

Abstract
Background
Mitochondrial DNA (mtDNA) mutations account for at least 5% of cases of postlingual, nonsyndromic hearing impairment. Among them, mutation A1555G is frequently found associated with aminoglycoside-induced and/or nonsyndromic hearing loss in families presenting with extremely variable clinical phenotypes. Biochemical and genetic data have suggested that nuclear background is the main factor involved in modulating the phenotypic expression of mutation A1555G. However, although a major nuclear modifying locus was located on chromosome 8p23.1 and regardless intensive screening of the region, the gene involved has not been identified.
Methods
With the aim to gain insights into the factors that determine the phenotypic expression of A1555G mutation, we have analysed in detail different genetic and genomic elements on 8p23.1 region (DEFA3 gene absence, CLDN23 gene and MRPS18CP2 pseudogene) in a group of 213 A1555G carriers.
Results
Family based association studies identified a positive association for a polymorphism on MRPS18CP2 and an overrepresentation of DEFA3 gene absence in the deaf group of A1555G carriers.
Conclusion
Although none of the factors analysed seem to have a major contribution to the phenotype, our findings provide further evidences of the involvement of 8p23.1 region as a modifying locus for A1555G 12S rRNA gene mutation.
Background
Mitochondrial DNA (mtDNA) mutations are an important cause of human disease and have been associated with many clinical abnormalities, including various forms of both syndromic and nonsyndromic hearing loss [1]. It has been reported that at least 5% of cases of postlingual, nonsyndromic hearing impairment are attributable to known mtDNA mutations, representing one of the most frequent causes of hearing impairment [2]. The most commonly reported nonsyndromic deafness-causing mtDNA mutations are a C insertion or deletion at position 961 [3-5], C1494T [6,7] and A1555G [8-12] in the 12S rRNA gene, and mutations A7445G [13-15], 7472insC [16,17], T7510C [18] and T7511C [4,19,20] in the tRNASer(UCN) gene.
In particular, the A1555G mutation has been associated with aminoglycoside-induced and/or nonsyndromic hearing loss in various families of different ethnic backgrounds [8-12]. Remarkably, in Spain A1555G accounts for about 15% of all familial and sporadic cases of hearing loss, irrespective of their mode of inheritance and age of onset [21]. The phenotype associated to A1555G mutation varies considerably among matrilineal relatives, ranging from severe deafness, to moderate progressive hearing loss or even completely normal hearing. Biochemical and genetic data suggest that nuclear background may be the main factor involved in modulating the phenotypic expression of the mutation [22-24]. Extensive genome wide search revealed that nuclear modifying factors are likely to be numerous, but a region in chromosome 8p23.1 has been proposed as a putative localization for a modifier locus [22,23,25-27]. However, the gene involved has not been identified yet.
Chromosome band 8p23.1 is known to be a frequent site of chromosomal rearrangements mediated by low copy repeats (LCRs) or segmental duplications (SDs). It has been described that as many as one in four individuals from the general population carry a 4.7 Megabase (Mb) inversion of the region [28-30]. A high density of genes are present in the region, and copy number variability (CNV) involving both α-defensin (DEFA1 and DEFA3) and β-defensin (DEFB4, DEFB103 and DEFB104) genes has been well detected and characterized [31-34].
The objective of the present work was to analyse in detail the contribution of different 8p23.1 genetic elements to the phenotypic variability observed in deaf patients with mitochondrial 12S rRNA A1555G mutation. The analysis has focused on three different genomic features: DEFA3 gene absence, claudin23 (CLDN23) mutational analysis and the putative function of a ribosomal mitochondrial protein pseudogene (MRPS18CP2). These genes were selected after an exhaustive screening of the region looking for candidates as genetic modifiers of A1555G associated phenotype. Defensins were chosen because of their close proximity to the positive linkage region and CLDN23 and MRPS18CP2 were selected on the basis of their putative biological function.
Methods
Patients and samples
Familial cases of sensorineural hearing loss have been collected from different Spanish clinical centres with the aim to study the molecular basis of hearing loss associated to mtDNA A1555G mutation. The analysis was performed on 213 patients, from 55 pedigrees with A1555G mutation and 336 Spanish controls. The Spanish control samples were unrelated blood donor controls, all of Caucasian origin. Informed consent was obtained from all participants prior to their participation in the study, in accordance with the Institutional Review Board and Ethic Committee.
Clinical information such as the severity and age of onset of hearing impairment, the exposure to some kind of ototoxic substances, specifically aminoglycosides, and any other medical diagnoses were evaluated from at least one member of each pedigree.
Detection of A1555G mutation
The detection of the A1555G mutation was either by PCR amplification of a 340-bp fragment (Forward 5'-GCTCAGCCTATATACCGCCATCTTCAGCAA-3' and Reverse 5'-TTTCCAGTACACTTACCATGTTACGACTTG-3'), followed by the digestion with restriction endonuclease HaeIII, or alternatively using Pyrosequencing™ technology (PSQ96MA) (Biotage AB, Sweden). A specific SNP assay was designed for Pyrosequencing (Forward 5'-CGACATTTAACTAAAACCCCTACGC-3', Reverse 5'-GTTGGGTGCTTTGTGTTAAGCT-3' and Sequencing 5'-CACTTACCATGTTACGACT-3' primers) and sequence identification was performed automatically by the SQA software.
DEFA3 determination
A PCR amplification assay followed by restriction enzyme digestion (PCR-RFLP) has been used to discriminate DEFA1 and DEFA3 gene alleles differing by a single nucleotide. A fragment of 304 bp around C3400A PSV was PCR amplified with fluorescently labelled primers (Forward 5'-TGAGAGCAAAGGAGAATGAG-3', Reverse 5'-GCAGAATGCCCAGAGTCTTC-3') and digested with HaeIII enzyme. About 2 μl of digestion product were added to 10 μl HiDi formamide containing ROX500 marker (Applied Biosystems) and run on an ABI 3100 capillary system (Applied Biosystems). Peaks were analysed using Genemapper software (Applied Biosystems).
Mutational screening
The genetic screening of CLDN23 gene and MRPS18CP2 pseudogene was performed by direct sequencing. The entire coding sequence of CLDN23 gene was PCR-amplified in two different fragments of 483 bp (Forward 5'-CCAGGAGGGAACTAGCCTAA-3' and Reverse 5'-AGCGAGGTGACCATGAGTG-3') and 679 bp (Forward 5'-GACGAGCCCAACTTCGTG-3' and Reverse 5'-AGGCAGATTTCCATCCACAC-3'). The MRPS18CP2 pseudogene was PCR amplified in a single fragment spanning 543 bp (Forward 5'-CTCTGTTTACAGAAGACCTGG-3', Reverse 5'-TTTTAATCTAAAATCCATGTAGCAAA-3'). The resulting PCR products were sequenced using an ABI PRISM® 3730 xl DNA Analyzer and ABI PRISM® BigDye Terminator v3.1 Sequencing Kit (Applied Biosystems).
Analysis of MRPS18CP2 expression
Analysis of MRPS18CP2 expression was assessed by RT-PCR. We used total RNA isolated from lymphoblastoid cell lines of general population subjects as well as total adult RNA from ovary, liver, spleen, lung, placenta, kidney, thymus, heart, skeletal muscle, testes, colon (Stratagene) and brain (Ambion). We employed 1 μg of total RNA for reverse transcription using SuperScript First Strand Synthesis System (Invitrogen). Reverse transcribed RNA was then PCR amplified using specific primers for MRPS18CP2 (Forward 5'-TGTTACAACCTTTAGGGTCCTTG-3', Reverse 5'-AGAGGTTGTTCACAATATAAAC-3').
Statistical analysis
To compare the proportion of DEFA3 absence in the different groups, between groups chi-square test was performed.
Family based association tests were performed using FBAT package [35]. FBAT decomposes large pedigrees into individual nuclear families which are treated as independent in most of the calculations. The analysis was performed with 111 nuclear families, which belong to 33 large pedigrees, from which we have detailed phenotypic information and were suitable for being analysed with FBAT package. Bonferroni correction was used to account for multiple testing, correcting for the number of tests performed by the FBAT software.
For all statistical analysis performed, subjects were classified as affected or unaffected according to the available clinical data. The phenotype of subjects with reported aminoglycoside exposure was considered unknown.
The DEFA3 absence analysis was performed independently from the CLDN23 and MRPS18CP2 tests, considering only two different possible genotypes: present (with at least one copy of DEFA3 gene) or absent (without DEFA3). Heterozygotes for DEFA3 absence were only annotated in those cases were the genotype could be inferred from the pedigree data.
Results
α-defensin cluster is located in the positive linkage region on chromosome 8p23.1
Bykhovskaya and colleagues identified chromosome 8p23.1 as a major modifying locus for hearing loss phenotype associated to A1555G mutation [26,27]. Neither a gene nor a genetic factor involved has been found, regardless of intensive screening of the region.
Genomic organization of chromosome 8p23.1 is characterized by the existence of blocks of segmental duplications flanking the region, which are known to mediate a 4.7 Mb inversion [28]. The microsatellite markers with highest lodscores in the linkage analysis [26,27] are located telomerically with respect to the inverted region and within a cluster of α-defensin genes (Figure 1).
Figure 1.

Schematic representation of the α-defensin gene cluster on human chromosome 8p23.1. The marker with higher lodscore in the linkage analysis is localized as well as all the genes in the region and the segmental duplication (positions are based on hg17, May 2004 genome assembly).
The α-defensin cluster consists of five α-defensin genes (DEFA6, DEFA4, DEFA1, DEFA3 and DEFA5), five α-defensin pseudogenes (DEFA8P, DEFA9P, DEFA10P, DEFA11P and DEFA7P) and one θ-defensin pseudogene (DEFT1P) [36]. Three copies of a 19-kb repeat unit or copy number variant (CNV) were identified within the α-defensin cluster, which correspond to the DEFA1A3 CNV (based on May 2004 genome assembly). Each of the 19-kb repeats contained a copy of the DEFA1 or DEFA3 genes, but DEFA3 gene is known to be completely absent in a significant proportion of the population [26,27,32,34]. The description of these genomic features is relevant for the search of genetic modifying factors for A1555G mutation. Both, the presence of the polymorphic inversion and CNVs involving the α-defensin gene cluster could influence the phenotypic manifestation of deafness linked to A1555G mutation.
With the aim to investigate the role of DEFA3 absence in the phenotypic manifestation of A1555G mutation, we analysed the absence of DEFA3 gene in a group of 55 hearing impaired families or sporadic subjects with A1555G mutation (213 subjects; 135 deaf and 78 hearing) and 336 unrelated blood donor controls, all of Caucasian origin. Twenty-one of the families analysed were previously included in the whole-genome linkage analysis performed by Bykhovskaya and colleagues [26,27]. In this study, the families with non-parametric lodscore (GeneHunter) above 0.8 were considered linked to chromosome 8p23.1, and below 0 unlinked. Using these criteria, seven of the families tested (55 subjects; 31 deaf and 24 hearing) were considered linked to 8p23.1 and 14 (48 subjects; 30 deaf and 18 hearing) considered unlinked.
The frequency of individuals lacking DEFA3 in a control population was determined. A group of 336 subjects were tested for the absence of DEFA3, and found 42 individuals in whom DEFA3 gene was absent (12.5%). No differences were found in the rate of DEFA3 absence between deaf and hearing subjects in any of the situations considered: whole set of families, index cases versus control population individuals or subjects from families linked to 8p23.1 region versus controls (Table 1). The data were also analysed using a family based association test[35] under a recessive mode of inheritance, as DEFA3 complete absence is the only situation which could be unambiguously determined with our assay. In this case, an over-representation of DEFA3 absence was found in the affected group (Z = 2.36; p = 0.018) (Table 2). No distinction between linked and unlinked families was possible in this case, because of lack of statistical power to perform the calculations, as FBAT is based on the analysis of large sample groups.
Table 1.
DEFA3 gene absence in A1555G patients and control subjects.
| SAMPLES | Phenotype | DEFA3 | NO DEFA3 | p-value* |
| A1555G carriers (n = 213) | Deaf (n = 135) | 115 (85%) | 20 (15%) | 0.678 |
| Hearing (n = 78) | 69 (88%) | 9 (12%) | ||
| A1555G carriers (n = 213) | Deaf & Hearing | 184 (86%) | 29 (14%) | 0.283 |
| A1555G index cases (n = 55) | Deaf | 45 (82%) | 10 (18%) | 0.697 |
| A1555G linked samples (n = 55) | Deaf & Hearing | 52 (95%) | 3 (5%) | 0.171 |
| Controls (n = 336) | 294 (87.5%) | 42 (12.5%) | ||
* Between groups chi-square p-value resulting from the comparison of deaf vs hearing carriers or carriers vs. control population subjects.
Table 2.
Family based association study of DEFA3 gene absence in A1555G families.
| Marker | Genotype | Freq | Fam# | S | E(S) | Var(S) | Z | P |
| DEFA3 | present | 0.689 | 9 | 8.00 | 6.83 | 2.69 | 0.71 | 0.477 |
| absent | 0.311 | 9 | 11.00 | 6.67 | 3.36 | 2.36 | 0.018** |
# Number of informative families; S, observed transmission of genotype to affected offspring;
E(S), expected transmission under Mendelian inheritance; Var(S), variance; P, two-tailed P value;
**Significant P value after Bonferroni correction (P < 0.025).
CLDN23 gene is not involved in the phenotypic manifestation of A1555G
Claudins are a multigene family consisting of more than 20 members. They function as cell adhesion molecules working at tight junctions. An important function in the inner ear has been postulated for several claudin genes [37,38]. Taking into account the function of other claudin family members and the fact that CLDN23 gene is located nearby (1.8 Mb) the defined linkage region in chromosome 8p23.1, it was selected for mutational screening as a modifier candidate gene for A1555G deafness phenotype.
Sequencing of the CLDN23 gene coding sequence and flanking regions in A1555G pedigrees resulted in the identification of eight sequence variants or polymorphisms, five of them already reported in public databases (Figure 2). Three of the changes resulted in an amino acid change, but none of them was identified in homozygosity, neither the variants were found to segregate with the phenotype in the pedigrees where they were identified. In addition, a deletion of 12 bp in the 5'UTR of the gene was identified in heterozygosity in one deaf sample. However, the pedigree was not informative enough to state whether it has a role in the deafness phenotype.
Figure 2.

Genetic variants identified in CLDN23 gene (A) and MRPS18CP2 pseudogene (B). The rs entry for the previously described SNPs or the nucleotide positions for the new identified SNPs are given. Arrows represent the position of the primers used for the PCR amplification of the corresponding genomic fragments.
Although none of the variants segregated with the deafness phenotype in the analysed families, to completely rule out the involvement of CLDN23 gene as a modifying factor for A1555G mutation, a family based association test was performed (Table 3). The test could be only performed for two of the variants, as the others were found in a small number of samples. No significant association was found for any of the SNPs comparing the expected vs. observed transmission of each possible genotype (Table 3).
Table 3.
Family based association study of CLDN23 and MRPS18CP2 in A1555G families.
| Gene | Marker | Genotype | Freq | Fam# | S | E(S) | Var(S) | Z | P |
| CLDN23 | rs9644774 | GG | 0.38 | 5 | 4.00 | 4.98 | 1.32 | -0.82 | 0.41 |
| GA | 0.38 | 10 | 7.00 | 7.40 | 2.93 | -0.24 | 0.81 | ||
| AA | 0.24 | 8 | 7.00 | 5.65 | 2.21 | 0.91 | 0.36 | ||
| rs11995449 | GG | 0.56 | 6 | 7.00 | 6.17 | 2.06 | 0.58 | 0.56 | |
| GA | 0.36 | 7 | 5.00 | 6.83 | 2.44 | -1.17 | 0.24 | ||
| AA | 0.08 | 2 | NA | ||||||
| MRPS18CP2 | rs4841072 | AA | 0.38 | 8 | 13.00 | 8.78 | 3.48 | 2.26 | 0.02* |
| AC | 0.31 | 9 | 6.00 | 9.28 | 4.03 | -1.63 | 0.10 | ||
| CC | 0.31 | 3 | NA | ||||||
| rs17154962 | CC | 0.85 | 5 | 4.00 | 6.28 | 2.00 | -1.60 | 0.10 | |
| CT | 0.15 | 5 | 7.00 | 4.55 | 2.17 | 1.67 | 0.09 | ||
| TT | 0.00 | 2 | NA | ||||||
SNPs with less than 5 informative families were excluded from the analysis. NA; not applicable.
# Number of informative families; S, observed transmission of genotype to affected offspring;
E(S), expected transmission under Mendelian inheritance; Var(S), variance; P, two-tailed P value;
*Significant p value < 0.05.
MRPS18C pseudogene located on 8p23.1 is expressed in humans
Pseudogenes, in the case of protein-coding genes, are gene copies that have lost the ability to code for a protein. A processed pseudogene, i.e. made through mRNA retrotransposition, derived from mitochondrial ribosomal protein S18C gene (MRPS18C) was identified 2 Mb centromeric from D8S1819, the marker with a highest positive linkagee score on chromosome 8p23.1. The MRPS18CP2 pseudogene on chromosome 8p23.1 spans 293 bp, corresponding to the whole coding region of exons 1, 2, 5 and 6 of MRPS18C gene, but lacking all introns and exons 3 and 4. MRPS18CP2 pseudogene shares 96,9% homology with MRPS18C nucleotide coding sequence. There are 13 nucleotide alterations and a 6 bp deletion compared to MRPS18C gene (Figure 3).
Figure 3.
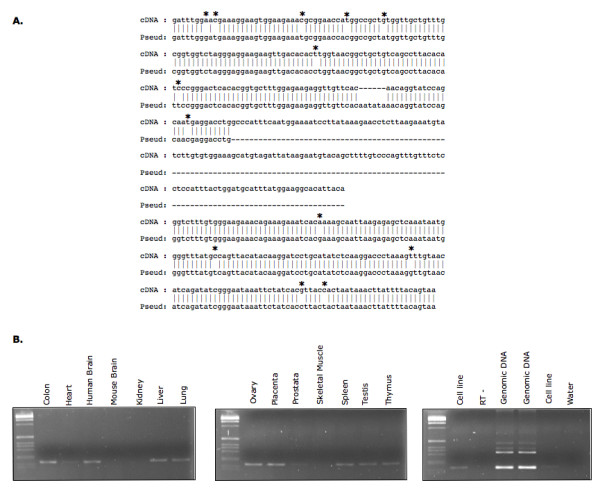
MRPS18CP2 sequence and expression analysis. (A) Alignment of MRPS18CP2 pseudogene with MRPS18C mRNA (GenBank accession number NM_016067). Asterisks indicate sequence changes between the gene mRNA and the chromosome 8p23.1 pseudogene. (B) RT-PCR experiments showing expression of a transcript containing MRPS18CP2 pseudogene in different tissues.
Despite lacking the original promoter, a processed pseudogene can occasionally be transcribed [39]. In the public databases, neither mRNAs nor ESTs are annotated for MRPS18CP2 pseudogene in chromosome 8p23.1. To check whether MRPS18CP2 is transcribed, its expression was assessed by RT-PCR experiments using total RNA from different human tissues, human lymphoblastoid cell lines and mouse brain. A transcript containing MRPS18CP2 was found to be expressed in all tested tissues, except for human kidney, human skeletal muscle and mouse brain (Figure 3).
Based on the physical localization of MRPS18CP2, its expression pattern and the function of its corresponding coding gene, MRPS18CP2, was selected for a genetic screening as a candidate to be involved in the phenotypic manifestation of A1555G mutation. The mutational screening of MRPS18CP2 pseudogene in A1555G pedigrees resulted in the identification of seven polymorphisms, three of them already reported in public databases (Figure 2). None of the SNPs segregate with the deafness phenotype in any of the A1555G pedigrees analysed. A family based association analysis was also performed for the two informative SNPs identified (Table 3). In the case of SNP rs4841072, an overtransmission of the AA genotype (Z = 2.26; p = 0.02) was found associated to the disease, although after Bonferroni correction statistical significance was no longer supported (Table 3).
Discussion
Large-scale chromosomal rearrangements, such as duplications, deletions and inversions, are now known to be common in the human genome [40]. The substrates for these common rearrangements are generally highly homologous sequences, known as segmental duplications or LCRs, which flank the rearranged genomic segment [41]. To take into account genomic structural variation is crucial in linkage studies of human diseases for different reasons. First, when a fixed marker order is assumed for all individuals in an inverted region, one tends to see spurious recombination events among inversion carriers and/or to find genotyping contradictions, which may lead to discard some observations. In addition, the polymorphic genomic structure of the rearranged regions, which apart from large-scale genomic rearrangements can include sequences that vary in copy number, might complicate the mapping of putative disease genes. Chromosome 8p23.1 is such a region where a common neutral inversion mediated by clusters of olfactory-receptor genes, is present in a variable proportion of subjects, depending on the population [28-30]. The position of a major nuclear modifier gene for the deafness phenotype linked to A1555G mtDNA mutation has been localized to chromosome 8p23.1 [27], but the identification of this gene has remained elusive. This lack of progress may be partially explained because of 8p23.1 genomic organization.
In an attempt to further study the putative genetic modifying factors for A1555G mutation, including those derived from the presence of segmental duplications, we have performed a detailed analysis of three 8p23.1 candidate genetic features: CLDN23 gene, MRPS18CP2 pseudogene and DEFA3 gene absence. CLDN23 gene and MRPS18CP2 pseudogene were selected based on their putative biological role in the inner ear, whereas DEFA3 gene absence was tested due to its close location to the marker with a higher lodscore.
Claudins are essential components of tight junctions [42] and therefore, they play important roles in the physiological function of the inner ear. Tight junctions are well developed in the epithelial cell layers that delineate the inner ear compartments containing perilymph and endolymph, to prevent intercellular leakage of solutes and ions [43]. In fact, mutation of the Claudin-14 gene was reported to cause human hereditary deafness [38] and Claudin-11 null mice exhibit severe deafness associated with low endocochlear potential [37]. In addition, at least 10 species of claudins are expressed in the inner ear [44].
Pseudogenes are non-functional sequences of genomic DNA originally derived from functional genes [45]. The human genome encodes at least 79 mitochondrial ribosomal proteins from which more than 100 pseudogenes have been identified [46]. Located on chromosome 8p23.1, there is MRPS18CP2, a processed pseudogene of mitochondrial ribosomal protein S18C (MRPS18C). Five other pseudogenes derived from MRPS18C gene are located in the human genome on chromosomes 3q26.1, 8p21.3, 12p13.31, 15q11.2 and 22q13.31 respectively [46]. Interestingly, the MRPS18C pseudogene on chromosome 15q11.2 is located only 1-Mb apart from a microsatellite marker, which gave a positive linkage score in the analysis performed by Bykovskaya and colleagues [26]. It has been postulated that pseudogenes may play regulatory roles for the genes from which they have been derived, such as serving as a source of antisense RNA [45]. Taking all these evidences into account and regardless that the functional role of pseudogenes is not clear, MRPS18CP2 was considered a good candidate.
None of the identified SNPs in either CLDN23 or MRPS18CP2 segregate with the phenotype in A1555G families, but as modifying factors are likely to be multiple [25,26], this observation did not provide enough evidence to completely discard their contribution in the A1555G deafness phenotype. Thus, a family-based association test was used to analyse the genotype data from CLDN23 gene and MRPS18CP2 pseudogene. Family-based association designs are particularly attractive, since they test for linkage as well as association, avoid spurious associations caused by admixture of populations, and are convenient for investigators interested in refining linkage findings in family samples [35]. With this approach, a weak positive association with a single SNP in MRPS18CP2 pseudogene was found. Although most of the analysed samples come from the same geographic area, founder effects do not account for the association found as it was previously reported [47,48].
These results, although have to be taken with caution, are of great interest as they may suggest a possible role for MRPS18CP2 pseudogene. Three sequence variants have been found for MRPS18 protein of the small mitochondrial ribosome subunit. In analogy to bacterial ribosomes, it is likely that each mitochondrial ribosome contains a single copy of MRPS18. Therefore, the presence of three different isoforms suggests that there is a heterogeneous population of mitochondrial ribosomes, which may have different decoding properties and may be subjected to a precise regulation of its expression [47]. The existence of MRPS18 pseudogenes could play a role in the regulation of each isoform expression, for example by blocking the expression of the corresponding gene. If this is demonstrated, it could explain the tissue specificity of A1555G homoplasmic mtDNA mutation, leading to a clinical phenotype confined in the cochlea. Thus, additional studies involving typing of additional SNPs in gene-coding and regulatory regions in additional A1555G families are needed, especially in the case of pseudogenes, whose putative biological function is still unclear.
CNVs have been proposed to have an important role in the pathological variation in the human population [49]. The DEFA1A3 CNV is located within the region previously described to contain a major modifying locus for mutation A1555G [32,34]. On the premise that the presence of a gene in multiple copies could have a dosage effect and therefore, contribute to genetic basis of some complex disorders, it is feasible that the copy number polymorphism of α-defensin cluster could be involved in the pathogenesis associated to the A1555G mutation. An overrepresentation of DEFA3 gene absence was found in deaf A1555G carriers. Defensins are small cationic peptides that form an important part of the innate immune system. It is difficult to establish a direct relationship between defensin function and A1555G deaf phenotype. However, as the distinction between DEFA1 and DEFA3 is based on the typing of a single SNP (C3400A), the differences in the rate of DEFA3 gene absence observed between deaf and hearing carriers of A1555G mutation could be considered as a positive association signal that confirms the localization of a modifier factor.
Conclusion
Both positive results found in MRPS18CP2 pseudogene and DEFA3 gene absence within the deaf group of A1555G carriers are weak associations, which do not demonstrate a role in the phenotype linked to A1555G mtDNA mutation. However, they provide further evidences of the involvement of 8p23.1 region as a modifying factor for A1555G mutation. Further analyses in additional families, as well as functional studies, which should shed light on the function of these genetic features, are needed in order to confirm or discard the associations found between 8p23.1 genes and A1555G hearing impairment.
Competing interests
The author(s) declare that they have no competing interests.
Authors' contributions
EB carried out the molecular genetic studies, participated in the statistical analysis and drafted the manuscript. JMM carried out the statistical analysis of the data and participated in the interpretation of the data. NFG participated in the design of the study and coordination. XE conceived the study and participated in its design and coordination and helped to draft the manuscript. All authors have read and approved the final version of the manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Acknowledgments
Acknowledgements
We thank the patients for participation in the study. This work was supported by "La Marató de TV3" (993610), "Instituto de Salud Carlos III, FIS-ISCIII" (G03/203, PI052347 and CIBER-CB06/02/0058) and "Generalitat de Catalunya" (2005SGR00008). The Spanish National Genotyping Center (CeGen) is founded by "Genoma España". EB is recipient of a fellowship from "Generalitat de Catalunya" (2003FI00066). JMM was supported by the CRG under project SAF2002-00799 (Spanish Ministry of Science and Education), and by a fellowship of the Danone Institute. NFG acknowledges support from NIH/NIDCD grant RO1DC01402.
Contributor Information
Ester Ballana, Email: ester.ballana@gmail.com.
Josep Maria Mercader, Email: josepmaria.mercader@crg.es.
Nathan Fischel-Ghodsian, Email: nathan.fischel@cshs.org.
Xavier Estivill, Email: xavier.estivill@crg.es.
References
- DiMauro S, Schon EA. Mitochondrial DNA mutations in human disease. Am J Med Genet. 2001;106:18–26. doi: 10.1002/ajmg.1392. [DOI] [PubMed] [Google Scholar]
- Jacobs HT, Hutchin TP, Kappi T, Gillies G, Minkkinen K, Walker J, Thompson K, Rovio AT, Carella M, Melchionda S, Zelante L, Gasparini P, Pyykko I, Shah ZH, Zeviani M, Mueller RF. Mitochondrial DNA mutations in patients with postlingual, nonsyndromic hearing impairment. Eur J Hum Genet. 2005;13:26–33. doi: 10.1038/sj.ejhg.5201250. [DOI] [PubMed] [Google Scholar]
- Yoshida M, Shintani T, Hirao M, Himi T, Yamaguchi A, Kikuchi K. Aminoglycoside-induced hearing loss in a patient with the 961 mutation in mitochondrial DNA. ORL J Otorhinolaryngol Relat Spec. 2002;64:219–222. doi: 10.1159/000058028. [DOI] [PubMed] [Google Scholar]
- Li R, Xing G, Yan M, Cao X, Liu XZ, Bu X, Guan MX. Cosegregation of C-insertion at position 961 with the A1555G mutation of the mitochondrial 12S rRNA gene in a large Chinese family with maternally inherited hearing loss. Am J Med Genet A. 2004;124:113–117. doi: 10.1002/ajmg.a.20305. [DOI] [PubMed] [Google Scholar]
- Bacino C, Prezant TR, Bu X, Fournier P, Fischel-Ghodsian N. Susceptibility mutations in the mitochondrial small ribosomal RNA gene in aminoglycoside induced deafness. Pharmacogenetics. 1995;5:165–172. doi: 10.1097/00008571-199506000-00005. [DOI] [PubMed] [Google Scholar]
- Wang Q, Li QZ, Han D, Zhao Y, Zhao L, Qian Y, Yuan H, Li R, Zhai S, Young WY, Guan MX. Clinical and molecular analysis of a four-generation Chinese family with aminoglycoside-induced and nonsyndromic hearing loss associated with the mitochondrial 12S rRNA C1494T mutation. Biochem Biophys Res Commun. 2006;340:583–588. doi: 10.1016/j.bbrc.2005.12.045. [DOI] [PubMed] [Google Scholar]
- Zhao H, Li R, Wang Q, Yan Q, Deng JH, Han D, Bai Y, Young WY, Guan MX. Maternally inherited aminoglycoside-induced and nonsyndromic deafness is associated with the novel C1494T mutation in the mitochondrial 12S rRNA gene in a large Chinese family. Am J Hum Genet. 2004;74:139–152. doi: 10.1086/381133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prezant TR, Agapian JV, Bohlman MC, Bu X, Oztas S, Qiu WQ, Arnos KS, Cortopassi GA, Jaber L, Rotter JI, et al. Mitochondrial ribosomal RNA mutation associated with both antibiotic-induced and non-syndromic deafness. Nat Genet. 1993;4:289–294. doi: 10.1038/ng0793-289. [DOI] [PubMed] [Google Scholar]
- Fischel-Ghodsian N, Prezant TR, Bu X, Oztas S. Mitochondrial ribosomal RNA gene mutation in a patient with sporadic aminoglycoside ototoxicity. Am J Otolaryngol. 1993;14:399–403. doi: 10.1016/0196-0709(93)90113-L. [DOI] [PubMed] [Google Scholar]
- Hutchin T, Haworth I, Higashi K, Fischel-Ghodsian N, Stoneking M, Saha N, Arnos C, Cortopassi G. A molecular basis for human hypersensitivity to aminoglycoside antibiotics. Nucleic Acids Res. 1993;21:4174–4179. doi: 10.1093/nar/21.18.4174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Estivill X, Govea N, Barcelo E, Badenas C, Romero E, Moral L, Scozzri R, D'Urbano L, Zeviani M, Torroni A. Familial progressive sensorineural deafness is mainly due to the mtDNA A1555G mutation and is enhanced by treatment of aminoglycosides. Am J Hum Genet. 1998;62:27–35. doi: 10.1086/301676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casano RA, Bykhovskaya Y, Johnson DF, Hamon M, Torricelli F, Bigozzi M, Fischel-Ghodsian N. Hearing loss due to the mitochondrial A1555G mutation in Italian families. Am J Med Genet. 1998;79:388–391. doi: 10.1002/(SICI)1096-8628(19981012)79:5<388::AID-AJMG11>3.0.CO;2-N. [DOI] [PubMed] [Google Scholar]
- Hutchin TP, Lench NJ, Arbuzova S, Markham AF, Mueller RF. Maternally inherited hearing impairment in a family with the mitochondrial DNA A7445G mutation. Eur J Hum Genet. 2001;9:56–58. doi: 10.1038/sj.ejhg.5200581. [DOI] [PubMed] [Google Scholar]
- Tekin M, Duman T, Bogoclu G, Incesulu A, Comak E, Fitoz S, Yilmaz E, Ilhan I, Akar N. Frequency of mtDNA A1555G and A7445G mutations among children with prelingual deafness in Turkey. Eur J Pediatr. 2003;162:154–158. doi: 10.1007/s00431-002-1129-z. [DOI] [PubMed] [Google Scholar]
- Hyslop SJ, James AM, Maw M, Fischel-Ghodsian N, Murphy MP. The effect on mitochondrial function of the tRNA Ser(UCN)/COI A7445G mtDNA point mutation associated with maternally-inherited sensorineural deafness. Biochem Mol Biol Int. 1997;42:567–575. doi: 10.1080/15216549700202971. [DOI] [PubMed] [Google Scholar]
- Hutchin TP, Navarro-Coy NC, Van Camp G, Tiranti V, Zeviani M, Schuelke M, Jaksch M, Newton V, Mueller RF. Multiple origins of the mtDNA 7472insC mutation associated with hearing loss and neurological dysfunction. Eur J Hum Genet. 2001;9:385–387. doi: 10.1038/sj.ejhg.5200640. [DOI] [PubMed] [Google Scholar]
- Verhoeven K, Ensink RJ, Tiranti V, Huygen PL, Johnson DF, Schatteman I, Van Laer L, Verstreken M, Van de Heyning P, Fischel-Ghodsian N, Zeviani M, Cremers CW, Willems PJ, Van Camp G. Hearing impairment and neurological dysfunction associated with a mutation in the mitochondrial tRNASer(UCN) gene. Eur J Hum Genet. 1999;7:45–51. doi: 10.1038/sj.ejhg.5200247. [DOI] [PubMed] [Google Scholar]
- Hutchin TP, Parker MJ, Young ID, Davis AC, Pulleyn LJ, Deeble J, Lench NJ, Markham AF, Mueller RF. A novel mutation in the mitochondrial tRNA(Ser(UCN)) gene in a family with non-syndromic sensorineural hearing impairment. J Med Genet. 2000;37:692–694. doi: 10.1136/jmg.37.9.692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapiro E, Feldmann D, Denoyelle F, Sternberg D, Jardel C, Eliot MM, Bouccara D, Weil D, Garabedian EN, Couderc R, Petit C, Marlin S. Two large French pedigrees with non syndromic sensorineural deafness and the mitochondrial DNA T7511C mutation: evidence for a modulatory factor. Eur J Hum Genet. 2002;10:851–856. doi: 10.1038/sj.ejhg.5200894. [DOI] [PubMed] [Google Scholar]
- Ishikawa K, Tamagawa Y, Takahashi K, Kimura H, Kusakari J, Hara A, Ichimura K. Nonsyndromic hearing loss caused by a mitochondrial T7511C mutation. Laryngoscope. 2002;112:1494–1499. doi: 10.1097/00005537-200208000-00030. [DOI] [PubMed] [Google Scholar]
- Ballana E, Morales E, Rabionet R, Montserrat B, Ventayol M, Bravo O, Gasparini P, Estivill X. Mitochondrial 12S rRNA gene mutations affect RNA secondary structure and lead to variable penetrance in hearing impairment. Biochem Biophys Res Commun. 2006;341:950–957. doi: 10.1016/j.bbrc.2006.01.049. [DOI] [PubMed] [Google Scholar]
- Guan MX, Fischel-Ghodsian N, Attardi G. A biochemical basis for the inherited susceptibility to aminoglycoside ototoxicity. Hum Mol Genet. 2000;9:1787–1793. doi: 10.1093/hmg/9.12.1787. [DOI] [PubMed] [Google Scholar]
- Guan MX, Fischel-Ghodsian N, Attardi G. Nuclear background determines biochemical phenotype in the deafness-associated mitochondrial 12S rRNA mutation. Hum Mol Genet. 2001;10:573–580. doi: 10.1093/hmg/10.6.573. [DOI] [PubMed] [Google Scholar]
- Guan MX. Molecular pathogenetic mechanism of maternally inherited deafness. Ann N Y Acad Sci. 2004;1011:259–271. doi: 10.1196/annals.1293.025. [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y, Shohat M, Ehrenman K, Johnson D, Hamon M, Cantor RM, Aouizerat B, Bu X, Rotter JI, Jaber L, Fischel-Ghodsian N. Evidence for complex nuclear inheritance in a pedigree with nonsyndromic deafness due to a homoplasmic mitochondrial mutation. Am J Med Genet. 1998;77:421–426. doi: 10.1002/(SICI)1096-8628(19980605)77:5<421::AID-AJMG13>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- Bykhovskaya Y, Estivill X, Taylor K, Hang T, Hamon M, Casano RA, Yang H, Rotter JI, Shohat M, Fischel-Ghodsian N. Candidate locus for a nuclear modifier gene for maternally inherited deafness. Am J Hum Genet. 2000;66:1905–1910. doi: 10.1086/302914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bykhovskaya Y, Yang H, Taylor K, Hang T, Tun RY, Estivill X, Casano RA, Majamaa K, Shohat M, Fischel-Ghodsian N. Modifier locus for mitochondrial DNA disease: linkage and linkage disequilibrium mapping of a nuclear modifier gene for maternally inherited deafness. Genet Med. 2001;3:177–180. doi: 10.1097/00125817-200105000-00005. [DOI] [PubMed] [Google Scholar]
- Giglio S, Broman KW, Matsumoto N, Calvari V, Gimelli G, Neumann T, Ohashi H, Voullaire L, Larizza D, Giorda R, Weber JL, Ledbetter DH, Zuffardi O. Olfactory receptor-gene clusters, genomic-inversion polymorphisms, and common chromosome rearrangements. Am J Hum Genet. 2001;68:874–883. doi: 10.1086/319506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giglio S, Calvari V, Gregato G, Gimelli G, Camanini S, Giorda R, Ragusa A, Guerneri S, Selicorni A, Stumm M, Tonnies H, Ventura M, Zollino M, Neri G, Barber J, Wieczorek D, Rocchi M, Zuffardi O. Heterozygous submicroscopic inversions involving olfactory receptor-gene clusters mediate the recurrent t(4;8)(p16;p23) translocation. Am J Hum Genet. 2002;71:276–285. doi: 10.1086/341610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugawara H, Harada N, Ida T, Ishida T, Ledbetter DH, Yoshiura K, Ohta T, Kishino T, Niikawa N, Matsumoto N. Complex low-copy repeats associated with a common polymorphic inversion at human chromosome 8p23. Genomics. 2003;82:238–244. doi: 10.1016/S0888-7543(03)00108-3. [DOI] [PubMed] [Google Scholar]
- Mars WM, Patmasiriwat P, Maity T, Huff V, Weil MM, Saunders GF. Inheritance of unequal numbers of the genes encoding the human neutrophil defensins HP-1 and HP-3. J Biol Chem. 1995;270:30371–30376. doi: 10.1074/jbc.270.51.30371. [DOI] [PubMed] [Google Scholar]
- Aldred PM, Hollox EJ, Armour JA. Copy number polymorphism and expression level variation of the human alpha-defensin genes DEFA1 and DEFA3. Hum Mol Genet. 2005;14:2045–2052. doi: 10.1093/hmg/ddi209. [DOI] [PubMed] [Google Scholar]
- Hollox EJ, Armour JA, Barber JC. Extensive normal copy number variation of a beta-defensin antimicrobial-gene cluster. Am J Hum Genet. 2003;73:591–600. doi: 10.1086/378157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linzmeier RM, Ganz T. Human defensin gene copy number polymorphisms: comprehensive analysis of independent variation in alpha- and beta-defensin regions at 8p22-p23. Genomics. 2005;86:423–430. doi: 10.1016/j.ygeno.2005.06.003. [DOI] [PubMed] [Google Scholar]
- Horvath S, Laird NM, Knapp M. The transmission/disequilibrium test and parental-genotype reconstruction for X-chromosomal markers. Am J Hum Genet. 2000;66:1161–1167. doi: 10.1086/302823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballana E, Gonzalez JR, Bosch N, Estivill X. Inter-population variability of DEFA3 gene absence: correlation with haplotype structure and population variability. BMC Genomics. 2007;8:14. doi: 10.1186/1471-2164-8-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gow A, Davies C, Southwood CM, Frolenkov G, Chrustowski M, Ng L, Yamauchi D, Marcus DC, Kachar B. Deafness in Claudin 11-null mice reveals the critical contribution of basal cell tight junctions to stria vascularis function. J Neurosci. 2004;24:7051–7062. doi: 10.1523/JNEUROSCI.1640-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilcox ER, Burton QL, Naz S, Riazuddin S, Smith TN, Ploplis B, Belyantseva I, Ben-Yosef T, Liburd NA, Morell RJ, Kachar B, Wu DK, Griffith AJ, Friedman TB. Mutations in the gene encoding tight junction claudin-14 cause autosomal recessive deafness DFNB29. Cell. 2001;104:165–172. doi: 10.1016/S0092-8674(01)00200-8. [DOI] [PubMed] [Google Scholar]
- Harrison PM, Zheng D, Zhang Z, Carriero N, Gerstein M. Transcribed processed pseudogenes in the human genome: an intermediate form of expressed retrosequence lacking protein-coding ability. Nucleic Acids Res. 2005;33:2374–2383. doi: 10.1093/nar/gki531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebat J, Lakshmi B, Troge J, Alexander J, Young J, Lundin P, Maner S, Massa H, Walker M, Chi M, Navin N, Lucito R, Healy J, Hicks J, Ye K, Reiner A, Gilliam TC, Trask B, Patterson N, Zetterberg A, Wigler M. Large-scale copy number polymorphism in the human genome. Science. 2004;305:525–528. doi: 10.1126/science.1098918. [DOI] [PubMed] [Google Scholar]
- Lupski JR. Genomic disorders: structural features of the genome can lead to DNA rearrangements and human disease traits. Trends Genet. 1998;14:417–422. doi: 10.1016/S0168-9525(98)01555-8. [DOI] [PubMed] [Google Scholar]
- Furuse M, Fujita K, Hiiragi T, Fujimoto K, Tsukita S. Claudin-1 and -2: novel integral membrane proteins localizing at tight junctions with no sequence similarity to occludin. J Cell Biol. 1998;141:1539–1550. doi: 10.1083/jcb.141.7.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wangemann P. Comparison of ion transport mechanisms between vestibular dark cells and strial marginal cells. Hear Res. 1995;90:149–157. doi: 10.1016/0378-5955(95)00157-2. [DOI] [PubMed] [Google Scholar]
- Kitajiri SI, Furuse M, Morita K, Saishin-Kiuchi Y, Kido H, Ito J, Tsukita S. Expression patterns of claudins, tight junction adhesion molecules, in the inner ear. Hear Res. 2004;187:25–34. doi: 10.1016/S0378-5955(03)00338-1. [DOI] [PubMed] [Google Scholar]
- Balakirev ES, Ayala FJ. Pseudogenes: are they "junk" or functional DNA? Annu Rev Genet. 2003;37:123–151. doi: 10.1146/annurev.genet.37.040103.103949. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Gerstein M. Identification and characterization of over 100 mitochondrial ribosomal protein pseudogenes in the human genome. Genomics. 2003;81:468–480. doi: 10.1016/S0888-7543(03)00004-1. [DOI] [PubMed] [Google Scholar]
- Cavdar Koc E, Burkhart W, Blackburn K, Moseley A, Spremulli LL. The small subunit of the mammalian mitochondrial ribosome. Identification of the full complement of ribosomal proteins present. J Biol Chem. 2001;276:19363–19374. doi: 10.1074/jbc.M100727200. [DOI] [PubMed] [Google Scholar]
- Torroni A, Cruciani F, Rengo C, Sellitto D, Lopez-Bigas N, Rabionet R, Govea N, Lopez De Munain A, Sarduy M, Romero L, Villamar M, del Castillo I, Moreno F, Estivill X, Scozzari R. The A1555G mutation in the 12S rRNA gene of human mtDNA: recurrent origins and founder events in families affected by sensorineural deafness. Am J Hum Genet. 1999;65:1349–1358. doi: 10.1086/302642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redon R, Ishikawa S, Fitch KR, Feuk L, Perry GH, Andrews TD, Fiegler H, Shapero MH, Carson AR, Chen W, Cho EK, Dallaire S, Freeman JL, Gonzalez JR, Gratacos M, Huang J, Kalaitzopoulos D, Komura D, MacDonald JR, Marshall CR, Mei R, Montgomery L, Nishimura K, Okamura K, Shen F, Somerville MJ, Tchinda J, Valsesia A, Woodwark C, Yang F, Zhang J, Zerjal T, Armengol L, Conrad DF, Estivill X, Tyler-Smith C, Carter NP, Aburatani H, Lee C, Jones KW, Scherer SW, Hurles ME. Global variation in copy number in the human genome. Nature. 2006;444:444–454. doi: 10.1038/nature05329. [DOI] [PMC free article] [PubMed] [Google Scholar]