

Abstract
Some studies of CFTR imply that channel activation can be explained by an increase in open probability (Po), whereas others suggest that activation involves an increase in the number of CFTR channels (N) in the plasma membrane. Using two-electrode voltage clamp, we tested for changes in N associated with activation of CFTR in Xenopus oocytes using a cysteine-substituted construct (R334C CFTR) that can be modified by externally applied, impermeant thiol reagents like [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET+). Covalent modification of R334C CFTR with MTSET+ doubled the conductance and changed the I-V relation from inward rectifying to linear and was completely reversed by 2-mercaptoethanol (2-ME). Thus, labeled and unlabeled channels could be differentiated by noting the percent decrease in conductance brought about by exposure to 2-ME. When oocytes were briefly (20 s) exposed to MTSET+ before CFTR activation, the subsequently activated conductance was characteristic of labeled R334C CFTR, indicating that the entire pool of CFTR channels activated by cAMP was accessible to MTSET+. The addition of unlabeled, newly synthesized channels to the plasma membrane could be monitored on-line during the time when the rate of addition was most rapid after cRNA injection. The addition of new channels could be detected as early as 5 h after cRNA injection, occurred with a half time of ∼24–48 h, and was disrupted by exposing oocytes to Brefeldin A, whereas activation of R334C CFTR by cAMP occurred with a half time of tens of minutes, and did not appear to involve the addition of new channels to the plasma membrane. These findings demonstrate that in Xenopus oocytes, the major mechanism of CFTR activation by cAMP is by means of an increase in the open probability of CFTR channels.
Keywords: trafficking, MTS reagents, labeling, mutagenesis, oocyte expression
INTRODUCTION
CFTR is the product of the gene that is mutated in cystic fibrosis, the most common, fatal genetic disorder in the Caucasian population. Mutations in the CFTR gene result in altered function in multiple organs including the lung, the pancreas, the intestine, the liver, the reproductive organs, and the sweat glands and ducts (Quinton 1999). CFTR functions as an anion-selective channel, and activation requires phosphorylation of the R-domain by PKA and the hydrolysis of ATP at the two nucleotide binding folds, NBF1 and NBF2 (Anderson et al. 1991; Cheng et al. 1991; Rich et al. 1991; Anderson and Welsh 1992; Winter et al. 1994; Sheppard and Welsh 1999). cAMP-induced, CFTR-mediated Cl− currents have been observed, not only in native tissues, but also in cells transfected with CFTR cDNA and in Xenopus oocytes injected with cRNA encoding CFTR (Bear et al. 1991; Drumm et al. 1991; Kartner et al. 1991; Tabcharani et al. 1991; Anderson et al. 1992; Sood et al. 1992), but the mechanism for the activation of Cl− channels by cAMP remains controversial. Some studies suggest that activation can be attributed to an increase in the open probabilities of CFTR channels resident in the plasma membrane (Denning et al. 1992; Dho et al. 1993; Prince et al. 1993; Santos and Reenstra 1994; Hug et al. 1997; Loffing et al. 1998; Moyer et al. 1998), whereas others suggest that cAMP can induce the insertion of CFTR channels into the plasma membrane from a submembranous compartment via vesicle fusion (Schwiebert et al. 1994; Howard et al. 1996; Tousson et al. 1996; Lehrich et al. 1998; Howard et al. 2000). In particular, recent studies using Xenopus oocytes report increases in membrane capacitance and antibody labeling that were interpreted as being indicative of cAMP-dependent, exocytotic delivery of CFTR to the plasma membrane (Takahashi et al. 1996; Peters et al. 1999; Weber et al. 1999).
We have used engineered cysteines to identify residues that lie within the anion-conducting pore of CFTR (see Smith et al. 2001 in this issue). The targeted amino acid residues were replaced with cysteine, which in turn, could be modified with highly polar, membrane impermeant derivatives of methanethiolsulfonate (MTS) reagents. One of these cysteine-substituted constructs (R334C) was readily modified by MTS reagents in the external bath and covalent modification gave rise to the changes in anion conduction that could be easily detected in the macroscopic I-V plots recorded in Xenopus oocytes, permitting us to distinguish modified from unmodified-channels. R334C CFTR, in conjunction with membrane impermeant thiol reagent, MTSET+ (Holmgren et al. 1996), offered an opportunity to test directly the hypothesis that activation of Cl− conductance in oocytes expressing CFTR is accompanied by an increase in channel number in the plasma membrane. We found that exposure of oocytes expressing R334C CFTR to MTSET+ for 20 s before activation resulted in labeling of the entire membrane pool of functional channels, suggesting that channels activated by cAMP are resident in the membrane before activation and that activation of Cl− conductance, therefore, is due largely to an increase in the open probability (Po) of CFTR channels.
MATERIALS AND METHODS
Mutagenesis
The CFTR mutants were generated using the QuickChangeTM site-directed mutagenesis kit from Stratagene. A dsDNA pBluescript vector with a CFTR insert and a pair of synthetic oligonucleotide primers containing the desired mutation were used in this procedure. The primers, each complimentary to the opposite strands of the vector, extend during the temperature cycling by means of Pfu DNA polymerase to generate a hybrid plasmid containing one mutated DNA strand and one wild-type parental strand. The final product is treated with DpnI, a restriction enzyme selectively digesting the methylated parental DNA strand. The newly synthesized DNA containing the desired mutation is not methylated, therefore, it is not susceptible to DpnI digestion. After Escherichia coli transformation, the colonies only contain the mutated plasmids. The sequences at the mutation region and in the whole PCR-generated region are confirmed by direct DNA sequencing.
In Vitro Transcription
The CFTR cRNAs for Xenopus oocyte injection were synthesized by using the in vitro transcription kit, mMessage Machine (Ambion, Inc.). The T7 RNA polymerase was used because the insert in the Bluescript CFTR clone is downstream from the T7 promoter. The Bluescript CFTR cDNA templates were prepared by digestion with XhoI, a restriction enzyme with a 5′ protruding end to avoid the problem of Nonspecific transcription associated with 3′ overhang ends. The XhoI site is located downstream from the CFTR insert, therefore, the run-off transcripts generated are of a defined size with the sequence the same as that of the mRNA. The transcription products were purified and the quality and quantity of the transcripts assessed on an agarose gel.
Oocyte Preparation
The protocols for preparing oocytes were similar to those previously described (Smit et al. 1993; Wilkinson et al. 1996). Briefly, oocytes were surgically removed from anesthetized Xenopus laevis toads and manually defolliculated after incubation in a collagenase-containing solution for 2–2.5 h. The oocytes were kept in an 18°C humidifier overnight in a modified Barth's Solution (MBSH) containing the following (in mM) 88 NaCl, 1 KCl, 0.82 MgSO4, 0.33 Ca(NO3)2·4H2O, 0.41 CaCl2·2H2O, 2.4 NaHCO3, 5 HEPES-Na, 5 HEPES-H+, and 150 mg/liter gentamicin. Oocytes were injected with 50 nl cRNA with or without the cRNA encoding human β2-adrenergic receptor. Typically, oocytes were used within 5 d after injection.
Electrophysiological Recordings
Electrophysiological recording methods were similar to those described by Mansoura et al. 1998. Briefly, individual oocytes were placed in the recording chamber and continuously perfused with frog Ringer's solution unless noted. The Ringer's solution contains the following (in mM): 98 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 2.5 HEPES-Na, and 2.5 HEPES-H+. The volume of the perfusion chamber used in the current study was ∼100 μl, and the flow rate to the chamber was ∼67 μl/s (4 ml/min). We estimated that the complete mixing time should be <20 s. The room temperature was between 21 and 24°C. The two-electrode voltage-clamp system (model TEVC-200, Dagan Corp.) and the pClamp data acquisition program (Axon Instruments, Inc.) were used for data acquisition. Oocytes were normally kept under open circuit condition in experimental chambers. At the time of interest, the membrane potential was ramped from −120 to +60 mV in a period of 1.8 s to construct the whole cell I-V plots.
Reagents
The CFTR Cl− channels were activated using a cocktail containing the phosphodiesterase inhibitor, isobutylmethyl xanthine (IBMX; RBI or Sigma-Aldrich), and the adenylate cyclase activator, forskolin, or the β-adrenergic agonist, isoproterenol (Sigma-Aldrich). Coinjected β-adrenergic receptor was used on occasion as an alternative to activate adenylate cyclase. Although the receptor has 10 native cysteines, three of which reside in the second extracellular loop, and five in the transmembrane domain, Javitch et al. 1997 showed that MTSEA+ had no effect on the binding of agonist or antagonist to wild-type β2-adrenergic receptor expressed in HEK293 cells. The majority of the experiments for this study were done using 10 μM isoproterenol and 1 mM IBMX as the stimulating cocktail (Isop + IBMX), but all the results were confirmed using 10 μM forskolin and 1 mM IBMX.
Other reagents included highly polar derivatives of MTS reagents, [2-(trimethylammonium)ethyl] methanethiosulfonate bromide (MTSET+) and sodium [2-sulfonatoethyl]methanethiosulfonate MTSES−) obtained from Toronto Research Chemicals, a reducing reagent, 2-mercaptoethanol (2-ME; Sigma-Aldrich), and Brefeldin A (BFA; Sigma-Aldrich), which blocks protein trafficking to cell membranes by causing the retention of proteins in the ER (Klausner et al. 1992).
Data Analysis
The data was analyzed using an analysis program developed in our laboratory and was presented in the form of I-V plots. The conductance reported here was calculated from the slope of the I-V plot at the reversal potential. Data are reported as mean ± SEM.
RESULTS
Modification of R334C CFTR by MTSET+ Is Stable but Reversible
The effects of MTSET+ and MTSES− modification on the conductance of R334C CFTR are documented in the companion paper (see Smith et al. 2001, in this issue) and were briefly summarized in Fig. 1. Expression of R334C CFTR in Xenopus oocytes gives rise to cAMP-activated Cl− conductance characterized by modest inward rectification, which is distinct from that seen with expression of wt CFTR that is characterized by modest outward rectification. Brief exposure of oocytes expressing R334C CFTR to MTSET+ results in an approximate doubling of the conductance and a change in the shape of the I-V plot to one that is linear. In contrast, application of MTSES− attenuates the conductance by ∼50% and enhances the inward rectification. Recordings from excised patches presented in the companion paper (see Smith et al. 2001, in this issue) also demonstrated that MTSET+ modification increased the single-channel conductance of R334C CFTR. The effect of MTSET+-modification was not spontaneously reversible, but was readily reversed by a reducing reagent such as 2-ME (see Fig. 3). These observations indicated that MTSET+-modified and -unmodified channels could be distinguished by their functional characteristics.
Figure 1.

I-V relationship of R334C CFTR is modified by MTSET+ and MTSES−. (A) I-V plots at steady-state activation. Oocytes were continuously perfused with a cocktail containing 10 μM isoproterenol and 1 mM IBMX (control). An ∼5-min exposure to 1 mM MTSET+ induced an approximate doubling of the conductance and a change in the shape of the I-V plot. (B) I-V plots obtained at steady-state activation (control) and after ∼5-min exposure to 1 mM MTSES− that attenuated the conductance by ∼50% and enhanced inward rectification.
Figure 3.
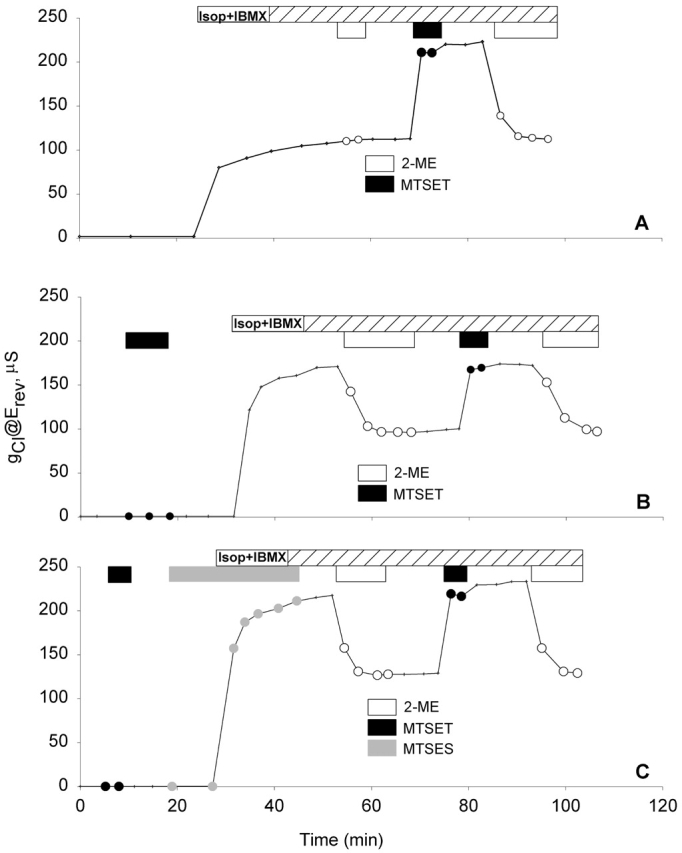
The entire membrane pool of R334C CFTR channels that were activated by cAMP was labeled with MTSET+ before the activation. Records of gCl versus time obtained from R334C CFTR expressing oocytes that were obtained from the same frog and assayed on the same day. Oocytes were always perfused with frog Ringer's unless noted (see materials and methods). After a control period, they were perfused with stimulatory cocktail containing 10 μM isoproterenol and 1 mM IBMX (Isop + IBMX). (A) A record of gCl versus time showing that at the steady-state activation, MTSET+ caused about a doubling of gCl. (B) A record of gCl versus time showing that the activated gCl at the steady state of an oocyte preexposed to MTSET+ was much higher than the gCl at unmodified condition as seen in A, and 2-ME reduced gCl to ∼50% of the maximum gCl. (C) A record of gCl versus time of showing that the exposure to MTSES− after prelabeling with MTSET+ had no effect on gCl before and after activation.
To test the stability of MTSET+ labeling, we performed a group of experiments in which the whole-cell conductance was monitored for up to 5 h after a 20-s exposure of oocytes to 100 μM or 1 mM MTSET+. The conductance (@Erev) was first obtained 30–40 min after the initial exposure of the oocytes to stimulatory cocktail when the activation of the Cl− conductance had attained a steady state (control). Each oocyte was then exposed to MTSET+ for 20 s. The MTSET+-containing stimulatory cocktail was then replaced with stimulatory cocktail lacking the thiol reagent, and the oocytes were continuously perfused for up to 5 h. The conductances measured at 2 min, 2 h, and 5 h after exposure of oocytes to MTSET+ were normalized to the steady-state conductance before exposure to MTSET+. The values displayed in Fig. 2 demonstrate that the MTSET+-induced conductance increase was identical at 2 min, 2 h, and 5 h after exposure to the reagent. The change in the shape of the I-V relation (unpublished data) was also characteristic of R334C CFTR as described in the accompanying paper (see Smith et al. 2001, in this issue) and Fig. 1 and did not change with time.
Figure 2.

Modification of R334C CFTR by MTSET+ was stable for at least 5 h. The conductance (gCl@Erev) was first obtained 30–40 min after the initial exposure of the oocytes to stimulatory cocktail when the activation of the Cl− conductance had attained a steady state (control). Each oocyte was exposed to 100 μM or 1 mM MTSET+ for 20 s. The MTSET+-containing stimulatory cocktail was then replaced with stimulatory cocktail lacking the thiol reagent, and the oocytes were continuously perfused for up to 5 h. The conductances measured at 2 min, 2 h, and 5 h after exposure of oocytes to MTSET+ were normalized to the steady-state conductance before exposure to MTSET+.
The Entire Membrane Pool of R334C CFTR Activated by cAMP Is Accessible to MTSET+ before Activation
The level of CFTR expression used in these studies was such that, before activation by stimulatory cocktail, the conductance of the oocyte membrane was similar to that seen in an oocyte that was not expressing R334C CFTR. The activated CFTR conductance was generally 50–100-fold greater than that of the background conductance, so that before activation, the product of the number of channels and the open probability (NPo) for the CFTR channels was of the order of one hundredth of that seen in the active state due to a very low value of Po, a low value of N or some combination of the two. Therefore, we refer to this condition as the “inactive state,” despite the fact that it could represent a large population of membrane-localized channels, each of which exhibits a very low, but nonzero, probability of opening.
Fig. 3 contains plots of the conductance (@Erev) versus time obtained from a group of experiments designed to determine if R334C CFTR channel can be modified by externally applied, impermeant thiol reagents in the active as well as the inactive state. As seen in Fig. 3 A, after the channels were activated by stimulatory cocktail containing 10 μM isoproterenol and 1 mM IBMX, exposure of the oocyte to 1 mM 2-ME had no effect on the conductance (gCl) before MTSET+ modification. Subsequently, a 5-min exposure to 100 μM MTSET+ caused about a doubling in gCl, and the effect was reversed by 2-ME.
The efficacy of MTSET+ modification of R334C CFTR did not depend on the state of activation of CFTR (Fig. 3 B). Before activation, an oocyte was exposed to 100 μM MTSET+ for about 5 min, and then to stimulatory cocktail. Afterwards, 1 mM 2-ME decreased gCl to ∼50% of the maximum value. A second exposure of the oocyte to MTSET+ and 2-ME induced the same response, suggesting that pre- and postactivation exposure to MTSET+ labeled the same population of R334C CFTR channels.
To determine if labeling of the entire pool of R334C CFTR channels could be attributed to nonreacted MTSET+ that might remain after washing and, thus, be present during channel activation, the channels were prelabeled with MTSET+ and then activated in the presence of MTSES− as illustrated in Fig. 3 C. If unlabeled channels were appearing at the surface during activation, then the relative abundance of MTSES− over that of MTSET+ (>1,000:1) would render it much more likely that negative charges would be added to the channel and gCl would be consequently reduced. However, the result was identical to that obtained in the absence of MTSES−, which is consistent with the notion that all of the channels in the activatable pool were labeled during the exposure to MTSET+ in the inactive state.
To determine if labeling with MTSET+ altered the process of R334C CFTR activation, we first labeled the channels with 100 μM MTSET+ after activation, then inactivated the labeled channels, and then reactivated them at a later time (Fig. 4). Fig. 4 is an example of four similar experiments. It can be seen that the labeled channels were completely inactivated by ∼40 min after the removal of stimulatory cocktail. Reactivation of R334C CFTR increased gCl to a level similar to that seen after the first modification, and 2-ME decreased gCl by ∼50%. The effects were reproduced by the second exposure to MTSET+ and 2-ME, indicating that MTSET+ did not interfere with the activation or inactivation of R334C CFTR.
Figure 4.

MTSET+ labeling did not affect the activation and inactivation process of R334C CFTR. A record of the conductance measured at the reversal potential throughout an experiment. After activation, an oocyte was exposed to 100 μM MTSET+, and then was inactivated without removing MTSET+. The oocyte was then reactivated, showing a gCl similar to the gCl before inactivation. The gCl was then reduced to ∼50% by 2-ME.
If the channels underwent rapid recycling, a 5-min exposure of oocytes to MTSET+ might be long enough to label channels that were in a submembranous pool but surfaced during this time, so we performed another group of experiments similar to that described in Fig. 2 C, in which the MTSET+ exposure time was reduced to 20 s. Fig. 5 contains the result of one of eight similar experiments conducted from day 4 to day 6 after RNA injection in which gCl was measured throughout a single experiment. Also shown are I-V plots corresponding to specific points of interest in Fig. 5 (B–D). The oocyte was exposed to 1 mM MTSET+ for 20 s in the inactive state, and was immediately perfused with a second thiol reagent, MTSES− (1 mM), for 5 min. Neither reagent affected the background conductance (Fig. 5 B, 1 and 2). In the continuous presence of MTSES−, the oocyte was then exposed to stimulatory cocktail. The shape of the I-V plot obtained at steady-state activation in the presence of MTSES− was characteristic of MTSET+-modified R334C CFTR (Fig. 5 C, 3), indicating that prelabeling with MTSET+ for 20 s prevented modification by MTSES− before and during activation. Furthermore, subsequent exposure to MTSET+ had no marked effect on the conductance or the shape of the I-V plot (Fig. 5 C, 5). Exposure of the oocyte to the reducing reagent (2-ME) decreased the conductance to about half the stimulated value, and changed the shape of the I-V plot to inward rectification typical of unmodified R334C (Fig. 5 C, 6), indicating that the positively charged, TEA group that was added in the inactive state was readily removed. After 2-ME treatment, the second exposure of the oocyte to MTSET+ produced similar results as described above (Fig. 5 D, 7). The results indicate that the modification of R334C CFTR by MTSET+ was complete, regardless of whether exposure to the reagent took place in the active or inactive state, confirming that the entire pool of CFTR channels was accessible to MTSET+ before activation.
Figure 5.

The entire membrane pool of R334C CFTR channels that were activated by cAMP was labeled with 20-s exposure to MTSET+ before the activation. (A) A record of the conductance measured at the reversal potential throughout an experiment. (B) Exposure to 1 mM MTSET+ or 1 mM MTSES− had no significant effects on the background conductance before activation of CFTR (1 and 2). (C) Modification by 1 mM MTSET+ for 20 s before activation prevented any further modification by MTSES+ or MTSET+ during or after activation (3–5), and 2-ME reversed the effect of MTSET+ (6). (D) Further modification by MTSET+ was possible after 2-ME treatment and was also reversed by 2-ME (7 and 8). The apparent lower values of Erev (∼17 mV) resulted from a shift in the tip potential of the recording electrode.
The Time Course of Addition of R334C CFTR Channels to the Plasma Membrane after cRNA Injection
After cRNA injection, new CFTR channels must be synthesized and inserted in the oocyte plasma membrane. To study the time course of the insertion of new channels, we recorded the level of whole-cell conductance in single oocytes for five consecutive days after cRNA injection. Each day the channels were maximally activated using stimulatory cocktail and were subsequently inactivated within two hours by perfusing with frog Ringer's solution. The oocytes were then returned to the incubator overnight and subjected to the same experimental manipulation the next day. Not all of the oocytes survived the entire experimental period. Those that survived for at least two consecutive days were used in the analysis. Thus, the conductance was normalized to the conductance measured on day 2 (48 h after RNA injection). Fig. 6 is a summary of the conductance of R334C CFTR in its inactive and active state during the first 5 d after cRNA injection. It can be seen that the rate of addition of new channels was most rapid between 24 and 48 h, and the total conductance leveled off after day 3. A small cAMP induced conductance (2.82 ± 0.27 μS, N = 4), greater than the background conductance (0.72 ± 0.30 μS), was observed 5–7 h after RNA injection.
Figure 6.

Time-dependent increase in the conductance of R334C CFTR in the first 5 d after cRNA injection. Oocyte conductance in both inactive and cAMP-activated states was measured on days 1, 2, 3, 4, and 5 (24, 48, 72, 96, and 120 h) after cRNA injection and the values were normalized to the conductance on day 2.
The Addition of New Channels via the Biosynthetic Pathway Can Be Monitored by Labeling Plasma Membrane Channels with MTSET+
If indeed the addition of the channels is most rapid between 24 and 48 h after cRNA injection, it should be possible to monitor the addition of new channels to the membrane by labeling the channels on the surface with MTSET+, and assaying the labeled fraction of the surface pool as a function of time. Shown in Fig. 7 A is one of four similar experiments performed on day 1 (24 h after cRNA injection). Exposure of an oocyte to 100 μM MTSET+ for 5 min after activation roughly doubled the conductance, as expected from previous experiments. After removal of stimulatory cocktail, the conductance returned to its background level within 2 h. Exposure of the oocyte to stimulating cocktail 4.5 h later induced a steady-state conductance ∼40% higher than that measured 4.5 h earlier. Subsequent exposure of the oocyte to 2-ME reduced the conductance. The decrease in the conductance (23.13 μS) was essentially the same as the original MTSET+-induced conductance (21.17 μS), as expected if the number of labeled channels remained constant during this period, and the entire increase in the total conductance was due to the addition of new (unlabeled) channels to the plasma membrane via the biosynthetic pathway. A second exposure to MTSET+ doubled the conductance as expected, indicating that both populations (old and new channels) were modifiable by the thiol reagent. However, when the same experiment was conducted on day 5 (Fig. 7 B, showing one of six similar experiments), MTSET+ had no additional effect when applied after the second activation, indicating that no new channels were added during the 4.5 h period. In some oocytes, such as the one shown, we observed run-down of conductance with time in experiments lasting several hours.
Figure 7.

The addition of new channels can be monitored by labeling preexisting channels with MTSET+. (A) A record of the conductance measured at the reversal potential throughout an experiment on day 1. The conductance was increased after 4.5 h due to the addition of new, unlabeled channels. (B) A record of the conductance measured at the reversal potential throughout an experiment on day 5. No increase in the conductance was observed during the 4.5-h period.
BFA Reduced the Fractional Conductance Contributed by Newly Added Channels
If the increase in conductance seen over a 4.5 h period on day 1 reflects the addition of new channels to the membrane, then blocking the trafficking of the new channels should prevent the increase in the conductance. Brefeldin A (BFA) has been shown to inhibit protein secretion at an early step in the secretory pathway by inhibiting the membrane traffic from the ER to the Golgi and enhancing the movement of Golgi membrane into the ER; the result being retention of proteins in the ER compartment (Klausner et al. 1992). Shown in Fig. 8 are two experiments done on day 1 and day 5, respectively, in which oocytes were exposed to BFA before activation and continuously exposed to BFA throughout the entire experiment. On day 1, BFA prevented the increase in conductance seen after 4.5 h in the absence of the drug, and there was no evidence of the appearance of unlabeled channels. Although the fractional decrease in the conductance induced by 2-ME (Fig. 8 A, time point 2) appeared to be somewhat smaller that the fractional increase in conductance induced by the first MTSET+ exposure (Fig. 8 A, time point 1), this was due to an increase in background conductance over the period. On day 5, no increase in conductance was seen in the presence of BFA (Fig. 8 B) as expected from the result obtained in the absence of the drug. This sort of long term experiment (>8 h) was often difficult to perform because the membrane of some oocytes became leaky after a few hours perfusion, but the results obtained from oocytes that survived (two from day 1 and three from day 5–6) indicated that BFA prevented the addition of new channels on day 1, and it had no marked effect on the function of channels resident in the plasma membrane.
Figure 8.

BFA prevented the addition of new channels on day 1, and had no effect on the conductance on day 5. Records of the conductance measured at the reversal potential throughout experiments. Oocytes were exposed to BFA throughout the entire experimental period. No increase in the conductance with time was observed after 4.5 h on either day 1 (A) or day 5 (B).
The two oocytes shown in Fig. 7 that were never exposed to BFA appeared to have a slower rate of deactivation than the two oocytes exposed to BFA shown in Fig. 8, but a comparison of 15 control and 5 BFA-treated oocytes disclosed that the time required for the activated conductance to return to background level varied from 50 to 200 min in both groups. Because oocytes often did not survive prolonged periods of recording in the experimental chamber, we characterized the effects of BFA on CFTR expression in a separate group of oocytes by adding BFA to the incubating solution (MBSH) beginning a few hours after cRNA injection and assaying the cAMP-activated Cl− conductance after either 24 or 48 h continuous exposure to BFA (Fig. 9). The oocytes used in this group of experiments were obtained from the same frog and were injected with R334C CFTR RNA at the same time. In the absence of BFA treatment, cAMP elevated Cl− conductance was well above the background conductance (gbkg) on day 1 and continued to increase on day 2. However, in oocytes treated with BFA, cAMP activated conductance was greatly reduced as expected if BFA attenuated the delivery of CFTR to the plasma membrane.
Figure 9.

Long-term BFA treatment prevented the addition of new channels. Some oocytes were kept in MBSH solution as usual (control). Some oocytes were transferred to the MBSH solution containing 5 μM BFA a few hours after cRNA injection and remained in the same solution thereafter (BFA-treated). The background conductance (gbkg) and cAMP-elevated Cl− conductance (gCl) were assayed at 24 and 48 h after cRNA injection.
DISCUSSION
In CFTR-expressing Oocytes cAMP Increases Cl− Conductance by Increasing the Open Probability of Channels
The results presented here are consistent with the notion that the entire, activatable pool of R334C CFTR in the Xenopus oocyte is accessible to MTSET+ during a 20-s exposure to a perfusion solution containing the reagent. The simplest interpretation of this result is that the entire activatable pool of CFTR channels resides in the plasma membrane, and that the process of activation is due solely to an increase in the Po of membrane-resident channels. This interpretation is consistent with previous functional studies of CFTR channels in which single-channel records were obtained from detached patches exposed to the catalytic subunits of PKA and ATP on the cytoplasmic side (Tabcharani et al. 1991; Baukrowitz et al. 1994; Gadsby et al. 1994; Hwang et al. 1994; Zeltwanger et al. 1999). Although fewer in number, there are also several studies in which cell-attached recording provided evidence of increases in Po associated with activation of CFTR (Fischer and Machen 1994; Hwang et al. 1994). The hypothesis that CFTR activation involves an increase in Po is also supported by studies showing that mutations in the R domain or the nucleotide binding folds alter the relation between Po and stimulating conditions (Carson et al. 1995; Wilkinson et al. 1996, Wilkinson et al. 1997).
However, there are two alternative interpretations of the MTSET+ labeling results that lead to quite different conclusions about the mechanism of activation of CFTR mediated Cl− conductance. First, if the thiol reagent used in these studies (MTSET+) can enter the oocyte, it is possible that exposing the cell to the reagent could lead to the labeling of CFTR proteins located in submembranous vesicles that might subsequently appear on the surface after activation. Second, if there is a pool of CFTR protein that is subject to rapid recycling between the plasma membrane and a submembranous pool of vesicles then, in principle, the entire pool could be labeled despite the fact that the label was only present in the extracellular bath for a short period of time.
MTSET+ Is Impermeant
The studies of Holmgren et al. 1996 and Yang et al. 1996 suggested that MTSET+ is not likely to cross the plasma membrane and label R334C CFTR protein that might exist in subplasma membrane vesicles. Holmgren et al. 1996 tested the membrane permeability to MTS reagents using three different membrane systems, liposomes, HEK293 cells, and Xenopus oocytes. They reported that MTSET+ and MTSES− were not permeant in liposomes at pH 7.4, whereas MTSEA+ (aminoethyl methanethiosulfonate) was. In HEK293 cells transfected with Shaker K+ channel, they tested the accessibility of a cysteine residue (391C) located in the intracellular loop between the transmembrane segments S4 and S5 of the Shaker K+ channel construct to cis- and trans application of MTS reagents in excised (inside-out and outside-out) patches. The authors showed that 391C mutant could only be modified by MTSET+ when applied from the intracellular side, whereas MTSEA+ modified the channel when applied from either side. In Xenopus oocytes expressing 391C Shaker K channel, recording of macroscopic currents indicated that extracellular MTSET+ did not modify the channel, whereas 2 mM extracellular application of MTSEA+ produced the typical response. We have confirmed this observation in oocytes (unpublished data).
The results of a study of the voltage-gated sodium channel by Yang et al. 1996 were also consistent with the notion that MTSET+ cannot reach cysteines by crossing the plasma membrane. The S4 segment of domain 4 of the voltage-gated sodium channel is thought to move outwardly upon depolarization. The authors showed that two basic residues in S4, referred to as R1 and R3, when substituted with cysteine, were accessible to extracellular MTSET+ (which altered the inactivation kinetics) only when the cell was depolarized. Under the same conditions, however, R3C was inaccessible to MTSET+ applied on the cytoplasmic side, but it became accessible immediately after repolarization. This result indicated that MTSET+ did not cross the membrane and modify reactive cysteines.
Recycling due to Constitutive Endocytosis and Exocytosis Is Not likely to Be Rapid Enough to Account for the Labeling of the Entire Pool of CFTR Channels in 20 s
In three mammalian cell lines, CFTR was reported to undergo rapid endocytosis when incubated at 37°C (Prince et al. 1994; Howard et al. 1996; Lukacs et al. 1997). These authors used similar approaches in which they first labeled the cell surface CFTR at 4°C, and then warmed up the cells to 37°C for various times to allow for endocytosis. The rate of endocytosis was determined by measuring the time-dependent, fractional internalization of CFTR or the amount of CFTR that remained on the surface. Prince et al. 1994 reported that in T84 cells, 50% of the surface CFTR was internalized in ∼1 min. They also reported that after ∼7.5 min incubation at 37°C, the internalized CFTR started to return to the surface. Lukacs et al. 1997 reported that in CHO cells, ∼15% of biotinylated cell-surface CFTR was internalized in 3 min. Howard et al. 1996 reported that in HeLa cells expressing a construct bearing an epitope tag in the fourth extracellular loop (M2–901 CFTR), most of the M2 antibody bound CFTR was internalized within 2 min.
If CFTR endocytosis in Xenopus oocytes proceeds at the highest rate reported for mammalian cells, then, in 20 s, the amount of recycled CFTR would be only ∼17% of the membrane channel pool. Because all of our experiments were conducted at ∼21–24°C, the rate of endocytosis is expected to be even less. Furthermore, we found no difference in the effectiveness of MTSET+ labeling when the exposure time was increased to 5 min. Therefore, it seems unlikely that the recycled CFTR channels contributed to the labeled channel pool under our experimental conditions.
If endocytosis of R334C CFTR were proceeding in Xenopus oocytes at the highest rate reported for mammalian cells (∼50%/min), then the maintenance of the steady-state, activated CFTR conductance commonly observed in oocytes would require an equally high exocytotic delivery rate to maintain the channel population. In this condition, however, just after a brief exposure to MTSET+, a decrease in conductance should be observed because labeled, high conductance channels would be predicted to leave the membrane, whereas unlabeled, low conductance channels were being added. This sort of behavior was never observed, suggesting that the rate of turnover of R334C CFTR is relatively slow in Xenopus oocytes.
The absence of detectable endocytosis in oocytes might be attributed to the reduced temperature or to some other differences between Xenopus oocytes and mammalian cells. In the case of the human low density lipoprotein receptor, however, not only can the protein be synthesized, glycosylated and transported to the cell surface in Xenopus oocytes, but it also undergoes rapid internalization similar to that observed in mammalian cells (Peacock et al. 1988). The observations of Peacock et al. 1988 suggest that the signals for glycosylation and endocytosis of the low density lipoprotein receptor are similar in Xenopus oocytes and mammalian cells.
Although the results reported here provide no evidence for a stimulation-dependent increase in the number of CFTR channels in the plasma membrane of oocytes, we cannot exclude a small contribution to activation via this mechanism. In any single experiment, we assume that we can measure the conductance with an accuracy of at least 1 μS. If we assume the unitary conductance of CFTR channel expressed in oocytes to be ∼5 pS and Po to be 0.5, then a conductance of 100 μS represents ∼40 million channels/oocyte and 1 μS would represent ∼400 thousand channels.
It must also be emphasized that all of the results reported here were obtained using a CFTR mutant (R334C), so that it is possible this mutation suppresses stimulation-dependent trafficking mechanism that is more prominent in the wild-type and M2–901 CFTR (see Mechanism of CFTR Activation). However, like other membrane proteins that undergo clathrin-mediated endocytosis, the internalization signal contained in the cytoplasmic tail of the wt CFTR (which was retained in R334C CFTR) was found to be sufficient for promoting endocytosis (Prince et al. 1999). It also seems unlikely that labeling of R334C CFTR with thiol reagents would cause the apparent low rate of recycling, given that biotinylated CFTR is efficiently endocytosed in mammalian cells (Prince et al. 1994; Lukacs et al. 1997).
Blocking the Insertion of Channels Did Not Affect Activation
After labeling with MTSET+, the appearance of unlabeled channels on the plasma membrane was observed during the first few days after the injection of R334C cRNA when the insertion of new channels via the biosynthetic pathway was expected. We inferred that the unlabeled channels were likely to be newly synthesized channels that were inserted into the membrane via vesicle trafficking mechanism because their appearance on the membrane was prevented by BFA treatment. On the other hand, blocking channel insertion with BFA did not alter activation or inactivation of surface CFTR.
The results presented here indicate that the insertion of new CFTR channels into the plasma membrane via the biosynthetic pathway occurred in the absence of agonist-dependent increase in cAMP. The endogenous cAMP level in Xenopus oocytes is expected to be much lower than that elicited by stimulatory cocktail. For example, Smith et al. 1987 reported that exposure of oocytes with intact follicular enclosure to forskolin or isoproterenol increased intracellular cAMP concentration by 15-fold. Therefore, if the delivery of CFTR channels to the plasma membrane via the biosynthetic pathway is cAMP-dependent, the cAMP level required is much less than that required for channel activation.
Mechanism of CFTR Activation
The results of several recent studies using either oocytes (Takahashi et al. 1996; Peters et al. 1999) or mammalian cells (Howard et al. 1996, Howard et al. 2000) have been interpreted as indicating that cAMP-dependent activation of CFTR Cl− conductance is due, in large part, to an increase in the number of CFTR channels (N) in the plasma membrane. Although mechanisms of activation involving increases in N or increases in Po are not mutually exclusive, the results presented here are consistent with the hypothesis that activation of CFTR conductance occurs largely, if not solely, by an increase in the Po of CFTR channels.
The most direct comparisons of the present results are with those of Takahashi et al. 1996 and Peters et al. 1999 who reported that the activation of either wt CFTR or M2–901 CFTR expressed in Xenopus oocytes was associated with an increase in membrane capacitance, Cm. The increase in Cm was correlated with an increase in Cl− conductance (gCl) and also with an increase in membrane area determined via electron microscopy and digital morphometry. However, Weber et al. 1999 used a frequency domain assay for Cm to study the effects of cAMP on gCl and Cm in oocytes expressing wild-type CFTR and obtained different results. They observed a correlation between increases in gCl and Cm, but found that in oocytes exposed to inhibitors of PKA (KT5720 or H8), cAMP increased gCl without affecting Cm. A similar dissociation of changes in gCl and Cm was seen in oocytes injected with the Ca2+ chelator, BAPTA. These results strongly suggest that, although increases in oocyte cAMP levels may lead to an increase in Cm, there is no obligatory relation between cAMP-induced increases in gCl and the increases in Cm. Hug et al. 1997 used a two-frequency, lock-in amplifier method to assay Cm in CHO cells expressing wt CFTR and found no change in Cm despite a sevenfold increase in gCl. Furthermore, Chen et al. 2001 recently demonstrated that large increases in membrane conductance can produce measurement errors that lead to apparent changes in Cm when none actually occur, suggesting that any correlation between increases in conductance and capacitance must be evaluated carefully. These investigators proposed that neither cAMP nor Ca2+ induced the delivery of CFTR to the plasma membrane of Calu-3 cells.
Peters et al. 1999 reported the results of a more direct assay of surface localization of M2–901 CFTR expressed in oocytes. They observed an increase in fluorescence intensity in oocytes treated with a cocktail of forskolin and IBMX, and interpreted the result as a confirmation of exocytotic delivery of CFTR protein to the plasma membrane induced by cAMP. Their data indicated that a near 10-fold increase in CFTR conductance was paralleled by a near sixfold increase in fluorescence intensity, as if the majority of cAMP-activated channels were newly inserted. This amount of channel insertion should be readily detectable by covalent labeling, but it was not evident in the studies reported here.
There are several potential problems associated with immunostaining. First, the protocol required overnight incubation of oocytes with the antibody at 4°C, so that it was not possible to determine if there was any temporal correlation between changes in gCl and staining intensity. It is also possible that the staining by M2 antibody may greatly underestimate the level of protein in unstimulated oocytes. In fact, Howard et al. 2000 recently reported that immunoprecipitation of M2–901 CFTR by M2 antibody was much less efficient than that by a COOH terminus antibody. Thus, the ratio of actual protein expression in the stimulated and unstimulated conditions could be dramatically smaller than that indicated. It is also possible that M2 antibody recognition of plasma membrane-localized M2–901 CFTR is in some way enhanced if the protein is in the active conformation. There are well-documented examples of conformation-dependent antibody binding to membrane proteins (Vassilev et al. 1988, Vassilev et al. 1989; Aoki 1992; Anthony and Azmitia 1997).
Howard et al. 2000 recently suggested that the mode of CFTR activation in cells might be dependent on the level of protein expression, such that at high expression levels an increase in Po predominates because the exocytotic pathway is “saturated.” This hypothesis cannot explain the discrepancy in the oocyte results, however, in as much as the level of expression used in covalent labeling studies was comparable to that used previously for determination of capacitance or antibody staining.
A survey of the literature reveals that the question of cAMP-induced delivery of CFTR to the plasma membrane of mammalian cells is controversial and that varying results have been reported, even for the same cell type. For example, in T84 cells, Prince et al. 1993 and Denning et al. 1992 using surface biotinylation and/or antibody staining reported no change in the amount of CFTR in the apical membrane after cAMP stimulation. Tousson et al. 1996 on the other hand, reported increases in fluorescent intensities toward the apical membranes of T84 cells when they examined changes in antibody staining patterns in cross sections of the cells. The interpretation of the latter studies is likely to be complicated by the difficulties in determining that channels marked by immunostaining or fluorescence actually reside within the plasma membrane. In MDCK cells, Moyer et al. 1998 used surface biotinylation and a green fluorescent protein–CFTR expression vector (green fluorescent protein linked to the NH2 terminus of CFTR) for labeling CFTR and observed no cAMP stimulated translocation of CFTR from an intracellular pool to cell surface. In contrast, Howard et al. 2000 reported in the same cell that when epitope-tagged, virally expressed CFTR (M2–901 CFTR) was expressed at low levels, forskolin increased immunostaining of the apical membrane. In HeLa cells, Denning et al. 1992 found no change in the amount of CFTR in the apical membrane after cAMP stimulation, whereas Howard et al. 1996 reported that a 10-min treatment with 10 μM forskolin caused about a twofold increase in surface fluorescence intensity in HeLa cells expressing M2–901 CFTR.
The covalent labeling technique used in the present study offers a relatively straightforward method for assaying time- and stimulation-dependent delivery of channels to the plasma membrane. It has the advantage of monitoring changes in real time, and it is a direct assay of functional channels that contain a reactive thiol, the modification of which is readily detectable. Snyder 2000 recently used a similar method to reveal cAMP-mediated translocation of the epithelial Na+ channel.
In mammalian cells the role of cAMP-induced plasma membrane delivery of CFTR in the activation of Cl− conductance remains to be tested directly. However, in oocytes, covalent labeling studies lead us to conclude that most, if not all cAMP-induced increase in gCl was the result of an increase in the Po of membrane localized CFTR channels.
Acknowledgments
We thank Dr. Steve Ernst and Dr. Caroline A. Enns for helpful discussion, Dr. William R. Skach for critical review of this manuscript, and Dr. G. Yellen for providing the 391C Shaker K+ construct. We thank Brian Dunckley, Chi-ching Wang, and Joshua Billingsley for their assistance in oocyte preparation and cRNA injection.
This work was supported by the National Institute for Diabetes, Digestive and Kidney Diseases (NIH DK45880) and the Center for Membrane Toxicology Studies at the Mount Desert Island Biological Laboratory.
Footnotes
Abbreviations used in this paper: BFA, Brefeldin A; IBMX, 3-isobutyl-methylxanthine; 2-ME, 2-mercaptoethanol; MTSEA, 2-(aminoethyl) methanethiosulfonate; MTS, methanethiolsulfonate; MTSES−, sodium[2-sulfonatoethyl] MTS; MTSET+, [2-(trimethylammonium) ethyl] MTS bromide; Po, open probability; wt CFTR, wild-type CFTR.
References
- Anderson M.P., Welsh M.J. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains. Science. 1992;257:1701–1704. doi: 10.1126/science.1382316. [DOI] [PubMed] [Google Scholar]
- Anderson M.P., Gregory R.J., Thompson S., Souza D.W., Paul S., Mulligan R.C., Smith A.E., Welsh M.J. Demonstration that CFTR is a chloride channel by alteration of its anion selectivity. Science. 1991;253:202–205. doi: 10.1126/science.1712984. [DOI] [PubMed] [Google Scholar]
- Anderson M.P., Sheppard D.N., Berger H.A., Welsh M.J. Chloride channels in the apical membrane of normal and cystic fibrosis airway and intestinal epithelia. Am. J. Physiol. 1992;263:L1–L14. doi: 10.1152/ajplung.1992.263.1.L1. [DOI] [PubMed] [Google Scholar]
- Anthony T.E., Azmitia E.C. Molecular characterization of antipeptide antibodies against the 5-HT1A receptorevidence for state-dependent antibody binding. Brain Res. Mol. Brain Res. 1997;50:277–284. doi: 10.1016/s0169-328x(97)00201-5. [DOI] [PubMed] [Google Scholar]
- Aoki C. Beta-adrenergic receptorsastrocytic localization in the adult visual cortex and their relation to catecholamine axon terminals as revealed by electron microscopic immunocytochemistry. J. Neurosci. 1992;12:781–792. doi: 10.1523/JNEUROSCI.12-03-00781.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baukrowitz T., Hwang T.C., Nairn A.C., Gadsby D.C. Coupling of CFTR Cl− channel gating to an ATP hydrolysis cycle. Neuron. 1994;12:473–482. doi: 10.1016/0896-6273(94)90206-2. [DOI] [PubMed] [Google Scholar]
- Bear C.E., Duguay F., Naismith A.L., Kartner N., Hanrahan J.W., Riordan J.R. Cl− channel activity in Xenopus oocytes expressing the cystic fibrosis gene. J. Biol. Chem. 1991;266:19142–19145. [PubMed] [Google Scholar]
- Carson M.R., Travis S.M., Welsh M.J. The two nucleotide-binding domains of cystic fibrosis transmembrane conductance regulator (CFTR) have distinct functions in controlling channel activity. J. Biol. Chem. 1995;270:1711–1717. doi: 10.1074/jbc.270.4.1711. [DOI] [PubMed] [Google Scholar]
- Chen P., Hwang T.-C., Gillis K.D. The relationship between cAMP, Ca2+, and transport of CFTR to the plasma membrane. J. Gen. Physiol. 2001;118:135–144. doi: 10.1085/jgp.118.2.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng S.H., Rich D.P., Marshall J., Gregory R.J., Welsh M.J., Smith A.E. Phosphorylation of the R domain by cAMP-dependent protein kinase regulates the CFTR chloride channel. Cell. 1991;66:1027–1036. doi: 10.1016/0092-8674(91)90446-6. [DOI] [PubMed] [Google Scholar]
- Denning G.M., Ostedgaard L.S., Cheng S.H., Smith A.E., Welsh M.J. Localization of cystic fibrosis transmembrane conductance regulator in chloride secretory epithelia. J. Clin. Invest. 1992;89:339–349. doi: 10.1172/JCI115582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dho S., Grinstein S., Foskett J.K. Plasma membrane recycling in CFTR-expressing CHO cells. Biochim. Biophys. Acta. 1993;1225:78–82. doi: 10.1016/0925-4439(93)90125-k. [DOI] [PubMed] [Google Scholar]
- Drumm M.L., Wilkinson D.J., Smit L.S., Worrell R.T., Strong T.V., Frizzell R.A., Dawson D.C., Collins F.S. Chloride conductance expressed by delta F508 and other mutant CFTRs in Xenopus oocytes. Science. 1991;254:1797–1799. doi: 10.1126/science.1722350. [DOI] [PubMed] [Google Scholar]
- Fischer H., Machen T.E. CFTR displays voltage dependence and two gating modes during stimulation. J. Gen. Physiol. 1994;104:541–566. doi: 10.1085/jgp.104.3.541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadsby D.C., Hwang T.C., Baukrowitz T., Nagel G., Horie M., Nairn A.C. Regulation of CFTR channel gating Jpn. J. Physiol. 44Suppl1994. S183 S192 [PubMed] [Google Scholar]
- Holmgren M., Liu Y., Xu Y., Yellen G. On the use of thiol-modifying agents to determine channel topology. Neuropharmacology. 1996;35:797–804. doi: 10.1016/0028-3908(96)00129-3. [DOI] [PubMed] [Google Scholar]
- Howard M., Jilling T., DuVall M., Frizzell R.A. cAmp-regulated trafficking of epitope-tagged CFTR. Kidney Int. 1996;49:1642–1648. doi: 10.1038/ki.1996.239. [DOI] [PubMed] [Google Scholar]
- Howard M., Jiang X., Stolz D.B., Hill W.G., Johnson J.A., Watkins S.C., Frizzell R.A., Bruton C.M., Robbins P.D., Weisz O.A. Forskolin-induced apical membrane insertion of virally expressed, epitope-tagged CFTR in polarized MDCK cells. Am. J. Physiol. Cell Physiol. 2000;279:C375–C382. doi: 10.1152/ajpcell.2000.279.2.C375. [DOI] [PubMed] [Google Scholar]
- Hug M.J., Thiele I.E., Greger R. The role of exocytosis in the activation of the chloride conductance in Chinese hamster ovary cells (CHO) stably expressing CFTR. Pflügers Arch. 1997;434:779–784. doi: 10.1007/s004240050465. [DOI] [PubMed] [Google Scholar]
- Hwang T.C., Nagel G., Nairn A.C., Gadsby D.C. Regulation of the gating of cystic fibrosis transmembrane conductance regulator C1 channels by phosphorylation and ATP hydrolysis. Proc. Natl. Acad. Sci. USA. 1994;91:4698–4702. doi: 10.1073/pnas.91.11.4698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javitch J.A., Fu D., Liapakis G., Chen J. Constitutive activation of the beta2 adrenergic receptor alters the orientation of its sixth membrane-spanning segment. J. Biol. Chem. 1997;272:18546–18549. doi: 10.1074/jbc.272.30.18546. [DOI] [PubMed] [Google Scholar]
- Kartner N., Hanrahan J.W., Jensen T.J., Naismith A.L., Sun S.Z., Ackerley C.A., Reyes E.F., Tsui L.C., Rommens J.M., Bear C.E. Expression of the cystic fibrosis gene in non-epithelial invertebrate cells produces a regulated anion conductance. Cell. 1991;64:681–691. doi: 10.1016/0092-8674(91)90498-n. [DOI] [PubMed] [Google Scholar]
- Klausner R.D., Donaldson J.G., Lippincott-Schwartz J. Brefeldin Ainsights into the control of membrane traffic and organelle structure. J. Cell Biol. 1992;116:1071–1080. doi: 10.1083/jcb.116.5.1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrich R.W., Aller S.G., Webster P., Marino C.R., Forrest J.N., Jr. Vasoactive intestinal peptide, forskolin, and genistein increase apical CFTR trafficking in the rectal gland of the spiny dogfish, Squalus acanthias. Acute regulation of CFTR trafficking in an intact epithelium. J. Clin. Invest. 1998;101:737–745. doi: 10.1172/JCI803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loffing J., Moyer B.D., McCoy D., Stanton B.A. Exocytosis is not involved in activation of Cl− secretion via CFTR in Calu-3 airway epithelial cells. Am. J. Physiol. 1998;275:C913–C920. doi: 10.1152/ajpcell.1998.275.4.C913. [DOI] [PubMed] [Google Scholar]
- Lukacs G.L., Segal G., Kartner N., Grinstein S., Zhang F. Constitutive internalization of cystic fibrosis transmembrane conductance regulator occurs via clathrin-dependent endocytosis and is regulated by protein phosphorylation. Biochem. J. 1997;328:353–361. doi: 10.1042/bj3280353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansoura M.K., Smith S.S., Choi A.D., Richards N.W., Strong T.V., Drumm M.L., Collins F.S., Dawson D.C. CFTRAnion binding as a probe of the pore. Biophys. J. 1998;74:1320–1332. doi: 10.1016/S0006-3495(98)77845-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moyer B.D., Loffing J., Schwiebert E.M., Loffing-Cueni D., Halpin P.A., Karlson K.H., II, Ismailov W., Guggino B., Langford G.M., Stanton B.A. Membrane trafficking of the cystic fibrosis gene product, cystic fibrosis transmembrane conductance regulator, tagged with green fluorescent protein in Madin-Darby canine kidney cells. J. Biol. Chem. 1998;273:21759–21768. doi: 10.1074/jbc.273.34.21759. [DOI] [PubMed] [Google Scholar]
- Peacock S.L., Bates M.P., Russell D.W., Brown M.S., Goldstein J.L. Human low density lipoprotein receptor expressed in Xenopus oocytes. Conserved signals for O-linked glycosylation and receptor-mediated endocytosis. J. Biol. Chem. 1988;263:7838–7845. [PubMed] [Google Scholar]
- Peters K.W., Qi J., Watkins S.C., Frizzell R.A. Syntaxin 1A inhibits regulated CFTR trafficking in xenopus oocytes. Am. J. Physiol. 1999;277:C174–C180. doi: 10.1152/ajpcell.1999.277.1.C174. [DOI] [PubMed] [Google Scholar]
- Prince L.S., Tousson A., Marchase R.B. Cell surface labeling of CFTR in T84 cells. Am. J. Physiol. 1993;264:C491–C498. doi: 10.1152/ajpcell.1993.264.2.C491. [DOI] [PubMed] [Google Scholar]
- Prince L.S., Workman R.B., Jr., Marchase R.B. Rapid endocytosis of the cystic fibrosis transmembrane conductance regulator chloride channel. Proc. Natl. Acad. Sci. USA. 1994;91:5192–5196. doi: 10.1073/pnas.91.11.5192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince L.S., Peter K., Hatton S.R., Zaliauskiene L., Cotlin L.F., Clancy J.P., Marchase R.B., Collawn J.F. Efficient endocytosis of the cystic fibrosis transmembrane conductance regulator requires a tyrosine-based signal. J. Biol. Chem. 1999;274:3602–3609. doi: 10.1074/jbc.274.6.3602. [DOI] [PubMed] [Google Scholar]
- Quinton P.M. Physiological basis of cystic fibrosisa historical perspective Physiol. Rev. 79Suppl1999. S3 S22 [DOI] [PubMed] [Google Scholar]
- Rich D.P., Gregory R.J., Anderson M.P., Manavalan P., Smith A.E., Welsh M.J. Effect of deleting the R domain on CFTR-generated chloride channels. Science. 1991;253:205–207. doi: 10.1126/science.1712985. [DOI] [PubMed] [Google Scholar]
- Santos G.F., Reenstra W.W. Activation of the cystic fibrosis transmembrane regulator by cyclic AMP is not correlated with inhibition of endocytosis. Biochim. Biophys. Acta. 1994;1195:96–102. doi: 10.1016/0005-2736(94)90014-0. [DOI] [PubMed] [Google Scholar]
- Schwiebert E.M., Gesek F., Ercolani L., Wjasow C., Gruenert D.C., Karlson K., Stanton B.A. Heterotrimeric G proteins, vesicle trafficking, and CFTR Cl− channels. Am. J. Physiol. 1994;267:C272–C281. doi: 10.1152/ajpcell.1994.267.1.C272. [DOI] [PubMed] [Google Scholar]
- Sheppard D.N., Welsh M.J. Structure and function of the CFTR chloride channel Physiol. Rev. 79Suppl1999. S23 S45 [DOI] [PubMed] [Google Scholar]
- Smit L.S., Wilkinson D.J., Mansoura M.K., Collins F.S., Dawson D.C. Functional roles of the nucleotide-binding folds in the activation of the cystic fibrosis transmembrane conductance regulator. Proc. Natl. Acad. Sci. USA. 1993;90:9963–9967. doi: 10.1073/pnas.90.21.9963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A.A., Brooker T., Brooker G. Expression of rat mRNA coding for hormone-stimulated adenylate cyclase in Xenopus oocytes. FASEB J. 1987;1:380–387. doi: 10.1096/fasebj.1.5.2824269. [DOI] [PubMed] [Google Scholar]
- Smith S.S., Liu X., Zhang Z.-R., Sun F., Kriewall T.E., McCarty N.A., Dawson D.C. CFTRcovalent and noncovalent modification suggests a role for fixed charges in anion conduction. J. Gen. Physiol. 2001;118:407–431. doi: 10.1085/jgp.118.4.407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder P.M. Liddle's syndrome mutations disrupt cAMP-mediated translocation of the epithelial Na(+) channel to the cell surface. J. Clin. Invest. 2000;105:45–53. doi: 10.1172/JCI7869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sood R., Bear C., Auerbach W., Reyes E., Jensen T., Kartner N., Riordan J.R., Buchwald M. Regulation of CFTR expression and function during differentiation of intestinal epithelial cells. EMBO J. 1992;11:2487–2494. doi: 10.1002/j.1460-2075.1992.tb05313.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabcharani J.A., Chang X.B., Riordan J.R., Hanrahan J.W. Phosphorylation-regulated Cl− channel in CHO cells stably expressing the cystic fibrosis gene. Nature. 1991;352:628–631. doi: 10.1038/352628a0. [DOI] [PubMed] [Google Scholar]
- Takahashi A., Watkins S.C., Howard M., Frizzell R.A. CFTR-dependent membrane insertion is linked to stimulation of the CFTR chloride conductance. Am. J. Physiol. 1996;271:C1887–C1894. doi: 10.1152/ajpcell.1996.271.6.C1887. [DOI] [PubMed] [Google Scholar]
- Tousson A., Fuller C.M., Benos D.J. Apical recruitment of CFTR in T-84 cells is dependent on cAMP and microtubules but not Ca2+ or microfilaments. J. Cell Sci. 1996;109:1325–1334. doi: 10.1242/jcs.109.6.1325. [DOI] [PubMed] [Google Scholar]
- Vassilev P., Scheuer T., Catterall W.A. Inhibition of inactivation of single sodium channels by a site-directed antibody. Proc. Natl. Acad. Sci. USA. 1989;86:8147–8151. doi: 10.1073/pnas.86.20.8147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vassilev P.M., Scheuer T., Catterall W.A. Identification of an intracellular peptide segment involved in sodium channel inactivation. Science. 1988;241:1658–1661. doi: 10.1126/science.241.4873.1658. [DOI] [PubMed] [Google Scholar]
- Weber W.M., Cuppens H., Cassiman J.J., Clauss W., Van Driessche W. Capacitance measurements reveal different pathways for the activation of CFTR. Pflügers Arch. 1999;438:561–569. doi: 10.1007/s004249900086. [DOI] [PubMed] [Google Scholar]
- Wilkinson D.J., Mansoura M.K., Watson P.Y., Smit L.S., Collins F.S., Dawson D.C. CFTRthe nucleotide binding folds regulate the accessibility and stability of the activated state. J. Gen. Physiol. 1996;107:103–119. doi: 10.1085/jgp.107.1.103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson D.J., Strong T.V., Mansoura M.K., Wood D.L., Smith S.S., Collins F.S., Dawson D.C. CFTR activationadditive effects of stimulatory and inhibitory phosphorylation sites in the R domain. Am. J. Physiol. 1997;273:L127–L133. doi: 10.1152/ajplung.1997.273.1.L127. [DOI] [PubMed] [Google Scholar]
- Winter M.C., Sheppard D.N., Carson M.R., Welsh M.J. Effect of ATP concentration on CFTR Cl− channelsa kinetic analysis of channel regulation. Biophys. J. 1994;66:1398–1403. doi: 10.1016/S0006-3495(94)80930-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., George A.L., Jr., Horn R. Molecular basis of charge movement in voltage-gated sodium channels. Neuron. 1996;16:113–122. doi: 10.1016/s0896-6273(00)80028-8. [DOI] [PubMed] [Google Scholar]
- Zeltwanger S., Wang F., Wang G.T., Gillis K.D., Hwang T.C. Gating of cystic fibrosis transmembrane conductance regulator chloride channels by adenosine triphosphate hydrolysis. Quantitative analysis of a cyclic gating scheme. J. Gen. Physiol. 1999;113:541–554. doi: 10.1085/jgp.113.4.541. [DOI] [PMC free article] [PubMed] [Google Scholar]