

Abstract
KATP channels, comprised of the pore-forming protein Kir6.x and the sulfonylurea receptor SURx, are regulated in an interdependent manner by adenine nucleotides, PIP2, and sulfonylureas. To gain insight into these interactions, we investigated the effects of mutating positively charged residues in Kir6.2, previously implicated in the response to PIP2, on channel regulation by adenine nucleotides and the sulfonylurea glyburide. Our data show that the Kir6.2 “PIP2-insensitive” mutants R176C and R177C are not reactivated by MgADP after ATP-induced inhibition and are also insensitive to glyburide. These results suggest that R176 and R177 are required for functional coupling to SUR1, which confers MgADP and sulfonylurea sensitivity to the KATP channel. In contrast, the R301C and R314C mutants, which are also “PIP2-insensitive,” remained sensitive to stimulation by MgADP in the absence of ATP and were inhibited by glyburide. Based on these findings, as well as previous data, we propose a model of the KATP channel whereby in the presence of ATP, the R176 and R177 residues on Kir6.2 form a specific site that interacts with NBF1 bound to ATP on SUR1, promoting channel opening by counteracting the inhibition by ATP. This interaction is facilitated by binding of MgADP to NBF2 and blocked by binding of sulfonylureas to SUR1. In the absence of ATP, since KATP channels are not blocked by ATP, they do not require the counteracting effect of NBF1 interacting with R176 and R177 to open. Nevertheless, channels in this state remain activated by MgADP. This effect may be explained by a direct stimulatory interaction of NBF2/MgADP moiety with another region of Kir6.2 (perhaps the NH2 terminus), or by NBF2/MgADP still promoting a weak interaction between NBF1 and Kir6.2 in the absence of ATP. The region delimited by R301 and R314 is not involved in the interaction with NBF1 or NBF2, but confers additional PIP2 sensitivity.
Keywords: nucleotide binding fold, inward rectifier K channel, SUR, adenine nucleotides, PIP2
INTRODUCTION
ATP-sensitive K (KATP) channels are regulated by multiple factors, among which ADP is believed to play a crucial role in their short-term adaptation to physiological stimuli (Ashcroft 1988). ADP regulates KATP channel activity via three different processes. In the absence of the sulfonylurea receptor, Kir6.2 channels show brief openings without long bursts, and both ADP and ATP inhibit channel activity by interacting directly with Kir6.2 (Tucker et al. 1997; John et al. 1998). When Kir6.2 is coexpressed with SUR1, KATP channel openings become longer and occur in bursts. In the presence of Mg, ADP (hereafter referred to as MgADP) counters the inhibition by ATP. In the absence of ATP, MgADP and other nucleotide diphosphates activate directly channel activity by promoting bursting behavior (Findlay 1988; Terzic et al. 1994).
The two nucleotide-binding folds of SUR1, NBF1 and NBF2, bind ATP, but NBF2 hydrolyzes ATP at a higher rate (Ueda et al. 1999; Bienengraeber et al. 2000). Based on these observations, Ueda et al. 1999 proposed a model whereby binding of MgADP to NBF2 stabilizes the binding of ATP to NBF1, which then stimulates channel activity. Upon removal of MgADP, the ATP–NBF1 interaction decreases, ATP dissociates from NBF1, and channel activity is thereby reduced. In addition, they showed that while sulfonylureas such as glyburide have no direct effect on nucleotide binding to either NBF1 or NBF2, they weaken the cooperativity between NBF2 and NBF1. As a result, MgADP no longer stabilizes the binding of ATP at NBF1 and thus ATP dissociates from NBF1, resulting in channel inhibition.
Shyng et al. 2000 have recently mutated positively charged residues in the Kir6.2 C tail and showed that distinct single point mutations can affect either activation by phosphatidylinositol bisphosphate (PIP2) or inhibition by ATP. As these mutations involved positive residues on Kir6.2, they proposed that the ATP and PIP2 effects involve electrostatic interaction between the negatively charged phosphate groups on ATP and PIP2 with the positively charged residues on Kir6.2. However, the positively charged residues interacting with ATP and PIP2 are different, suggesting two separate regulatory domains. They also suggested that PIP2 interacts with two distinct regions on Kir6.2 located in the proximal C tail between R176 and K222 and in the distal C tail between R301 and R314.
We recently reported that MgADP and PIP2 regulate Kir6.2+SUR1 channels in a similar manner (Ribalet et al. 2000): both activate channel activity and decrease the channel sensitivity to ATP. In addition, PIP2 prevented reactivation by MgADP and, conversely, MgADP prevented stimulation by PIP2 when applied before channel rundown. Based on these observations, we suggested that PIP2 and a moiety formed by the binding of MgADP to NBF2 of SUR compete for channel regulation by interacting with similar positive residues on Kir6.2. To test this hypothesis, here we investigate channel reactivation by MgADP of “PIP2-insensitive” mutants previously identified (Shyng et al. 2000), specifically R176C, R177C, R301C, and R314C. We find that R176C and R177C show no glyburide sensitivity and no MgADP-dependent reactivation, which is consistent with a lack of interaction with NBF1. In contrast, mutants R301C and R314C are sulfonylurea-sensitive and also exhibit MgADP-dependent stimulation (in the absence of ATP), suggesting preserved interactions with NBF1 as well as NBF2.
Based on these results and our present data, we propose a model of the KATP channel whereby in the presence of ATP, the R176 and R177 residues on Kir6.2 are the primary site of interaction with NBF1 on SUR1 promoting channel opening by counteracting the inhibition by ATP. This interaction is facilitated by binding of MgADP to NBF2 and blocked by binding of sulfonylureas to SUR1. In the absence of ATP, since KATP channels are not blocked by ATP, the counteracting effect of NBF1 interacting with R176/R177 is not required for channel opening. In this case, one of two possibilities may explain channel activation by MgADP. Either a direct stimulatory interaction takes place between the NBF2/MgADP moiety and another region of Kir6.2, perhaps the NH2 terminus, which controls Kir6.2 channel kinetics (Reimann et al. 1999); or NBF2/MgADP still promotes channel stimulation via NBF1 in the absence of bound ATP, albeit to a weaker extent than when ATP is bound to NBF1. The region delimited by R301 and R314 is not involved in the interaction with NBF1 or NBF2, but interacts with PIP2.
MATERIALS AND METHODS
The techniques for cDNA expression and patch-clamp recording have been recently described in detail (John et al. 1998) and are only briefly outlined here.
Molecular Biology and cDNA Expression in HEK293 Cells
In most cases, HEK293 cells were transfected with cDNA for Kir6.2 mutants linked to GFP at the COOH terminus (Kir6.2-GFP), so that insertion of the constructs into the plasma membrane could be investigated. Our previous findings that linkage to GFP did not affect the kinetics or adenine nucleotide sensitivity of wild-type Kir6.2+SUR1 channels (John et al. 1998) justify this approach. However, for Kir6.2 mutant channels, such as R176C+SUR1, exhibiting MgADP and sulfonylurea sensitivities different from wild-type channels, experiments were also performed in the absence of GFP to rule out any possible effect of GFP on the MgADP- or glyburide-sensitive regulatory mechanisms. We observed that the R176C mutant had similar behaviors with and without GFP, excluding the possibility that GFP interferes with the coupling of the Kir6.2 mutant R176C to SUR1. All wild-type cDNAs were subcloned into the vector pCDNA3amp (Invitrogen). cDNAs used to make the GFP chimeras were subcloned into the pEGFP vector (CLONTECH). Both vectors use the CMV promoter. Single site Kir6.2 mutations were constructed using the QuickChange technique (Stratagene). All mutants were in the Kir6.2-GFP backbone. R176C and R177C were provided by Dr. Z. Fan (University of Tennessee, Memphis, TN) and subcloned into the Kir6.2-GFP backbone. The transfections were performed using the calcium phosphate precipitation method (Graham and van der Eb 1973). Expression of proteins linked to GFP was detected as early as 12 h after transfection. Patch-clamp experiments were started ∼30 h after transfection. HEK293 cells were cultured in DME high glucose medium supplemented with 10% (vol/vol) FCS, 100 U/ml penicillin, 100 U/ml streptomycin, and 2 mM glutamine and divided once a week by treatment with trypsin.
Patch-clamp Methods
Currents were recorded in HEK293 cells using the inside-out patch-clamp configuration, with the pipette solution containing the following (in mM): 140 KCl, 10 NaCl, 1.1 MgCl2, and 10 HEPES, pH adjusted to 7.2 with KOH. The bath solution consisted of the following (in mM): 140 KCl, 10 NaCl, 1.1 MgCl2, 10 HEPES, 5 EGTA, and 0.5 CaCl2, pH adjusted to 7.2 with KOH. ATP was added directly to the bath as MgATP. PIP2, purchased from Boehringer Mannheim, was sonicated immediately before use.
The data, filtered at 2 kHz with an 8-pole Bessel filter, was recorded with a patch-clamp amplifier (model EPC 7; List) and recorded on videotape at a fixed frequency of 44 kHz after digitization with a digital audio processor. For analysis, the data was sampled at a rate of 5.5 kHz. When discrete current steps could be resolved, channel activity was expressed as NPo. To express channel activity as a function of ATP concentration, NPo was estimated at each ATP concentration from data samples of 15 s duration. Since channel activity varied widely from patch to patch, NPo values were normalized to values measured in the absence of ATP. When single channels could not be resolved, steady-state current values were used instead of NPo to assess the effects of adenine nucleotides and other interventions on channel activity.
RESULTS
Effects of PIP2 and ADP on the Kir6.2 Mutants R176C and R177C (Linked to Green Fluorescent Protein [GFP])
The R176A and R177A mutations in Kir6.2 channels have been found to markedly diminish and slow reactivation by PIP2, suggesting a reduced affinity for PIP2 in these mutants (Fan and Makielski 1997; Baukrowitz et al. 1998; Shyng and Nichols 1998). Because R176A and R177A typically yield very small or no currents in excised patches (Shyng et al. 2000), we studied the R176C and R177C mutations, which in general yielded larger currents, to investigate channel regulation by PIP2, adenine nucleotide, and sulfonylureas. Whereas the R176C mutant consistently yielded measurable K+ current (although of variable amplitude), the R177C mutant yielded current in only 55% of the patches. Nonetheless, in the “silent” patches, channel insertion into the plasma membrane was normal as indicated by GFP labeling of the plasma membrane (see Fig. 10). This observation corroborates a previous report by Shyng et al. 2000, suggesting that R177A mutant channels may be inserted into the plasma membrane, while remaining closed or nonfunctional.
Figure 10.
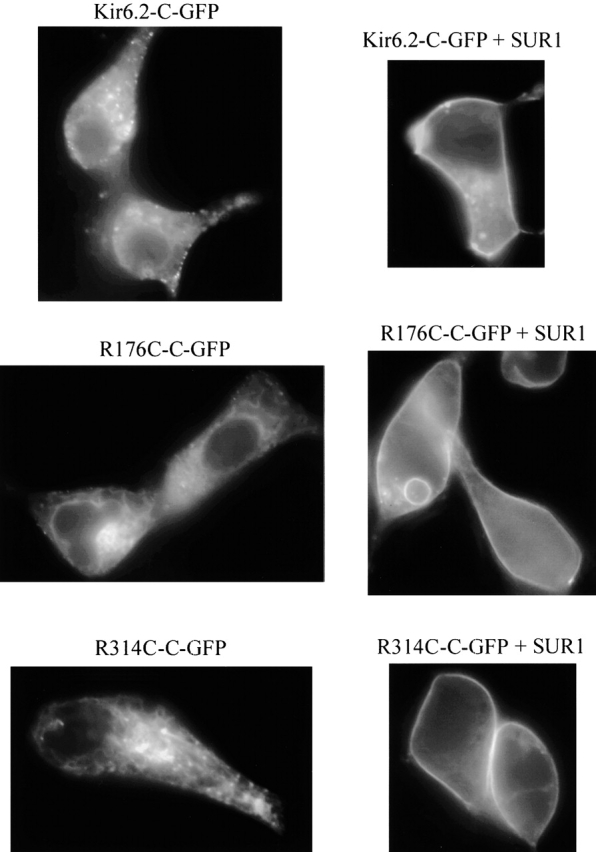
Effects of SUR1 on the insertion into the plasma membrane of Kir6.2 wild-type, R176C, and R314C mutants linked to GFP. Three sets of images obtained from different transfection experiments representative of the distribution of GFP linked to the C tail of Kir6.2 wild-type, Kir6.2-GFP, (top images), R176C-GFP mutant (middle images), and R314C-GFP mutant (bottom images). In each dataset, the left image was obtained in the absence of SUR1, and the right image was obtained with SUR1 coexpression. All data without SUR1 showed strong fluorescent labeling of intracellular lamellar structure tentatively identified as the ER. Almost no fluorescence was associated with the plasma membrane. In contrast, when the pore-forming protein was coexpressed with SUR1, bright uniform fluorescence was associated with the plasma membrane and the intracellular lamellar structure had almost subsided. These data together with the high level of channel activity, recorded in the latter case, suggest that SUR1 facilitates similar plasma insertion of wild-type Kir6.2-GFP and R176C-GFP as well as R314C-GFP mutants. Plasma membrane insertion in the presence of SUR1 was also observed with the Kir6.2 mutants R177C-GFP and R301C-GFP.
Fig. 1 compares the effects of PIP2 and MgADP on wild-type Kir6.2+SUR1 channels and the Kir6.2 mutant R176C. After brief exposure to 10 μM Ca2+ to induce rundown, PIP2 had potent reactivating effects on wild-type Kir6.2+SUR1 (Fig. 1 A), but not on the mutant R176C+SUR1 (Fig. 1 B), which only partially and slowly responded to PIP2, as reported previously (Baukrowitz et al. 1998; Fan and Makielski 1997; Shyng and Nichols 1998). Moreover, unlike wild-type channels, which were reactivated by MgADP by an average of 46 ± 12.5% (n = 5 patches) after ATP-induced inhibition (Fig. 1 C), ATP-inhibited R176C+SUR1 mutant channels were almost insensitive to reactivation by MgADP (average response 6 ± 5.4%, n = 9 patches; Fig. 1 D). Similarly, in R177C+SUR1 mutant channels, MgADP failed to reactivate channel activity after ATP inhibition (unpublished data). The lack of reactivation by MgADP in these mutants was similar to Kir6.2 channels expressed in the absence of SUR1, suggesting functional uncoupling of Kir6.2 and SUR1. It is unlikely that the loss of MgADP-dependent reactivation caused by the R176C or R177C mutation was due to lowering of the MgADP sensitivity since concentrations as high as 400 μM MgADP did not stimulate the activity of ATP-inhibited mutant channels. Since regulation by MgADP involves the nucleotide-binding fold NBF2 of SUR (Nichols et al. 1996; Gribble et al. 1997; Ueda et al. 1999), we conclude that the effects of MgADP involve an interaction between the positively charged residues R176 and R177 on Kir6.2 and SUR1, with NBF2 in its MgADP-bound conformation.
Figure 1.
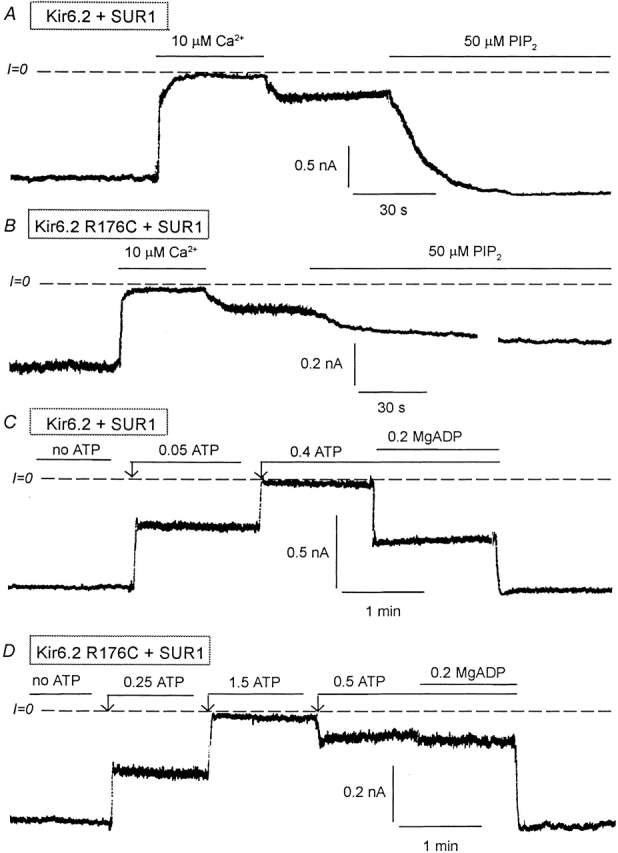
Effects of PIP2 and MgADP on Kir6.2 wild-type and R176C mutant (linked to GFP) coexpressed with SUR1. (A) Representative inward currents from excised inside-out patches with wild-type Kir6.2+SUR1. Brief application of 10 μM Ca2+ (for ∼1 min) to the cytoplasmic surface of an excised inside-out patch rapidly inhibited channel activity, which recovered only minimally after removal of Ca2+. PIP2 application led to full recovery of channel activity after brief Ca2+-induced rundown. Dashed lines indicate zero current level. (B) A similar experiment performed with the R176C mutant + SUR1 show a similar inhibitory effect due to Ca2+, but the response to PIP2 was very slow and, in this case, reached 42% of control after 8 min. The trace was interrupted for 3 min during the application of PIP2. (C) Wild-type Kir6.2+SUR1 inward currents were suppressed by increasing ATP concentrations. In the presence of 400 μM ATP, addition of 200 μM MgADP caused a robust stimulation of channel activity, indicating functional coupling between Kir6.2 and SUR1. (D) A similar inside-out patch experiment performed with the R176C mutant + SUR1 shows that in the presence of 500 μM ATP, addition of 200 μM MgADP had little or no effect indicating functional uncoupling of Kir6.2 and SUR1. In this case we first applied 1.5 mM ATP to show complete inhibition by ATP and we reduced the ATP level to 0.5 mM so that the effects of MgADP could be compared with that obtained with the wild-type channel.
ATP Sensitivity of the R176C and R177C Mutants (Linked to GFP) Expressed Alone and with SUR1
We and others have previously reported that coupling of SUR1 to Kir6.2 not only confers MgADP-dependent reactivation, but also increases the channel sensitivity to ATP (Tucker et al. 1997; John et al. 1998). To further investigate how the R176C and R177C mutations affect functional coupling between the two KATP channel subunits, we studied the ATP sensitivity of the mutant channels expressed alone and with SUR1. We previously reported that, in the absence of SUR1, the IC50 for Kir6.2 channel inhibition by ATP is close to 165 μM (John et al. 1998). By comparison, the IC50 for R176C channel inhibition by ATP was close to 230 μM (Fig. 2B and Fig. C) and not significantly different from that measured in wild-type Kir6.2. This result together with the observation that single R176C channel openings are very brief (Fig. 2 A) suggest that the mutation R176C has no major effects on either the intrinsic channel kinetics or the ATP sensitivity of Kir6.2 when expressed alone. Fig. 3 compares the sensitivities to ATP of Kir6.2 wild-type with R176C mutant channels coexpressed with SUR1. Wild-type Kir6.2+SUR1 channels were more sensitive to inhibition by ATP with an IC50 averaging 55 μM, compared with 223 μM for R176C+SUR1 mutant channels. Similarly, in R177C+SUR1, the IC50 for ATP inhibition was increased to 217 ± 35 μM.
Figure 2.
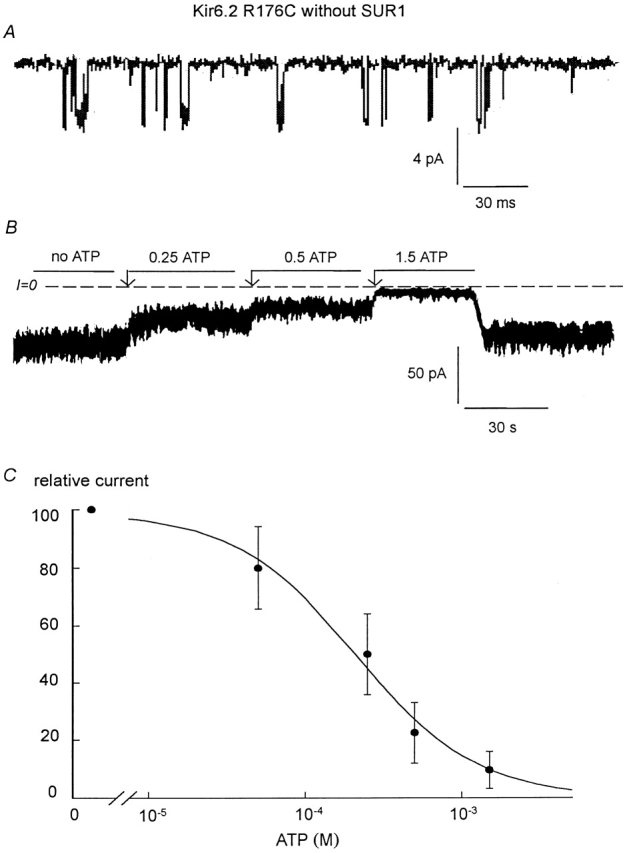
ATP sensitivity of Kir6.2 R176C (linked to GFP) expressed alone. (A) Single-channel kinetics of the R176C mutant showing similar low open probability (Po < 0.15) as wild-type Kir6.2 (John et al. 1998). Channel openings are downward. (B and C) ATP sensitivity of Kir6.2 R176C in an inside-out patch. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 near 250 μM. (C) Plot of channel ATP sensitivity for Kir6.2 R176C. The data points (•) were fit with the equation y = 1/(1 + (k/[ATP])n), which yielded k = 234 μM and n = 1.13 (for n = 3).
Figure 3.
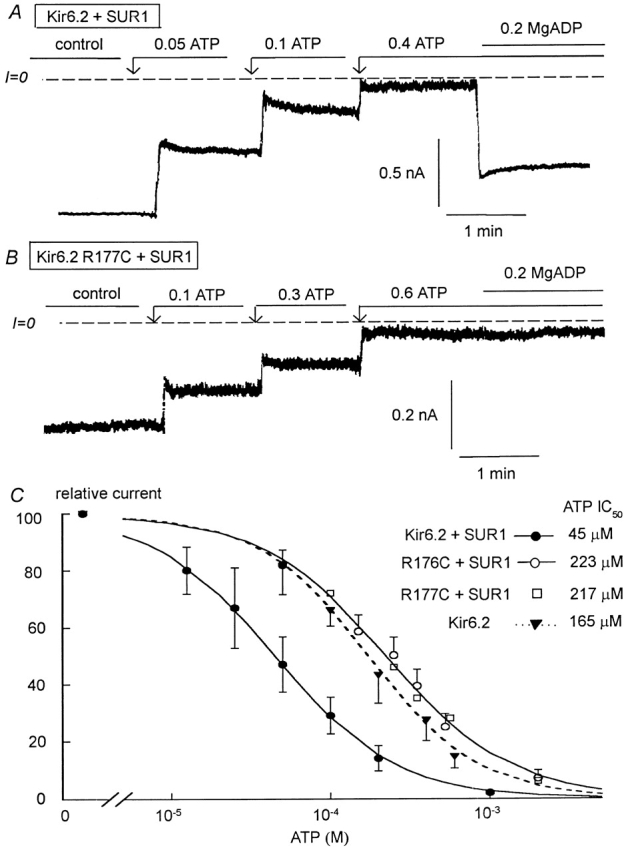
ATP sensitivity of Kir6.2 wild-type, R176C, and R177C mutants (linked to GFP) coexpressed with SUR1. (A and B) ATP sensitivity in representative inside-out patches with wild-type and R177C Kir6.2, respectively. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 near 50 and 200 μM for Kir6.2 wild-type + SUR1 and R177C+SUR1, respectively. (C) Plot of channel ATP sensitivity for Kir6.2 wild-type + SUR1 (•), R176C+SUR1 (O), and R177C+SUR1 (□). The data points were fit with the equation y = 1/(1 + (k/[ATP])n), which yielded k = 45 μM and n = 1.16 (•) and k = 223 μM and n = 1.1 (O). An ATP dose–response previously obtained for wild-type Kir6.2 alone (▾) has been added for comparison purposes in this case k = 165 μM and n = 1.25.
The plot in Fig. 3 C compares the ATP sensitivity of Kir6.2 alone with that of R176C+SUR1 and R177C+ SUR1. The two types of sensitivity were not significantly different. Therefore, it is reasonable to postulate that the shift in ATP sensitivity observed with the R176C and R177C mutants coexpressed with SUR1 is not due to a change in intrinsic ATP sensitivity of Kir6.2, but rather to the functional uncoupling of Kir6.2 from SUR1. This is consistent with the proposal of Shyng et al. 2000 that positively charged residues in two different Kir6.2 locations are involved in interactions with either PIP2 or ATP.
Sensitivity of Kir6.2+SUR1 to Sulfonylurea and Adenine Nucleotide
To further explore whether the lower ATP sensitivity of R176C+SUR1 and R177C+SUR1 was due to functional uncoupling from SUR1, we next investigated the sensitivity of Kir6.2+SUR1 and R176C(R177C)+SUR1 to the sulfonylurea (glyburide), which has been shown to disrupt the regulatory interaction between Kir6.2 and SUR1 (Ueda et al. 1999).
Fig. 4 and Fig. 5 illustrate the effects of glyburide on wild-type Kir6.2+SUR1 channels. Glyburide caused increased ATP sensitivity and loss of reactivation by MgADP, mimicking the effects of channel rundown previously reported (Ribalet et al. 2000). In five patches with an average IC50 for channel inhibition by ATP of 115 μM (at the high end of the typically observed range), reactivation of ATP-inhibited channels by MgADP averaged 45% (Fig. 4 A). 200 nM glyburide caused channel activity to decrease slowly, with the average inhibition reaching 45 ± 7% of control within ∼1 min. During application as well as after the subsequent washout of glyburide, channel sensitivity to ATP increased dramatically, with the IC50 shifting to 13 μM (Fig. 4 C). In addition, there was a complete loss of MgADP-dependent channel reactivation (Fig. 4 B), indicating functional uncoupling of Kir6.2 and SUR1. After functional uncoupling by glyburide, MgADP became inhibitory (Fig. 4 B, bottom trace), with an IC50 for channel inhibition averaging 96 ± 23 μM.
Figure 4.

Effects of glyburide on the ATP sensitivity of Kir6.2 (linked to GFP) + SUR1. (A) ATP sensitivity in a representative inside-out patch with wild-type Kir6.2+SUR1. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 near 100 μM. 200 μM MgADP was then added to the patch in the presence of ATP to show functional coupling between Kir6.2 and SUR1. In this series of experiments, patches with low ATP sensitivity were selected to better show the effect of glyburide on ATP sensitivity. B shows the effect of 0.2 μM glyburide on inward current. (C) After glyburide, the ATP sensitivity increased dramatically with the IC50 shifting from near 100 μM before glyburide to ≅15 μM after glyburide. After glyburide MgADP did not reactivate ATP-inhibited channels, indicating functional uncoupling. C demonstrates that after glyburide-induced uncoupling ADP becomes inhibitory. In this case, the IC50 for channel inhibition by ADP is close to 150 μM. (D) Plot of channel ATP sensitivity for Kir6.2 wild-type + SUR1 before (▴) and after (•) glyburide. In this case, a fit of the data points yielded k = 115 μM and n = 1.18 (▴) and k = 13 μM and n = 1.15 (•).
Figure 5.
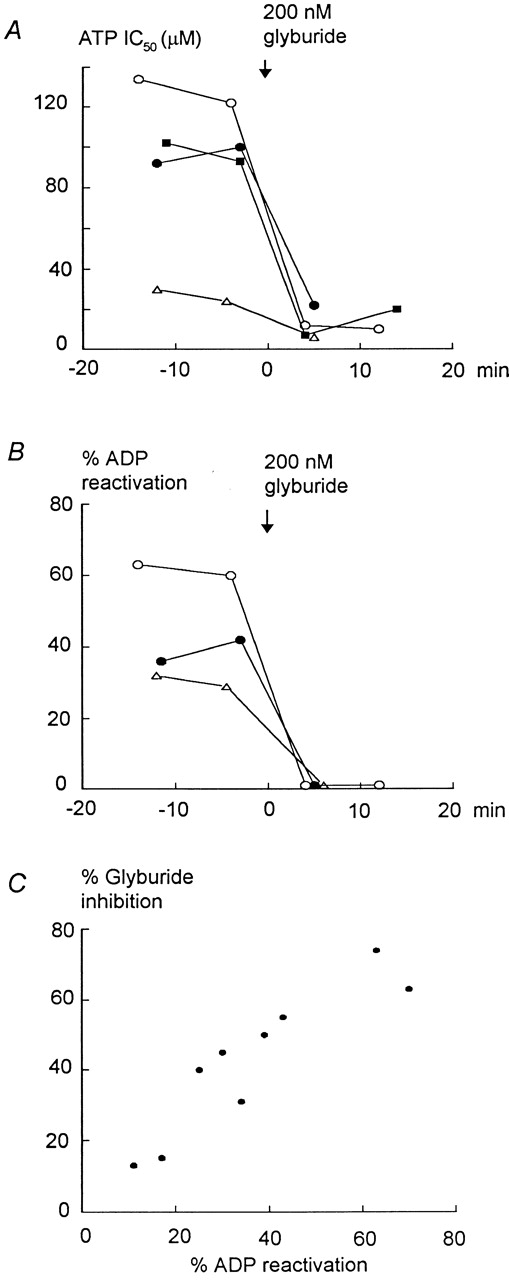
Effects of glyburide and rundown on ATP sensitivity and MgADP-dependent reactivation of Kir6.2 (linked to GFP) + SUR1. (A) The ATP sensitivity of wild-type Kir6.2+SUR1 channels was tested every 7–8 min after patch excision. Channel rundown was minimized by excising the membrane patch into a 200-μM MgADP solution. MgADP was then removed before assessing inhibition by ATP. Under these conditions, rundown caused a small and gradual increase in ATP sensitivity, which was negligible compared with the profound and almost irreversible effect due to glyburide. This effect was independent of the ATP sensitivity before addition of the sulfonylurea and always reached the same IC50, close to 10 μM, after addition of glyburide. B shows the concomitant loss in MgADP-dependent reactivation of ATP-inhibited channels. In this case, the effect of rundown on the reactivation by MgADP was also minimal and addition of glyburide caused complete and irreversible loss of the MgADP-dependent effect. These data also indicate that the degree of MgADP-dependent reactivation could be independent, at least right after patch excision, of the channel sensitivity to ATP, MgADP having similar reactivating effects on channels with high (IC50 = 2 5 μM) and low (IC50 = 90 μM) sensitivity to ATP. This did not hold after ATP sensitivity had increased after prolonged rundown (unpublished data). C illustrates the close correlation between the degrees of MgADP-dependent reactivation and inhibition by glyburide, supporting our hypothesis whereby sulfonylureas cause functional uncoupling of Kir6.2 and SUR1.
Channel rundown also causes an increase in ATP sensitivity and loss in MgADP-dependent reactivation (Ribalet et al. 2000). Data presented in Fig. 5 exclude that the effects of glyburide on ATP and MgADP sensitivity were due to channel rundown. In these experiments, rundown was initially minimized by excising the membrane patch into a 200-μM MgADP solution. After MgADP removal, inhibition by ATP and reactivation by MgADP were assessed at least twice before application of 200 nM glyburide. Fig. 5 A shows that, in these patches, the dramatic 5- to 10-fold increase in ATP sensitivity cannot be mistaken with the slow increase in ATP sensitivity evoked by rundown. A dataset obtained in a patch with high initial ATP sensitivity (Fig. 5 A, triangles) is also shown to illustrate the similar response to glyburide of such channels. Fig. 5 B shows the corresponding complete loss in MgADP-dependent channel reactivation due to glyburide in the same patches. Fig. 5 C illustrates the close correlation between the level of reactivation by MgADP of ATP-inhibited channels and the percent inhibition evoked by glyburide. This close correlation supports our hypothesis whereby sulfonylureas cause functional uncoupling, or loss of MgADP-dependent reactivation, of the two KATP channel subunits.
Adenine Nucleotide and Sulfonylurea Sensitivity of the Kir6.2 Mutants R176C and R177C (with and without GFP)
Fig. 6 depicts the effect of glyburide on the ATP sensitivity of R176C+SUR1 mutant channels (tested in six patches). Before glyburide, the IC50 for channel inhibition by ATP was 223 μM, and no channel stimulation occurred in response to MgADP (Fig. 6 A). 0.5 μM glyburide had negligible effects on channel activity (average inhibition 5.5 ± 3%, n = 7) or channel sensitivity to ATP (Fig. 6 B). After glyburide, the IC50 for channel inhibition by ATP was 184 μM, which was not significantly different from that recorded before glyburide (Fig. 6B and Fig. C). A similar lack of glyburide sensitivity was observed with R177C+SUR1 mutant channels (unpublished data), supporting the hypothesis that the R176C and R177C mutations functionally uncouple Kir6.2 and SUR1.
Figure 6.
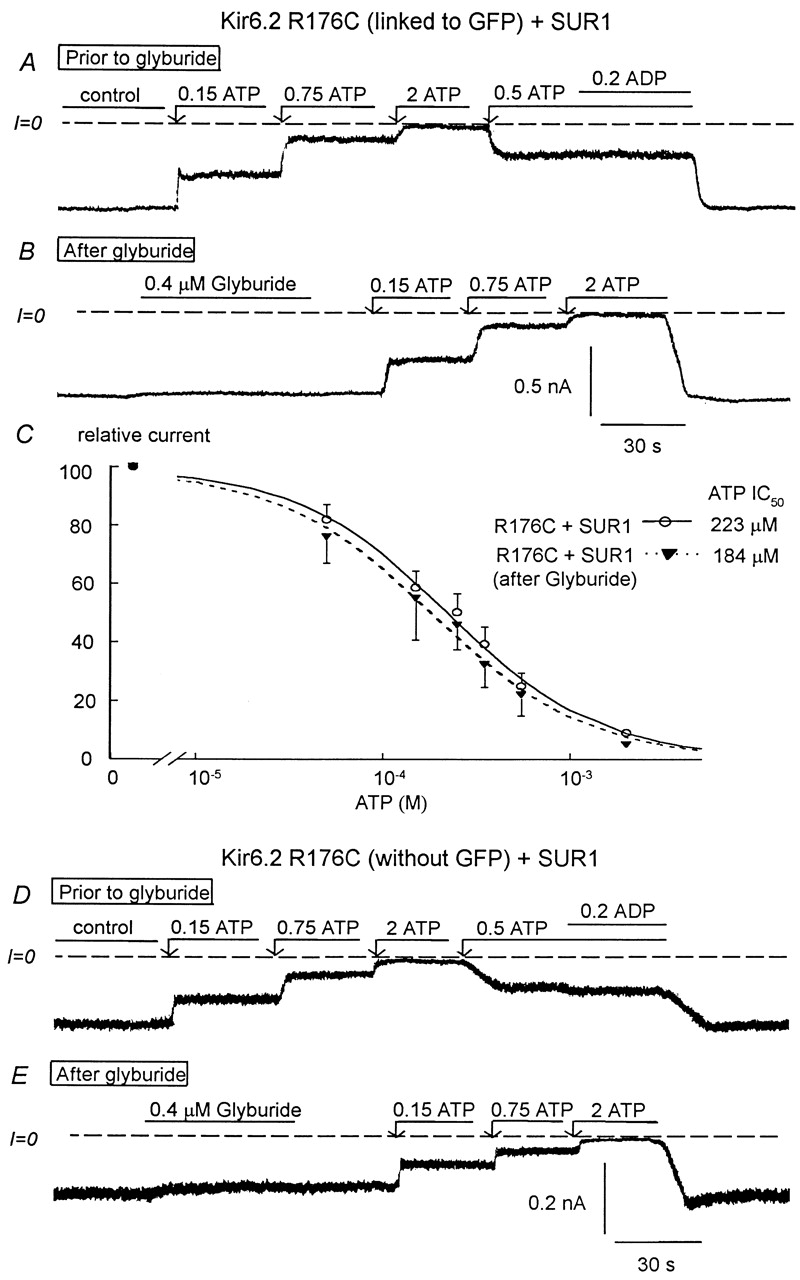
Effects of glyburide on the ATP sensitivity of the R176C mutant (linked or not linked to GFP) coexpressed with SUR1. A–C show data obtained with R176C linked to GFP. D and E show similar data obtained with a GFP-free R176C mutant. (A) ATP sensitivity in an inside-out patch with R176C+SUR1. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 for channel inhibition near 200 μM. Addition of 200 μM MgADP in the presence of ATP had no effect, indicating functional uncoupling between Kir6.2 and SUR1. B shows that 0.4 μM glyburide had no significant effect on inward current and after glyburide the ATP sensitivity remained unchanged with the IC50 remaining close to 200 μM. (C) Plot of channel ATP sensitivity for Kir6.2 R176C+SUR1 before (▾) and after (O) glyburide. In this case, a fit of the data points yielded k = 223 μM and n = 1.1 (▾; n = 5) and k = 184 μM and n = 1.05 (O; n = 5). D and E show data obtained with R176C without GFP and coexpressed with SUR1, which are similar to that in A and B, respectively. Before glyburide (D), the IC50 for channel inhibition by ATP was close to 200 μM and MgADP had almost no stimulatory effect. Glyburide had almost no inhibitory effect (E), and the ATP sensitivity remained unchanged after application of the sulfonylurea.
To demonstrate that the lack of MgADP and sulfonylurea sensitivity and low ATP sensitivity in R176C and R177C mutants was not due to GFP interfering with channel regulation, experiments were performed with the R176C mutant not linked to GFP (Fig. 6D and Fig. E). When coexpressed with SUR1, the GFP-free R176C mutant duplicated the data obtained with the GFP-linked mutant. Before glyburide (Fig. 6 D), the IC50 for channel inhibition by ATP averaged 245 ± 50 μM (n = 3) and MgADP had almost no stimulatory effect on ATP-inhibited channels. In addition, glyburide had negligible effects on channel activity (average inhibition 8 ± 4%), and the IC50 for channel inhibition by ATP after glyburide was 193 ± 37 μM (Fig. 6 C), which is not significantly different from that obtained before glyburide. These data rule out that linkage to GFP accounts for the lack of MgADP and sulfonylurea sensitivity observed with the R176C or R177C mutants. Because the NBFs of SUR1 are required to confer sulfonylurea sensitivity to KATP channels (Ueda et al. 1999), the loss of glyburide sensitivity shown here provides additional support for the hypothesis that Kir6.2 residues R176 and R177 are involved in interactions with the NBFs or related structures in SUR1, which confer functional coupling between Kir6.2 and SUR1.
Adenine Nucleotide and Sulfonylurea Sensitivity of the Kir6.2 Mutants R301C and R314C (Linked to GFP)
In addition to R176 and R177, a second domain in the COOH terminus of Kir6.2 containing positively charged residues and stretching from R301 to R314 has been reported to be involved in interaction with PIP2 (Shyng et al. 2000). When the Kir6.2 mutants R301C and R314C were coexpressed with SUR1, channel activity in the excised patch configuration ran down rapidly within less than a minute after ATP was removed from the bath. After rundown, PIP2 partially restored channel activity. In five patches, the activity increased by ∼12-fold, but the level of channel activity reached at steady state was only 18% of that measured before rundown. In addition, recovery of channel activity occurred slowly, reaching steady state within 6 ± 2 min. These data are consistent with a reduced sensitivity to PIP2. Similar results were obtained with the R314C+ SUR1 mutant channels.
We then examined how the mutations R301C and R314C affected the channel sensitivity to ATP (Fig. 7). Fast rundown could be partially alleviated by adding 200 μM MgADP to the bath before patch excision. Furthermore, upon addition of ATP to the bath, rundown stopped (Fig. 7 A). Under these conditions, the channel sensitivity to ATP could be estimated. In the presence of 200 μM MgADP, the measured IC50 for R301C+SUR1 mutant channel inhibition by ATP was 205 μM (Fig. 6 A). Upon removal of ATP, there was a potent recovery of channel activity (previously termed refreshment) followed by pronounced rundown. Because addition of MgADP shifts ATP sensitivity by about fivefold (Ribalet et al. 2000), these data suggest that, in the absence of MgADP, the predicted ATP sensitivity of the R301C mutants coexpressed with SUR1 might be ∼40 μM, which is similar to that of wild-type Kir6.2 channels coexpressed with SUR1. We estimated the channel sensitivity in the absence of MgADP, and in three patches the IC50 for channel inhibition by ATP averaged 32 μM (Fig. 7 B). Coexpression of the Kir6.2 mutant R314C with SUR1 exhibited the same behavior as R301C. The activity ran down rapidly upon ATP removal and the IC50 for inhibition by ATP in the presence of 200 μM MgADP was close to 200 μM.
Figure 7.
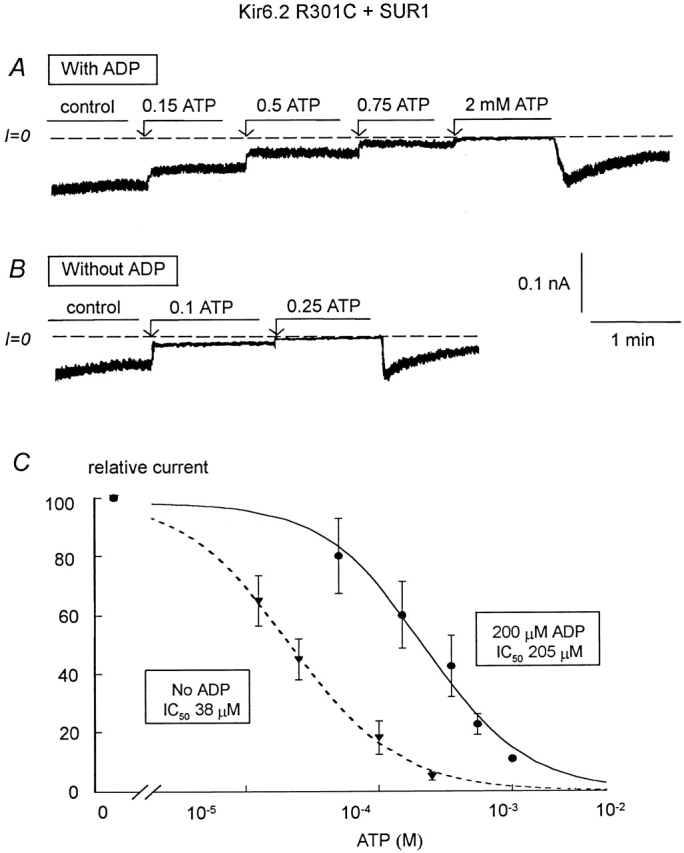
ATP sensitivity of the R310C and R314C mutants (linked to GFP) coexpressed with SUR1. (A) ATP sensitivity in an inside-out patch with R301C+SUR1 in the presence of 200 μM MgADP to slow channel rundown. Inward currents were progressively suppressed by increasing ATP concentrations, with the IC50 for channel inhibition near 200 μM. Note the inhibition of channel rundown in the presence of ATP. Upon removal of ATP, there was a potent recovery of channel activity followed by prominent rundown. (B) In the absence of ADP, channel rundown was more pronounced, however, ATP could still stop rundown, but after removal of ATP channel rundown was almost complete. The IC50 for channel inhibition by ATP was close to 40 μM in this case. (C) Plot of channel ATP sensitivity for R301C+SUR1 with (•) and without (▾) 200 μM MgADP. In this case, a fit of the data points yielded k = 205 μM and n = 1.14 (•; n = 5) and k = 38 μM and n = 1.2 (▾; n = 3).
Direct evidence for intact functional coupling of R301C and R314C channels to SUR1 was indicated by their response to MgADP in the absence of ATP. In the absence of ATP, MgADP stimulates wild-type KATP channel activity after channel rundown (Terzic et al. 1994), and this process requires intact coupling of the pore-forming protein Kir6.2 with SUR. Because the activity of R301C and R314C mutants rapidly ran down, we tested the effects of MgADP on channel activity 1–2 min after patch excision in the absence of ATP. As depicted in Fig. 8 A, MgADP stimulated R301C+SUR1 mutant channel activity significantly, with the maximum effect at ∼200 μM. At higher concentrations, MgADP became inhibitory, with channel activity completely blocked near 10 mM. A similar pattern of stimulation and inhibition by ADP was observed with the R314C+SUR1 mutant channels. Stimulation of the R301C and R314C mutants by MgADP further suggests that these two mutants are functionally coupled to SUR1 and more precisely to NBF2, which binds MgADP (Ueda et al. 1999).
Figure 8.
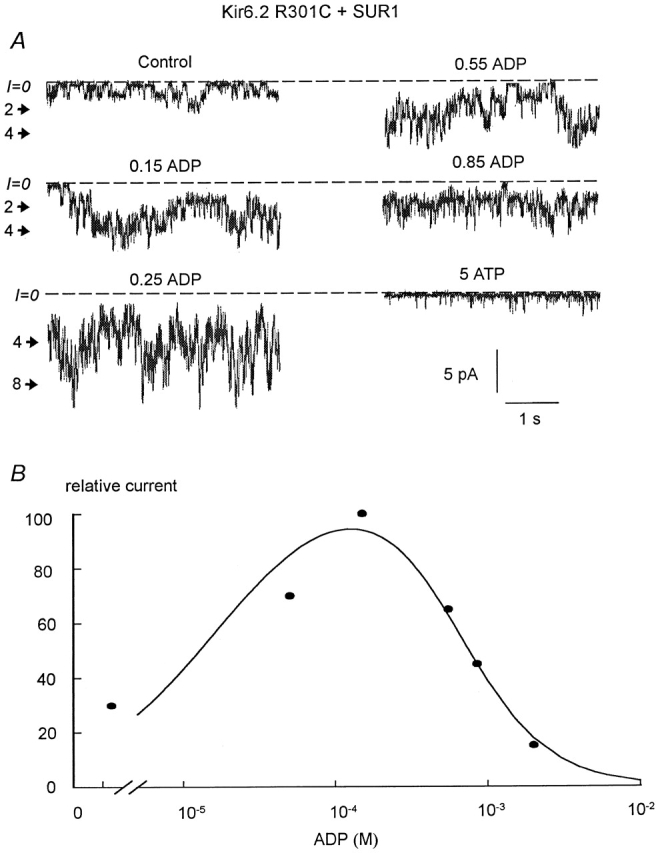
Stimulation and inhibition by ADP of the R301C mutant (linked to GFP) coexpressed with SUR1. (A) Current traces obtained in an inside-out patch with R301C+SUR1, inward currents are shown as downward deflections. The dashed line indicates the zero current, and the number on the side of the traces indicates the open channel levels. These data show that maximum channel activity was reached near 250 μM and maximum inhibition occurred at 5 mM. (B) Plot of data points obtained from the traces in A. The curve represents a fit to the experimental data with an equation comprising two Hill functions. In this case, the half-stimulatory concentration was 22 μM, and the IC50 for inhibition by ADP was 727 μM.
Finally, we investigated the effect of glyburide on R301C and R314C mutants coexpressed with SUR1. Fig. 9 shows that R301C+SUR1 mutant channels were inhibited by glyburide, which is consistent with functional coupling of R301C and SUR1. In Fig. 9 A, MgADP was present throughout the application of glyburide. Inhibition by glyburide was irreversible, with no reactivation of the inhibited channel after glyburide removal. As with wild-type Kir6.2+SUR1 channels, glyburide also significantly increased the ATP sensitivity of R301C+SUR1 mutant channels (Fig. 9 B), with the IC50 for channel inhibition by ATP decreasing to 21 μM in the presence of 200 μM MgADP (Fig. 9; n = 2). This IC50 value for channel inhibition by ATP after glyburide treatment is very close to that obtained for wild-type Kir6.2+SUR1 channel in the absence of MgADP (≅12 μM), indicating that MgADP has no effect on the R301C+SUR1 mutant channel sensitivity to ATP after functional uncoupling due to glyburide treatment.
Figure 9.
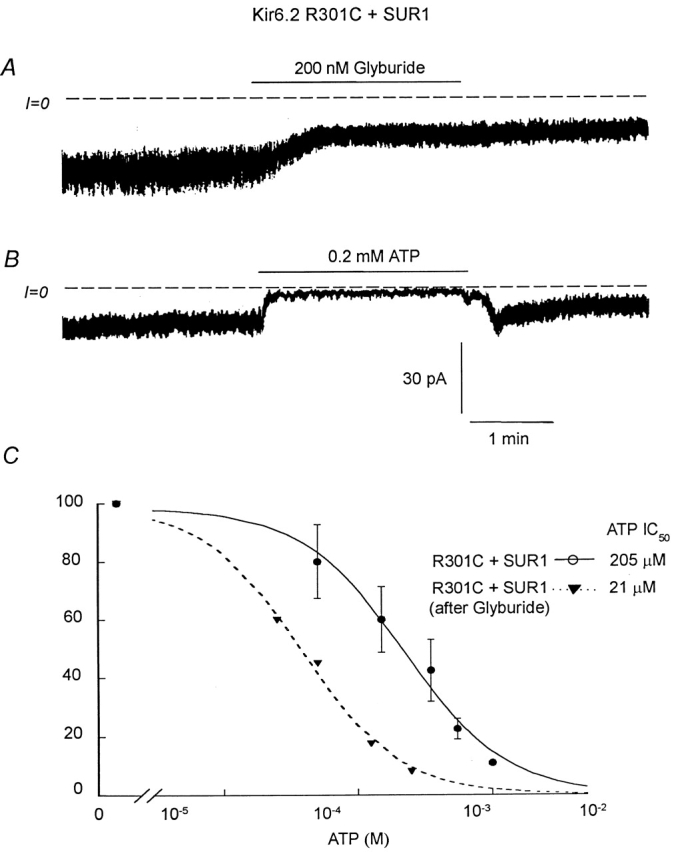
Effects of glyburide on the ATP sensitivity of R301C+SUR1. (A) Shows the effect of 0.2 μM glyburide on inward current in an inside-out patch with R301C+SUR1 channels. In this case, 200 μM ADP was present throughout the experiment. B shows that, after glyburide, 200 μM ATP almost completely blocked channel activity, suggesting increased ATP sensitivity. (C) Plot of channel ATP sensitivity for R301C+SUR1 before (•) and after (▾) 200 nM glyburide. In this case, a fit of the data points yielded k = 205 μM and n = 1.14 (•; n = 5) and k = 21 μM and n = 1.08 (▾; n = 2).
However, while glyburide completely and irreversibly blocked MgADP-dependent reactivation of ATP-inhibited wild-type channels, at least part of the stimulatory effect of MgADP in the absence of ATP remained, supporting the hypothesis that the two effects of ADP involve separate processes even though both require binding of MgADP to NBF2. Similar results were obtained with R314C+SUR1 mutant channels. Thus, unlike the R176C and R177C mutants, the R301C and R314C mutants remained functionally coupled to SUR1, exhibiting a sensitivity to adenine nucleotides and sulfonylureas similar to wild-type channels.
Insertion of Kir6.2 into the Plasma Membrane Requires Physical, but Not Functional Coupling to SUR1
It has been suggested that functional coupling between Kir6.2 and SUR via NBFs may be important for facilitating KATP channel insertion into the plasma membrane (Sharma et al. 1999), although this hypothesis has been challenged (Schwappach et al. 2000). To address whether intact functional coupling between Kir6.2 and SUR1 is required for proper plasma membrane insertion, we used Kir6.2 constructs (Kir6.2-C-GFP) with green fluorescent protein (GFP) linked to the C terminus (John et al. 1998). As we previously reported (John et al. 1998), in the absence of SUR1, the fluorescence emitted by the Kir6.2-C-GFP was associated with intracellular lamellar structures tentatively identified with the ER. When Kir6.2-C-GFP was coexpressed with SUR1, however, the fluorescence appeared at the cell periphery, which is consistent with efficient plasma membrane insertion, since this pattern was associated electrophysiologically with a high KATP current density. Fig. 10 shows that, like wild-type Kir6.2-C-GFP, the mutant R176C-C-GFP remained associated with intracellular structures in the absence of SUR1, but when coexpressed with SUR1, it gave a strong peripheral labeling pattern consistent with efficient plasma membrane insertion. Similar results were obtained with R177C as well as R301C and R314C. Since R176C and R177C are not functionally coupled to SUR1, these findings support the hypothesis that functional coupling between Kir6.2 and SUR1 is not critical for proper channel trafficking.
DISCUSSION
Regulation of inward rectifier K channels in the Kir superfamily by PIP2 involves positively charged residues in the C tail region (Fan and Makielski 1997; Baukrowitz et al. 1998; Shyng and Nichols 1998). In Kir6.2, these residues span 2 distinct domains encompassing residues R176 to R222 and R301 to R314. Many single residue mutations markedly reduce reactivation by PIP2 (Shyng et al. 2000), suggesting that phosphate groups of PIP2 interact with multiple residues. PIP2 causes channel stimulation and decreased ATP sensitivity by acting directly on Kir6.2. Although MgADP interacts directly with SUR rather than Kir6.2, it has similar effects to PIP2, stimulating channel activity and decreasing ATP sensitivity (Ribalet et al. 2000). We previously showed that PIP2 prevented channel reactivation by MgADP, while addition of MgADP before channel rundown prevented channel activation by PIP2 (Koster et al. 1999; Ribalet et al. 2000). Based on these results, we postulated that PIP2 and a MgADP-bound region of SUR may compete for the same Kir6.2 regulatory sites. To test this hypothesis we investigated the effect of MgADP on PIP2-insensitive mutants. The data presented here show that the mutations R176C and R177C, but not R301C and R314C, prevented MgADP-dependent channel stimulation as well as sulfonylurea-induced inhibition. Thus, these two regions appear to have different roles: the former is involved in functional coupling between Kir6.2 and SUR1, and the latter, in PIP2 regulation. The former region identified by Shyng et al. 2000, which includes R176 and R177, is near other PIP2-sensitive residues, such as R192, R201, and R221 that could also play a role in functional coupling with SUR1. We did not include a study of these residues because they have additional effects on ATP sensitivity and may be, as previously suggested (Drain et al. 1998), part of a region that interacts with ATP to inhibit channel activity. Studies of these residues will be the subject of a subsequent report.
Mechanism of Antagonism of ATP Inhibition by MgADP
Ueda et al. 1999 recently proposed that reactivation of ATP-inhibited channels by MgADP is due to MgADP binding to NBF2, which stabilizes binding of ATP to NBF1. In their model, the ATP bound to NBF1 stimulates KATP channels by counteracting the inhibitory effect of ATP bound to Kir6.2. Thus, ATP has an inhibitory as well as a stimulatory role, the latter enhanced by MgADP binding to NBF2. In line with this model, our data suggests that the positively charged residues R176 and R177 are sites on Kir6.2 at which the NBF1/ATP moiety interacts to stimulate channel activity. One possibility is that this interaction occurs electrostatically via the negatively charged phosphate groups of the ATP molecule bound to NBF1. Channel regulation by PIP2 has been proposed to occur by an analogous mechanism, with the phosphate groups of PIP2 interacting with similar positively charged residues on Kir6.2 (Fan and Makielski 1997; Baukrowitz et al. 1998; Shyng and Nichols 1998). However, we cannot rule out that binding of ATP to NBF1 relieves ATP inhibition of Kir6.2 by another mechanism, such as an allosteric effect.
Ueda et al. 1999 also proposed that glyburide-induced channel block is not due to a direct effect on the affinity of adenine nucleotides for NBFs, but rather to a weakening of intramolecular cooperativity between NBF2 bound to MgADP and NBF1 bound to ATP. Because glyburide prevents MgADP from stabilizing the binding of ATP to NBF1, ATP is released from NBF1 and channel activity decays. Our data showing that mutations R176C and R177C render the channel glyburide-insensitive further supports the hypothesis whereby channel opening depends on interaction between the NBF1/ATP moiety and R176 and R177 to activate ATP-inhibited channels.
Mechanism of KATP Channel Stimulation by MgADP in the Absence of ATP
In the absence of SUR, full-length Kir6.2 channels exhibit brief openings without long bursts, and bursting behavior requires coexpression with SUR (John et al. 1998). Also, Kir6.2+SUR1 channels show a fast phase of rundown due to decreased bursting, whereas fast rundown does not occur in Kir6.2 channels expressed without SUR1 (Ribalet et al. 2000). MgADP, as well as other nucleotide diphosphates, reactivate KATP channels after rundown in the absence of ATP by promoting increased bursting (Terzic et al. 1994), and fast rundown can be prevented by MgADP (Ribalet et al. 2000). Even though Ueda et al. 1999 did not address this issue, the stimulatory effect of MgADP observed in the absence of ATP may imply a direct interaction between the NBF2/MgADP moiety and Kir6.2 sufficient to evoke bursting. Alternatively, in the absence of ATP, NBF1 may still interact weakly with R176 and R177 when stimulated by the NBF2/MgAPD moiety. At the present time, we do not distinguish between these possibilities. In either case, fast rundown in the absence of ATP would be due to the loss of MgADP from NBF2, and channel reactivation by MgADP (or other nucleotide diphosphates) is due to binding of exogenous MgADP or nucleotide diphosphates to NBF2 when reapplied intracellularly.
Interestingly, after treatment with glyburide, MgADP continued to activate R301C and R314C mutants in the absence of ATP. In contrast, the ability of MgADP to stimulate ATP-inhibited channels was irreversibly hindered by glyburide, suggesting that MgADP stimulates channel activity via two independent processes. These observations can be explained by the model of Ueda et al. 1999 if it is assumed that glyburide primarily inhibits the cooperativity between NBF2 and NBF1, causing NBF1 to release its ATP molecule. Thus, after glyburide, MgADP still can bind to NBF2 and stimulate channel activity in the absence of ATP, but its ability to promote the NBF1/ATP moiety's interaction with R176 and R177 is limited because NBF1 cannot retain ATP as effectively. As a consequence, binding of MgADP to NBF2 can no longer counteract the inhibition by ATP.
Interaction of PIP2 with R301 and R314
The mutations R301C and R314C yielded mutants that were glyburide-sensitive and stimulated by MgADP, suggesting that this region of Kir6.2 does not interact with either the NBF1/ATP or NBF2/MgADP moieties. Therefore, this region probably stimulates Kir6.2 activity by another additional mechanism. The low channel activity and fast rundown observed with these mutants after patch excision in the absence of ATP suggest this site may be responsible for a “tonic” effect of PIP2 that is necessary for channel opening in the absence of ATP.
Sensitivity to ATP
In the presence of SUR1, the R176C and R177C mutants exhibited a low sensitivity to ATP, with an IC50 for ATP inhibition near 200 μM. Because the ATP sensitivity of the wild-type Kir6.2 and that of the R176C mutant expressed alone was also close to this value, we propose that the low ATP sensitivity of R176C(R177C)+SUR1 is not due to a decrease in ATP affinity of the Kir6.2 mutant, but rather to functional uncoupling of Kir6.2 from SUR1. The ATP sensitivity of the R301C and R314C mutants was also low (≅200 μM), but this sensitivity was recorded in the presence of 200 μM MgADP, which shifts the ATP sensitivity by as much as five-fold (Ribalet et al. 2000). In the absence of MgADP, the ATP sensitivity of R301C and R314C mutants was not significantly different from that of wild-type Kir6.2+ SUR1 channels (≅35 μM). These observations support the hypothesis of Nichols and co-workers (Shyng et al. 2000) whereby the Kir6.2 residues that interact with ATP to confer ATP-dependent inhibition are distinct from the residues that confer PIP2 sensitivity. We now add that the residues that confer MgADP-dependent reactivation of ATP-inhibited channels are different from the residues involved in ATP inhibition.
Glyburide also caused dramatic increase in ATP sensitivity, in addition to the loss of MgADP-dependent reactivation, suggesting functional uncoupling of Kir6.2 and SUR. Assuming that ATP binds preferentially to the closed state (Trapp et al. 1998; Fan and Makielski 1999; Enkvetchakul et al. 2000), this increased ATP sensitivity may be accounted for by the concomitant decrease in channel activity, even though, a change in ATP affinity cannot be excluded. Interestingly, glyburide increased the ATP sensitivity of the Kir6.2 mutants R301C and R314C, but not that of R176C and R177C. These results further support the hypothesis that NBF1/ATP interacts with the R176 to R222 region, but not with the R310 to R314 region.
Based on the hypothesis whereby both NBF1/ATP and PIP2 interact with the same R176/R177 to stimulate channel activity and decrease the ATP sensitivity, we propose the following scheme to account for the low ATP sensitivity of Kir6.2 expressed alone and the higher ATP sensitivity of Kir6.2+SUR1 measured in excised patches (Tucker et al. 1997; John et al. 1998). In intact cells, in the absence of SUR, PIP2 interacts with R176/R177 to lower the channel ATP sensitivity, whereas in the presence of SUR and MgADP, low ATP sensitivity results from NBF1–ATP interaction with R176/R177, which prevents the effect of PIP2 on this site. Upon patch excision, dissociation of MgADP from the Kir6.2+SUR1 complex, in the absence of exogenous MgADP, is fast compared with the hydrolysis of PIP2 and fast rundown is responsible for the higher ATP sensitivity of Kir6.2+ SUR1. After rundown and functional uncoupling, R176 and R177 are free to interact with PIP2 and the ATP sensitivity of the Kir6.2+SUR1 complex may then approach that of PIP2-regulated Kir6.2.
Functional Coupling and Plasma Membrane Insertion
Based on the mutation of residues in NBF2 of SUR, it has been suggested that the impaired channel activity observed with coexpression of these SUR mutants with wild-type Kir6.2 was due to impaired channel insertion into the plasma membrane (Sharma et al. 1999). Thus, functional coupling between Kir6.2 and SUR1 may play a role in channel insertion, presumably as a quality control mechanism. However, our data obtained with the Kir6.2 mutants linked to GFP do not support this hypothesis, since Kir6.2 mutants lacking MgADP and sulfonylurea sensitivity when coexpressed with SUR1, nevertheless, showed normal trafficking to the plasma membrane and suggest the presence of a second type of coupling between Kir6.2 and SUR1, i.e., “physical” coupling which is responsible for channel insertion into the plasma membrane. A region of the Kir6.2 C tail comprised between residues 208 and 279 and the M1 transmembrane domain may play a role in this coupling with SUR (Giblin et al. 1999; Schwappach et al. 2000).
Summary and Conclusions
We have extended the model of Ueda et al. 1999 to propose that functional coupling of SUR1 to Kir6.2 is mediated by the interaction of NBF1 in SUR1 with the positively charged residues R176 and R177 in C tail of Kir6.2. This interaction requires binding of ATP to NBF1 and increases channel activity by promoting bursting behavior, as shown previously. Thus, ATP has a direct inhibitory effect on Kir6.2 and a stimulatory effect mediated via binding to NBF1. MgADP, by binding to NBF2, stabilizes the latter, and causes reactivation of ATP-inhibited channels. Sulfonylureas are proposed to inhibit channel activity by weakening the cooperativity between the NBF2/MgADP moiety and the NBF1/ATP moiety, perhaps by promoting release of ATP from NBF1. In the absence of ATP, ATP-induced inhibition does not take place and interaction of NBF1 with R176/R177 is not required for channel opening. Under these conditions, channel stimulation by MgADP may be explained by either NBF2/MgADP moiety directly stimulating channel opening by interacting with another region of Kir6.2, or permitting a weak ATP-independent interaction of NBF1 with R176 and R177. In addition, tonic stimulation by PIP2 interacting with other positively charged residues in the Kir6.2 C tail, such as R301 and R314, may be necessary to facilitate channel opening.
Acknowledgments
The authors would like to thank Dr. Y. Lu for her expert assistance in preparing the various DNA constructs.
This work was supported by a research grant from the American Diabetes Association to B. Ribalet, by National Institutes of Health grants R37 HL60025 and SCOR in Sudden Cardiac Death P50 HL52319, and Laubisch and Kawata Endowments to J.N. Weiss, and by a Grant-in-Aid from the American Heart Association (Western States Affiliate) to S.A. John.
Footnotes
Abbreviations used in this paper: KATP, ATP-sensitive K; PIP2, phosphatidylinositol bisphosphate.
References
- Ashcroft F.M. Adenosine 5′-triphosphate-sensitive potassium channels. Annu. Rev. Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Baukrowitz T., Schulte U., Oliver D., Herlitze S., Krauter T., Tucker S.J., Ruppersberg J.P., Fakler B. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 1998;282:1141–1144. doi: 10.1126/science.282.5391.1141. [DOI] [PubMed] [Google Scholar]
- Bienengraeber M., Alekseev A.E., Abraham M.R., Carrasco A.J., Moreau C., Vivaudou M., Dzeja P.D., Terzic A. ATPase activity of the sulfonylurea receptora catalytic function of the KATP channel complex. FASEB J. 2000;14:1943–1952. doi: 10.1096/fj.00-0027com. [DOI] [PubMed] [Google Scholar]
- Drain P., Li L., Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 1998;95:13953. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D., Loussouarn G., Makhina E., Shyng S.L., Nichols C.G. The kinetic and physical basis of KATP channel gatingtoward a unified molecular understanding. Biophys. J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 1997;272:5388–5395. doi: 10.1074/jbc.272.9.5388. [DOI] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 1999;114:251–269. doi: 10.1085/jgp.114.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay I. Effects of ADP upon the ATP-sensitive K+ channel in rat ventricular myocytes. J. Membr. Biol. 1988;101:83–92. doi: 10.1007/BF01872823. [DOI] [PubMed] [Google Scholar]
- Giblin J.P., Leany J.L., Tinker A. The molecular analysis of ATP-sensitive potassium channels. Determinants on the pore subunit. J. Biol. Chem. 1999;274:22652–22659. doi: 10.1074/jbc.274.32.22652. [DOI] [PubMed] [Google Scholar]
- Graham F.L., van der Eb A.J. Transformation of rat cells by DNA of human adenovirus 5. Virology. 1973;52:456–467. doi: 10.1016/0042-6822(73)90163-3. [DOI] [PubMed] [Google Scholar]
- Gribble F.M., Tucker S.J., Ashcroft F.M. The essential role of Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- John S.A., Monck J.R., Weiss J.N., Ribalet B. The sulfonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K channels linked to the green fluorescent protein in human embryonic kidney cells (HEK 293) J. Physiol. 1998;510:333–345. doi: 10.1111/j.1469-7793.1998.333bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koster J.C., Sha Q., Nichols C.G. Sulfonylurea and K+-channel opener sensitivity of KATP channelsfunctional coupling of Kir6.2 and SUR1 subunits. J. Gen. Physiol. 1999;114:203–213. doi: 10.1085/jgp.114.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C.G., Shyng S.-L., Nestorowicz A., Glaser B., Clement J.P., IV, Gonzalez G., Aguilar-Bryan L., Permutt M.A., Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Ribalet B., John S.A., Weiss J.N. Regulation of cloned ATP-sensitive K channels by phosphorylation, MgADP, and phosphatidylinositol bisphosphate (PIP2)a study of channel rundown and reactivation. J. Gen. Physiol. 2000;116:391–409. doi: 10.1085/jgp.116.3.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reimann F., Tucker S.J., Proks P., Ashcroft F.M. Involvement of the N-terminus of Kir6.2 in coupling to the sulfonylurea receptor. J. Physiol. 1999;518:325–336. doi: 10.1111/j.1469-7793.1999.0325p.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwappach B., Zerangue N., Jan Y.N., Jan L.Y. Molecular basis for KATP channel assemblytransmembrane interactions mediate association of a K+ channel with an ABC transporter. Neuron. 2000;26:155–167. doi: 10.1016/s0896-6273(00)81146-0. [DOI] [PubMed] [Google Scholar]
- Sharma N., Crane A., Clement J.P., IV, Gonzalez G., Babenko A.P., Bryan J., Aguilar-Bryan L. The C terminus of SUR1 is required for trafficking of KATP channels. J. Biol. Chem. 1999;274:20628–20632. doi: 10.1074/jbc.274.29.20628. [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Nichols C.G. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 1998;282:1138–1141. doi: 10.1126/science.282.5391.1138. [DOI] [PubMed] [Google Scholar]
- Shyng S.-L., Cukras C.A., Hardwood J., Nichols C.G. Structural determinant of PIP2 regulation of inward rectifier KATP channels. J. Gen. Physiol. 2000;116:599–607. doi: 10.1085/jgp.116.5.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzic A., Findlay I., Hosoya Y., Kurachi Y. Dualistic behavior of ATP-sensitive K+ channels toward intracellular nucleotide diphosphates. Neuron. 1994;12:1049–1058. doi: 10.1016/0896-6273(94)90313-1. [DOI] [PubMed] [Google Scholar]
- Trapp S., Proks P., Tucker S.J., Ashcroft F.M. Molecular analysis of ATP-sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Zhao C., Trapp S., Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K channels in the absence of the sulfonylurea receptor. Nature. 1997;387:179–182. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Ueda K., Komine J., Matsuo M., Seino S., Amachi T. Cooperative binding of ATP and MgADP in the sulfonylurea receptor is modulated by glibenclamide. Proc. Natl. Acad. Sci. USA. 1999;96:1268–1272. doi: 10.1073/pnas.96.4.1268. [DOI] [PMC free article] [PubMed] [Google Scholar]