5 and CXCR4, and these chemokine receptors can mediate CD4-independent entry and infection of target cells (Hill et al, 1997; Hesselgesser et al, 1997). Human immunodeficiency virus-1 gp120 is neurotoxic (for recent reviews see Kaul et al, 2001, 2005; Nath, 2002; Albright et al, 2003; Persidsky and Gendelman, 2003; Peruzzi et al, 2005); it causes cytotoxicity in human endothelial cells derived from the umbilical vein (Huang et al, 1999), lung and brain (Kanmogne et al, 2001, 2002, 2005a, Kanmogne et al, b). Cytopathic effects in virus-infected cells or virus shedding result in the release of HIV-1 gp120 protein and peptide products into the serum (ranging from 12 to 92 ng/ml) (Oh et al, 1992) and brain tissue (Jones et al, 2000).
We previously showed that HIV-1 gp120 elicits endothelial toxicity (Kanmogne et al, 2002) leading to disruption and downregulation of TJ proteins in human brain microvascular endothelial cells (HBMECs) (Kanmogne et al, 2005a). The main goal of the current study was to delineate the mechanisms of gp120-induced endothelial cell dysfunction. We demonstrated that gp120 from CCR5- and CXCR4-tropic viruses diminished integrity, increased permeability and monocyte migration in laboratory BBB models through CCR5/CXCR4 receptor-mediated protein kinase C (PKC) activation and [Ca2 +]i release.
Materials and methods
Reagents and Antibodies
Phospho-PKC antibodies and horseradish peroxidase-conjugated secondary antibodies were purchased from Cell Signaling Technology (Beverly, MA, USA). Protein kinase C antibodies and Transwell inserts (Cyclopore® polyethylene terephthalate membrane) were obtained from BD Bioscience (San Jose, CA, USA). Protein kinase C inhibitors were purchased from Calbiochem (San Diego, CA, USA). Gradient gels and nitrocellulose membranes were purchased from Bio-Rad (Hercules, CA, USA). CCR5, CXCR4 and CCR3 antibodies were either purchased from R&D Systems (Minneapolis, MN, USA) or obtained from the AIDS Research and Reference Reagent Program, NIAID, NIH. All other reagents were purchased from Sigma (St Louis, MO, USA).
Cell Culture and Human Immunodeficiency Virus-1 gp120
Primary HBMECs were isolated from the temporal cortex of brain tissue obtained during surgical removal of epileptogenic foci in adult patients using procedures described previously (Miller and Borchardt, 1992) and were supplied by Dr M Witte (University of Arizona). Routine evaluation for von Willebrand factor (vWF), Ulex europeus lectin, and CD31 demonstrated that cells were > 99% pure. Freshly isolated cells were cultured on collagen- and fibronectin-coated six-well or 96-well plates, 75 cm2 flasks, and 24-well tissue culture inserts. Cells at passage 1 to 4 were used in this study. All compounds and inhibitors were used in nontoxic concentrations as determined by the 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide cell viability assay (24 to 48 h exposure). Human immunodeficiency virus-1 gp120 proteins purified from the macrophage-tropic HIV-1 strain Bal (gp120Bal or R5 gp120) was obtained from the AIDS Research and Reference Reagent Program, NIAID, NIH. The HIV-1 gp120 protein from lymphocyte-tropic HIV-1 strain MN (gp120MN or X4 gp120) was purchased from ImmunoDiagnostics, Inc. (Woburn, MA, USA). Both HIV-1 gp120 proteins were tested and were free of endotoxin contamination.
Transendothelial Electrical Resistance
Electrical resistance across the endothelial cell monolayer was measured by an EVOM voltmeter (World Precision Instrument, Sarasota, FL, USA) as previously described (Ma et al, 1999; Haorah et al, 2005a). The BBB model was constructed by seeding 2 × 104 HBMECs in the upper chamber of 24-well tissue culture inserts (0.4 μm pore size). Cells were cultured for 7 to 10 days before use. Confluent cells were treated with 0.1, 1, 10 and 100 ng/ml gp120 and transendothelial electrical resistance (TEER) recorded at 1, 2, 4 and 24 h. After the 24-h measurement, media containing gp120 was replaced by fresh culture media, and TEER was recorded 1 and 2 h later. Electrical resistance of the blank inserts (150 Ωcm2) with media alone was subtracted from the TEER readings obtained from inserts with confluent endothelial cell monolayers. The resulting TEER values (300 to 570 Ωcm2) represented the resistance (‘tightness’) of the endothelial cell monolayers. Ethanol-treated endothelial cells (50 mmol/L) served as positive control (Haorah et al, 2005a, b).
Monocyte Migration in blood–Brain Barrier Models
Monocytes were obtained one day prior to migration from HIV-1, HIV-2 and hepatitis B seronegative donor leukopaks, and were separated by countercurrent centrifugal elutriation as previously described (Gendelman et al, 1988). These cells were identified as > 98% pure monocytes by Wright staining and CD68 immunostaining (at 1:50 dilution, Dako, Carpentaria, CA, USA). For endothelial cell culture, 2 × 104 HBMECs were seeded in the upper chamber of 24-well tissue culture inserts (3 μm pore size) and cultured for 7 to 10 days. From the TEER experiments, we determined that 4 h was the optimum time for gp120-induced alterations in BBB tightness. Therefore, for migration experiments, cells were treated for 4 h with 0.1, 1, 10 and 100 ng/ml gp120. Untreated cells and cells treated for 4 h with heat-inactivated gp120 served as controls. Transendothelial electrical resistance values were recorded before (0-h control) and after the 4-h gp120 treatment to determine gp120-induced changes in BBB tightness. Monocyte migration experiments were performed immediately after the 4-h TEER measurement; 105 monocytes were placed in the upper chamber of each insert and allowed to migrate for 2 h (37°C, 5% CO2). The migrated monocytes in the lower chamber were stained with a macrophage marker (CD68) and counted as described (Haorah et al, 2005b). Each treatment condition was performed in triplicate. To mimic the chemotactic gradient seen in HIV-1 encephalitis (Persidsky et al, 1999), migration was carried out in response to the chemokine, monocyte chemotactic protein-1 (MCP-1, 10 to 30 ng/ml) applied to the lower chamber.
Immunofluorescent Microscopy
Confluent HBMECs monolayers on glass coverslips were washed with phosphate-buffered saline (PBS) and fixed in methanol/acetone (1:1) for 20 min at −20°C. Cells were permeabilized with 0.1% triton X-100 and blocked for nonspecific binding with 3% bovine serum albumin in PBS (10 min at 4°C). Cells were immunostained with antibodies to the HIV-1 co-receptors CXCR4, CCR5, CCR3 or vWF (endothelial cell marker). Incubation with primary antibody was done for 1 h at room temperature followed by staining (1 h in the dark at room temperature) with respective secondary antibodies coupled with Alexa-488 and Alexa-568 (1:50, Molecular Probes, Eugene, OR, USA). Stained cell monolayers were mounted in Immunomount (Molecular Probes) and examined using a fluorescent microscope (E800 Nikon, Melville, NY, USA) connected to a color MagnaFire digital camera (Optronics, Goleta, CA, USA).
Calcium Imaging
Human brain microvascular endothelial cells were grown to 50% confluence on 25-mm glass coverslips and loaded with the Ca2 + sensitive dye, Fura-2 AM (7-μmol/L) in HEPES buffer (10 mmol/L HEPES, 140 mmol/L NaCl, 5 mmol/L KCl, 5 mmol/L Dextrose, 1 mmol/L MgCl2, 2 mmol/L CaCl2, pH 7.4) for 1 h at room temperature. After loading, cells were washed three times to remove extracellular Fura-2 and incubated 15 mins in HEPES buffer for further de-esterification of Fura-2 AM. Cells were then mounted on the stage of a Nikon TE2000 inverted microscope and imaged through a × 40 oil objective. Cells were pulse stimulated four times at a frequency of 0.1 Hz. At 30 s after the last stimulation, cells were exposed to gp120 protein (0.1, 1, 10 or 100 ng/ml). The rate and amplitude of Ca2 + release were recorded using a dual-excitation fluorescence photomultiplier system (Image Master Fluorescence Microscope, Photon Technology International, Birmingham, NJ, USA). For blocking experiments, Fura-2-loaded cells were incubated with CCR5 antibodies (10 μg/ml) or the CXCR4 antagonist AMD3100 (1 μg/ml) for 10 min before gp120 application.
Protein Extraction and Western Blot Analysis
Human immunodeficiency virus-1 gp120-treated and control HBMECs were lysed in a mammalian total protein extraction reagent (M-PER®, Pierce) containing 1 × Protease Inhibitor Cocktail (Sigma). Lysates were kept on ice for 20 mins and centrifuged 30 mins at 9000 g to remove insoluble materials. Total protein concentration in the resulting supernatant was measured using the bicinchoninic acid assay (Pierce, Rockford, IL, USA). Twenty five micrograms of protein were fractionated in a 4% to 15% gradient gel and electrophoretically transferred onto nitrocelluose membranes. Membranes were blocked for 1 h with SuperBlock® T-20 (Pierce), blotted 2 h or overnight with the appropriate primary antibodies, 1 h with the secondary antibody, washed and visualized using SuperSignal® West Pico Substrate (Pierce). For Western blot with phospho-PKC antibodies, membranes were stripped using Restore™ Western Blot Stripping Buffer (Pierce) and re-blotted with antibodies to PKC isoforms, then stripped again and re-blotted with β-actin antibody. Results were expressed as ratio of relative intensity of the target protein to that of the internal standard, β-actin, or to that of the corresponding (total) PKC isoform.
Permeability Assay
Human brain microvascular endothelial cells were seeded on 24-well tissue culture inserts (0.4 μm pore size), grown to confluence and exposed to gp120 for 4 to 12 h. To detect changes in monolayer permeability, 1 ml assay medium was added to each lower well and 200 μl medium containing 100-μmol/L fluorescein isothiocyanate (FITC)-dextran (MW 40000) was added in the upper well of each insert. These volumes have been reported to ensure similar hydrostatic pressure on both sides of the endothelial cell monolayer (Quan and Godfrey, 1998). After 1, 2 and 3 h incubation (37°C, 5% CO2), 100 μl medium was removed from each lower well and fluorescence measured using a microplate reader (Spectramax, Gemini, Molecular Devices) with excitation at 488 nm; emission at 525 nm. All samples were tested in triplicate. Background fluorescence of the assay medium was subtracted from all values. Permeability was assayed with FITC-dextrans of different sizes (MW 10,000 and 70,000); however, optimal results were obtained with FITC-dextran of MW 40,000. A standard curve derived from serial dilution of the 100-μmol/L FITC-dextran stock was used to estimate the permeability coefficient.
Statistical Analysis
Statistical analysis was performed by one- or two-way analysis of variance (ANOVA) and the General Linear Model (GLM) procedure using the SAS program, followed by Dunnet Multiple Comparison Test. Both statistical procedures test differences among several means for significance without increasing Type I error rate (Klockars and Gilbert, 1986). Threshold significance level was 0.05.
Results
Human Brain Microvascular Endothelial Cells Express CCR5 and CXCR4
The ability of HIV-1 to infect target cells or cause alteration of their function depends on the cellular membrane expression of CD4, CCR5 or CXCR4. We evaluated the expression of three major HIV-1 co-receptors (CCR5, CXCR4 and CCR3 chemokine receptors) on HBMECs. Both CCR5 and CXCR4 were detected on HBMECs, but these cells lacked CCR3 expression (Figure 1). Strong staining for vWF confirmed the endothelial nature of these cells.
Figure 1.
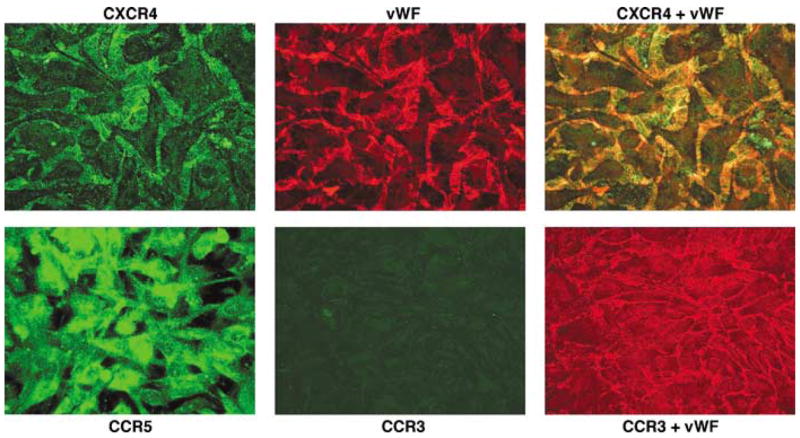
Primary HBMECs expressed CCR5 and CXCR4 chemokine receptors, but they lack CCR3. Confluent HBMECs were fixed and stained with antibodies to CXCR4, CCR5, CXCR3 and von Willebrand factor (vWF). Original magnification: × 400.
Human Immunodeficiency Virus-1 gp120 Decreases Blood–Brain Barrier Tightness
To assess the integrity of the BBB model following gp120 exposure, TEER was measured in controls and gp120-treated HBMEC monolayers. At 2 h exposure of HBMECs to gp120 (0.1 ng/ml) from R5-or X4-tropic viruses significantly decreased BBB tightness (Figure 2, P < 0.01). At 2 h exposure to R5 gp120 at 0.1, 1, 10 and 100 ng/ml diminished TEER by 14, 13.8, 14 and 15.47%, respectively (Figure 2A, P < 0.001). Similarly, a 2-h application of X4 gp120 at concentrations of 0.1, 1, 10 and 100 ng/ml decreased TEER by 8 to 10.56% (Figure 2B). Longer gp120 exposure (4 to 24 h) further decreased TEER. A 4-h application of R5 gp120 concentrations of 0.1 to 100 ng/ml decreased TEER by 11.75% to 19.7% (Figure 2A). At 4-h exposure of cells to X4 gp120 (0.1 to 100 ng/ml) decreased TEER by 8.4% to 14% (Figure 2B). Gp120 withdrawal for 2 h restored BBB integrity to almost basal (control) levels (Figure 2A and 2B, P < 0.01). Similar results were obtained even after 24 h gp120 exposure (data not shown).
Figure 2.
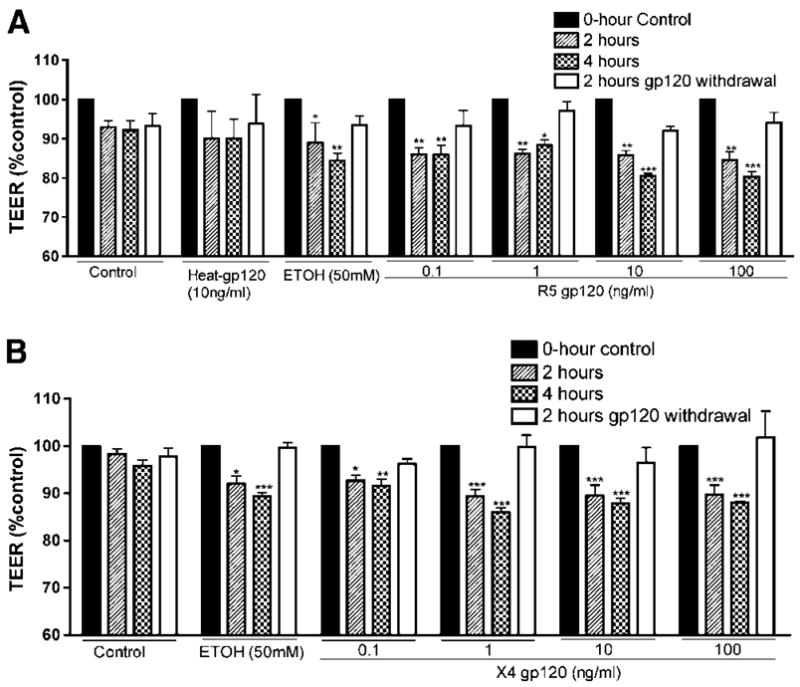
Blood–brain barrier integrity is diminished by gp120. R5 gp120 (from the M-tropic HIV-1 strain Bal) (A) and X4 gp120 (from the T-tropic HIV-1 strain MN) (B) decrease blood–brain barrier tightness, and gp120 withdrawal restores it. Negative controls consist of untreated cells (control) and cells treated with heat-inactivated gp120 (heat-gp120). Endothelial cells exposed to 50 mmol/L ethanol (EtOH) served as positive controls. Each experimental condition was performed in triplicate and for each time-point, three independent TEER (Ωcm2) measurements were recorded. The results are expressed as mean percent of controls at initial time point±s.e.m. (n = 3) (*P < 0.05, **P < 0.01, ***P < 0.001). This figure is representative of four independent experiments.
Human Immunodeficiency Virus-1 gp120 Enhances Monocyte Migration Across the Blood–Brain Barrier Models
Next, we examined whether gp120-induced alterations in BBB integrity would be associated with changes in monocyte traffic across the barrier. Following 4 h gp120 treatment, 105 monocytes were placed on top of the BBB constructs, and 2 h later monocytes that migrated across the monolayer were stained and counted. Exposure of HBMECs to R5 gp120 and X4 gp120 resulted in a dose-dependent increase in monocyte migration (Figure 3). R5 gp120 at 0.1, 1, 10 and 100 ng/ml increased the number of migrated monocytes 4.3-, 2.6-, 6.4- and 11-fold, respectively (Figure 3A, P < 0.01). Similarly, 0.1, 1, 10 and 100 ng/ml of X4 gp120 increased the number of migrated monocytes 6.3-, 10-, 10.8- and 16.6-fold, respectively (Figure 3C). Heat-inactivated gp120 had no effect on cell migration. Enhanced migration paralleled diminished tightness of HBMEC monolayers (Figure 3B and 3D). Since both BBB integrity and leukocyte migration across BBB are regulated by myosin light chain kinase (MLCK) activation in HBMECs (Haorah et al, 2005a), we assessed the effects of MLCK inhibitor, ML-7 on gp120 mediated BBB dysfunction. ML-7 did not change the TEER but it significantly diminished monocyte migration (two- to three-fold) and permeability to FITC-dextran induced by R5 and X4 gp120 in BBB models (Figure 3 and 4). Migration in response to MCP-1 (10 ng/ml) or MCP-1 (10 ng/ml) plus gp120 (100 ng/ml) served as positive controls. Monocyte chemotactic protein-1 increased the number of migrated monocytes by 5- to 12-fold (Figure 3A and 3C), and there was no additive effect of combination of MCP-1 and gp120.
Figure 3.
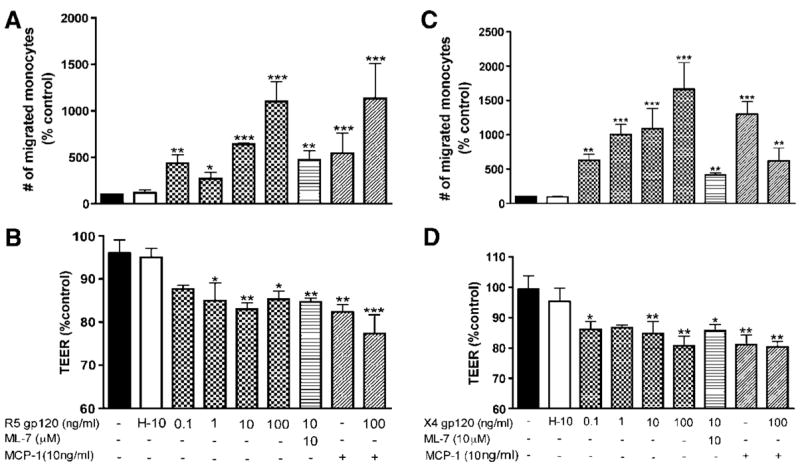
Enhanced monocyte migration across endothelial monolayers induced by R5 gp120 (A) and X4 gp120 (C) paralleled decrease in BBB tightness (B, D). Human brain microvascular endothelial cells were exposed to gp120 (0.1 to 100 ng/ml) and gp120 plus ML-7 (10-μmol/L, MLCK inhibitor) for 4 h followed by monocytes application. After 2 h, migrated monocytes were stained with CD68 antibody and counted. Each experimental condition was performed in triplicate. Inhibition of MLCK partially blocked gp120-enhanced monocyte migration. Untreated cells and HBMECs treated with 10 ng/ml heat-inactivated gp120 (H-10) served as negative controls. Transendothelial electrical resistance and migration in response to MCP-1 (10 ng/ml) or MCP-1 plus gp120 served as positive controls. Results were expressed as mean percent control±s.e.m. (n = 3) (*P < 0.05, **P < 0.004, ***P < 0.0008). These results are representative of three independent experiments.
Figure 4.
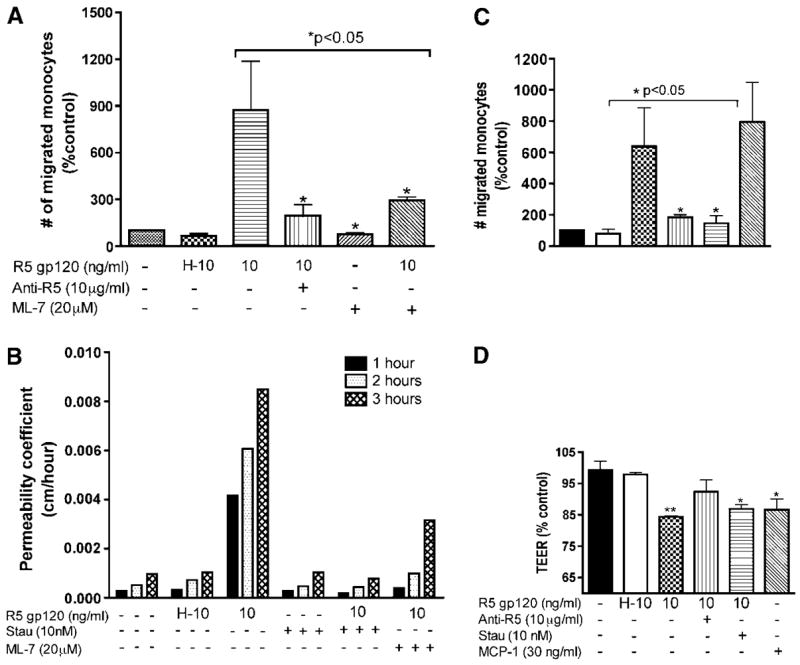
Blocking of chemokine receptors or inhibition of MLCK or PKC prevented gp120-induced effects on BBB in vitro. (A) Increased monocyte migration induced by R5 gp120 was blocked by CCR5 antibody and MLCK inhibitor (*P < 0.05 as compared to gp120-treated cells). (B) Exposure to R5 gp120 (12 h) increased permeability of HBMEC monolayers to FITC-dextran; the gp120 induced increase in permeability was blocked by staurosporine (10 nmol/L, PKC inhibitor) and ML-7. (C and D) R5 gp120 decreases TEER and increases monocyte migration. CCR5 antibody prevented the decrease in TEER (D) and increase in monocyte migration (C) induced by gp120. Staurosporine also blocked the increase in monocyte migration induced by gp120. Untreated cells and HBMECs treated with 10 ng/ml heat-inactivated gp120 (H-10) served as negative controls. Transendothelial electrical resistance and migration in response to MCP-1 (30 ng/ml) served as positive controls. For (A), (C) and (D), results were expressed as mean percent control±s.e.m. (n = 3) (*P < 0.05, **P < 0.01). These results are representative of three independent experiments.
Direct evidence of chemokine receptor involvement in BBB impairment by gp120 was obtained using CCR5 blocking antibodies. Pretreatment with CCR5 antibody prevented the decrease in TEER induced by gp120 (Figure 4D) and resulted in a 4.7-to 5.6-fold decrease of gp120-enhanced cell migration (Figure 4C and 4A, respectively). Increased BBB permeability induced by R5 gp120 was diminished by inhibitors of MLCK and PKC (staurosporine). R5 gp120 increased BBB permeability to FITC-dextran; the increased permeability was partially blocked by ML-7 or completely prevented by staurosporine (Figure 4B). Similarly, pretreatment with ML-7 and staurosporine resulted respectively in a 3.3- and 5.7-fold decrease of gp120-enhanced monocyte migration (Figure 4A and 4C).
Human Immunodeficiency Virus-1 gp120 Induces [Ca2 +]i Release in Human Brain Microvascular Endothelial Cells
It has been shown that CXCR4 and CCR5 ligands induced Ca2 + flux in cells bearing CXCR4 and CCR5 chemokine receptors (Alfano et al, 1999; Le et al, 2001; Zhang et al, 2003). To explore calcium involvement in gp120-induced BBB dysfunction, we performed Ca2 + imaging analysis of HBMECs after exposure to R5 or X4 gp120. Both gp120 proteins induced [Ca2 +]i release in HBMECs. R5 gp120 at concentrations of 1, 10 and 100 ng/ml increased Ca2 + flux by 45%, 210% and 100%, respectively (Figure 5A), while 0.1 ng/ml had no effect. Based on these results, 10 ng/ml gp120 was used in subsequent experiments. [Ca2 +]i release induced by R5 gp120 was completely blocked by anti-CCR5 antibodies (Figure 5B). X4 gp120 (10 ng/ml) increased [Ca2 +]i flux by 40% (Figure 5C) that was partially blocked by the CXCR4 antagonist, AMD3100 (Figure 5D).
Figure 5.
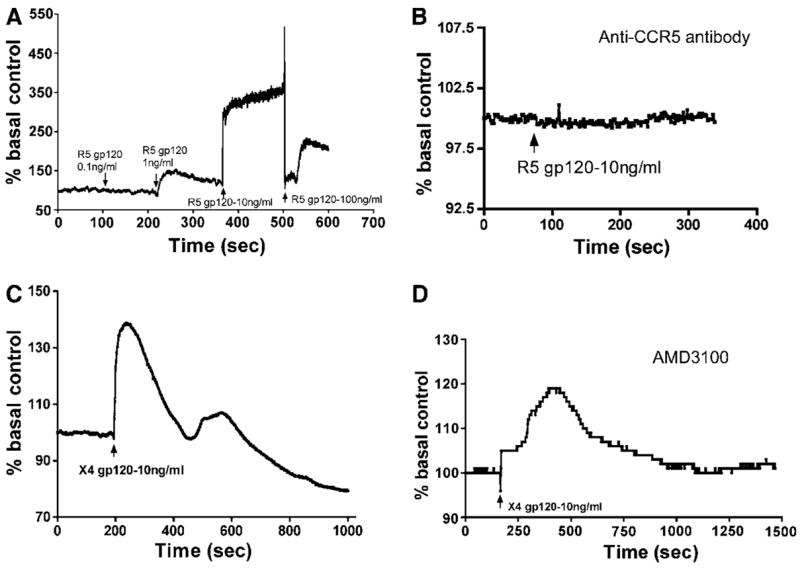
Gp120 induced [Ca2 +]i flux in brain endothelium. R5 gp120 (A) or X4 gp120 (C and D) induced intracellular calcium release. Gp120-induced Ca2 + release was blocked by CCR5 antibody (B) and diminished by the CXCR4 antagonist AMD3100 (D). This figure is representative of five independent experiments using primary HBMECs from five different donors.
Human Immunodeficiency Virus-1 gp120 Cause Phosphorylation of PKC-ζ/λ, PKC-α/βII and PKC(pan)-βII) in Human Brain Microvascular Endothelial Cells
Both [Ca2 +]i release and PKC activation mediate TJ assembly and barrier permeability (Anderson et al, 1988; Citi, 1992; Stevenson and Begg, 1994; Ballabh et al, 2004). We tested the activity of 10 known PKC isoforms (PKC-α, PKC-β, PKC-γ, PKC-δ, PKC-ε, PKC-η, PKC-θ, PKC-ι, PKC-λ and PKD/PKC-μ) in gp120-treated HBMECs. Three PKC isoforms (PKC-α, PKC-βII and PKC-λ) were consistently expressed in these cells (Figure 6). X4 gp120 effects on PKC activity were detected by Western blot using antibodies to eight phosphorylated PKC isoforms including pPKC(pan) (βII Ser660), pPKC-α/βII (Thr638/641), pPKC-δ (Thr505), pPKC-δ/θ (Ser643/676), pPKC-ζ/λ (Thr410/403), pPKC-θ (Thr538), pPKD/PKC-μ (Ser744/748) and pPKD/PKC-μ (Ser916). X4 gp120 (100 ng/ml) induced phosphorylation of PKC-ζ/λ, PKC-α/βII and PKC(pan)-βII. Exposure of HBMECs to gp120 for 5 to 120 min increased levels of pPKC-ζ/λ 19.5- to 46.6-fold (P < 0.01, Figure 6A). Similarly, the levels of pPKC-α/βII were increased 4.4- to 77.6-fold following 5 to 120 min exposure of HBMECs to gp120 (P < 0.01, Figure 6B). The most dramatic increase following gp120 application was seen in pPKC(pan)- βII (95 to 818 times, P < 0.001, Figure 6C). Phospho-PKD/PKC-μ (Ser744/748) was detected in primary HBMECs in very small quantities (data not shown).
Figure 6.
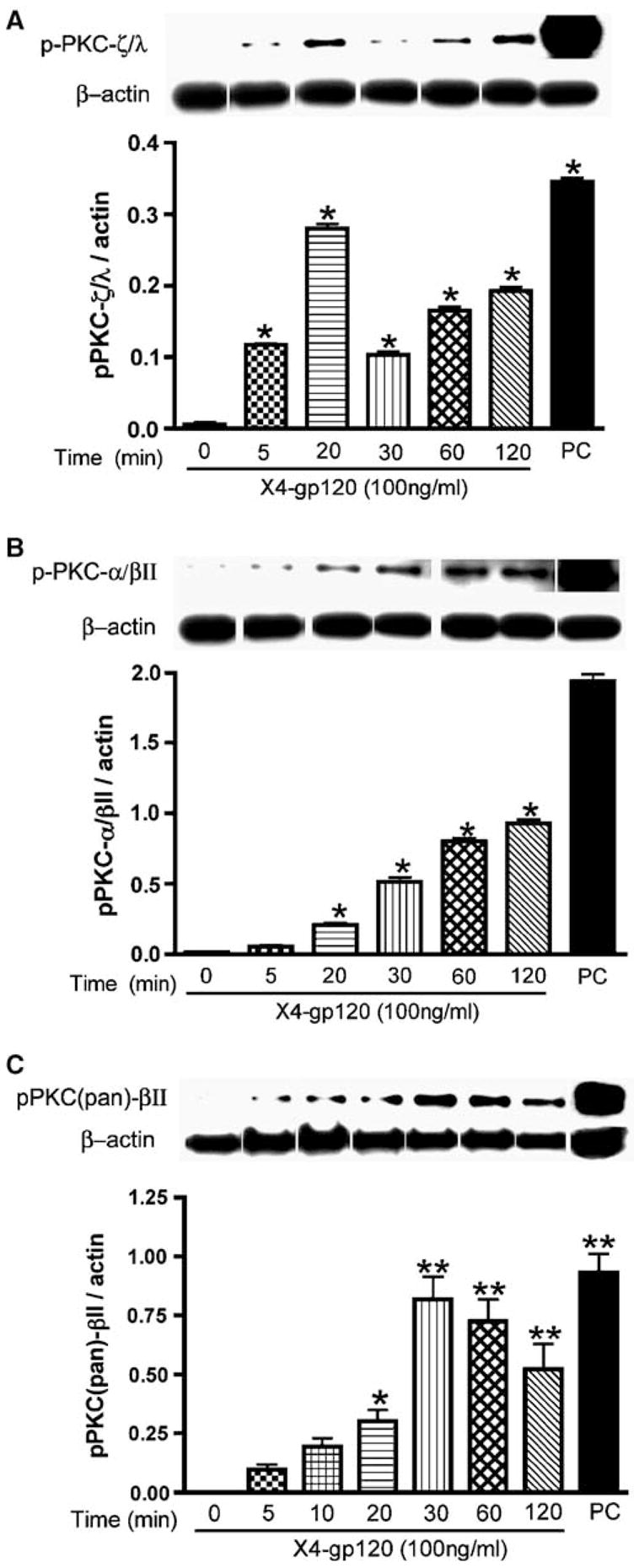
Exposure of HBMECs to X4 gp120 induced PKC phosphorylation. PhosphoPKC-ζ/λ (Thr638/641) (A), PKC-α/βII (Thr638/641) (B) and PKC(pan)-βII (Ser660) (C) were detected with respective antibodies. Following Western blot analysis with phospho-PKC each blot was stripped and probed with β-actin antibody. Relative intensity was expressed as ratio of arbitrary volume integrated densitometric units of the target protein to that of β-actin. Results are expressed as mean values ±s.e.m. (n = 3) (*P < 0.01, **P < 0.001, compared to 0-h control). Positive controls (PC) were protein extracts from rat cerebrum.
Gp120-Mediated Protein Kinase C Activation was Blocked by Protein Kinase C Inhibitors
To further elucidate the signaling pathway and second messengers involved in gp120-induced BBB dysfunction, six inhibitors were used to block PKC activation induced by X4 gp120: U-73122 (phospholipase C (PLC) inhibitor), R59949 (Diacylglycerol kinase (DAGK) inhibitor), Go6850 (competitive inhibitor for the ATP-binding site of PKC), Go6979 (selective inhibitor of calcium-dependent PKC-α and PKC-β isozymes), calphostin-C (compete at the PKC binding site of diacylglycerol and phorbol esters), and staurosporine (broad spectrum inhibitor of PKC). Staurosporine, Go6850 and Go6979 blocked gp120-induced PKC phosphorylation. An 84% reduction of PKC-α/βII phosphorylation was achieved by Go6850 (P < 0.01). Go6850 also reduced total PKC-α by 51.5% (Figure 7A). Go6979 and calphostin-C partially blocked gp120-induced phosphorylation of PKC-α/βII (by 38% and 43%, respectively, P < 0.01). Phospholipase C and DAGK inhibitors did not block gp120-induced phosphorylation of PKC-α/βII (Figure 7A). Go6979 reduced gp120-induced phosphorylation of PKC-ζ/λby 92% (P < 0.01). Go6850 and calphostin-C slightly reduced gp120-induced phosphorylation of PKC-ζ/λ; PLC and DAGK inhibitors had no effect (Figure 7B). Staurosporine also blocked gp120-induced phosphorylation of PKC-ζ/λ by 62.8% (P < 0.01, Figure 7C).
Figure 7.
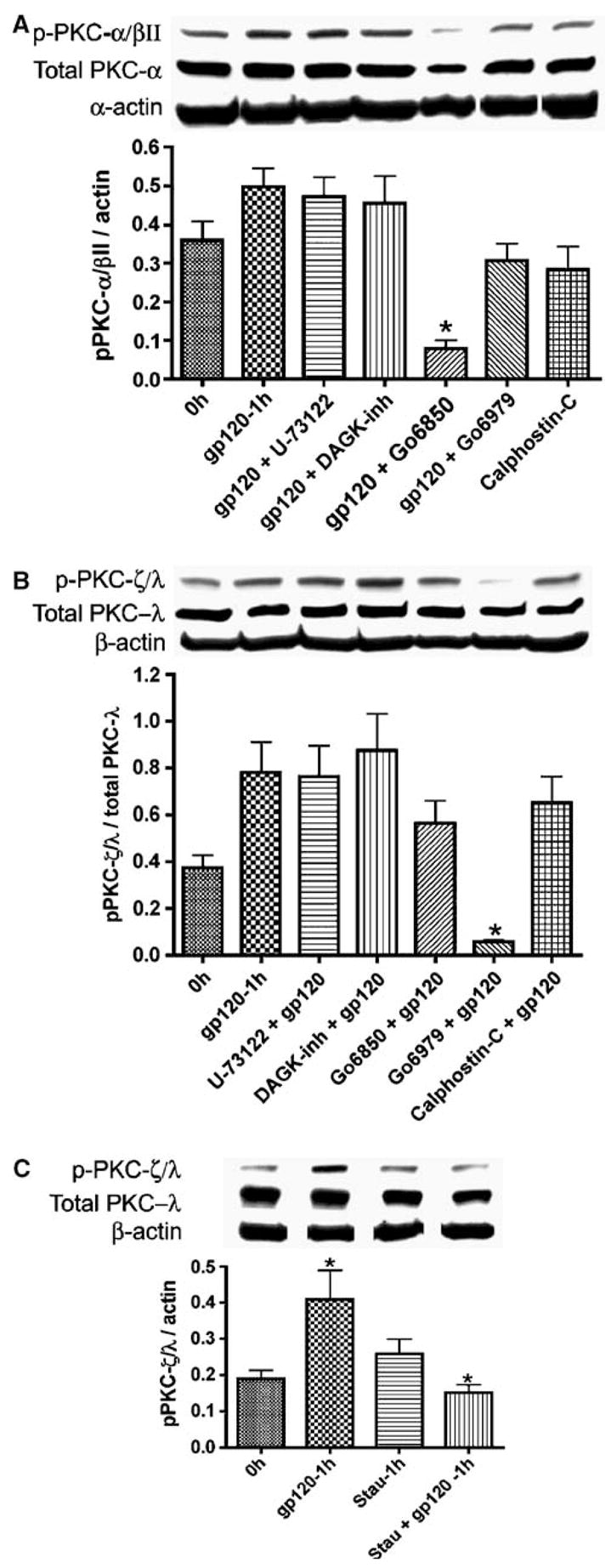
PKC inhibitors blocked PKC activation induced by X4 gp120. HBMECs were exposed 1 h to gp120 (100 ng/ml) or gp120 and the following PKC inhibitors: staurosporine, calphostin-C, Go6979, Go6850, diacylglycerol kinase (DAGK) inhibitor, or U-73122 (phospholipase C inhibitor) and Western blot analysis performed as described in Materials and methods. Following Western blot analysis with phospho-PKC-ζ/λ (B, C) and phospho-PKC-α/βII (A), the blots were stripped and probed with antibodies to PKC-λand PKC-α, respectively, then stripped again and probed with β-actin antibody. Relative intensity is expressed as ratio of arbitrary volume integrated densitometric units of the target protein to that β-actin or to that of the corresponding (total) PKC isoform. Results were expressed as mean values±s.e.m. (n = 3) (*P < 0.05, **P < 0.01, compared to gp120-treated cells).
Discussion
Blood–brain barrier compromise is commonly observed in HIV-1-infected patients, and it correlates with the intensity of cognitive impairment (Gendelman et al, 1998; Avison et al, 2004). Disease manifestations are associated with the numbers of immune activated macrophages and microglia, cells that affect neurodegeneration and BBB integrity (Navia and Price 2005). Blood–brain barrier breakdown may enhance entrance of toxins, free virus, infected and/or activated monocytes and lymphocytes into the CNS, spreading HIV-1 infection to brain macrophages and microglia (Burger et al, 1997; Dallasta et al, 1999; Kanmogne et al, 2002, 2005a). Dysfunction of brain endothelium caused by HIV-1 plays an important role in AIDS neuropathogenesis (Persidsky and Gendelman, 2003; Toborek et al, 2005), but the exact mechanisms allowing HIV-1 to invade the brain are poorly understood. We previously showed that HIV-1 gp120 was cytotoxic to brain endothelium (Kanmogne et al, 2002), causing downregulation and disruption of TJ proteins in HBMECs (Kanmogne et al, 2005a). The current study defines the mechanisms of gp120-induced BBB impairment. Human brain microvascular endothelial cells express the CCR5 and CXCR4 chemokine receptors (Figure 1). In addition to CD4, CCR5/CXCR4 are used as co-receptors by HIV-1 to infect target cells and can mediate CD4-independent infection (Hill et al, 1997; Hesselgesser et al, 1997). Human brain microvascular endothelial cell do not express CD4 receptor (Kanmogne, unpublished data). It is likely that CCR5 and CXCR4 receptors mediate gp120-induced effects on human brain endothelium, since R5 and X4 gp120 decreased tightness (8% to 19%) and enhanced monocyte migration (2.6- to 16-fold) across the BBB models. Significantly, the gp120-induced TEER decrease was reversible, and gp120 withdrawal restored BBB tightness (even after 24 h exposure). These changes were mediated via chemokine receptors, since CCR5 blocking antibodies and CXCR4 antagonists blocked gp120-mediated BBB dysfunction.
Our previous findings showed that HIV-1 gp120 downregulates and disrupts TJ proteins in HBMECs (Kanmogne et al, 2005a). Diminished BBB integrity could be mediated via alteration in TJ and adherent junctions (AJ), leading to enhanced permeability and monocyte migration across HBMECs. Disassembly and opening of junctional complexes are often associated with contraction of cytoskeletal actin and myosin filaments and can be mediated by activation of kinases such as MLCK, PKC and Rho kinase (for recent reviews see van Hinsbergh and van Nieuw Amerongen, 2002; Feldman et al, 2005). Myosin light chain kinase and Rho kinase regulate the phosphorylation of MLC and TJ proteins (van Hinsbergh and van Nieuw Amerongen, 2002; Feldman et al, 2005; Haorah et al, 2005a; Persidsky et al, 2006). A rise in [Ca2 +]i activates key signaling pathways, which mediate cytoskeletal reorganization (through MLC-dependent contraction) and disassembly of AJ (Tiruppathi et al, 2002). The Ca2 +-dependent PKC isoform, PKC-α, plays a critical role in initiating endothelial cell contraction and AJ disassembly (Tiruppathi et al, 2002). [Ca2 +]i and PKC regulate TJ assembly and increases in BBB permeability, and PKC-ζ was shown to co-localize with ZO-1 at TJ (Stuart and Nigam, 1995). Molecules modulating BBB permeability often act through alteration of [Ca2 +]i (reviewed by Ballabh et al, 2004). [Ca2 +]i changes play an important role in restoring TJ assembly and BBB structure, increasing TEER and promoting ZO-1 migration from intracellular sites to the plasma membrane (Anderson et al, 1988; Stevenson and Begg, 1994). We demonstrate here that R5 and X4 gp120 (at 1 or 10 ng/ml, levels observed in serum of HIV-1 infected patients) induce [Ca2 +]i flux in HBMECs. Extrapolation of these results in vivo suggests that free circulating or viral-associated gp120 via HBMECs binding will deplete endothelial [Ca2 +]i stores, causing cytoskeletal/TJ/AJ disorganization, enhanced BBB permeability, and increased leukocyte trafficking.
It has been demonstrated that modulation of BBB permeability induced by alteration of [Ca2 +]i is mediated through heterotrimeric G proteins and PKC signaling (Stevenson and Begg, 1994; Ballabh et al, 2004). The involvement of Ca2 + and protein kinases in the modulation of cell–cell junction assembly and function has also been demonstrated in other cell types. Incubation of MDCK epithelial cells with low Ca2 +induces disruption of cell–cell contact, change in cell shape, redistribution of TJ proteins from the periphery to the cytoplasm, and decreased transepithelial resistance (Citi, 1992). Protein kinase C isoforms play a vital role in cell signaling (Sandoval et al, 2001; Verin et al, 2000) and as such mediate microvascular hyperpermeability (reviewed by Mehta and Malik, 2006). Activation and inhibition of PKC can result in increases and decreases, respectively, in permeability (Alexander et al, 1998; Chen et al, 1998; Huang and Yuan, 1997). Our data are in agreement with these studies and underscore the role of Ca2 +and PKC in gp120-induced BBB dysfunction. Our data demonstrate that gp120 stimulated [Ca2 +]i release and activated three PKC isoforms (PKC-ζ/λ, PKC-α/β II and PKC(pan)-βII), decreased TEER, increased permeability, and enhanced monocyte migration across the endothelial cell monolayers. The broad spectrum PKC inhibitor, staurosporine blocked gp120-induced increased permeability and PKC activation. Significantly, two PKC inhibitors (Go6979 and Go6850) acting at the Ca2 +and ATP binding site completely blocked gp120-mediated activation of PKC-ζ/λand PKC-α/βII, while PLC and DAGK inhibitors had no effect. This suggests that gp120 induces its toxic effect on HBMECs primarily at the Ca2 +and ATP binding sites of PKC, not through the PLC or DAG pathways. Other studies using epithelial cells showed that direct activation of PKC with phorbol esters caused cell–cell dissociation, redistribution of cytoskeletal proteins (Schliwa et al, 1984) and a drop in transepithelial resistance (Ojakian, 1981).
Studies in epithelial cells also showed that increase in TJ permeability coincided with serine/threonine phosphorylation of PKC and tyrosine dephosphorylation of TJ proteins (reviewed by Feldman et al, 2005). Our data demonstrated that gp120 induced phosphorylation of PKC-α/βII (at threonine residues), PKC-ζ/λ (at threonine residues), and PKC(pan)-βII (at serine residues), and correlated with increased permeability. Interestingly, the HIV-1 Tat protein also changed functional properties of the BBB (including expression of TJ proteins), however, PKC did not mediate these alterations in brain endothelial cells (Andras et al, 2005; Pu et al, 2005). Myosin light chain kinase in endothelial cells has a unique 922-amino-acid NH2-terminal domain containing consensus sites that could be phosphorylated by multiple protein kinases, including cAMP-dependent protein kinase A (Garcia et al, 1997; Verin et al, 1998), PKC (Bogatcheva et al, 2003; Satpathy et al, 2004), and Ca2 +-dependent protein kinase II (Verin et al, 1998). We demonstrated that MLCK inhibitor also blocked the increase in HBMEC permeability induced by gp120, which suggest that gp120-induced BBB dysfunction involves cytoskeletal alteration.
Human brain microvascular endothelial cells express functional CCR5 and CXCR4 receptors since anti-CCR5 antibodies inhibited [Ca2 +]i release induced by R5 gp120 and the CXCR4 antagonist, AMD3100 partially blocked [Ca2 +]i release caused by X4 gp120. Blocking CCR5 antibodies inhibited the increase in monocyte migration induced by R5 gp120. Taken together, these data suggest that gp120 binds to CCR5 and CXCR4 receptors on HBMECs and induces BBB dysfunction. Other studies have shown the involvement of chemokine receptors and PKC in gp120-mediated effects on human smooth muscle cells and endothelial cells of other origin. HIV-1 gp120 induced apoptosis in human umbilical vein endothelial cells through CCR5 and CXCR4 receptors, and PKC agonists and antagonists, as well as CXCR4 antibodies blocked gp120-induced apoptosis (Huang and Bond, 2000). Furthermore, gp120 also increased PKC activity in endothelial cells, and CXCR4 antibodies blocked PKC activation (Huang and Bond, 2000). A synthetic peptide corresponding to amino acid 414 to 434 in the V4-C4 region of gp120 induced Ca2 + release and chemotaxis in human monocytes and neutrophils; activation of monocytes by this peptide resulted in downregulation of cell surface expression of CCR5 and CXCR4 in a PKC-dependent manner (Deng et al, 1999). In summary, our data suggest that HIV-1 gp120 can activate PKC and cause [Ca2 +]i release in HBMECs, leading to MLCK activation, disruption of TJ, decreased BBB tightness and increased monocyte migration across the barrier. Our study suggests that circulating and viral-associated gp120 can alter the functional properties of the BBB, promoting infiltration of HIV-1-infected cells in the brain. Thus, gp120 likely plays an important role in BBB impairment and the development of HIV-1 associated dementia.
Acknowledgments
We thank Ms Robin Taylor and Adrian Koesters for excellent editorial support and Dr Mark Thomas for critical reading of the manuscript. This work was supported in part by NIH Grants 1K01 MH068214 (GDK), R37NS36136 (HEG) and RO1 MH65151 (YP).
References
- Albright AV, Soldan SS, Gonzalez-Scarano F. Pathogenesis of human immunodeficiency virus-induced neurological disease. J NeuroVirol. 2003;9:222–7. doi: 10.1080/13550280390194073. [DOI] [PubMed] [Google Scholar]
- Alexander JS, Jackson SA, Chaney E, Kevil CG, Haselton FR. The role of cadherin endocytosis in endothelial barrier regulation: involvement of protein kinase C and actin-cadherin interactions. Inflammation. 1998;22:419–33. doi: 10.1023/a:1022325017013. [DOI] [PubMed] [Google Scholar]
- Alfano M, Schmidtmayerova H, Amella CA, Pushkarsky T, Bukrinsky M. The B-oligomer of pertussis toxin deactivates CC chemokine receptor 5 and blocks entry of M-tropic HIV-1 strains. J Exp Med. 1999;190:597–605. doi: 10.1084/jem.190.5.597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Stevenson BR, Jesaitis LA, Goodenough DA, Mooseker MS. Characterization of ZO-1, a protein component of the tight junction from mouse liver and Madin–Darby canine kidney cells. J Cell Biol. 1988;106:1141–9. doi: 10.1083/jcb.106.4.1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andras IE, Pu H, Tian J, Deli MA, Nath A, Hennig B, et al. Signaling mechanisms of HIV-1 Tat-induced alterations of claudin-5 expression in brain endothelial cells. J Cereb Blood Flow Metab. 2005;25:1159–70. doi: 10.1038/sj.jcbfm.9600115. [DOI] [PubMed] [Google Scholar]
- Avison MJ, Nath A, Greene-Avison R, Schmitt FA, Greenberg RN, Berger JR. Neuroimaging correlates of HIV-associated BBB compromise. J Neuroimmunol. 2004;157:140–6. doi: 10.1016/j.jneuroim.2004.08.025. [DOI] [PubMed] [Google Scholar]
- Ballabh P, Braun A, Nedergaard M. The blood–brain barrier: an overview structure, regulation and clinical implications. Neurobiol Dis. 2004;16:1–13. doi: 10.1016/j.nbd.2003.12.016. [DOI] [PubMed] [Google Scholar]
- Bjorndal A, Deng H, Jansson M, Fiore JR, Colognesi C, Karlsson A, et al. Coreceptor usage of primary HIV-1 isolates varies according to biological phenptype. J Virol. 1997;71:7478–87. doi: 10.1128/jvi.71.10.7478-7487.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogatcheva NV, Verin AD, Wang P, Birukova AA, Birukov KG, Mirzopoyazova T, et al. Phorbol esters increase MLC phosphorylation and actin remodeling in bovine lung endothelium without increased contraction. Am J Physiol Lung Cell Mol Physiol. 2003;285:L415–26. doi: 10.1152/ajplung.00364.2001. [DOI] [PubMed] [Google Scholar]
- Burger DM, Boucher CA, Meenhorst PL, Kraayeveld CL, Portegies P, Mulder JW, et al. HIV-1 RNA levels in the cerebrospinal fluid may increase owing to damage to the blood–brain barrier. Antiviral Ther. 1997;2:113–7. [PubMed] [Google Scholar]
- Chen CC, Wang JK, Lin SB. Antisense oligonucleotides targeting protein kinase C-alpha, -beta, or -delta but not eta inhibit lipopolysaccharide-induced nitric oxide synthase expression in RAW 264.7 macrophages: involvement of a nuclear factor B-dependent mechanism. J Immunol. 1998;161:6206–14. [PubMed] [Google Scholar]
- Citi S. Protein kinase inhibitors prevent junction dissociation induced by low extracellular calcium in MDCK epithelial cells. J Cell Biol. 1992;117:169–78. doi: 10.1083/jcb.117.1.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor RI, Sheridan KE, Ceradini D, Choe S, Landau NR. Change in coreceptor use correlates with disease progression in HIV-1-infected individuals. J Exp Med. 1997;185:621–8. doi: 10.1084/jem.185.4.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dallasta LM, Pisarov LA, Esplen JE, Werley JV, Moses AV, Nelson JA, et al. blood–brain barrier tight junction disruption in HIV encephalitis. Am J Pathol. 1999;155:1915–27. doi: 10.1016/S0002-9440(10)65511-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng H, Liu R, Ellmeier W, Choe S, Unutmaz D, Burkhart M, et al. Identification of a major co-receptor for primary isolates of HIV-1. Nature. 1996;381:661–6. doi: 10.1038/381661a0. [DOI] [PubMed] [Google Scholar]
- Deng X, Ueda H, Su SB, Gong W, Dunlop NM, Goa JL, et al. A synthetic peptide derived from HIV type 1 gp120 downregulates the expression and function of chemokine receptors CCR5 and CXCR4 in monocytes by activating the 7-transmembrane G-protein-coupled receptor FPRL1/LXA4R. Blood. 1999;94:1165–73. [PubMed] [Google Scholar]
- Dore GJ, Correll PK, Li Y, Kaldor JM, Cooper DA, Brew BJ. Changes to AIDS dementia complex in the era of HAART. AIDS. 1999;13:1249–53. doi: 10.1097/00002030-199907090-00015. [DOI] [PubMed] [Google Scholar]
- Dragic T, Litwin V, Allaway GP, Martin SR, Huang Y, Nagashima KA, et al. HIV-1 entry into CD4+ cells is mediated by the chemokine receptor CC-CKR5. Nature. 1996;381:667–73. doi: 10.1038/381667a0. [DOI] [PubMed] [Google Scholar]
- Feldman GJ, Mullin JM, Ryan MP. Occludin: structure, function and regulation. Adv Drug Deliv Rev. 2005;57:883–917. doi: 10.1016/j.addr.2005.01.009. [DOI] [PubMed] [Google Scholar]
- Garcia JG, Lazar V, Gilbert-McClain LI, Gallagher PJ, Verin AD. Myosin light chain kinase in endothelium: molecular cloning and regulation. Am J Respir Cell Mol Biol. 1997;16:489–94. doi: 10.1165/ajrcmb.16.5.9160829. [DOI] [PubMed] [Google Scholar]
- Gendelman HE, Orenstein JM, Martin MA, Ferrua C, Mitra R, Phipps T, et al. Efficient isolation and propagation of HIV on recombinant colony-stimulating factor 1-treated monocytes. J Exp Med. 1988;167:1428–41. doi: 10.1084/jem.167.4.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gendelman HE, Zheng J, Coulter CL, Ghorpade A, Che M, Thylin M, et al. Suppression of inflammatory neurotoxins by highly active antiretroviral therapy in HIV-associated dementia. J Infect Dis. 1998;178:1000–7. doi: 10.1086/515693. [DOI] [PubMed] [Google Scholar]
- Haorah J, Heilman D, Knipe B, Chrastil J, Leibhart J, Ghorpade A, et al. Ethanol-induced activation of myosin light chain kinase leads to dysfunction of tight junction and blood–brain barrier compromise. Alcohol Clin Exp Res. 2005a;29:999–1009. doi: 10.1097/01.alc.0000166944.79914.0a. [DOI] [PubMed] [Google Scholar]
- Haorah J, Knipe B, Leibhart J, Ghorpade A, Persidsky Y. Alcohol-induced oxidative stress in brain endothelial cells causes blood–brain barrier dysfunction. J Leukoc Biol. 2005b;78:1223–32. doi: 10.1189/jlb.0605340. [DOI] [PubMed] [Google Scholar]
- Hawkins BT, Davis TP. The blood–brain barrier/neurovascular unit in health and disease. Pharmacol Rev. 2005;57:173–85. doi: 10.1124/pr.57.2.4. [DOI] [PubMed] [Google Scholar]
- Hesselgesser J, Halks-Miller M, DelVecchio V, Peiper SC, Hoxie J, Kolson DL, et al. CD4-independent association between HIV-1 gp120 and CXCR4: Functional chemokine receptors are expressed in human neurons. Curr Biol. 1997;7:112–21. doi: 10.1016/s0960-9822(06)00055-8. [DOI] [PubMed] [Google Scholar]
- Hill CM, Deng H, Unutmaz D, Kewalramani VN, Bastiani L, Gorny MK, et al. Envelope glycoproteins from HIV type 1 and 2 and SIV can use human CCR5 as a coreceptor for viral entry and make direct CD4-dependent interactions with this chemokine receptor. J Virol. 1997;71:6296–304. doi: 10.1128/jvi.71.9.6296-6304.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang MB, Bond VC. Involvement of protein kinase C in HIV-1 gp120 induced apoptosis in primary endothelium. J Acquir Immune Defic Syndr. 2000;25:375–89. doi: 10.1097/00042560-200012150-00001. [DOI] [PubMed] [Google Scholar]
- Huang MB, Hunter M, Bond VC. Effect of extracellular human immunodeficiency virus type 1 glycoprotein 120 on primary human vascular endothelial cell cultures. AIDS Res Hum Retroviruses. 1999;15:1265–77. doi: 10.1089/088922299310160. [DOI] [PubMed] [Google Scholar]
- Huang Q, Yuan Y. Interaction of PKC and NOS in signal transduction of microvascular hyperpermeability. Am J Physiol Heart Circ Physiol. 1997;273:H2442–51. doi: 10.1152/ajpheart.1997.273.5.H2442. [DOI] [PubMed] [Google Scholar]
- Jellinger KA, Setinek U, Drlicek M, Bohm G, Steurer A, Lintner F. Neuropathology and general autopsy findings in AIDS during the last 15 years. Acta Neuropathol (Berl) 2000;100:213–20. doi: 10.1007/s004010000245. [DOI] [PubMed] [Google Scholar]
- Jones MV, Bell JE, Nath A. Immunolocalization of HIV envelope gp120 in HIV encephalitis with dementia. AIDS. 2000;14:2709–13. doi: 10.1097/00002030-200012010-00010. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Kennedy RC, Grammas P. Analysis of human lung endothelial cells for susceptibility to HIV type 1 infection, coreceptor expression and cytotoxicity of gp120 proteins. AIDS Res Hum Retroviruses. 2001;17:45–53. doi: 10.1089/088922201750056771. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Kennedy RC, Grammas P. HIV-1 gp120 proteins and gp120 peptides are toxic to brain endothelial cells and neurons: possible pathway for HIV entry into the brain and HIV-associated dementia. J Neuropathol Exp Neurol. 2002;61:992–1000. doi: 10.1093/jnen/61.11.992. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Primeaux C, Grammas P. HIV-1 gp120 proteins alter tight junction protein expression and brain endothelial cell permeability: implications for the pathogenesis of HIV-associated dementia. J Neuropathol Exp Neurol. 2005a;64:498–505. doi: 10.1093/jnen/64.6.498. [DOI] [PubMed] [Google Scholar]
- Kanmogne GD, Primeaux C, Grammas P. Induction of apoptosis and endothelin-1 secretion in primary human lung endothelial cells by HIV-1 gp120 proteins. Biochem Biophys Res Commun. 2005b;333:1107–15. doi: 10.1016/j.bbrc.2005.05.198. [DOI] [PubMed] [Google Scholar]
- Kaul M, Garden GA, Lipton SA. Pathways to neuronal injury and apoptosis in HIV-associated dementia. Nature. 2001;410:988–94. doi: 10.1038/35073667. [DOI] [PubMed] [Google Scholar]
- Kaul M, Zheng J, Okamoto S, Gendelman HE, Lipton SA. HIV-1 infection and AIDS: consequences for the central nervous system. Cell Death Differ. 2005;12:878–92. doi: 10.1038/sj.cdd.4401623. [DOI] [PubMed] [Google Scholar]
- Klockars AJ, Gilbert S. Quantitative applications in the social sciences series #61. Thousand Oaks, CA: Sage Publications; 1986. Multiple comparisons. [Google Scholar]
- Le Y, Wetzel MA, Shen W, Gong W, Rogers TJ, Henderson EE, et al. Desensitization of chemokine receptor CCR5 in dendritic cells at the early stage of differentiation by activation of formyl peptide receptors. Clin Immunol. 2001;99:365–72. doi: 10.1006/clim.2001.5021. [DOI] [PubMed] [Google Scholar]
- Ma TY, Nguyen D, Bui V, Nguyen H, Hoa N. Ethanol modulation of intestinal epithelial tight junction barrier. Am J Physiol. 1999;276:G965–74. doi: 10.1152/ajpgi.1999.276.4.G965. [DOI] [PubMed] [Google Scholar]
- Masliah E, DeTeresa RM, Mallory ME, Hansen LA. Changes in pathological findings at autopsy in AIDS cases for the last 15 years. AIDS. 2000;14:69–74. doi: 10.1097/00002030-200001070-00008. [DOI] [PubMed] [Google Scholar]
- McArthur JC. HIV dementia: an evolving disease. J Neuroimmunol. 2004;157:3–10. doi: 10.1016/j.jneuroim.2004.08.042. [DOI] [PubMed] [Google Scholar]
- Mehta D, Malik AB. Signaling mechanisms regulating endothelial permeability. Physiol Rev. 2006;86:279–367. doi: 10.1152/physrev.00012.2005. [DOI] [PubMed] [Google Scholar]
- Miller DA, Borchardt RT. Application of cultured endothelial cells of the brain microvasculature in the study of the blood–brain barrier. J Tissue Culture Methods. 1992;14:217–24. [Google Scholar]
- Nath A. Human immunodeficiency virus (HIV) proteins in neuropathogenesis of HIV dementia. J Infect Dis. 2002;186:S193–8. doi: 10.1086/344528. [DOI] [PubMed] [Google Scholar]
- Navia BA, Price RW. An overview of the clinical and biological feature of the AIDS dementia complex. In: Gendelman HE, Grant I, Everall IP, Lipton SA, Swindells S, editors. The Neurology of AIDS. 2. New York: Oxford University Press; 2005. pp. 339–56. [Google Scholar]
- Nottet HSLM, Persidsky Y, Sasseville VG, Nukuna AN, Bock P, Zhai QH, et al. Mechanisms for the transendothelial migration of HIV-1-infected monocytes into the brain. J Immunol. 1996;156:1284–95. [PubMed] [Google Scholar]
- Oh SK, Cruikshank WW, Raina J, Blanchard GC, Adler WH, Walker J, et al. Identification of HIV-1 envelope glycoprotein in the serum of AIDS and ARC patients. J AIDS. 1992;5:251–6. [PubMed] [Google Scholar]
- Ojakian GK. Tumor promoter-induced changes in the permeability of epithelial tight junctions. Cell. 1981;23:95–103. doi: 10.1016/0092-8674(81)90274-9. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Ghorpade A, Rasmussen J, Limoges J, Liu XJ, Stins M, et al. Microglial and astrocyte chemokine regulate monocyte migration through the blood–brain barrier in HIV encephalitis. Am J Pathol. 1999;155:1599–611. doi: 10.1016/S0002-9440(10)65476-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persidsky Y, Zheng J, Miller D, Gendelman HE. Mononuclear phagocytes mediate blood–brain barrier compromise and neuronal injury during HIV-associated dementia. J Leukocyte Biol. 2000;68:413–22. [PubMed] [Google Scholar]
- Persidsky Y, Gendelman HE. Mononuclear phagocyte immunity and the neuropathogenesis of HIV-1 infection. J Leukocyte Biol. 2003;74:691–701. doi: 10.1189/jlb.0503205. [DOI] [PubMed] [Google Scholar]
- Persidsky Y, Heilman D, Haorah J, Persidsky R, Weber G, Shimokawa H, et al. Rho-mediated regulation of tight junctions during monocyte migration across blood–brain barrier in HIV-1 encephalitis (HIVE) Blood. 2006;107 doi: 10.1182/blood-2005-11-4721. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peruzzi F, Bergonzini V, Aprea S, Reiss K, Sawaya BE, Rappaport J, et al. Cross talk between growth factors and viral and cellular factors alters neuronal signaling pathways: implications for HIV-associated dementia. Brain Res Rev. 2005;50:114–25. doi: 10.1016/j.brainresrev.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Pu H, Tian J, Andras IE, Hayashi K, Flora G, Henning B, et al. HIV-1 Tat protein-induced alterations of ZO-1 expression are mediated by redox-regulated ERK 1/2 activation. J Cereb Blood Flow Metab. 2005;25:1325–35. doi: 10.1038/sj.jcbfm.9600125. [DOI] [PubMed] [Google Scholar]
- Quan X, Godfrey HP. In vitro study of cytokine-mediated activation of endothelial cell permeability using Falcon®cell culture inserts. Becton Dickinson and Company. Tech Bull. 1998;413:1–3. [Google Scholar]
- Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca2+ signaling and PKC activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol. 2001;533:433–45. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satpathy M, Gallagher P, Lizotte-Waniewski M, Srinivas SP. Thrombin-induced phosphorylation of the regulatory light chain of myosin II in cultured bovine corneal endothelial cells. Exp Eye Res. 2004;79:477–86. doi: 10.1016/j.exer.2004.06.018. [DOI] [PubMed] [Google Scholar]
- Schliwa M, Nakamura T, Porter KR, Euteneuer U. A tumor promoter induces rapid and coordinated reorganization of actin and vinculin in cultured cells. J Cell Biol. 1984;99:1045–59. doi: 10.1083/jcb.99.3.1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevenson BR, Begg DA. Concentration-dependent affects of cytochalasin D on tight junctions and actin filaments in MDCK epithelial cells. J Cell Sci. 1994;107:367–75. doi: 10.1242/jcs.107.3.367. [DOI] [PubMed] [Google Scholar]
- Stuart RO, Nigam SK. Regulated assembly of tight junctions by protein kinase C. Proc Natl Acad Sci USA. 1995;92:6072–6. doi: 10.1073/pnas.92.13.6072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiruppathi C, Minshall RD, Paria BC, Vogel SM, Malik AB. Role of Ca2+ signaling in the regulation of endothelial permeability. Vasc Pharmacol. 2002;39:173–85. doi: 10.1016/s1537-1891(03)00007-7. [DOI] [PubMed] [Google Scholar]
- Toborek M, Lee YW, Flora G, Pu H, Andras IE, Wylegala E, et al. Mechanisms of the blood–brain barrier disruption in HIV-1 infection. Cell Mol Neurobiol. 2005;25:181–99. doi: 10.1007/s10571-004-1383-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vago L, Bonetto S, Nebuloni M, Duca P, Carsana L, Zerbi P, et al. Pathological findings in the central nervous system of AIDS patients on assumed antiretroviral therapeutic regimens: retrospective study of 1597 autopsies. AIDS. 2002;16:1925–8. doi: 10.1097/00002030-200209270-00009. [DOI] [PubMed] [Google Scholar]
- van Hinsbergh VW, van Nieuw Amerongen GP. Intracellular signaling involved in modulating human endothelial barrier function. J Anat. 2002;200:549–60. doi: 10.1046/j.1469-7580.2002.00060.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verin AD, Liu F, Bogatcheva N, Borbiev T, Hershenson MB, Wang P, Garcia JGN. Role of Ras-dependent ERK activation in phorbol ester-induced endothelial cell barrier dysfunction. Am J Physiol Lung Cell Mol Physiol. 2000;279:L360–70. doi: 10.1152/ajplung.2000.279.2.L360. [DOI] [PubMed] [Google Scholar]
- Verin AD, Gilbert-McClain LI, Patterson CE, Garcia JG. Biochemical regulation of the nonmuscle myosin light chain kinase isoform in bovine endothelium. Am J Respir Cell Mol Biol. 1998;19:767–76. doi: 10.1165/ajrcmb.19.5.3126. [DOI] [PubMed] [Google Scholar]
- Zhang N, Hodge D, Rogers TJ, Oppenheim JJ. Ca2+-independent protein kinase Cs mediate heterologous desensitization of leukocyte chemokine receptors by opioid receptors. J Biol Chem. 2003;278:12729–36. doi: 10.1074/jbc.M300430200. [DOI] [PubMed] [Google Scholar]