

Abstract
Threonine 37 is conserved among all the members of the old yellow enzyme (OYE) family. The hydroxyl group of this residue forms a hydrogen bond with the C-4 oxygen atom of the FMN reaction center of the enzyme [Fox, K. M. & Karplus, P. A. (1994) Structure 2, 1089–1105]. The position of Thr-37 and its interaction with flavin allow for speculations about its role in enzyme activity. This residue was mutated to alanine and the mutant enzyme was studied and compared with the wild-type OYE1 to evaluate its mechanistic function. The mutation has different effects on the two separate half-reactions of the enzyme. The mutant enzyme has enhanced activity in the oxidative half-reaction but the reductive half-reaction is slowed down by more than one order of magnitude. The peaks of the absorption spectra for enzyme bound with phenolic compounds are shifted toward shorter wavelengths than those of wild-type OYE1, consistent with its lower redox potential. It is suggested that Thr-37 in the wild-type OYE1 increases the redox potential of the enzyme by stabilizing the negative charge of the reduced flavin through hydrogen bonding with it.
Old yellow enzyme (OYE, EC 1.6.99.1), originally isolated from brewer’s bottom yeast (1), was the first enzyme found to contain flavin as prosthetic group (2). It is a mixture of homo- and heterodimers of ≈45 kDa, the monomeric units derived from two different genes (3). Each of the subunits has a noncovalently bound FMN. OYE binds phenolic compounds tightly, and the resulting complexes have specific absorbance bands in the long wavelength range (500–800 nm) (4). The enzyme can be reduced by NADPH and relatively less efficiently by NADH (5, 6). Molecular oxygen is a substrate for the oxidative half-reaction, but it is unlikely to be the physiological oxidant because of the slow reaction rate and the undesirable product, H2O2. Recently, it has been found that a large number of α,β-unsaturated aldehydes and ketones (6, 7), including quinones, serve as more efficient substrates, in which the olefinic bond is reduced. In the reaction of OYE with α,β-unsaturated cyclic ketones in the absence of reduced pyridine nucleotide, the enzyme flavin can be reduced by one molecule of the substrate and reoxidized by another molecule of the substrate. The net result is the catalytic dismutation of the unsaturated cyclic ketone to saturated ketone and aromatic product in 1:1 ratio (7). OYE catalyzes this host of reactions by means of the shuttling of electrons in a manner typical of redox active flavoproteins. Catalysis occurs by both the spatial coupling of the redox pair and by enzyme-induced alterations to increase reactivity of the reactants.
Several isoforms of OYE have been found in brewer’s bottom yeast (3) and Saccharomyces cerevisiae (8, 9). One of these isoforms, OYE1 from S. carslbergensis, has been cloned and expressed in Escherichia coli. (8) and its crystal structure solved at a resolution of 2.0 Å (10), revealing several important characteristics around the active site (Fig. 1). Substrates for both the reductive and oxidative half-reactions as well as phenolic ligands are spatially coupled by stacking interactions on the si-face of the flavin. The stacking interactions between the π systems of the oxidized FMN and phenolic ligands provide explanations for the charge-transfer absorption. Further binding interactions are provided by residues His-191 and Asn-194, which were shown by construction and characterization of His-191-to-Asn and Asn-194-to-His mutants to interact with the phenolic oxygen of the ligand through hydrogen bonding (11). The hydrogen-bonding residues are, by analogy, proposed to alter electron distribution to increase the reactivity of oxidative half-reaction substrates. Further, by its proximity and location (12) and by construction of the Tyr-196-to-Phe mutant, Tyr-196 was shown to be a general acid, providing a proton to the α-position of the α,β-unsaturated substrate coincident with or after hydride transfer from the reduced flavin to the β-position of the substrates (13).
Figure 1.

The FMN binding site of OYE1. Hydrogen bonds are shown as dashed lines and the distance between the atom attached to the donor hydrogen and the acceptor is shown in Å (Protein Data Bank ID code 1OYB). In this projection, the viewer is looking down on the si-face of the flavin. In this figure, H-atoms have been eliminated; N-atoms are displayed in blue, O-atoms in red, flavin C-atoms in orange, the P-atom in purple, and amino acid C-atoms in green.
In the crystal structure, Thr-37 is hydrogen bonded with the C4 carbonyl oxygen of FMN through its side-chain hydroxyl group (10). Such hydrogen bonding is likely to play a role in controlling the properties of the enzyme flavin. In the Tyr-129-to-Phe mutant of spinach glycolate oxidase, the hydrogen bond formed by this side-chain hydroxyl group of Tyr-129 and the flavin C4 carbonyl oxygen was demonstrated to influence significantly several properties of the enzyme-bound flavin (14).
To investigate the possible role of the hydrogen bond in catalysis, a Thr-37- to-Ala mutation of OYE1 was constructed and expressed. The properties of the mutant enzyme were examined in detail through direct measurement of redox potential, binding of phenolic ligands, rapid reaction studies of both the reductive and oxidative half-reactions, and catalytic turnover.
MATERIALS AND METHODS
Yeast extract and tryptone were from Difco. E. coli strain BL21 (DE3) was from Novagen. Ampicillin, isopropyl 1-thio-β-d-galactopyranoside (IPTG), and XbaI were from Boehringer Mannheim. PMSF, NADPH, and NADH were from Sigma, and the various aldehyde and ketone oxidative substrates were from Aldrich. MscI was from New England Biolabs. All other materials used in mutagenesis were purchased from sources described previously (15).
Mutagenesis for T37A-OYE1.
The mutagenesis reaction was performed on the OYE1 gene cloned into the pET expression system as described previously (8). The primer selection method is outlined in Fig. 2. XbaI and ApaI were selected as the unique restriction sites on either side of the region to be mutated. Sequencing/PCR primers were designed to flank the region defined by the two wild-type restriction sites. PCR was conducted on a Perkin–Elmer GeneAmp PCR system 2400 apparatus. The first PCR reaction was carried out as previously described (15) to generate a 251-bp fragment containing the desired mutation within the mutagenic primer. The product was purified by using a PCR Purification Kit (Qiagen) and concentrated. The second PCR was conducted under similar conditions to PCR1 with the substitution of PCR2 primer and the PCR1 reaction product for the primers in the reaction (16). Cycling conditions were altered to allow for increased annealing time for the larger primer. The product was purified as described above. The PCR fragment was cloned into the pGEM-T PCR vector as described previously. By using the unique ApaI and XbaI restriction sites, the cassette was replaced into the wild-type vector (15). The final plasmid of T37A-OYE1-pET was confirmed by digestion with MscI and sequencing.
Figure 2.

Primer selection for mutagenesis. In the mutagenic primer, the underlined codon includes the italicized substitution that encodes the Thr-37 to Ala mutation. The solid arrows describe the endpoints for the PCR1 reaction product; the dashed arrows describe the endpoints for the PCR2 reaction. The mutagenesis primer contains the desired Thr-37 to Ala mutation with a TGG → CGG substitution in the Thr-37 codon that introduces an analytically useful MscI restriction site (ACC/GGT).
E. coli strain BL21-DE3 harboring the pET-OYE1-T37A plasmid was used to express the mutant enzyme by induction with 400 μM IPTG 8 hr after 1% inoculation of LB/Ampicillin media. The cells were allowed to grow overnight before being harvested. Purification of OYE1-T37A was conducted as described previously (9, 17).
Unless otherwise noted, all experiments were conducted at 25°C in 50 mM KPI buffer (pH 7.0).
Extinction Coefficient Measurement.
The extinction coefficient of the enzyme-bound flavin was determined by dissociation from the protein with SDS, as described previously (11, 13).
Ligand Binding Studies.
Titration of OYE1-T37A with phenolic ligands was conducted by addition of small amounts of ligand stock solution to the oxidized enzyme in 1 ml quartz cuvettes. The titration process was recorded by following the absorbance changes with a Varian Cary 3 UV/Vis spectrophotometer. Dissociation constants were calculated from the amount of spectral perturbation on addition of nonsaturating concentrations of ligand, compared with that at saturating concentrations (11, 13).
Redox Potential Measurement.
The redox potential of OYE1-T37A was measured by titrating the oxidized enzyme in an anaerobic cuvette with NADPH or NADH. The titration was recorded by following the spectrum with a Hewlett–Packard 8452A Diode Array spectrophotometer. In a separate experiment, the xanthine/xanthine oxidase reducing system (18) was incubated with the oxidized OYE1-T37A in the presence of 4.5 μM benzyl viologen as one-electron mediator in the anaerobic cuvette. The spectra during reduction were recorded with a Varian Cary 3 spectrophotometer.
Stopped-Flow Studies of Half-Reactions.
The reductive half-reaction of the T37A mutant enzyme with NADPH and the oxidative half-reaction with 11 substrates were studied with a Kinetic Instruments stopped-flow spectrophotometer (19). The reaction of reduced wild-type or mutant T37A OYE with various substrates was conducted with a Hi-Tech stopped-flow spectrophotometer. The system was made anaerobic by incubation overnight with an anaerobic solution of 3,4-dihydroxybenzoate and protocatechuate-3,4-dioxygenase (19). All buffer and substrate solutions were made anaerobic by 15 min bubbling with argon before use. For the reaction with oxygen, solutions of various oxygen concentrations were obtained by bubbling 50 mM phosphate buffer with standardized O2-N2 gas mixtures from Matheson. The enzyme solution was made anaerobic in a tonometer by alternate vacuum and purging with argon. In the study of the oxidative half-reaction, the enzyme was reduced by adding an NADPH generating system, containing glucose-6-phosphate, NADP+ and an appropriate amount of glucose-6-phosphate dehydrogenase from a side arm after the tonometer had been made anaerobic. The amount of glucose 6-phosphate dehydrogenase from Leuconostoc mesenteroides was adjusted so that complete reduction of the OYE took more than 10 min, in order not to interfere with the oxidation reaction being studied. Analysis was conducted by fitting data to exponential equations by using the Marquardt algorithm (20) with program a, developed by C. J. Chiu, R. Chung, J. Diverno, and D. P. Ballou at the University of Michigan (Ann Arbor, MI).
Turnover Reactions.
Enzyme-monitored turnover reaction of OYE1-T37A and its substrates, NADPH and oxygen, was conducted with a Hi-Tech stopped-flow spectrophotometer (21). The enzyme in air-equilibrated solution was mixed with air-equilibrated NADPH solutions of various concentrations, all in excess of that of O2 (260 μM). The concentration of NADPH was taken as constant in each reaction, but that of oxygen decreased with the progress of the reaction. The absorbance at 450 nm, monitoring the concentration of oxidized enzyme, was used to calculate observed turnover numbers as a function of residual O2 concentration. When 2-cyclohexenone was used instead of molecular oxygen as substrate, both the tonometer of enzyme and the syringe that contained 400 μM 2-cyclohexenone and excess NADPH were made anaerobic before reaction.
RESULTS
Mutagenesis, Expression, and Purification.
Mutagenesis was conducted by a modified version of the megaprimer method for site-directed mutagenesis (16), which calls for PCR-based generation of a lengthy mutant fragment, subsequently used as a primer in a second round of PCR extending the mutant fragment to the other side of the mutation. Because the method inherently carries with it a high risk of mutation, sequencing of the entire insert region taken from PCR was conducted to confirm the presence of the desired sequence. Mutagenesis was confirmed additionally by digestion with MscI (data not shown).
Spectroscopic Properties.
The peaks of absorbance of the mutant enzyme are shifted from those of the wild-type OYE1, 462 nm and 384 nm, to 452 nm and 376 nm, respectively. The extinction coefficient of the peak, ɛ452 =11,900 M−1 cm−1, is about the same as that of the wild-type enzyme (ɛ462 = 11, 700 M−1 cm−1) (11) (Fig. 3). Like the wild-type OYE, the mutant enzyme is devoid of fluorescence from the bound flavin.
Figure 3.

Spectral comparison of oxidized and p-chlorophenol-bound wild-type OYE1 and the T37A mutant. The spectral data are scaled so the wild-type absorbance 462 nm agrees with the T37A absorbance at 452 nm, corresponding to 15 μM wild-type enzyme. The solid lines and open symbols are for the T37A mutant enzyme; the dashed lines and closed symbols represent the wild-type OYE1 spectra. The spectra with long wavelength absorbance marked by squares are the result of addition of saturating (500 μM) p-chlorophenol to the enzyme.
Ligand Binding.
Like the wild-type enzyme, oxidized OYE1-T37A binds phenolic compounds to form charge-transfer complexes with strong absorbance in the long wavelength range (Fig. 3). In this type of complex, the phenolate ion acts as electron donor while the oxidized FMN in the enzyme is the acceptor. Four phenolic ligands with various substitutions at the para position to the hydroxyl group were tested for binding to the T37A mutant enzyme. The dissociation constants (Kd) are moderately greater than those obtained for binding of the same ligands to OYE1. The extinction coefficients of the absorbance peaks for the complexes are essentially the same as those of OYE1 bound with the corresponding ligands (Table 1). However, the peaks of absorbance for the charge-transfer complexes formed are all blue shifted (Fig. 3, Table 1), with the wavenumber increased by 890–1,450 cm−1 from those of OYE1 (11).
Table 1.
Comparison of binding of phenolic compounds by wild-type OYE1 and the T37A mutant
| Ligands | OYE1-wild type
|
OYE1-T37A
|
||
|---|---|---|---|---|
| Kd | CTmax (ɛ) | Kd | CTmax (ɛ) | |
| μM | M−1 cm−1 | μM | M−1cm−1 | |
| p-chlorophenol* | 1 | 645 nm | 13.7 | 590 nm |
| (4,400) | (4,300) | |||
| p-cyanophenol* | 0.08 | 576 nm | 1.4 | 548 nm |
| (4,500) | (4,900) | |||
| p-hydroxy-benzaldehyde* | 0.2 | 585 nm | 1.2 | 548 nm |
| (3,500) | (4,100) | |||
| p-methoxyphenol† | 66 | 700 nm | 348 | 640 nm |
| (3,700) | (4,100) | |||
Data for the wild-type OYE1 are from ref. 11.
Data for the wild-type enzyme are from OYE purified from Brewers’ Bottom Yeast (V.M., unpublished).
Redox Potential Measurement.
In earlier studies of OYE where the native FMN was replaced by flavin analogues, the energy of the long-wavelength transition (wavenumber of the absorbance peak) was shown to correlate linearly with redox potential of the enzyme (22, 23). The redox potential of OYE1-T37A was measured by titrating the oxidized enzyme with NADPH in an anaerobic cuvette. At the chemical equilibrium of this reaction, the redox potential of NADP+/NADPH equals that of the enzyme. According to the Nernst equation,
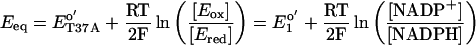 |
where Eeq refers to the redox potential of both couples at the equilibrium. In this equation, ET37Ao′ and E1o′ represent the redox potential of the mutant enzyme and that of NADP+/NADPH, respectively, at equal concentration of electron donor and acceptor, 25°C, pH 7.0. The value of E1o′ was taken as −320 mV (24). The relative amount of reduced and oxidized OYE1-T37A was calculated from the absorbance at 452 nm and ET37Ao′ was determined as −263 mV, 33 mV lower than that of the wild-type enzyme (Fig. 4 Upper). To eliminate the possibility that the enzyme may have different affinities for NADPH and NADP+ that would influence the measured value of the redox potential, NADH was also used instead of NADPH for titration. The measured value, in this case, was −265 mV. These values and the wavenumbers of charge-transfer complexes by using p-chlorophenol and p-methoxyphenol as ligands fit well with the previously demonstrated linear relationship of the redox potential and the charge-transfer complex maxima of OYE with artificial flavin replacements (22, 23) (Fig. 4 Lower).
Figure 4.

Redox potential measurement of OYE1-T37A. (Upper) Oxidized enzyme (35 μM, 1.2 ml) was titrated in an anaerobic cuvette with NADPH (closed circles) or NADH (open circles). The redox potential is acquired as the value of log ([Ox]/[Red]) when log ([NAD(P)+]/[NAD(P)H]) = 0. (Lower) Correlation of the absorbance peak positions of the charge-transfer complex formed by OYE1 and p-chlorophenol and the redox potential of the coenzyme. The open circles represent the results of wild-type enzyme with flavin analogues bound (refs. 22, 23). The result with T37A is shown as the closed circle.
When the xanthine/xanthine oxidase system, which transfers single electrons through formation of the reactive benzyl viologen radical, was used to reduce OYE1-T37A, no formation of semiquinone was detectable during the whole reduction process of the enzyme (data not shown). These results differ from those found with wild-type OYE1, where approximately 60% thermodynamic stabilization of the flavin semiquinone is observed (23).
Reductive Half-Reaction.
Given the fact that the mutant OYE1 has a lower redox potential, the reductive half-reaction with NADPH is expected to be slower than with OYE1. This expectation was confirmed by stopped-flow experiments. When oxidized OYE1-T37A was reduced by NADPH under anaerobic conditions, the rate constant (kred) was only 8% that of the wild-type enzyme (11) (Table 2). Because of the slower reduction, the buildup of the long-wavelength absorbing charge-transfer complex between the enzyme and NADPH, which occurs before hydride transfer, can be observed clearly (Fig. 5). The rate constant of this step (kCT) is essentially unaffected by the mutation. As with the phenolic ligands, the binding affinity of NADPH is not dramatically changed in the mutant.
Table 2.
Reductive half-reaction of OYE1-T37A by NADPH vs. the wild-type enzyme
| OYE1-T37A | OYE1-wild type | Ratio (T37A/wild type) | |
|---|---|---|---|
| Kd, μM | 110 | 100 | 1.1 |
| kCT, s−1 | 290 | 340 | 0.85 |
| kred, s−1 | 0.43 | 5.1 | 0.084 |
Data for the wild-type OYE1 are from ref. 11.
Figure 5.

Reductive half-reaction of T37A-OYE1. Oxidized enzyme (final concentration 15 μM) was reacted anaerobically with NADPH (final concentration 100 μM) in the stopped-flow apparatus. The absorbance change was recorded at 450 nm (dashed line) and 550 nm (solid line).
Oxidative Half-Reaction.
The kinetics of the oxidative half-reaction for the wild-type and mutant enzymes were determined with a series of substrates (Table 3). Several trends are readily apparent in the data. For every one of these oxidants, the rate constant is larger than that for the reaction with wild-type OYE1 (11, 13) (Table 3). The most significant difference is with molecular oxygen, whose reaction with the reduced enzyme is accelerated by a factor of 26. The results are what could be expected, because the lower redox potential of the T37A mutant enzyme indicates decreased electrophilicity of the reduced flavin, with the result that it should lose electrons faster and easier than the wild-type enzyme. Like the reductive substrate, NADPH, the dissociation constants for the oxidative substrates are not appreciably affected by the mutation (Table 3).
Table 3.
Oxidative half-reaction of OYE-T37A vs. the wild-type enzyme
|
Kd, μM
|
kox, s−1
|
Ratio of kox (T37A/Wild type) | |||
|---|---|---|---|---|---|
| T37A | Wild type | T37A | Wild type | ||
| 2-Cyclohexenone* (CHX) (1) | 28 | 32 | 298 | 102 | 2.9 |
| 3-Methyl-2-CHX (2) | 50 | 64 | 0.17 | 0.062 | 2.7 |
| 1,2-Cyclohexanedione (3) | 570 | 520 | 100 | 32 | 3.1 |
| Cinnamaldehyde† (CNA) (4) | 105 | 125 | 77 | 17 | 4.5 |
| α-methyl-trans-CNA (5) | 11 | 8.6 | 256 | 34 | 7.5 |
| 3-Penten-2-one (6) | 590 | 430 | 50.9 | 15.6 | 3.3 |
| Mesityl oxide (7) | 250 | ≥1,000 | 0.261 | ∼0.03 | 9.0 |
| 3-Methyl-2-butenal (8) | 47 | 70 | 3.0 | 1.7 | 1.8 |
| Crotonaldehyde (M−1 s−1) (9) | — | — | 3.4 × 105 | 1.6 × 105 | 2.1 |
| Molecular oxygen* (M−1 s−1) | — | — | 1.0 × 105 | 3.8 × 103 | 26 |
| 1-Nitrocyclohexene† (M−1 s−1) (11) | — | — | 2.5 × 106 | 6.1 × 105 | 4.1 |
The Kd values and rate constants given were obtained by analysis of at least four duplicate reactions at each concentration of oxidant studied and which varied by less than 5%. The values given therefore have corresponding associated error limits.
Data for the wild-type enzyme are from ref. 11.
Data for the wild-type enzyme are from ref. 13.
 |
In addition to comparison between wild-type and the mutant enzyme, comparisons between oxidative substrates are quite telling. In agreement with earlier results from turnover studies (7), the increased reactivities of crotonaldehyde and 3-methyl-2-butenal, compared with those of 3-penten-2-one and mesityl oxide, respectively, are suggestive of aldehydes being better oxidative substrates than ketones. A several-orders-of-magnitude decrease in kox occurs with β substitution, as is seen by comparing 3-methyl-2-cyclohexenone to 2-cyclohexenone, mesityl oxide to 3-penten-2-one and 3-methyl-2-butenal to crotonaldehyde. Previous mechanistic work has demonstrated that reduction of the double bond of α, β-unsaturated carbonyl compounds proceeds by means of transfer of hydride from reduced flavin to the β carbon (7) and transfer of a proton to the α carbon from Tyr196 (13). The data suggest that substitution likely slows the rate of hydride transfer both by steric hindrance and by increasing electron density of the β carbon. Substitution at the α carbon in α-methyl-cinnamaldehyde proves to make it a better oxidant than cinnamaldehyde and suggests a lack of steric interaction with the α position. The increasing reactivity may be explained by substitution leading to decreased energy difference between π and π* orbitals in α-methyl-cinnamaldehyde, as is reflected by the higher wavelength maxima of the chromophore. Alternatively, the increased electron density at the α position could aid in speeding proton transfer from Tyr-196.
Several individual reactants are notable as well. 1,2-Cyclohexanedione proved to be an excellent substrate for both the oxidative and reductive half-reactions. When high concentrations of this substrate are mixed with reduced enzyme, after oxidation is complete, a steady-state turnover is achieved with concomitant buildup of a long-wavelength absorbance. The resulting charge-transfer complex of oxidized enzyme is in agreement with the ligand-binding spectrum observed with catechol, suggesting that 1,2-cyclohexanedione undergoes a dismutation reaction, analogous to that observed with 2-cyclohexenone, generating 2-hydroxy-cyclohexanone and catechol in 1:1 ratio in the anaerobic steady state.
In the oxidation of the T37A mutant by 3-methyl-2-cyclohexenone, the spectrum of the enzyme changes in the dead time of the stopped-flow reaction as shown in Fig. 6. The new spectrum appears to be that of the Michaelis complex formed by the reduced enzyme and the substrate, 3-methyl-2-cyclohexenone. A similar complex has been observed with the reduced Tyr-196-to-Phe mutant enzyme and 2-cyclohexenone (13). The dissociation constant obtained by plotting the initial change of absorbance at 452 nm vs. substrate concentration is 89 μM (insert of Fig. 6), similar to that obtained through analysis of the reaction rate, 50 μM (Table 3). A similar phenomenon was observed in the reaction of reduced wild-type OYE1 with α-methyl-trans-cinnamaldehyde (data not shown).
Figure 6.

The Michaelis complex intermediate of 3-methyl-2-cyclohexenone and reduced OYE1-T37A. The reduced enzyme was mixed with the substrate (final concentrations 12 μM and 200 μM, respectively), and the dead time absorbances (3 ms) at various wavelengths (open circles) are shown and proposed to be the spectrum of the Michaelis complex. The spectra of the reduced (open squares) and oxidized (open triangles) mutant enzyme, scaled to the same concentration, are also shown. (Insert) The absorbance change at 452 nm during the dead time of the stopped-flow experiments is plotted against the final concentration of the 3-methyl-2-cyclohexenone.
Steady-State Kinetics of OYE1-T37A.
The steady-state kinetics of OYE1-T37A were measured in enzyme-monitored turnover experiments in which the oxidized enzyme and varying concentrations of oxidant were mixed with excess NADPH in the stopped-flow apparatus. The absorbance of the flavin at 450 nm was recorded and analyzed as described in ref. 20. The concentration of the oxidant changes during the reaction process, while that of NADPH is regarded as constant. These experiments yielded sets of parallel Lineweaver–Burk plots, from which the kinetic data were determined. The turnover numbers acquired by using molecular oxygen (0.49 s−1) and 2-cyclohexenone (0.38 s−1) are essentially the same as the rate constant of the reductive half-reaction (0.43 s−1), although both of them are good oxidative substrates for the mutant enzyme.
DISCUSSION
Solution of the crystal structure of OYE1 (10) makes it possible to pinpoint the residues critical for its catalytic function. Several residues of this enzyme have been mutated to reveal their roles in the reaction, including His-191, Asn-194 (11), and Tyr-196 (13). Thr-37 studied in this paper is different from these residues in that it interacts with the flavin molecule directly through hydrogen bonding (10) (Fig. 1). The redox potential of the Thr-37A mutant enzyme is considerably lower than that of the wild-type enzyme (Fig. 4). The hydrogen bond formed by the side-chain hydroxyl group of Thr-37 and the C4 oxygen of the FMN tends to withdraw electrons from the flavin ring, therefore enhancing the electrophilicity of the oxidized enzyme and stabilizing the negatively charged reduced flavin. When the residue is mutated to Ala, the hydroxyl group and its function cease to exist. It could then be predicted that the energy of the charge-transfer transition would be influenced by the mutation in the same way as found previously by substitution of artificial flavins that have redox potentials different from that of FMN (22, 23). This expectation is in accord with the experimental results that the maxima of the charge-transfer complexes formed by phenolic compounds are all blue shifted compared with those with the wild-type OYE1. The correlation with p-chlorophenol as charge-transfer ligand is shown in Fig. 4 Lower. A similar correlation is found with p-methoxyphenol (not shown).
The oxidized mutant enzyme is reduced slowly by the xanthine/xanthine oxidase reducing system. The spectrum of semiquinone was not observable during the reaction process, which lasted about 2 hr. This result contrasts with the ≈60% thermodynamic stabilization of the anionic flavin radical with OYE in similar experiments and is consistent also with the previously constructed relationship for the redox potential of FMNox/FMNHsq⋅ and the peak position of the charge-transfer complex (22). The relationship predicts that the potential of the Eox/Esq couple in the T37A mutant enzyme would be lower than −400 mV and therefore that the semiquinone form of the flavin would not be a thermodynamically stable species.
Thermodynamically, the drop of redox potential because of the mutation will change the chemical equilibrium of the half-reactions in which the flavin acquires or loses electrons. The change is reflected kinetically. The reduction rate of the oxidized mutant enzyme by NADPH is decreased to about 8% of that of the wild-type enzyme (Table 2). On the other hand, reduced T37A-OYE1 reacts faster with all the 11 oxidative substrates tested than does the wild-type enzyme. A faster reaction is expected because the lower redox potential of the enzyme renders the reduced species less favorable and could result in faster release of electrons to oxidant substrates (Table 3).
Other characteristics of OYE1 are essentially unchanged in the mutant enzyme. The extinction coefficients of the absorbance peaks are about the same for the wild-type and T37A mutant enzymes. This result is an indication that the structure of OYE1 is not disturbed by the single mutation from Thr to Ala. In the binding experiments, the dissociation constants for the phenolic compounds, as well as the extinction coefficients of the charge-transfer complexes, are only moderately altered in the mutant enzyme (Table 1). These results suggest that the side chain of Thr-37 is not involved directly in interacting with the phenolic ligands, in distinction to His-191 and Asn-194, where mutation of these residues results in orders of magnitude weaker binding (11). In the stopped-flow experiments for half-reactions, the dissociation constants for both NADPH and all the oxidative substrates remain unaffected in the T37A OYE1 (Tables 2 and 3). Even an important detail in the reductive half-reaction is not changed, the rate of constant for the formation of the transfer complex between NADPH and oxidized flavin (Fig. 3 and Table 2). All these results suggest that the main influence of the mutation is the decrease in redox potential, because of loss of the hydrogen bond between the Thr-37 hydroxyl and the C4 carbonyl of the flavin. It is unlikely that Thr-37 is involved directly in lowering the free energy of the transition state of the reaction because the rate constants in the two half-reactions are shifted in different directions.
Although the oxidation is sped up in the mutant OYE1, the rate of catalytic turnover is decreased because of the slower reduction by NADPH, the rate-limiting step. The enzyme seems to have evolved to obtain the redox potential most favorable for the overall reaction. In this aspect, the residue Thr-37 and its hydrogen bonding with the C4 oxygen of FMN seem to play an important role in controlling the environment and hence the nature of the flavin. A similar situation is also seen with spinach glycolate oxidase (14), in which Tyr-129 interacts with and influences the flavin coenzyme through hydrogen bonding with its C4 carbonyl oxygen.
Acknowledgments
This work was supported in part by U.S. Public Health Service Grant GM 11106.
ABBREVIATION
- OYE
old yellow enzyme
References
- 1.Warburg O, Christian W. Biochem Z. 1933;266:377–411. [Google Scholar]
- 2.Theorell H. Biochem Z. 1935;275:344–346. [Google Scholar]
- 3.Stott K, Saito K, Thiele D J, Massey V. J Biol Chem. 1993;268:6097–6106. [PubMed] [Google Scholar]
- 4.Abramovitz A S, Massey V. J Biol Chem. 1976;251:5327–5336. [PubMed] [Google Scholar]
- 5.Massey V, Schopfer L M. J Biol Chem. 1986;261:1215–1222. [PubMed] [Google Scholar]
- 6.Schopfer L M, Massey V. In: A Study of Enzymes. Kuby S A, editor. Boca Raton, FL: CRC; 1990. pp. 249–267. [Google Scholar]
- 7.Vaz A D N, Chakraborty S, Massey V. Biochemistry. 1995;34:4246–4256. doi: 10.1021/bi00013a014. [DOI] [PubMed] [Google Scholar]
- 8.Saito K, Thiele D J, Davio M, Lockridge O, Massey V. J Biol Chem. 1991;266:20720–20724. [PubMed] [Google Scholar]
- 9.Niino Y S, Chakraborty S, Brown B J, Massey V. J Biol Chem. 1995;270:1983–1991. doi: 10.1074/jbc.270.5.1983. [DOI] [PubMed] [Google Scholar]
- 10.Fox K M, Karplus P A. Structure (London) 1994;2:1089–1105. [PubMed] [Google Scholar]
- 11.Brown B J, Deng Z, Karplus A, Massey V. J Biol Chem. 1998;273:32753–32762. doi: 10.1074/jbc.273.49.32753. [DOI] [PubMed] [Google Scholar]
- 12.Karplus P A, Fox K M, Massey V. FASEB J. 1995;9:1518–1526. doi: 10.1096/fasebj.9.15.8529830. [DOI] [PubMed] [Google Scholar]
- 13.Kohli R M, Massey V. J Biol Chem. 1998;273:32763–32770. doi: 10.1074/jbc.273.49.32763. [DOI] [PubMed] [Google Scholar]
- 14.Macheroux P, Kieweg V, Massey V, Sàderlind E, Stenberg K, Lindqvist Y. Eur J Biochem. 1993;213:1047–1054. doi: 10.1111/j.1432-1033.1993.tb17852.x. [DOI] [PubMed] [Google Scholar]
- 15.Kohli R M. BioTechniques. 1998;25:184–188. doi: 10.2144/98252bm01. [DOI] [PubMed] [Google Scholar]
- 16.Sarkar G, Sommer S S. BioTechniques. 1990;8:404–407. [PubMed] [Google Scholar]
- 17.Abramovitz A S, Massey V. J Biol Chem. 1976;251:5321–5326. [PubMed] [Google Scholar]
- 18.Massey V. In: Flavins and Flavoproteins 1990. Curti B, Ronchi S, Zanetti G, editors. New York: Walter de Gruyter; 1991. pp. 59–66. [Google Scholar]
- 19.Beaty N B, Ballou D P. J Biol Chem. 1981;256:4611–4618. [PubMed] [Google Scholar]
- 20.Bevington P R. Data Reduction and Error Analysis for the Physical Sciences. New York: McGraw–Hill; 1969. [Google Scholar]
- 21.Gibson Q H, Swoboda B E P, Massey V. J Biol Chem. 1964;239:3927–3934. [PubMed] [Google Scholar]
- 22.Stewart R C, Massey V. J Biol Chem. 1985;260:13639–13647. [PubMed] [Google Scholar]
- 23.Murthy Y V S N, Massey V. J Biol Chem. 1998;273:8975–8982. doi: 10.1074/jbc.273.15.8975. [DOI] [PubMed] [Google Scholar]
- 24.Clark W M. Oxidation-Reduction Potentials. Baltimore: Williams & Wilkins; 1960. p. 490. [Google Scholar]