

Abstract
Mutations of the CFTR, a phosphorylation-regulated Cl− channel, cause cystic fibrosis. Activation of CFTR by PKA stimulation appears to be mediated by a complex interaction between several consensus phosphorylation sites in the regulatory domain (R domain). None of these sites has a critical role in this process. Here, we show that although endogenous phosphorylation by PKC is required for the effect of PKA on CFTR, stimulation of PKC by itself has only a minor effect on human CFTR. In contrast, CFTR from the amphibians Necturus maculosus and Xenopus laevis (XCFTR) can be activated to similar degrees by stimulation of either PKA or PKC. Furthermore, the activation of XCFTR by PKC is independent of the net charge of the R domain, and mutagenesis experiments indicate that a single site (Thr665) is required for the activation of XCFTR. Human CFTR lacks the PKC phosphorylation consensus site that includes Thr665, but insertion of an equivalent site results in a large activation upon PKC stimulation. These observations establish the presence of a novel mechanism of activation of CFTR by phosphorylation of the R domain, i.e., activation by PKC requires a single consensus phosphorylation site and is unrelated to the net charge of the R domain.
Keywords: chloride channel, PKA, ABC proteins, R domain
INTRODUCTION
Cystic fibrosis results from mutations in the CFTR, a 1,480–amino acid protein that functions as a Cl−-selective ion channel in a variety of epithelial cells. CFTR has two topologically equivalent halves, each consisting of a transmembrane domain (six membrane-spanning helices) followed by a nucleotide-binding domain. The cytoplasmic regulatory domain (R domain) links these two halves. The membrane-spanning helices form the anion-conductive pore, the nucleotide-binding domains bind and hydrolyze ATP and are involved in channel gating, and the R domain contains consensus phosphorylation sites for PKA and PKC (for review see Seibert et al. 1997).
CFTR channels are gated by exposure to PKA and ATP, both in excised patches and after purification and reconstitution in planar lipid bilayers (for review see Seibert et al. 1997). Although it is well established that phosphorylation of the R domain is essential for activation of CFTR, the precise molecular mechanism of this activation has proven elusive, and is thought to involve several sites and complex interactions (for reviews see Chang et al. 1993; Gadsby and Nairn 1999; Sheppard and Welsh 1999; Ma 2000).
PKA-mediated activation of human CFTR (hCFTR) requires endogenous phosphorylation by PKC (Cohn et al. 1991; Jia et al. 1997; Vankeerberghen et al. 1999). However, PKC-mediated phosphorylation, per se, has only a small effect on hCFTR activity. In contrast, we discovered that Necturus CFTR can be activated by either PKA- or PKC-mediated phosphorylation (Copello et al. 1993). The same phenomenon occurs with Xenopus CFTR (XCFTR), as shown in this study.
The goal of the present work was to determine the molecular bases for the differences in the activation of hCFTR and XCFTR by PKC stimulation. Our results show that, in contrast with the complexity of PKA-mediated activation, the stimulation of XCFTR by PKC depends on a single PKC consensus phosphorylation site in the R domain. Confirming the importance of the critical site in XCFTR, engineering an equivalent site in hCFTR makes this molecule responsive to PKC stimulation.
MATERIALS AND METHODS
cRNA Preparation
Full-length hCFTR and XCFTR cDNAs were subcloned into the oocyte expression vector pOCYT7 (gift from Dr. Nancy Wills, University of Texas Medical Branch; Mo et al. 1999). This expression vector contains the Xenopus 5′ and 3′ β-globin untranslated regions to boost expression in oocytes (Mo et al. 1999). In brief, human CFTR (a gift from Dr. Lap-Chee Tsui, The Hospital for Sick Children, Toronto, Canada), was cut with XmaI and SalI and ligated into pOCTY7 cut with the same restriction enzymes. Xenopus CFTR (a gift from Drs. Margaret Price and Michael Welsh, University of Iowa, Iowa City, IA; Price et al. 1996), was cloned into the KpnI and SalI sites of pOCYT7. The resulting vectors were linearized with XhoI and used as templates for T7-directed capped cRNA synthesis (mMessage mMachine; Ambion).
Oocyte Preparation and cRNA Injection
Xenopus laevis oocytes were isolated and prepared using a well-documented protocol (Sharon et al. 1997). In brief, ovarian lobes were surgically removed from frogs anesthetized with tricaine methanesulfonate (1 g/liter). Stage-V and -VI oocytes were isolated by manual dissection and defolliculated using enzymatic treatment with 1 mg/ml type I collagenase in Barth's solution ([in mM] 88 NaCl, 1.0 KCl, 1.0 CaCl2, 1.0 MgCl2, and 10 HEPES/NaOH, pH 7.40) containing 10 μg/ml penicillin, 10 μg/ml streptomycin, and 100 μg/ml gentamicin sulfate, for 16 h (all incubations were performed at 16°C). The enzymatic treatment was followed by a 2-h incubation in calcium-free Barth's solution. Treated oocytes were transferred to Barth's solution. Oocytes were injected with 47 nl of CFTR cRNA (5–10 ng) or sterile water 24–72 h before analysis, using a Nanoject autoinjector (Drummond Scientific). The 8-Br-cAMP–activated currents in the mutant H667R-hCFTR were very high; thus, cRNA injection was reduced to 1–2.5 ng/oocyte.
Two-electrode Voltage Clamp
Oocytes were bathed in the HEPES-buffered solution ND96 ([in mM] 96 NaCl, 2 KCl, 1.0 MgCl2, 1.8 CaCl2, and 5 HEPES/NaOH, pH 7.40). All experiments were performed at 22–24°C. Borosilicate microelectrodes were pulled with a horizontal puller (P-97; Sutter Instruments), filled with 3 M KCl, and had tip resistances of 0.5–1.5 MΩ when immersed in ND96 solution. A voltage-clamp amplifier (model OC-725C; Warner Instruments) was used to measure whole-oocyte conductance. Voltages were referenced to the bath. Membrane currents were filtered at 1.0 kHz, digitized, stored, and analyzed with pCLAMP version 8.0 (Axon Instruments). I-V relationships were obtained by current measurements 400 ms after changing the potential from a holding value of −30 mV to test values ranging from −100 to +30 mV, in 10-mV steps, with 100-ms intervals between pulses. Oocyte conductance was determined in the consistently linear range between the reversal potential and 30 mV. The membrane conductances of unstimulated oocytes (i.e., no 8-Br-cAMP or PMA) after cRNA injection were not different from those of water-injected oocytes (Table ). In hCFTR- and XCFTR-expressing oocytes, membrane conductances were directly proportional to the amount of cRNA injected (not shown) and time after injection (Table ). In all experiments detailed here, supra-maximal concentrations of either cAMP cocktail (250 μM 8-Br-cAMP and 25 μM forskolin) or PMA (250 nM) were used to stimulate maximally PKA or PKC, respectively. For the time course experiments, differences between the current at −30 mV (the holding potential) and 0 mV were measured every 5 s. Halide selectivity sequences were determined from the changes in reversal potential after substituting NaCl with the corresponding sodium-halide salt for 2 min before the I-V plots. Corrections for liquid junction potentials were as previously described (Vanoye et al. 1997).
Table 1.
Conductances of Xenopus Oocytes Expressing CFTR
| Unstimulated | cAMP-stimulated | |||
|---|---|---|---|---|
| 24 h | 48 h | 72 h | ||
| Mock | 2.9 ± 1.7 (11) | 2.7 ± 1.4 (4) | 2.9 ± 1.6 (4) | 3.2 ± 2.1 (3) |
| hCFTR | 4.2 ± 3.8 (35) | 426 ± 60 (16) | 685 ± 131 (13) | 1,147 ± 217 (6) |
| XCFTR | 3.1 ± 2.1 (34) | 214 ± 50 (14) | 1,009 ± 268 (14) | 2,028 ± 217 (6) |
Xenopus laevis oocytes were injected with water (mock), hCFTR cRNA, or XCFTR cRNA, and their membrane conductances were measured at 24, 48, or 72 h after injection with and without stimulation with cAMP cocktail. Conductances are expressed in microSiemens. Each result represents the mean ± SEM of n experiments (numbers in parentheses).
Whole-cell Patch Clamp
Whole cell patch-clamp studies were performed essentially as previously described (Vanoye et al. 1997). COS-1 cells transiently transfected with hCFTR or XCFTR cDNA into the pTracer-CMV vector (Invitrogen) were studied after 2–3 d. The bath solution contained (in mM): 140 NMDG, 0.5 MgCl2, 1.3 CaCl2, and 10 HEPES/NaOH, pH 7.4. The pipet solution contained (in mM): 140 NMDG, 1.2 MgCl2, 1 EGTA, 2 ATP 10 mM HEPES/NaOH, pH 7.2. Patch pipets (2–4 MΩ tip resistance in nearly symmetric NMDG-Cl) were pulled with a horizontal pipet puller (model P-97; Sutter Instruments). Negative pressure was used to rupture the membrane patch after obtaining a gigaohm seal, and currents were measured in the whole-cell configuration with an amplifier (model Axopatch 200A; Axon Instruments). The holding voltage (Vm) was −60 mV. I-V relationships were constructed using 400-ms voltage pulses between −80 to +80 mV, at 10-mV intervals. PClamp6 (Axon Instruments) was used for generation of the pulses, data collection, and analysis.
Human-Xenopus R Domain Chimera Construction
A chimera, in which residues 607–811 of the hCFTR were replaced by the corresponding XCFTR residues, was engineered by a combination of site-directed mutagenesis and PCR. Site-directed mutagenesis was used to introduce MluI and KasI unique sites at the 5′ and 3′ ends of the R domain hCFTR cDNA sequence with silent mutations. The QuickChange site-directed mutagenesis kit (Stratagene) was used to insert the unique restriction sites. Two mutagenic oligonucleotide primers, each complementary to opposite strands of the vector, were used for each mutagenesis reaction. The wild-type hCFTR was used as a template. The primers used were as follows (only sense primer is shown, restriction sites underlined): 5′-TGGCTAACAAAACGCG TATTTTGGTCAC-3′ (MluI site) and 5′-GGATATATATTCAAG GCGCCTATCTCAAGAAAC-3′ (KasI site). The reaction mixture was heated at 95°C for 45 s and subjected to 12 cycles for 30 s at 95°C, 30 s at 55°C, and 10 min at 72°C. Cycling was finished by a 10-min incubation at 70°C. The PCR reaction was treated with DpnI to digest the DNA template, and then used to transform Escherichia coli. The XCFTR R domain cDNA was amplified by PCR, introducing MluI and KasI sites that were used to exchange the hCFTR and XCFTR R domain sequences. The PCR primers were as follows (MluI and KasI sites underlined): 5′-GGGACGCG TATTTTAGTTACATCT AAAGTCG-3′ (forward) and 5′-TTTGAAGTGGATATATATAATAGGCGCCGCG-3′ (reverse).
PKC Consensus Site Mutagenesis
The QuickChange site-directed mutagenesis kit was used, as described above, to make point mutations. To remove consensus phosphorylation sites, Ser or Thr were substituted with Ala. The primers also contained silent mutations to introduce unique restriction sites for primary screening. The following primers were used (only sense primer is shown, with mutations underlined): 5′-TAATAACTGAGGCCCTGAGACGATGCT-3′ (Thr665 to Ala, plus addition of HaeIII site), 5′-GTCAAGAATAAAGCTTTTAAGCAGG-3′ (Ser686 to Ala, plus addition of HindIII site), 5′-TGGGGATTTCGCTGAGAAAAGAAAGAG-3′ (Ser694 to Ala, plus addition of DdeI site), and 5′-CAAGAAAAACTGCAGTTCGTAAAATG-3′ (Ser790 to Ala, plus addition of PstI site). For construction of the H667R-hCFTR mutant, the primer used was 5′-AGACCTTGCGCCGTTTCTCA-3′ (construct screened with HhaI, underlined). The wild-type XCFTR or hCFTR in the pOCYT7 vector (see above) was used as a template.
Oocyte cAMP Levels
Intracellular cAMP was determined 24–48 h after cRNA injection. Oocytes were incubated for 20 min in the presence of either 250 nM PMA to activate PKC or 25 μM forskolin to activate PKA. At the end of the incubation period, cells were lysed by sonication. All samples were heated to 95°C for 5 min and centrifuged at 12,000 g for 15 min at 4°C. The supernatants were assayed using a cAMP enzyme immunoassay kit (Amersham Pharmacia Biotech).
Western Blot Analysis
Western blot analysis was performed with the affinity-purified rabbit anti-CFTR antibody α1468 (provided by Dr. Jonathan Cohn, Duke University, Durham, NC; Cohn et al. 1991; Torres et al. 1996). The epitope consists of the COOH-terminal 13 amino acids of human CFTR. The last 12 amino acids are identical in hCFTR and XCFTR. Enriched plasma membranes were prepared by membrane biotinylation and streptavidin affinity purification (Pajor et al. 1998). In brief, membrane proteins were labeled by incubating the oocytes with 0.5 mg/ml biotin (EZ-Link Sulfo-NHS-Biotin; Pierce Chemical Co.) for 30 min at room temperature in Barth's solution. Oocytes were washed three times with 4 ml of ice-cold PBS to remove unbound biotin, resuspended in Barth's solution, and lysed at 4°C by sonication with an ultrasonic processor (model GE501; Sonics and Materials). Yolk and debris were collected by centrifugation at 1,000 g for 5 min at 4°C. The supernatant was gently mixed for 1 h at 4°C with 50 μl of streptavidin attached to beads (ImmunoPure immobilized streptavidin; Pierce Chemical Co.). Bound membranes were collected by centrifugation at 1,000 g for 2 min at 4°C. The pellet was washed twice with PBS, resuspended in electrophoresis sample buffer, and subjected to 10% SDS-PAGE electrophoresis (NuPage Bis-Tris; Novex). Western blots were performed as previously described (Han et al. 1996). The primary antibody concentration was 1 μg/ml, and the secondary antibody was a horseradish peroxidase goat anti–rabbit. CFTR detection was performed by enhanced chemiluminescence (Amersham Pharmacia Biotech).
Solutions and Drugs
To activate PKA, intracellular cAMP was elevated using a cAMP cocktail containing 250 μM 8-Br-cAMP and 25 μM forskolin (Sigma-Aldrich). To activate PKC, 250 nM PMA (Sigma-Aldrich) was added to the bath. Stock solutions of these compounds were prepared in water (8-Br-cAMP), dimethylsulfoxide (PMA), or ethanol (forskolin), and diluted to the desired final concentration in ND96 solution immediately before use. At the concentrations used, the vehicles had no effects on the CFTR currents.
Statistical Analysis
Data are expressed as the means ± SEM. Differences between means were compared by either paired or unpaired two-tailed t tests, as appropriate. Statistical significance was ascribed to P < 0.05.
RESULTS
Differential Activation of Human and Xenopus CFTR by PKC
We expressed human and Xenopus CFTR orthologues in Xenopus oocytes because these cells are capable of appropriate posttranslational processing, express CFTR at high levels, and have no significant endogenous cAMP- or PMA-activated Cl− currents (Cunningham et al. 1992). Injection of either human or Xenopus CFTR cRNA into Xenopus oocytes resulted in functional CFTR expression in the plasma membrane, as shown by the stimulation of CFTR chloride currents in response to 8-Br-cAMP and forskolin (Fig. 1). The current in water-injected oocytes was insensitive to PKA agonists (Fig. 1 A; Bear et al. 1991; Yamazaki et al. 1999). The currents are mediated by CFTR based on their halide selectivities (i.e., Br− ≈ Cl− > I− > F− for hCFTR and I− > Cl− > Br− > F− for XCFTR [Fig. 1C and Fig. D]), and their reversal potentials (in NaCl-based bath solution) near the Cl− equilibrium potential (about −30 mV; Cunningham et al. 1992).
Figure 1.
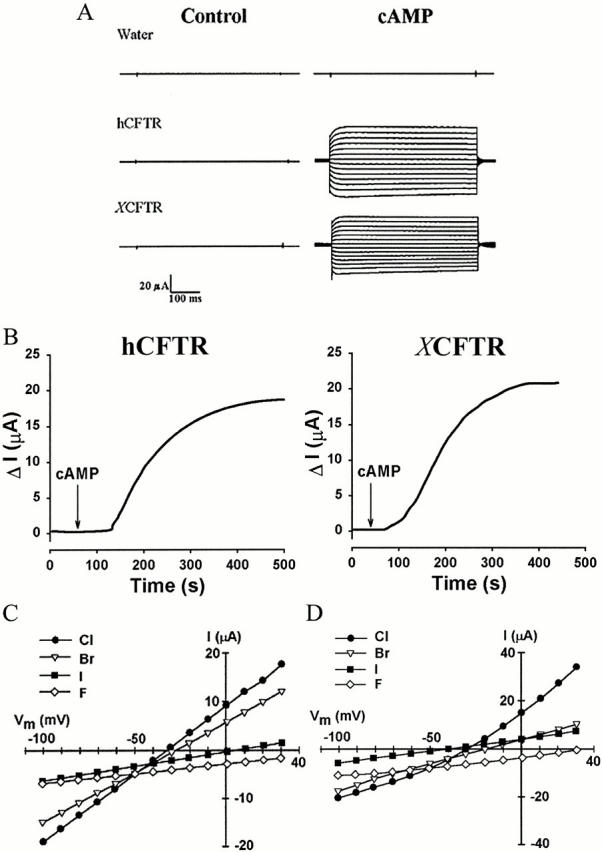
Activation of CFTR Cl− conductance by PKA stimulation. The two-electrode voltage-clamp technique was used to measure ionic currents before and ∼30 min after exposure to a cAMP cocktail consisting of 250 μM 8-Br-cAMP and 25 μM forskolin (cAMP). (A) Representative currents obtained in water-injected oocytes and oocytes expressing hCFTR or XCFTR. (B) Time course of hCFTR and XCFTR activation by cAMP. ΔI is the current measured at 0 mV minus the current at −30 mV (holding potential). (C) Halide-selectivity sequence of cAMP-activated hCFTR. I-V relationships were obtained from an oocyte in the constant presence of cAMP cocktail by substituting NaCl in each solution with the corresponding sodium halide salt for 2 min before the I-V plots. Currents were measured at 400 ms after the start of voltage pulses ranging from −100 to 30 mV at 10-mV intervals. (D) Halide-selectivity sequence obtained from a XCFTR-expressing oocyte. Injection of hCFTR and XCFTR cRNAs elicits anion currents with the expected halide selectivity.
To determine the effect of PKC-mediated phosphorylation on the activation of the two CFTR orthologues, oocytes expressing hCFTR or XCFTR were exposed to PMA. I-V relationships, current time course, and conductance data in Fig. 2 show that, in hCFTR-expressing oocytes, PMA alone elicits only a small fraction of the conductance observed after the subsequent application of the cAMP cocktail. Exposure to PMA resulted, as previously observed by others (Berger et al. 1993; Yamazaki et al. 1999), in a transient hCFTR conductance (Fig. 2 C) that amounted to 28 ± 7%, followed by a sustained conductance of only 13 ± 3% of the total conductance observed with stimulation of both PKC and PKA (Fig. 2 A, C, and E). Higher PMA concentrations did not increase the currents further. Relative to the current elicited by 250 nM PMA, the current after 500 nM PMA was 103 ± 5% (n = 4), and after 1 μM it was 89 ± 9% (n = 2).
Figure 2.
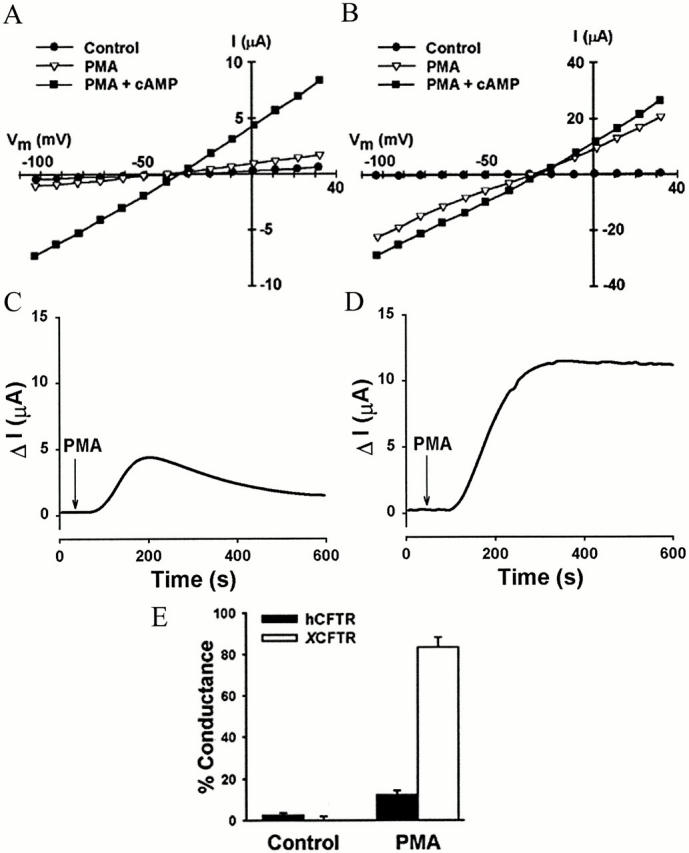
Activation of CFTR Cl− conductance by PKC. (A) Representative I-V plot obtained from an oocyte injected with hCFTR. (B) I-V plot obtained from an oocyte injected with XCFTR. (C) Time course of hCFTR activation by PMA. (D) Time course of XCFTR activation by PMA. (E) Summary of the maximal conductance elicited by PMA. Data were normalized to the maximal conductance obtained with PMA + cAMP. Data shown are from 10 to 12 hCFTR- and XCFTR-expressing oocytes, respectively. I-V plots in PMA-treated oocytes were obtained approximately 15 min after exposure to 250 nM PMA. Next, the cells were exposed to the cAMP cocktail in the continuous presence of PMA, and I-V plots were recorded approximately 10 min later. The results show that PMA is highly effective in activating XCFTR, but not hCFTR. See Fig. 1 for additional details.
In contrast, in XCFTR-expressing oocytes, PMA activated 79 ± 8% of the conductance seen after stimulation of both PKA and PKC (Fig. 2B, Fig. D, and Fig. E). The PMA-activated conductance was sustained for over 30 min, in the continued presence of PMA, and was reversible after extended washout, like the XCFTR conductance elicited by PKA stimulation (not shown). In these experiments, higher 8-Br-cAMP concentrations did not increase the currents further. Relative to the current elicited by 250 μM 8-Br-cAMP, the current after 500 μM 8-Br-cAMP was 98 ± 12% (n = 3), and after 1 mM 8-Br-cAMP it was 96 ± 2% (n = 3).
The above results demonstrate that exposure to PMA activates XCFTR to a level similar to that obtained after PKA stimulation. To determine whether the effect of PMA is due to activation of PKC, we tested the inactive phorbol analogue 4α-PMA on XCFTR-expressing oocytes. Application of 4α-PMA (250 nM) did not change oocyte membrane conductance, whereas subsequent exposure to 4β-PMA caused an increase in conductance to 86 ± 11% of the value after exposure to the cAMP cocktail and 4β-PMA combined, similar to the results in Fig. 2. Further, application of 4β-PMA to water- or hCFTR-injected oocytes, as well as application of 4α-PMA after 4β-PMA in the XCFTR-injected oocytes, had no effect on the oocyte conductances (data not shown).
Another possibility is that PMA increases XCFTR conductance via an elevation of cAMP. Cross-talk between the PKA and PKC signaling systems has been demonstrated in a number of systems (Gao et al. 1999). To explain our data, the cross-talk would have to take place in XCFTR-, but not in hCFTR-expressing oocytes. To test this possibility, intracellular cAMP was measured under control conditions (in ND96 medium) and after exposure to either 250 nM PMA or 25 μM forskolin. As seen in Fig. 3, a 20-min exposure to PMA did not increase intracellular cAMP in oocytes injected with water or expressing either hCFTR or XCFTR. In contrast, forskolin increased Xenopus oocytes intracellular cAMP level by more than 80-fold.
Figure 3.

Effects of PMA and forskolin on cAMP levels. Data are from unstimulated oocytes and oocytes exposed for 20 min to either PMA (250 nM) or forskolin (25 μM). Forskolin, but not PMA, increased oocytes cAMP (P < 0.01, n = 3 for each group of oocytes).
Finally, the difference between hCFTR and XCFTR sensitivity to PMA could be in principle explained by the expression system used. One or more cell-specific components in Xenopus oocytes could allow XCFTR, but not hCFTR, to be directly or indirectly sensitive to PKC-mediated phosphorylation. This question was addressed by experiments in which either XCFTR or hCFTR was expressed in COS-1 cells. In four experiments in cells transfected with XCFTR cDNA, PMA elicited a conductance of 55 ± 3% of that produced by 8-Br-cAMP or forskolin. In contrast, in COS-1 cells expressing hCFTR, the increase in conductance elicited by PMA was on average 21 ± 4%. Taken together, the experiments in Xenopus oocytes and COS-1 cells demonstrate that the difference in PKC-mediated activation of human and Xenopus orthologues is inherent to the CFTR molecules and not to the host cells.
The R Domain Accounts for the Differences in the Responses of Human and Xenopus CFTR to PKC Activation
To test for a possible role of the R domain, a chimera was constructed in which residues 607– 811 of hCFTR (Fig. 4 and Fig. 5 A) were replaced with the equivalent residues of XCFTR. This chimera (HXH-CFTR) contains the NH2- and COOH-terminal transmembrane domains and nucleotide-binding domains from hCFTR. It should exhibit the halide-selectivity sequence of hCFTR, because previous experiments with human/Xenopus chimeras revealed that the halide selectivity sequence of hCFTR and XCFTR depends on the NH2-terminal transmembrane domain (Price et al. 1996). In addition, if the R domain from XCFTR is the region responsible for the regulation by PKC-mediated phosphorylation, then HXH-CFTR should be sensitive to PKC agonists, like XCFTR.
Figure 4.

Alignment of the sequences of human, Xenopus and Necturus CFTR R domains. The number below each consensus sequence refers to the Ser/Thr position of the full-length hCFTR sequence. Arrows above indicate that the labeled residue has been shown to be phosphorylated by either PKA or PKC in human CFTR (Cheng et al. 1991; Picciotto et al. 1992).
Figure 5.
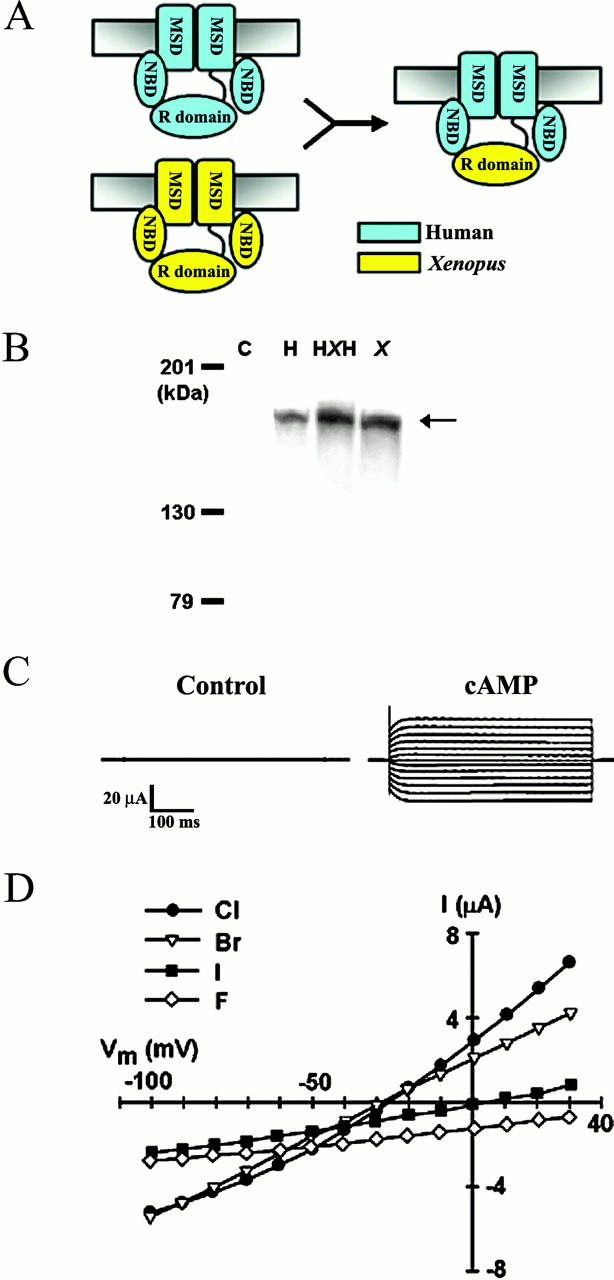
Design and expression of the human-Xenopus chimera (HXH-CFTR). (A) Schematic representation of hCFTR, XCFTR, and HXH-CFTR, showing the location of the membrane-spanning domain (MSD), nucleotide-binding domain (NBD), and the R domain. (B) Plasma membrane expression of hCFTR, XCFTR, and HXH-CFTR chimera. Western blots of biotinylated membranes injected with water [C], HXH-CFTR [HXH], hCFTR [H], or XCFTR [X]; there were five oocytes per condition. The arrow denotes the expected size of full-length CFTR (∼170 kD). (C) Representative currents from an oocyte expressing HXH-CFTR. (D) Halide selectivity sequences of cAMP-activated HXH-CFTR. The results show that PKA stimulation elicits a near-linear current with reversal potential near ECl and halide selectivity identical to that of wild-type hCFTR (Br− = Cl− > I− > F−). See Fig. 1 for additional details.
HXH-CFTR was fully processed and inserted in the plasma membranes of Xenopus oocytes as shown by Western blot analysis of plasma membranes (Fig. 5 B). Electrophysiological studies showed no appreciable current in the absence of kinase stimulation (Fig. 5 C and 6 A), as shown above for wild-type hCFTR and XCFTR (Fig. 1 and Fig. 2). The PKA agonist 8-Br-cAMP reversibly elicited a large, near-linear current, with a reversal potential of approximately −30 mV (Fig. 6A and Fig. C) and halide-selectivity sequence identical to that of wild-type hCFTR (Fig. 5 D).
Figure 6.

Stimulation of HXH-CFTR by PKC-mediated phosphorylation. (A) I-V plot from an oocyte expressing HXH-CFTR. (B) I-V plot from an oocyte expressing hCFTR. (C) Time course of HXH-CFTR activation by PMA. Priming denotes the addition of 50 μM 8-Br-cAMP. (D) Comparison of CFTR activation by PMA in oocytes expressing either wild-type hCFTR or XCFTR, or the HXH-CFTR chimera. Data were obtained in oocytes primed with 50 μM cAMP (see text), and were normalized to the current elicited by PMA and cAMP cocktail together. Data shown are from 12, 12, and 15 experiments in hCFTR-, XCFTR-, and HXH-CFTR-expressing oocytes, respectively. The results show that the HXH-CFTR chimera is fully sensitive to PMA stimulation. See Fig. 1 for additional details.
The response to PMA of oocytes expressing HXH-CFTR was similar to those of oocytes expressing either hCFTR or XCFTR, in a clearly bimodal distribution, without intermediate responses. A basal level of PKA-mediated phosphorylation, insufficient to activate XCFTR by itself, is required for the PMA effect (Yamazaki et al. 1999; Button et al. 2000). Since oocyte endogenous cAMP levels from oocyte to oocyte (on average 300 nmol/oocyte) are quite variable, we reasoned that the variability in activation of HXH-CFTR by PMA alone could result from differences in intracellular cAMP. In oocytes with low PKA activity, HXH-CFTR may not be phosphorylated enough to be activated by PMA alone. To “standardize” the level of PKA activity, we exposed the cells to 8-Br-cAMP at a concentration insufficient to activate the current by itself (Fig. 6). We call this “priming.” In all 15 experiments, application of PMA to primed oocytes expressing HXH-CFTR elicited full current activation (102 ± 8% of the current observed with the subsequent addition of the cAMP cocktail; Fig. 6A and Fig. C), which is not different from the results in oocytes expressing wild-type XCFTR (Fig. 2 C). However, priming did not increase the activation of the wild-type hCFTR by PKC-mediated phosphorylation (Fig. 6B and Fig. C). From these results, we conclude that the R domain is the target for the activation of XCFTR by PKC stimulation, and that the Xenopus R domain is sufficient to confer the sensitivity of CFTR to PKC stimulation.
Conserved Phosphorylation Sites Are Not Involved in the Response to PKC Activation
As shown in Fig. 4, the R domain of XCFTR contains at least seven consensus sites for phosphorylation by PKC. Four of the phosphorylatable residues are also present in hCFTR (Thr605, Ser686, Ser707, and Ser790), and two of them (Ser686 and Ser790) are phosphorylated in vitro in hCFTR (Picciotto et al. 1992). The R domain of XCFTR contains two additional sites (Thr665 and Ser694) also present in the N. maculosus CFTR homologue, but absent in hCFTR (Fig. 4). Like the Xenopus CFTR, the PKC agonist PMA activates the Necturus homologue of CFTR (Copello et al. 1993). The role of the “conserved” and “unique” phosphorylation sites in the stimulation of XCFTR by PMA was explored by substituting serine or threonine of the consensus sequences with alanine. In these experiments, priming was carried out as explained above.
Substitution of the two conserved serine residues known to be phosphorylated (Ser686 and Ser790; Picciotto et al. 1992) with alanine residues (S686A/S790A-XCFTR; Fig. 7A and Fig. C) did not affect the activation by PMA. The unstimulated oocytes displayed no significant current, and the subsequent application of 8-Br-cAMP elicited a large, linear, reversible current, with a reversal potential close to ECl (approximately −30 mV; Fig. 7 A) and a halide-selectivity sequence identical to that of XCFTR (I− > Cl− > Br− > F−; Fig. 1 C). These results indicate that the currents are mediated by CFTR, and agree with the notion that the ion pore is formed by the transmembrane segments (Price et al. 1996). In seven experiments, PMA stimulated the current to 92 ± 11% of the response to the combination of 8-Br-cAMP and PMA (Fig. 7A and Fig. C). This observation indicates that Ser686 and Ser790 are not necessary for the activation of XCFTR by PKC stimulation.
Figure 7.

Knockout of the unique PKC consensus phosphorylation sites prevents full activation of XCFTR by PMA. (A) Representative I-V relationships from an oocyte expressing the double knockout of conserved PKC consensus phosphorylation sites (S686A/S790A-XCFTR). (B) I-V plot from a cell expressing the double knockout of unique PKC consensus phosphorylation sites (T665A/S694A-XCFTR). (C) Summary of results from experiments identical to those in A (n = 4) and B (n = 6). The results indicate that at least one of the conserved PKC consensus sites is necessary for full activation by PMA. See Fig. 1 and Fig. 2 for additional details.
Thr665 Is Essential for the Response to PKC Activation of XCFTR
To investigate the roles of the unique PKC consensus sites (Thr665 and Ser694), we expressed the mutant lacking both sites (T665A/S694A-XCFTR) in Xenopus oocytes. This mutant was also well expressed, did not exhibit basal current, and was sensitive to agonists of PKA-mediated phosphorylation (Fig. 7 B). However, PMA elicited a small current (Fig. 7B and Fig. C) that was not different from that measured in oocytes expressing wild-type hCFTR. The lack of response to PMA could not be overcome by a higher concentration (750 nM, data not shown). This result suggests that at least one of the two unique sites in the R domain of XCFTR is critical for activation by PKC-mediated phosphorylation. Although the degrees of activation of this mutant by PKA and PKC were the same as in the wild-type hCFTR, its halide-selectivity sequence (I− > Cl− > Br− > F−; Fig. 1) was identical to that of the wild-type XCFTR, as expected.
To ascertain the roles of the two conserved sites, we mutated Thr665 and Ser694 separately and tested the effect of PMA in primed oocytes. When S694A-XCFTR was stimulated with PMA, there was full current activation (Fig. 8A and Fig. C). In contrast, in oocytes expressing T665A-XCFTR, PMA failed to produce full current activation (Fig. 8B and Fig. C). Currents in cRNA-injected oocytes vary from cell to cell. Thus, it is possible that the reduced activation by PMA of XCFTR mutants containing the Thr665 to Ala mutation is not absolute, but relative to the activation by PKA stimulation (i.e., the mutation increases the activation of CFTR by cAMP). The data in Fig. 9 indicate that this is not the case because the cAMP-activated currents are not different among oocytes expressing wild-type XCFTR, S686A/S790A-XCFTR, or T665A/S694A-XCFTR. These results prove that Thr665 is the residue necessary for the activation of CFTR by PKC.
Figure 8.

Thr665 is critical for activation of XCFTR by PKC-mediated phosphorylation. (A) Representative I-V plot from an oocyte expressing a single knockout of the unique PKC consensus phosphorylation site containing Ser694 (S694A-XCFTR). (B) I-V plot from an oocyte expressing a single knockout of the unique PKC consensus phosphorylation site containing Thr665 (Thr665/A-XCFTR). (C) Summary of results from experiments identical to those in A (n = 6) and B (n = 14). The results denote a critical role of Thr665 in the stimulation of XCFTR by PMA. See Fig. 1 and Fig. 2 for additional details.
Figure 9.

The mutation Thr665 to Ala does not increase cAMP-activated XCFTR currents. Time course of the currents after stimulation with the cAMP cocktail in oocytes injected with wild-type XCFTR, S686A/S790A-XCFTR, or T665A/S694A-XCFTR cRNAs. Currents were measured at 30 mV, 20 min after exposure to the cAMP cocktail. The data show that the Thr665 to Ala mutation has no significant effect on the level of cAMP-activated currents.
Engineering a PKC Consensus Phosphorylation Site that Includes Thr665 Confers PKC-sensitivity to hCFTR
The hCFTR is unique among all known CFTR homologues in that it lacks a PKC consensus site at the location equivalent to residue 665 in XCFTR. As shown in Fig. 4, this is due to the presence of His667 instead of arginine or lysine. Since Thr665 is present in hCFTR, we decided to determine whether the formation of a PKC consensus phosphorylation site in hCFTR by substituting His with Arg at position 667 is sufficient to confer the Xenopus phenotype to hCFTR. The mutant H667R-hCFTR was expressed in Xenopus oocytes and tested for activation by PKC stimulation. In oocytes primed with 8-Br-cAMP, as described above, PMA produced a large activation of the conductance, amounting to 68 ± 6% (n = 6) of the current elicited by the combination of PMA and cAMP cocktail (Fig. 10). The PMA-dependent conductance in the presence of priming was significantly greater than that in wild-type hCFTR, although perhaps smaller than the analogous value in HXH-CFTR. This demonstrates that the phosphorylation site at Thr665 is sufficient to explain CFTR activation by PKC-mediated phosphorylation.
Figure 10.
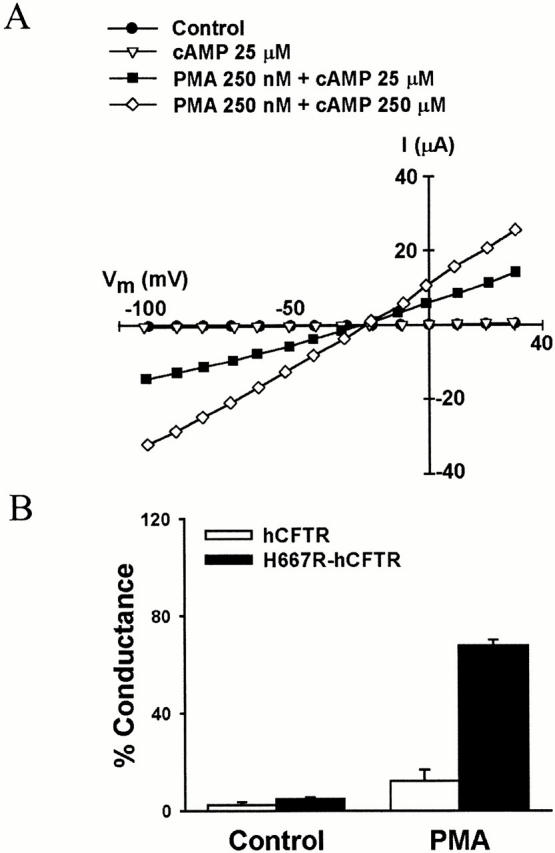
PMA produces a large increase in conductance in a mutant hCFTR (H667R-hCFTR) with an engineered PKC consensus phosphorylation sequence that includes Thr665. (A) Representative I-V plot from an oocyte expressing H667R-hCFTR. (B) Summary of results from experiments such as those in A. Oocytes were primed with 25 μM 8-Br-cAMP. Data were obtained from six experiments. The results indicate that the engineered PKC consensus site is sufficient to confer a large response to PKC stimulation.
DISCUSSION
The phosphorylation of serines and threonines by cAMP-dependent kinases is a conserved step in the activation of CFTR Cl− channels from several species (Seibert et al. 1997). It is clear that at least four of the dibasic PKA consensus sites in the R domain are important for activation (Chang et al. 1993; Rich et al. 1993).
PKC stimulation alone produced only a small activation of hCFTR expressed in Xenopus oocytes, a result consistent with those previously reported using either heterologous expression systems or native CFTR-expressing cells (Berger et al. 1993; Yamazaki et al. 1999). In contrast, PKC stimulation of XCFTR elicited a large Cl− conductance, similar in magnitude to that activated by PKA agonists. Further, experiments with expression of either hCFTR or XCFTR in COS-1 cells yielded similar results to those in Xenopus oocytes. Thus, activation of CFTR by PKC is a property of the amphibian CFTR isoforms.
A Single PKC Consensus Phosphorylation Site in the R Domain Is Needed for the Activation of Xenopus CFTR by PKC Stimulation
The chimera in which the R domain of XCFTR replaced the R domain of hCFTR exhibited the halide-selectivity sequence of hCFTR, as expected from previous studies indicating that the ion selectivity of CFTR is determined by the NH2-terminal transmembrane spanning domain (Price et al. 1996). In addition, the HXH-CFTR chimera was sensitive to PKC stimulation, indicating that the R domain is the target for the activation of XCFTR by PKC stimulation, and that the Xenopus R domain is sufficient to confer sensitivity of CFTR to PKC stimulation. These experiments also indicate that the interaction between the R domain and other regions of CFTR is preserved in the HXH chimera. Although the human and Xenopus R domains exhibit only 65% amino acid sequence identity, the Xenopus R domain does interact with the nucleotide-binding domains and/or transmembrane domains of hCFTR, eliciting channel activation in response to PKA stimulation.
The mutagenesis experiments presented in this paper indicate that the PKC consensus phosphorylation site including Thr665 is required for the activation of the channel by PKC stimulation. In hCFTR, Thr665 is conserved, but the PKC consensus phosphorylation site is missing. Insertion of this site in the H667R-hCFTR mutant resulted in a large increase in current in response to PKC activation. The experiments on this mutant and the HXH chimera were different in some respects (e.g., priming concentration of 8-Br-cAMP and amount of cRNA injected), making a quantitative comparison of the responses to PKC activation difficult. In any event, the results indicate that the PKC consensus phosphorylation site that includes Thr665 is critical for the response to PMA. If the activation of the H667R-hCFTR mutant is in fact less than that of the HXH chimera, then one would conclude also that the phosphorylation of Thr665, or its functional effect, involves other residues of the R domain of CFTR.
PKC-mediated activation of hCFTR requires endogenous phosphorylation by PKA (Yamazaki et al. 1999). Activation of XCFTR by PMA occurred in all oocytes in the absence of priming, whereas HXH-CFTR needed priming (Fig. 2 and Fig. 4). This difference could be the result of either a lower PKA-mediated phosphorylation of HXH-CFTR at basal cAMP levels, or differences in the interaction of the R domain of XCFTR with other hCFTR domains. Oocytes expressing a chimera consisting of the R domain and COOH-terminal half of XCFTR and the NH2-terminal half of hCFTR did not require priming, whereas a CFTR molecule consisting of the NH2-terminal half and R domain of XCFTR and the COOH-terminal half of hCFTR did (Button et al. 2000). Additional studies will be needed to establish the bases of the need for priming of the HXH-CFTR.
Our data suggest, but do not prove, that the stimulation of XCFTR by PMA is directly mediated by phosphorylation of Thr665 by PKC. This is the simplest explanation of our results because of the following reasons. First, the inactive phorbol ester 4α-PMA is ineffective, ruling out most nonspecific effects of PMA. Second, PMA treatment does not increase cAMP levels in oocytes, ruling out an indirect effect via PKA activation. Third, cGMP is ineffective in Necturus gallbladder epithelial cells (Heming et al. 1994), suggesting no role of the cGMP-dependent kinase in the regulation of amphibian CFTR. Fourth, substitution of Thr665 in a PKC consensus phosphorylation site of XCFTR abolishes the response to PMA. Finally, addition of a PKC consensus phosphorylation site in hCFTR confers sensitivity of this molecule to PMA. Based on these observations, we hypothesize that phosphorylation of Thr665 by PKC is responsible for the activation of XCFTR by PMA. The phosphorylation level of CFTR depends on the rates of phosphorylation and dephosphorylation. Thus, it is conceivable that PMA may inhibit a phosphatase that dephosphorylates Thr665, and this inhibition solely or in part could account for the increase in phosphorylation of Thr665. Resolution of the precise mechanism of XCFTR activation will require additional experiments.
The Mechanism of CFTR Activation by Phosphorylation of the R Domain
It is clear that hCFTR is activated by PKA-mediated phosphorylation of residues located mainly in the R domain (Anderson et al. 1991; Tabcharani et al. 1991; Nagel et al. 1992). Phosphorylation of specific residues can modulate CFTR either positively or negatively (Wilkinson et al. 1997), but mutagenesis studies indicate that no single site is responsible for the activation (Cheng et al. 1991; Chang et al. 1993; Rich et al. 1993). Therefore, it is thought that the increase in CFTR channel open probability by PKA activation is a consequence of either a change in the net charge of the R domain (Ostedgaard et al. 2000) or a more complicated phenomenon involving multiple phosphorylation consensus sites (Gadsby and Nairn 1999; Baldursson et al. 2000). In contrast, our data indicate that a specific residue (Thr665) is necessary for the activation of the XCFTR channel by PMA.
It has been assumed that exon 13 of hCFTR (residues 591–830) codes for the R domain (Cheng et al. 1991). However, recent experimental evidence suggests that exon 13 includes three distinct regions. The first region is the last portion of the first nucleotide-binding domain (Chan et al. 2000), based on the homology between CFTR and the ATP-binding subunit of the histidine permease (Hung et al. 1998). The second region is the NH2-terminal region of the R domain, which contains few consensus phosphorylation sites, but includes Thr665, the critical residue for activation of XCFTR by PKC stimulation. This region is somewhat structured and phosphorylation by PKA probably changes its secondary structure (Dulhanty and Riordan 1994). The third region is the COOH-terminal region of the R domain, which contains most PKA and PKC phosphorylation sites. This region is mostly made of random coils, and PKA-mediated phosphorylation has no effect on its secondary structure, which was assessed by circular dichroism spectroscopy (Ostedgaard et al. 2000). Our results are compatible with the notion that activation of CFTR by PKC stimulation involves the more structured NH2-terminal region of the R domain and occurs independently of the net charge. Consistent with this view, a chimera in which part of the COOH-terminal portion of the R domain of hCFTR was replaced with the linker domain of MDR1 was fully activated by PKA (Vankeerberghen et al. 1999). Although the linker region of MDR1 has three sites known to be phosphorylated by PKC, stimulation of this kinase did not increase channel activity significantly (Vankeerberghen et al. 1999). These observations support the conclusion that activation of CFTR by PKC stimulation depends on the specific location of the phosphorylated residue. The demonstration that a single residue is critical for CFTR activation by PKC stimulation provides basic and novel information that will serve for future studies aimed at understanding CFTR regulation at the structural level.
Acknowledgments
We thank Drs. B.N. Christensen, S.A. Lewis, and S.A. Weinman for comments on preliminary versions of this manuscript, Dr. G. Zhang and Mr. S. Govani for help with the experimental work, and Ms. L. Durant for secretarial help.
This work was supported by National Institutes of Health grants No. DK-38734 (to L. Reuss) and No. CA-72783 (to G. Altenberg). B. Button is a Jeane Kempner Fellow.
Footnotes
Abbreviations used in this paper: hCFTR, human CFTR; HXH-CFTR, CFTR chimera containing the transmembrane domains and nucleotide-binding domains of hCFTR and the regulatory domain of Xenopus CFTR; R domain, regulatory domain; XCFTR, Xenopus CFTR.
In one experiment, there was no response to PMA. If this experiment is included in the calculation, the PMA-stimulated conductance is still significantly increased, i.e., 47 ± 4% of the 8-Br-cAMP-stimulated current. A negative result was also observed in 1 of 16 experiments in Xenopus oocytes expressing XCFTR. A possible explanation for the occasional absence of response to PMA is that the cells had low endogenous cAMP levels (see next page).
References
- Anderson M.P., Berger H.A., Rich D.P., Gregory R.J., Smith A.E., Welsh M.J. Nucleoside triphosphates are required to open the CFTR chloride channel. Cell. 1991;67:775–784. doi: 10.1016/0092-8674(91)90072-7. [DOI] [PubMed] [Google Scholar]
- Baldursson O., Berger H.A., Welsh M.J. Contribution of R domain phosphoserines to the function of CFTR studied in Fischer rat thyroid epithelia. Am. J. Physiol. 2000;279:L835–L841. doi: 10.1152/ajplung.2000.279.5.L835. [DOI] [PubMed] [Google Scholar]
- Bear C.E., Duguay F., Naismith A.L., Kartner N., Hanrahan J.W., Riordan J.R. Cl− channel activity in Xenopus oocytes expressing the cystic fibrosis gene. J. Biol. Chem. 1991;266:19142–19145. [PubMed] [Google Scholar]
- Berger H.A., Travis S.M., Welsh M.J. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by specific protein kinases and protein phosphatases. J. Biol. Chem. 1993;268:2037–2047. [PubMed] [Google Scholar]
- Button B., Altenberg G.A., Reuss L. Role of the Xenopus CFTR R-domain in the activation by PKC FASEB J. 14 2000. A336(Abstr.) [Google Scholar]
- Chan K.W., Csanady L., Seto-Young D., Nairn A.C., Gadsby D.C. Severed molecules functionally define the boundaries of the cystic fibrosis transmembrane conductance regulator's NH2-terminal nucleotide binding domain. J. Gen. Physiol. 2000;116:163–180. doi: 10.1085/jgp.116.2.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang X.B., Tabcharani J.A., Hou Y.X., Jensen T.J., Kartner N., Alon N., Hanrahan J.W., Riordan J.R. Protein kinase A (PKA) still activates CFTR chloride channel after mutagenesis of all 10 PKA consensus phosphorylation sites. J. Biol. Chem. 1993;268:11304–11311. [PubMed] [Google Scholar]
- Cheng S.H., Rich D.P., Marshall J., Gregory R.J., Welsh M.J., Smith A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Cohn J.A., Melhus O., Page L.J., Dittrich K.L., Vigna S.R. CFTRdevelopment of high-affinity antibodies and localization in sweat gland. Biochem. Biophys. Res. Commun. 1991;181:36–43. doi: 10.1016/s0006-291x(05)81378-6. [DOI] [PubMed] [Google Scholar]
- Copello J., Heming T.A., Segal Y., Reuss L. cAMP-activated apical membrane chloride channels in Necturus gallbladder epithelium. Conductance, selectivity, and block. J. Gen. Physiol. 1993;102:177–199. doi: 10.1085/jgp.102.2.177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cunningham S.A., Worrell R.T., Benos D.J., Frizzell R.A. cAMP-stimulated ion currents in Xenopus oocytes expressing CFTR cRNA. Am. J. Physiol. 1992;262:C783–C788. doi: 10.1152/ajpcell.1992.262.3.C783. [DOI] [PubMed] [Google Scholar]
- Dulhanty A.M., Riordan J.R. Phosphorylation by cAMP-dependent protein kinase causes a conformational change in the R domain of the cystic fibrosis transmembrane conductance regulator. Biochemistry. 1994;33:4072–4079. doi: 10.1021/bi00179a036. [DOI] [PubMed] [Google Scholar]
- Gadsby D.C., Nairn A.C. Control of CFTR channel gating by phosphorylation and nucleotide hydrolysis. Physiol. Rev. 1999;79:S77–S107. doi: 10.1152/physrev.1999.79.1.S77. [DOI] [PubMed] [Google Scholar]
- Gao Z., Chen T., Weber M.J., Linden J. A2B adenosine and P2Y2 receptors stimulate mitogen-activated protein kinase in human embryonic kidney-293 cells. Cross-talk between cyclic AMP and protein kinase C pathways. J. Biol. Chem. 1999;274:5972–5980. doi: 10.1074/jbc.274.9.5972. [DOI] [PubMed] [Google Scholar]
- Han E.S., Vanoye C.G., Altenberg G.A., Reuss L. P-glycoprotein-associated chloride currents revealed by specific block by an anti-P-glycoprotein antibody. Am. J. Physiol. 1996;270:C1370–C1378. doi: 10.1152/ajpcell.1996.270.5.C1370. [DOI] [PubMed] [Google Scholar]
- Heming T.A., Copello J., Reuss L. Regulation of cAMP-activated apical membrane chloride conductance in gallbladder epithelium. J. Gen. Physiol. 1994;103:1–18. doi: 10.1085/jgp.103.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung L.W., Wang I.X., Nikaido K., Liu P.Q., Ames G.F., Kim S.H. Crystal structure of the ATP-binding subunit of an ABC transporter. Nature. 1998;396:703–707. doi: 10.1038/25393. [DOI] [PubMed] [Google Scholar]
- Jia Y., Mathews C.J., Hanrahan J.W. Phosphorylation by protein kinase C is required for acute activation of cystic fibrosis transmembrane conductance regulator by protein kinase A. J. Biol. Chem. 1997;272:4978–4984. doi: 10.1074/jbc.272.8.4978. [DOI] [PubMed] [Google Scholar]
- Ma J. Stimulatory and inhibitory functions of the R domain on CFTR chloride channel. News Physiol. Sci. 2000;15:154–158. doi: 10.1152/physiologyonline.2000.15.3.154. [DOI] [PubMed] [Google Scholar]
- Mo L., Hellmich H.L., Fong P., Wood T., Embesi J., Wills N.K. Comparison of amphibian and human CIC-5similarity of functional properties and inhibition by external pH. J. Membr. Biol. 1999;168:253–264. doi: 10.1007/s002329900514. [DOI] [PubMed] [Google Scholar]
- Nagel G., Hwang T.-C., Nastiuk K.L., Nairn A.C., Gadsby D.C. The protein kinase A-regulated cardiac Cl− channel resembles the cystic fibrosis transmembrane conductance regulator. Nature. 1992;360:81–84. doi: 10.1038/360081a0. [DOI] [PubMed] [Google Scholar]
- Ostedgaard L.S., Baldursson O., Vermeer D.W., Welsh M.J., Robertson A.D. A functional R domain from cystic fibrosis transmembrane conductance regulator is predominantly unstructured in solution. Proc. Natl. Acad. Sci. USA. 2000;97:5657–5662. doi: 10.1073/pnas.100588797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajor A.M., Sun N., Valmonte H.G. Mutational analysis of histidine residues in the rabbit Na+/dicarboxylate co-transporter NADC-1. Biochem. J. 1998;331:257–264. doi: 10.1042/bj3310257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto M.R., Cohn J.A., Bertuzzi G., Greengard P., Nairn A.C. Phosphorylation of the cystic fibrosis transmembrane conductance regulator. J. Biol. Chem. 1992;267:12742–12752. [PubMed] [Google Scholar]
- Price M.P., Ishihara H., Sheppard D.N., Welsh M.J. Function of Xenopus cystic fibrosis transmembrane conductance regulator (CFTR) Cl− channels and use of human-Xenopus chimeras to investigate the pore properties of CFTR. J. Biol. Chem. 1996;271:25184–25191. doi: 10.1074/jbc.271.41.25184. [DOI] [PubMed] [Google Scholar]
- Rich D.P., Berger H.A., Cheng S.H., Travis S.M., Saxena M., Smith A.E., Welsh M.J. Regulation of the cystic fibrosis transmembrane conductance regulator Cl− channel by negative charge in the R domain. J. Biol. Chem. 1993;268:20259–20267. [PubMed] [Google Scholar]
- Seibert F.B., Loo T.W., Clarke D.M., Riordan J.R. Cystic fibrosischannel, catalytic, and folding properties of the CFTR protein. J. Bioenerg. Biomembr. 1997;29:429–442. doi: 10.1023/a:1022478822214. [DOI] [PubMed] [Google Scholar]
- Sharon D., Vorobiov D., Dascal N. Positive and negative coupling of the metabotropic glutamate receptors to a G protein–activated K+ channel, GIRK, in Xenopus oocytes. J. Gen. Physiol. 1997;109:477–490. doi: 10.1085/jgp.109.4.477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheppard D.N., Welsh M.J. Structure and function of the CFTR chloride channel. Physiol. Rev. 1999;79:S23–S45. doi: 10.1152/physrev.1999.79.1.S23. [DOI] [PubMed] [Google Scholar]
- Tabcharani J.A., Chang X.-B., Riordan J.R., Hanrahan J.W. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- Torres R.J., Altenberg G.A., Cohn J.A., Reuss L. Polarized expression of cAMP-activated chloride channels in isolated epithelial cells. Am. J. Physiol. 1996;271:C1574–C1582. doi: 10.1152/ajpcell.1996.271.5.C1574. [DOI] [PubMed] [Google Scholar]
- Vankeerberghen A., Lin W., Jaspers M., Cuppens H., Nilius B., Cassiman J.J. Functional characterization of the CFTR R domain using CFTR/MDR1 hybrid and deletion constructs. Biochemistry. 1999;38:14988–14998. doi: 10.1021/bi991520d. [DOI] [PubMed] [Google Scholar]
- Vanoye C.G., Altenberg G.A., Reuss L. P-glycoprotein is not a swelling-activated Cl− channelpossible role as a Cl− channel regulator. J. Physiol. 1997;502:249–258. doi: 10.1111/j.1469-7793.1997.249bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D.J., Strong T.V., Mansoura M.K., Wood D.L., Smith S.S., Collins F.S., Dawson D.C. CFTR activationadditive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. 1997;273:L127–L133. doi: 10.1152/ajplung.1997.273.1.L127. [DOI] [PubMed] [Google Scholar]
- Yamazaki J., Britton F., Collier M.L., Horowitz B., Hume J.R. Regulation of recombinant cardiac cystic fibrosis transmembrane conductance regulator chloride channels by protein kinase C. Biophys. J. 1999;76:1972–1987. doi: 10.1016/S0006-3495(99)77356-X. [DOI] [PMC free article] [PubMed] [Google Scholar]