

Abstract
Many cells express ryanodine receptors (RyRs) whose activation is thought to amplify depolarization-evoked elevations in cytoplasmic Ca2+ concentration ([Ca2+]i) through a process of Ca2+-induced Ca2+ release (CICR). In neurons, it is usually assumed that CICR triggers net Ca2+ release from an ER Ca2+ store. However, since net ER Ca2+ transport depends on the relative rates of Ca2+ uptake and release via distinct pathways, weak activation of a CICR pathway during periods of ER Ca accumulation would have a totally different effect: attenuation of Ca2+ accumulation. Stronger CICR activation at higher [Ca2+]i could further attenuate Ca2+ accumulation or trigger net Ca2+ release, depending on the quantitative properties of the underlying Ca2+ transporters. This and the companion study (Hongpaisan, J., N.B. Pivovarova, S.L. Colgrove, R.D. Leapman, and D.D. Friel, and S.B. Andrews. 2001. J. Gen. Physiol. 118:101–112) investigate which of these CICR “modes” operate during depolarization-induced Ca2+ entry in sympathetic neurons. The present study focuses on small [Ca2+]i elevations (less than ∼350 nM) evoked by weak depolarization. The following two approaches were used: (1) Ca2+ fluxes were estimated from simultaneous measurements of [Ca2+]i and ICa in fura-2–loaded cells (perforated patch conditions), and (2) total ER Ca concentrations ([Ca]ER) were measured using X-ray microanalysis. Flux analysis revealed triggered net Ca2+ release during depolarization in the presence but not the absence of caffeine, and [Ca2+]i responses were accelerated by SERCA inhibitors, implicating ER Ca2+ accumulation, which was confirmed by direct [Ca]ER measurements. Ryanodine abolished caffeine-induced CICR and enhanced depolarization-induced ER Ca2+ accumulation, indicating that activation of the CICR pathway normally attenuates ER Ca2+ accumulation, which is a novel mechanism for accelerating evoked [Ca2+]i responses. Theory shows how such a low gain mode of CICR can operate during weak stimulation and switch to net Ca2+ release at high [Ca2+]i, a transition demonstrated in the companion study. These results emphasize the importance of the relative rates of Ca2+ uptake and release in defining ER contributions to depolarization-induced Ca2+ signals.
Keywords: calcium signaling, endoplasmic reticulum, caffeine, ryanodine, electron probe X-ray microanalysis
INTRODUCTION
Calcium is an important signaling ion, and changes in Ca2+ concentration ([Ca2+]) regulate diverse processes in many cellular compartments. In excitable cells, depolarization-induced Ca2+ entry increases [Ca2+]i, leading to secondary changes in [Ca2+] within organelles such as mitochondria and ER that regulate specific Ca2+-sensitive targets within these organelles (Pozzan et al. 1994; Berridge 1998). Although mitochondria accumulate Ca2+ in response to depolarization-evoked [Ca2+]i elevations (Babcock and Hille 1998), the ER has been described as either a Ca2+ source or sink, in some cases even in the same cell type (Friel and Tsien 1992a; Kuba 1994; Verkhratsky and Shmigol 1996; Toescu 1998). Such disparate modes of net ER Ca2+ transport are expected to have very different effects on cytoplasmic and intraluminal Ca2+ signals, and on the processes they regulate. Nevertheless, the conditions that favor net Ca2+ uptake versus net release by the ER are not well understood. The direction of net ER Ca2+ transport depends on the relative rates of Ca2+ uptake and release via distinct transport pathways. ER Ca2+ uptake is regulated by sarco- and endoplasmic reticulum Ca ATPase (SERCA)* pumps, whereas passive Ca2+ release is regulated, at least in part, by Ca2+ release channels that are gated by elevations in [Ca2+]i. When the rate of Ca2+ uptake exceeds the rate of passive Ca2+ release, the ER would act as a Ca2+ sink, whereas if the converse is true, it would act as a Ca2+ source. One process by which depolarization-evoked Ca2+ entry is thought to trigger net ER Ca2+ release is CICR (Berridge 1998).
The machinery required for CICR is present in a variety of neurons (for review see Kuba 1994). For example, sympathetic neurons contain a Ca store that sequesters Ca via a thapsigargin (Tg)-sensitive uptake system and is discharged by caffeine in a ryanodine-sensitive manner (Lipscombe et al. 1988; Thayer et al. 1988; Friel and Tsien 1992a; Friel 1995), arguing that it expresses both SERCAs and ryanodine-sensitive Ca2+ release channels, (also known as ryanodine receptors, RyRs). Observations suggesting that [Ca2+]i-dependent activation of RyRs can amplify depolarization-induced [Ca2+]i elevations in these and other neurons include: (1) acceleration of [Ca2+]i responses in the presence of caffeine in a ryanodine-inhibitable manner (Friel and Tsien 1992a; Usachev and Thayer 1997); (2) slowing of [Ca2+]i responses after treatment with ryanodine (Friel and Tsien 1992a; Hua et al. 1993; Shmigol et al. 1995; Peng 1996); (3) a supralinear relationship between the amount of Ca2+ that enters the cells during depolarization and the size of the resulting [Ca2+]i elevation (Hua et al. 1993; Llano et al. 1994; Shmigol et al. 1995); and (4) facilitation of depolarization-evoked [Ca2+]i responses at high [Ca2+]i (Hua et al. 1993; Llano et al. 1994). The first observation implicates CICR in the presence of caffeine, but the others have been taken as support for a [Ca2+]i- and ryanodine-sensitive amplification system that operates even in the absence of caffeine.
Activation of a CICR pathway is usually assumed to trigger net Ca2+ release from the ER that amplifies depolarization-induced [Ca2+]i elevations. However, theory indicates that even when such a pathway is present, small [Ca2+]i elevations above the resting level may stimulate net Ca2+ uptake by the ER (referred to hereafter as Ca2+ accumulation). This would occur if the rate of Ca2+ uptake increases more steeply with [Ca2+]i than the rate of Ca2+ release. In this case, weak activation of RyRs would increase the rate of passive Ca2+ release and, as a result, lower the rate of Ca2+ accumulation. This is an interesting mode of CICR since, like net CICR, it would tend to increase the impact of Ca2+ entry on [Ca2+]i, but unlike net CICR, it would occur in the context of a rise in intraluminal [Ca2+] concentration.
This and the companion study (see Hongpaisan et al. 2001, in this issue) address four fundamental questions about neuronal RyR-mediated CICR, using sympathetic neurons as a model system: (1) What organelle is responsible for CICR? (2) How does CICR contribute to changes in the intracellular distribution of Ca during weak depolarization in the absence of CICR modifiers like caffeine? (3) How do these changes vary as the stimulus-evoked rise in [Ca2+]i becomes larger? (4) What are the spatiotemporal properties of CICR? This study examines the relationship between changes in [Ca2+]i and [Ca]ER during weak depolarization, while the companion study (see Hongpaisan et al. 2001, in this issue) examines how stimulus-evoked changes in [Ca]ER vary with [Ca2+]i and with proximity to the plasma membrane. Some of these results have been presented previously in abstract form (Albrecht and Friel 1997; Hongpaisan et al. 1999).
MATERIALS AND METHODS
Cell Dissociation and Culture
Bullfrog sympathetic neurons were prepared as described previously (Colegrove et al. 2000a). All procedures conform to guidelines established by our Institutional Animal Care and Use Committees.
Cytosolic Calcium Measurements
To measure [Ca2+]i, cells were incubated with 3 μM Fura-2 AM in normal Ringer's for 40 min at room temperature with gentle agitation followed by rinsing. The composition of normal Ringer's was (in mM): 128 NaCl, 2 KCl, 2 CaCl2, 10 HEPES, 10 glucose, pH adjusted to 7.3 with NaOH. Fura-2 AM was dispensed from a 1-mM stock solution in DMSO containing 25% (wt/wt) pluronic F127 (BASF Corporation). Cells were then washed with normal Ringer's and placed on the stage of an inverted microscope (Nikon Diaphot TMD) and superfused continuously (∼5 ml/min). Recordings began ∼20 min after washing away Fura-2 AM, permitting de-esterification of the Ca2+ indicator. With this loading procedure, there is little compartmentalization of fura-2 based on the low residual fluorescence observed after cells are dialyzed with dye-free internal solution under whole-cell conditions, and the loss of fluorescence after permeabilization of the plasma membrane with digitonin (Lipscombe et al. 1988). Solution changes (∼200 ms) were made using a system of microcapillaries (Drummond microcaps, 20 μl) mounted on a micromanipulator. Fluorescence measurements were performed as described in Colegrove et al. 2000a.
Voltage Clamp
Simultaneous measurements of depolarization-evoked [Ca2+]i elevations and voltage-sensitive Ca2+ currents (ICa) were made under voltage clamp in Fura-2 AM loaded cells using the perforated patch technique. Patch pipettes (1–2 MΩ) were pulled (Sutter Instruments P-97), coated with Sylgard, fire-polished, and the tips were filled with a solution containing (in mM): 125 CsCl, 5 MgCl2, 10 HEPES, pH 7.3 with CsOH. After filling tips, pipettes were back-filled with the same solution supplemented with amphotericin B dispensed from concentrated aliquots (12 mg/100 μl DMSO) to give a final concentration of 480 μg/ml. After they were prepared, amphotericin B-containing internal solutions were kept on ice and used within 2 h. Upon achieving a high resistance seal, series resistance declined over 5–10 min to <10 MΩ. Cells were exposed to an extracellular solution containing (in mM): 130 TEACl, 10 HEPES, 10 glucose, 2 CaCl2, 1 MgCl2, pH 7.3 with TEAOH. Currents were measured with an Axopatch 200A voltage clamp (Axon instruments) using series resistance compensation (∼90%) and were filtered at 5 kHz. Cells were held at −70 mV and depolarized to −35 mV while current and fluorescence intensity were measured at 5 kHz for 0.2 s before and after changes in voltage, and at 4–5 Hz otherwise and saved on a laboratory computer. Currents were corrected for a linear leak based on responses to small hyperpolarizing voltage steps.
Measurement of [Ca]ER
Total Ca concentrations within structurally identified cisternae of ER ([Ca]ER) were measured by energy-dispersive X-ray (EDX) microanalysis of freeze-dried cryosections obtained from rapidly frozen ganglia, as described previously (Pozzo-Miller et al. 1997; Pivovarova et al. 1999). In brief, dispersed ganglia were frozen by impact against a LN2-cooled metal block (modified Life Cell CF100); subsequently, cryosections were prepared (nominal thickness ∼80 nm) and analyzed using instrumentation described in the companion paper (see Hongpaisan et al. 2001, in this issue). For each experimental condition, individual [Ca]ER measurements were taken from random somatic regions from multiple cells and averaged, so that the concentrations reported should closely approximate true spatial averages. A probe size of 63 nm was used; smaller probes yielded essentially similar results, confirming that this probe was adequate to determine ER content without contamination from adjacent cytosol. Measurements are given in units of millimoles per kilogram dry weight, from which estimated concentrations in millimoles per liter of hydrated tissue are obtained after multiplying by the estimated ratio of dry weight to total wet weight within the ER (∼0.28; Pozzo-Miller et al. 1997). Resting [Ca]ER was measured in cells from ganglia that were incubated in normal Ringer's, and depolarization-induced changes in [Ca]ER were measured after transferring ganglia to 30 mM K+ Ringers (equimolar substitution for Na+) for 45 or 120 s. To assess the effect of ryanodine on resting [Ca]ER, ganglia were transferred to normal Ringer's plus 1 μM ryanodine supplemented with 10 mM caffeine; after five minutes, ganglia were transferred to normal Ringers plus 1 μM ryanodine without caffeine for one additional minute before rapid freezing. Measurements of depolarization-evoked changes in [Ca2+]i in fura-2–loaded cultured cells show that this protocol is sufficient to inhibit caffeine-induced [Ca2+]i transients and modify depolarization-induced [Ca2+]i elevations (not shown). Effects of ryanodine on evoked changes in [Ca]ER were assessed by treating ganglia with ryanodine as described above and then exposing them to 30 K+ in the presence of ryanodine.
Measurement of Ca2+ Fluxes
The Ca2+ fluxes responsible for changes in [Ca2+]i during and after depolarization-evoked Ca2+ entry were determined based on simultaneous measurements of [Ca2+]i and ICa. The total Ca2+ flux (Jtotal) and the component of Jtotal representing Ca2+ entry through voltage-sensitive Ca2+ channels (JICa) were estimated as described below. These measurements made it possible to estimate the net Ca2+ flux (JΣ) representing the combined activity of all other Ca2+ transport systems, including Ca2+ extrusion across the plasma membrane and uptake and release by organelles such as the ER and mitochondria. The sign of JΣ provides information about Ca2+ release from intracellular stores: when JΣ is negative, the rate of net Ca2+ release exceeds the combined rate of Ca2+ clearance; when JΣ is positive, net Ca2+ release, if it occurs, must be slower than Ca2+ clearance.
The total cytosolic Ca2+ flux per unit volume was measured (Jtotal, in nanomolars per second) by taking the time derivative of [Ca2+]i at each sample time ti during the period of low frequency sampling according to ([Ca2+]i(ti + Δt/2) − [Ca2+]i(ti − Δt/2))/Δt, where Δt (400–500 ms) is twice the sampling interval. During the periods of high frequency sampling immediately after depolarization and repolarization, the flux was determined by measuring the slope of a fitted exponential function. Before calculating the fluxes, the [Ca2+]i measurements were smoothed four to five times with a binomial filter.
Jtotal was dissected into two components representing the rate of Ca2+ entry through voltage-sensitive Ca2+ channels (JICa) and the composite net flux representing transport by all other systems, (JΣ = Jtotal − JICa). JICa is the rate of Ca2+ entry per unit cytoplasmic volume divided by the ratio (κT i) of changes in total cytoplasmic Ca concentration (bound plus free) that accompany small changes in [Ca2+]i (Neher and Augustine 1992; Colegrove et al. 2000a). The rate of Ca2+ entry per unit cytoplasmic volume was calculated from ICa/2Fvi, where F is the Faraday constant and vi is the cytoplasmic volume estimated from the measured membrane capacitance assuming hemispherical geometry to approximate the shape of adherent cells. To estimate κT i, it was reasoned that during the initial moments following depolarization, before [Ca2+]i has changed sufficiently to perturb basal Ca2+ transport, [Ca2+]i should rise at a rate that depends only on the rate of Ca2+ entry, the cytosolic volume, and κT i. Accordingly, κT iwas estimated as the average ratio of ICa/2Fvi to Jtotal during the early period of depolarization (from 0.24 to 1.00 s). During this time, Jto tal was insensitive to pharmacological interventions that dramatically modified Ca2+ transport by the caffeine-sensitive store (see Fig. 2 C, compare left and right panels, and Fig. 4A and Fig. B), indicating that it is dominated by Ca2+ entry. As expected, reducing the intracellular fura-2 concentration (by reducing loading times) systematically lowered κT iand increased the magnitudes of Jtotal, JICa, and JΣ. However, this did not change the signs of these fluxes, indicating that Ca2+ buffering by fura-2 did not influence the direction of JΣ.
Figure 2.
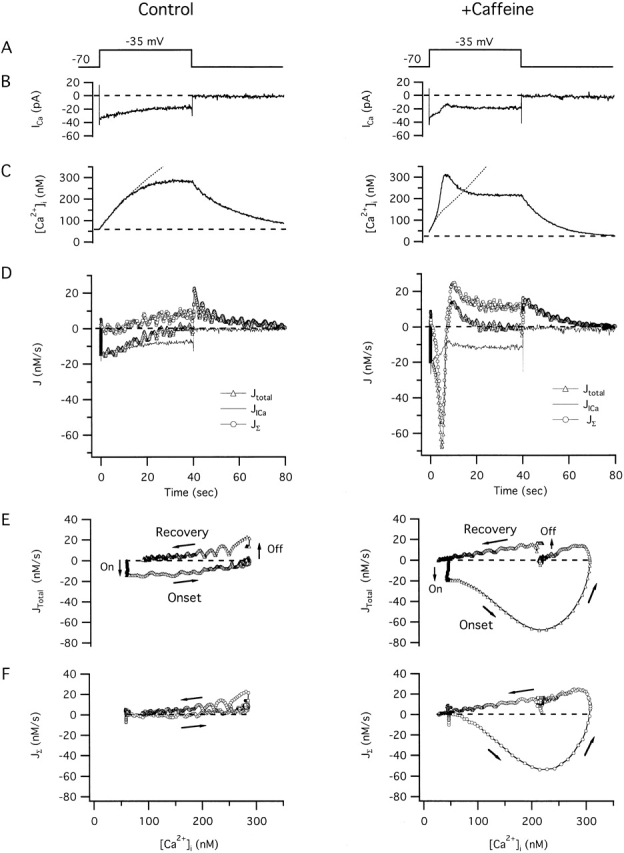
Two components of the total Ca2 + flux responsible for depolarization-induced changes in [Ca2 +]i and their modification by caffeine. Responses elicited under voltage clamp from the same cell before (left column) and during (right column) exposure to 5 mM caffeine. Panels show (A) voltage protocol, (B) ICa, (C) [Ca2+]i, (D) Jtotal (triangles) and its components JICa (continuous curve) and JΣ (circles) all plotted on the same time scale, whereas E and F show Jtotal and JΣ plotted against [Ca2+]i during and after depolarization. In E and F, arrows indicate the direction of the flux trajectories as [Ca2+]i changes during the response onset and recovery. Caffeine elicited a transient [Ca2+]i rise (not shown) when applied between the two depolarization-evoked [Ca2+]i responses illustrated here, as in Fig. 1. Dotted traces in C show integrated JICa, indicating that [Ca2+]i initially rises at a rate that is proportional to ICa, but then rises more slowly than the integrated flux in the absence of caffeine, and more rapidly in its presence. Fluxes were calculated as described in materials and methods. Cell ma4460.
Figure 4.
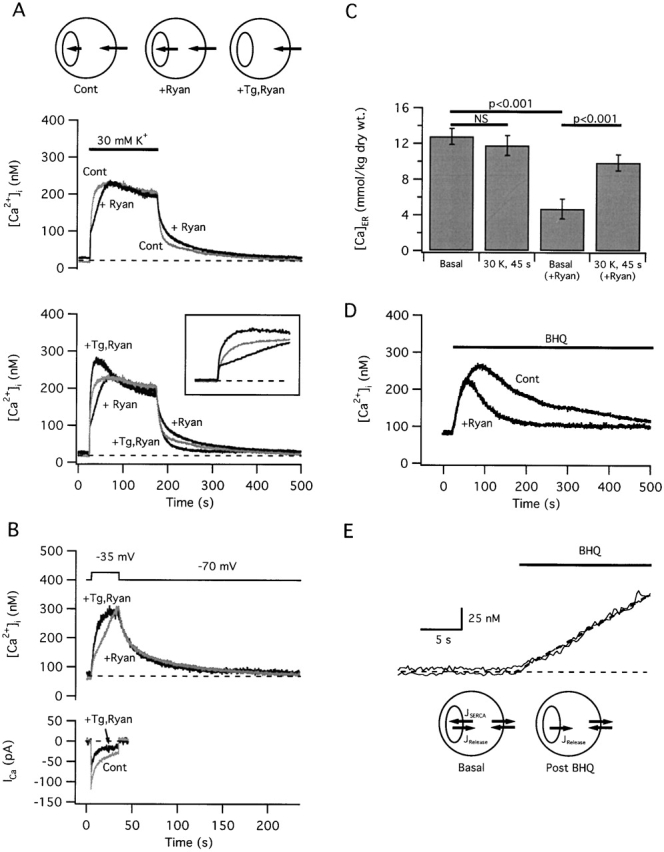
Effects of ryanodine on depolarization-induced changes in [Ca2 +]i and [Ca]ER. (A, top traces) Comparison between [Ca2+]i responses elicited by 30 mM K+ depolarization before (light trace) and after (dark trace) treatment with ryanodine (1 μM) to inhibit CICR. After recording a control response to 30 mM K+, cells were exposed to ryanodine, and then to caffeine in the presence of ryanodine, which elicited a transient [Ca2+]i elevation (not shown). After washing out caffeine, all subsequent high K+-induced [Ca2+]i responses were modified as illustrated and responsiveness to caffeine was abolished (not shown). (Bottom traces) Comparison between the upper traces and a final response elicited from the same cell after exposure to Tg (100 nM) which reversed the effect of ryanodine and speeded the rise in [Ca2+]i compared with the control response. This response was elicited after the Tg-induced [Ca2+]i transient was complete and resting [Ca2+]i was restored (not shown). Inset shows the initial period of these responses on an expanded time scale. Cell maac53. (B) Tg-induced reversal of ryanodine's effect on [Ca2+]i response kinetics does not reflect drug-induced changes in ICa; in the example shown, [Ca2+]i rises more rapidly after Tg treatment even though ICa underwent considerable rundown during the period between the first and second responses. Cell ma5030. (C) Comparison between resting and depolarization-induced changes in [Ca]ER in the presence and absence of ryanodine. Ryanodine reduces resting [Ca]ER but enhances ER Ca accumulation during 45 s 30 mM K+ depolarization. (D) The reversible SERCA inhibitor t-BuBHQ (10 μM) elicits a [Ca2+]i transient whose initial rate is not influenced by ryanodine but whose amplitude and duration are altered as expected based on the ryanodine-induced reduction in [Ca]ER. Cell maaa65. (E) The initial portions of the responses in D are shown on an expanded scale to illustrate the insensitivity of the initial rate to ryanodine. Diagram below shows the relationship between ER and plasma membrane Ca2+ fluxes under basal conditions (left) and just after inhibiting SERCAs by exposure to t-BuBHQ. Our interpretation of these results is illustrated in A (top).
Pharmacological Manipulation of CICR
Ryanodine was used as a tool to evaluate how CICR contributes to stimulus-evoked [Ca2+]i elevations. At low concentrations (≤1 μM), ryanodine inhibits Ca2+-dependent RyR channel gating and increases channel open probability. At high concentrations (>100 μM), it causes channel block (for reviews see Coronado et al. 1994; Berridge et al. 1995; Sutko et al. 1997; Zucchi and Roncha-Testoni 1997). To inhibit CICR, cells were exposed to ryanodine (1 μM) and then transiently to caffeine (10 mM) in the continued presence of ryanodine. Under these conditions, caffeine elicits a transient rise in [Ca2+]i like that observed in control cells, but unlike control cells, responsiveness to caffeine is not restored after caffeine is removed (Thayer et al. 1988). Caffeine opens RyRs by increasing their sensitivity to [Ca2+]i (Rousseau et al. 1988), and ryanodine is thought to inhibit caffeine responsiveness by irreversibly modifying RyRs so that they are insensitive to Ca2+ (Rousseau et al. 1987) or have greatly increased Ca2+ sensitivity (Chen et al. 2001). Ryanodine was used in conjunction with caffeine because ryanodine preferentially interacts with the open channel, causing ryanodine-induced RyR modifications to be use-dependent.
Simulations
Rate equations describing Ca2+ extrusion across the plasma membrane (Colegrove et al. 2000b) and Ca2+ uptake and release by the ER were incorporated into a system of differential equations (see ) that was solved numerically using a fourth-order Runge-Kutta routine (Boyce and DiPrima 1969) written in Igor Pro (Wavemetrics, Inc.). Step size was 50 ms; further reductions in step size did not noticeably alter the results.
Reagents and Data Analysis
Fura-2 AM was obtained from Molecular Probes, ryanodine was obtained from RBI, t-BuBHQ was purchased from Calbiochem, and unless indicated otherwise, all other compounds were obtained from Sigma-Aldrich. Population results are expressed as mean ± SEM and statistical significance was assessed using t test.
RESULTS
Modulation of Depolarization-evoked [Ca2+]i Responses by the Caffeine-sensitive Store
Fig. 1 shows [Ca2+]i responses elicited under three different conditions illustrating how the caffeine-sensitive store can influence the impact of Ca2+ entry on [Ca2+]i. Experiments were performed under voltage clamp (perforated patch conditions) so that components of the total Ca2+ flux representing Ca2+ entry and Ca2+ transport by other systems could be distinguished (see next section). Recordings were made using a test potential (−35 mV) close to the membrane potential established during exposure to 30 mM K+ to facilitate comparison with previous results obtained using this K+ concentration to stimulate Ca2+ entry (Friel and Tsien 1992a). We first show how net Ca2+ transport by the store can influence depolarization-evoked [Ca2+]i responses under conditions that favor net Ca2+ release or Ca2+ accumulation. Flux measurements are then described that provide information about CICR under control conditions and when it is modified by caffeine.
Figure 1.
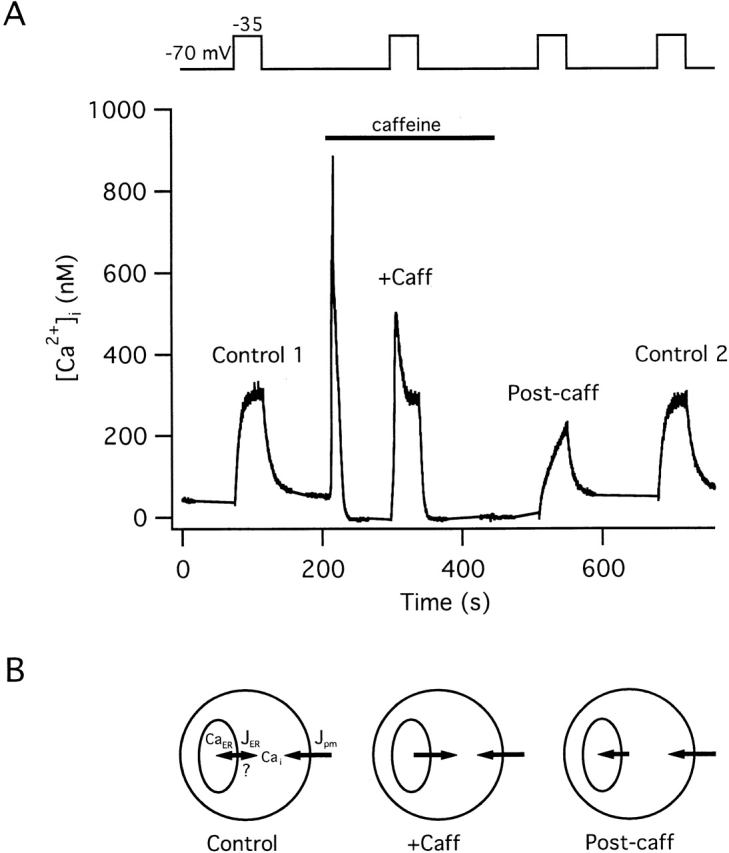
Effects of Ca2 + release and uptake by the caffeine-sensitive store on responses elicited by weak depolarization. [Ca2+]i responses elicited from a representative cell by voltage clamp depolarization under control conditions (Control 1), during continuous exposure to 5 mM caffeine (+Caff), after removing caffeine to initiate store replenishment (Post-caff), and finally after allowing sufficient time for the store to refill (Control 2). In the presence of caffeine, depolarization-induced [Ca2+]i responses are amplified, whereas during the period of replenishment after caffeine removal, responses are blunted; a final depolarization elicits a response like the control. Top trace indicates membrane potential. Exposure to caffeine at the holding potential (−70 mV) (between first and second depolarizations) elicited a large [Ca2+]i transient; the small reversible reduction in basal [Ca2+]i seen in the presence of caffeine is due, at least in part, to an effect of caffeine on fura-2 fluorescence independent of changes in [Ca2+]i (Friel and Tsien 1992a; Muschol et al. 1999). Cell ma4441. (B) Diagrams show schematically the relationship between the net Ca2+ flux across the plasma membrane (Jpm) and between the cytosol and ER (JER) during [Ca2+]i elevations elicited in the presence of caffeine (+Caff) and following caffeine washout (Post-caff). This study investigates the direction of net ER Ca2+ transport under control conditions.
During membrane depolarization, [Ca2+]i rises toward a steady level (Fig. 1, Control 1) and recovers after repolarization. Subsequent exposure to caffeine elicits a large [Ca2+]i transient reflecting Ca2+ release from an intracellular store (Friel and Tsien 1992a). Several observations provide information about the mechanisms of Ca2+ uptake and release by this store. Caffeine is ineffective after treatment with the SERCA inhibitors thapsigargin (Tg > 20 nM), 2,5-di-(t-butyl)-1,4-hydroquinone (t-BuBHQ, 10–100 μM) or cyclopiazonic acid (100 μM; unpublished data), each of which elicits a transient [Ca2+]i rise in the absence of extracellular Ca2+, indicating that Ca2+ uptake by the store requires SERCA activity. Caffeine is also ineffective after treatment with ryanodine (Thayer et al. 1988; Friel and Tsien 1992a), arguing that RyRs serve as the caffeine-sensitive Ca2+ release pathway. Evidence will be presented in the accompanying study directly demonstrating that the caffeine-sensitive store is the ER.
When cells are depolarized in the continued presence of caffeine (Fig. 1, +Caff), [Ca2+]i responses are dramatically amplified, showing an accelerated onset leading to a prominent [Ca2+]i spike. The recovery that follows repolarization is also accelerated compared with the control response. Two observations suggest that amplification is caused by Ca2+-induced release: (1) it is not observed after treatment with ryanodine (1 μM; Friel and Tsien 1992a); and (2) in the presence of caffeine, the total Ca2+ flux elicited by depolarization consists of two kinetically distinct components. One component resembles the flux elicited by depolarization in the absence of caffeine, whereas the other resembles the flux underlying caffeine-induced Ca2+ release (Friel and Tsien 1992b; Usachev and Thayer 1997).
When caffeine is washed out, the store refills at a rate that depends on the availability of cytosolic Ca2+ and the rate of net Ca2+ entry across the plasma membrane (Friel and Tsien 1992a). If cells are depolarized during this period of replenishment (Post-caff), the rise in [Ca2+]i is much slower than the control, even though the underlying Ca2+ current is similar (not shown). Evidence has been presented previously (Friel and Tsien 1992a) that the onset is slow because a portion of the Ca2+ entering through Ca2+ channels is taken up by the store as it refills. After allowing sufficient time for store replenishment, a final depolarization elicits a [Ca2+]i response (Fig. 1, Control 2) resembling the first control response. These observations are representative of four cells studied under voltage clamp and are consistent with previous results obtained from cells depolarized with 30 mM K+ (Friel and Tsien 1992a). Thus, in the presence of caffeine, Ca2+ release via a ryanodine-sensitive pathway can amplify depolarization-induced [Ca2+]i elevations; conversely, after being discharged, Ca2+ accumulation by the store can attenuate these responses (see diagrams, Fig. 1, bottom).
How does the caffeine-sensitive store contribute to [Ca2+]i dynamics when Ca2+ transport is not modified by caffeine? It has been proposed that in these and other neurons, depolarization-induced [Ca2+]i responses are amplified by CICR, even in the absence of caffeine. The main goal of the present study was to evaluate this possibility in a weak stimulus regime where [Ca2+]i is low (less than ∼350 nM). The companion study (see Hongpaisan et al. 2001, in this issue) examines the case where stronger stimuli raise [Ca2+]i to progressively higher levels.
Components of the Total Cytosolic Ca2+ Flux
[Ca2+]i rises during depolarization and declines after repolarization because there is a net cytoplasmic Ca2+ flux: inward during the onset and outward during the recovery. At each instant in time, this flux depends on the rate of stimulated Ca2+ entry and on the rate of endogenous Ca2+ transport representing, at a minimum, Ca2+ extrusion across the plasma membrane, and Ca2+ uptake and release by the ER and mitochondria. Given a measurement of the total Ca2+ flux and the rate of stimulated Ca2+ entry, the endogenous net flux can be estimated. Based on the sign of this flux, it is possible to place limits on the relative rates of net Ca2+ release from the caffeine-sensitive store and Ca2+ clearance by other transport systems.
The total cytosolic Ca2+ flux (Jtotal, measured in nanomolars/second) can be determined at each instant in time by measuring the time derivative of [Ca2+]i; this gives the rate at which Ca2+ leaves or enters the cytosol (e.g., in nanomoles/second) divided by the cytosolic volume and a buffering factor (κT i, see materials and methods). Jtotal can be separated into two components representing the net Ca2+ flux through voltage-sensitive Ca2+ channels (JICa), and the composite net flux representing endogenous Ca2+ transport (JΣ). JICa was calculated from the measured Ca2+ current (ICa), the estimated cytosolic volume, and κT i as described in materials and methods, whereas JΣ was calculated from Jtotal − JICa. By convention, inward fluxes that raise [Ca2+]i are negative and outward fluxes that lower [Ca2+]i are positive.
Fig. 2 illustrates how the interplay between JICa and JΣ defines Jtotal during and after weak depolarization before (left) and during (right) exposure to 5 mM caffeine from an experiment like that illustrated in Fig. 1. In the absence of caffeine (left column), depolarization elicits an inward Ca2+ current (B) causing [Ca2+]i to rise (C) toward a nearly steady level. Fig. 2 D shows the time courses of Jtotal and its components during and after depolarization. During depolarization, Jtotal (Fig. 2 D, triangles) is an inward flux whose magnitude increases rapidly and then declines toward zero as [Ca2+]i approaches a steady level of ∼250 nM. After repolarization, Jtotal rapidly becomes an outward flux and then declines toward zero as [Ca2+]i approaches its prestimulation level.
The temporal properties of Jtotal are defined by the interplay between JICa and JΣ. The initial negative-going deflection of Jtotal after depolarization reflects rapid activation of JICa (continuous curve), whereas the later decay reflects the slow development of an opposing outward flux (JΣ, circles) that nearly balances JICa by the end of the depolarization, accounting for the decline in Jtotal and the approach of [Ca2+]i to a steady value. Importantly, JΣ is positive under these conditions of stimulation, indicating that if the stimulus triggers net Ca2+ release from the caffeine-sensitive store, the rate of release must be slower than the rate of Ca2+ clearance by all other transport systems. Following repolarization, Ca2+ channel deactivation causes JICa to fall rapidly to zero, unmasking the outward flux JΣ that causes [Ca2+]i to decline.
Fig. 2 E plots Jtotal versus [Ca2+]i during the response onset and recovery, showing the abrupt negative-going transition that follows depolarization (“On” arrow) and the decline to zero as [Ca2+]i approaches a new steady level after depolarization, as well as the abrupt positive-going transition after repolarization (“Off” arrow) followed by a decline to zero as [Ca2+]i returns to its prestimulation value. Fig. 2 F plots JΣ against [Ca2+]i, showing that this flux depends weakly on [Ca2+]i, and for a given [Ca2+]i level, has similar values during the onset and recovery, indicating that JΣ, and the collective activity of the underlying transporters, do not depend strongly on ICa or voltage.
In the presence of caffeine (Fig. 2, right column), the Ca2+ fluxes underlying the [Ca2+]i response are strikingly different. Although initially Jtotal resembles the control flux, it becomes an explosively increasing inward flux, reaching nearly −70 nM/s after which it declines and changes sign to become a transient outward flux before finally approaching zero. The large transient inward flux is responsible for the upstroke and overshoot during the [Ca2+]i response, and the transient outward flux is responsible for the [Ca2+]i decay from its peak to the steady level. The complex kinetics of Jtotal cannot be explained by caffeine-induced changes in JICa since this flux is similar to the control flux, except for a small but consistent depression when [Ca2+]i is highest. This depression may represent [Ca2+]i-dependent inhibition of ICa, a contaminating outward current carried by Cs+ through incompletely blocked Ca2+-activated K+ channels, or a combination of the two. Caffeine did not systematically influence the impact of ICa on Jtotal during the initial period of depolarization: κT i= 263.5 ± 31.4 in the presence of caffeine and 244.2 ± 38.8 in the control (four cells, NS).
The temporal properties of JΣ account for the complicated dynamics of Jtotal and [Ca2+]i during depolarization in the presence of caffeine. JΣ consists of a large, inward spike, a transient outward component, and a steady-state component similar to that seen in the absence of caffeine. Since JΣ is negative during the upstroke of the [Ca2+]i spike, the rate of Ca2+ release must exceed the rate of Ca2+ clearance, and therefore the rise in [Ca2+]i would be expected to continue even if the cell were repolarized during this phase of the response (Usachev and Thayer 1997). When repolarization occurs after [Ca2+]i stabilizes and JΣ is positive, [Ca2+]i declines toward the prestimulation level. Jtotal and JΣ are plotted against [Ca2+]i for comparison with the control response (Fig. 2E and Fig. F, right). In the presence of caffeine, JΣ follows a continuous trajectory without abrupt changes in magnitude at the instants of depolarization and repolarization. This argues that in the presence of caffeine, as in the control, JΣ is not very sensitive to voltage but is controlled by other variables, such as [Ca2+]i and the free Ca concentration within the caffeine-sensitive store. Since JΣ follows a trajectory during and after depolarization like that followed by Jtotal during caffeine-induced Ca2+ release (Friel and Tsien 1992b), it appears that the component of Jtotal responsible for response amplification in the presence of caffeine is similar to the flux responsible for caffeine-induced Ca2+ release, namely, CICR. Similar results were obtained in each of four cells using the same stimulus protocol.
To summarize, depolarization elicits a rise in [Ca2+]i whose temporal properties reflect the interplay between voltage-sensitive Ca2+ entry and a composite net Ca2+ flux representing all other functional Ca2+ transport pathways. Under control conditions, the composite flux is outwardly directed, opposes the effects of Ca2+ entry on [Ca2+]i, shows little or no hysteresis, and is kinetically simple. In the presence of caffeine, the composite flux is biphasic, amplifies the effects of Ca2+ entry, shows strong hysteresis, and consists of a rapid transient inward component followed by a transient outward flux, having an overall trajectory resembling Jtotal after exposure to caffeine, even though the proximal stimulus is membrane depolarization, not caffeine. These observations lead to our first two important conclusions. First, when Ca2+ entry is stimulated in the presence of caffeine (5 mM), net Ca2+ release from the store occurs at a sufficiently high rate that it overwhelms available Ca2+ clearance systems, causing JΣ to be an inward net flux. Second, if Ca2+ entry triggers net Ca2+ release from the store in the absence of caffeine, the rate of release must be less than the rate of Ca2+ clearance by other transport systems; otherwise, JΣ would be an inward flux.
During Weak Depolarization, the Caffeine-sensitive (ER) Store Accumulates Calcium
Does the caffeine-sensitive store release net Ca2+ in response to depolarization-induced Ca2+ entry in the absence of caffeine? To examine this point, cells were depolarized before and after treatment with thapsigargin (Tg), which discharges the caffeine-sensitive store and elicits a transient [Ca2+]i rise in these cells (Friel 1995). If Ca2+ entry normally triggers net Ca2+ release, then after Tg treatment and depletion of the store, Ca2+ entry should elicit a [Ca2+]i rise that is slower than the control. Just the opposite was observed: depolarization-evoked [Ca2+]i elevations were faster after Tg (Fig. 3 A, top). This was quantified by calculating the time for [Ca2+]i to rise from 20–80% of its peak value during depolarization, which after Tg treatment was reduced to 67 ± 8% of the control value (n = 10, P < 0.005). Acceleration of the depolarization-induced [Ca2+]i responses could not be explained by Tg-induced enhancement of ICa (Fig. 3 A, bottom).
Figure 3.
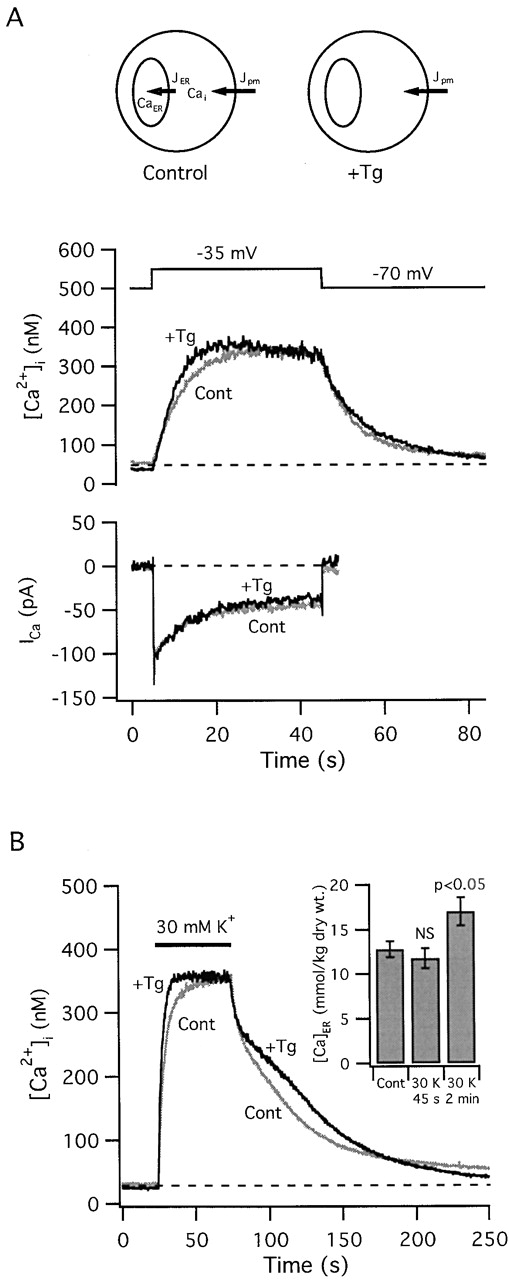
The thapsigargin-sensitive store accumulates Ca2 + during weak depolarization. (A) [Ca2+]i responses elicited by step depolarizations from −70 to −35 mV before (light trace) and after exposure to Tg (200 nM; dark trace). In each case, depolarization elicited similar voltage-sensitive Ca2+ currents (bottom), but [Ca2+]i increased more rapidly and reached its peak earlier after Tg treatment, supporting the conclusion that the Tg-sensitive store accumulates Ca2+ during these stimuli. Cell ma4620. Tg elicited a transient [Ca2+]i elevation between these responses (not shown). (B) Response acceleration after Tg treatment was also seen in cells during exposure to 30 mM K+, which depolarizes Vm to approximately the same potential (−35 mV). This cell also illustrates a slow phase of recovery that was observed in some cells after Tg treatment under these conditions of stimulation. Inset to B shows collected results from EDX microanalysis demonstrating that exposure to 30 mM K+ (2 min.) elevates [Ca]ER. Diagrams in A (top) summarize the finding that during periods of Ca2+ entry the ER accumulates Ca2+, which reduces the total cytoplasmic Ca2+ flux, an effect that is overcome by inhibiting Ca2+ uptake with Tg.
Treatment with Tg similarly modified [Ca2+]i responses elicited by 30 mM K+ depolarization (Fig. 3 B), leading to a reduction in the 20–80% rise time to 74% of the control value (n = 9, P < 0.05). After Tg treatment, some cells exhibited a plateau during the recovery (Fig. 3 B). The plateau appears to reflect Ca2+ release from mitochondria that become loaded during depolarization because it is not observed under conditions where mitochondrial Ca2+ release via the Na+/Ca2+ exchanger is inhibited, e.g., during exposure to CGP 37157 (3/3 cells, not shown; Colegrove et al. 2000a) or under voltage clamp using pipette solutions that lack Na+ (Fig. 3 A), or in cells that respond to weak depolarization with small [Ca2+]i elevations (less than ∼250 nM; see Fig. 4 A) that should be relatively ineffective in stimulating mitochondrial Ca2+ accumulation (Colegrove et al. 2000a). One possible explanation is that by inhibiting ER Ca2+ accumulation and accelerating the rise in [Ca2+]i, Tg lengthens the period during which [Ca2+]i is at levels that support stronger mitochondrial Ca2+ uptake, leading to increased loading and a more prominent plateau during the recovery.
These results suggest that the caffeine-sensitive pool accumulates Ca2+ when [Ca2+]i is elevated during weak depolarization. This was demonstrated directly by measuring [Ca]ER before and after exposure to 30 mM K+ by EDX microanalysis (Fig. 3 B, inset). [Ca]ER was increased significantly from its resting value during a 2 min exposure to 30 mM K+; the increase corresponds to a rise from ∼3.6 to ∼4.8 mmol/liter wet tissue. No change in [Ca]ER was detected at 45 s, possibly because average [Ca]ER measurements before and after stimulation were necessarily performed in different cell populations, whereas ratios of 20–80% rise times were determined in individual cells and then averaged. As a result, [Ca]ER comparisons are more sensitive to cell-to-cell variability. A possibility that is consistent with our data is that [Ca]ER rises continuously during weak depolarization, being large enough at 120 s to be distinguished from basal [Ca]ER measurements in a different cell population, but not at 45 s. Simulations supporting this possibility are presented below (see Fig. 5 A, third panel from top).
Figure 5.

Simulated changes in ci and cER induced by Ca2 + entry. A–C illustrate simulated effects of Ca2+ entry (top row) on the concentrations of cytosolic Ca2+ (ci, second row) and intraluminal Ca2+ (cER, third row); bottom row shows how the total Ca2+ permeability of the internal pool depends on ci. Components of the net Ca2+ flux across the plasma membrane (JICa, Jextru) and of the net ER Ca2+ flux (JSERCA, JRelease) are defined in the and illustrated in Fig. 7. (A) Simulated responses under four conditions that can be compared with results described in this study. Under control conditions (Cont), Ca2+ entry increases ci but at a rate that is attenuated by Ca2+ accumulation, which causes cER to rise from a high basal level. After inhibiting the uptake pathway (+Tg), cER remains low and ci increases more rapidly during stimulation than in the control. After inhibiting CICR and increasing Pbasal to model the effects of ryanodine (+Ryan), ci rises more slowly and cER rises more rapidly from a lower resting level. Finally, after increasing the maximal rate of CICR and its ci sensitivity (+Caff), the rise in ci triggers net Ca2+ release so that cER declines and the ci rise is accelerated, leading to a transient ci overshoot. For the control response, kleak= 1.5 × 10−7 s−1 co = 2 mM, Vmax,extru = 25 nM/s, EC50,extru = 386 nM, nextru = 2.4, Vmax,SERCA = 70 nM/s, EC50,SERCA = 700 nM, nSERCA = 1, Pbasal = 1.78 × 10−5 s−1, Pmax,RyR = 9 × 10−4 s−1, EC50,RyR = 1 μM, nRyR = 1 and γER = 0.01. To simulate responses after treatment with Tg, Vmax,SERCA was set to zero while all other parameters had their control values. To simulate responses in the presence of ryanodine, Pmax,RyR was set to zero and Pbasal was increased to 3 × 10−4 s−1. To simulate the effects of caffeine, Pmax,RyR was increased to 9 × 10−3 s−1, EC50,RyR was reduced to 250 nM, and nRyR was increased to 3. Ca2+ entry was represented by fitting a triple exponential function to a representative JICa measurement obtained during a 40 s depolarization and extrapolating in time; tail currents were not included. (B) Simulated responses elicited by stimuli of increasing strength (curves 1–5) using control parameters from A. (C) Simulated responses to a fixed stimulus illustrating the effect of increasing the ci-sensitive permeability, Pmax,RyR (curves 1–12) with EC50,RyR = 500 nM and nRyR = 3. The effect of these changes on total permeability (PER) is shown at bottom.
To summarize, our results indicate that Ca2+ accumulation by the ER reduces the total cytoplasmic Ca2+ flux during periods of Ca2+ entry and slows depolarization-evoked [Ca2+]i elevations in a Tg-sensitive manner (Fig. 3 A, top, diagrams). This leads to our third important conclusion: if a ryanodine-sensitive CICR pathway is activated by the small [Ca2+]i elevations elicited during weak depolarization, the rate at which Ca2+ is released by this pathway must be slower than the rate of Ca2+ uptake.
Ryanodine Slows Depolarization-evoked [Ca2+]i Elevations by Enhancing ER Ca Accumulation
The results presented so far indicate that when [Ca2+]i rises to levels below ∼350 nM during weak depolarization, the caffeine-sensitive store accumulates Ca. Nevertheless, [Ca2+]i responses elicited by such stimuli are sensitive to ryanodine in a way that implicates the activation of a CICR pathway. After treatment with ryanodine, caffeine-induced Ca2+ release is inhibited and stimulus-induced [Ca2+]i elevations are slowed (Fig. 4 A; also see Friel and Tsien 1992a), showing a 264 ± 21% increase in the 20–80% rise time compared with control responses in the same cells (21 cells). Slower [Ca2+]i elevations would account for the observation that responses induced by brief stimuli are smaller after treatment with ryanodine (Hua et al. 1993; Shmigol et al. 1995; Peng 1996; Sandler and Barbara 1999). At the concentrations used, ryanodine does not inhibit voltage-sensitive Ca2+ currents or change the initial rate at which [Ca2+]i rises after depolarization (Friel and Tsien 1992a; Fig. 4 A, bottom, inset). Moreover, ryanodine has no detectable effect after treatment with Tg (5/5 cells), indicating that it specifically influences Ca2+ transport by a Tg-sensitive pool. One possible explanation for the slower [Ca2+]i elevations observed after treatment with ryanodine is that depolarization normally triggers net Ca2+ release, and that by increasing the Ca2+ permeability of the ER, ryanodine depletes the store, thereby preventing net Ca2+ release. However, this is incompatible with the observation described above that the store accumulates Ca2+ during weak depolarization. Another possibility is that ryanodine prevents Ca2+-dependent activation of a CICR pathway that normally accelerates depolarization-induced [Ca2+]i elevations, but the same observations preclude net CICR. How can these findings be explained?
Interpretation of the kinetic effects of ryanodine on depolarization-evoked [Ca2+]i elevations requires information about how this compound modifies Ca2+ handling by the ER in these experiments. It has been shown previously that at high concentrations (≥10 μM), ryanodine enhances Ca2+ accumulation by cardiac sarcoplasmic reticulum in a way that is consistent with a reduction in sarcoplasmic reticulum Ca2+ permeability (Jones et al. 1979; Sutko et al. 1997). If ryanodine slows depolarization-induced [Ca2+]i elevations in sympathetic neurons by a similar mechanism, then Tg should overcome this effect, causing responses to be faster than the controls (Fig. 3). Alternatively, if ryanodine renders the ER so leaky that Ca2+ becomes passively distributed between the ER and cytoplasm, active Ca2+ accumulation could not occur and Tg would have no additional effect. Fig. 4 A (bottom) compares the [Ca2+]i responses from A (top) with a subsequent response elicited from the same cell after treatment with Tg in the continued presence of ryanodine. After Tg, the rise in [Ca2+]i was greatly accelerated, with the 20–80% rise time falling to 36 ± 4% of that observed after treatment with ryanodine in the same cells (n = 21). Acceleration of the [Ca2+]i rise does not reflect drug-induced changes in ICa (Fig. 4 B); in the example shown, [Ca2+]i rises more rapidly after Tg treatment despite partial rundown of ICa during the period between depolarizations. To test directly if ryanodine increases the rate of ER Ca accumulation during stimulation, a comparison was made between [Ca]ER after a 45-s exposure to 30 mM K+ in control and ryanodine-treated cells. Although depolarization of this duration did not change [Ca]ER detectably in control cells (Fig. 4 C, compare first and second bars), it increased [Ca]ER twofold in ryanodine-treated cells (Fig. 4 C, compare third and fourth bars), demonstrating that ryanodine increases the average rate of ER Ca accumulation during stimulation.
Is enhanced Ca accumulation a consequence of reduced ER Ca2+ permeability? Such an effect by itself would cause basal [Ca]ER to increase, but a ∼63% reduction was observed (from 12.8 ± 0.9 to 4.7 ± 1.1 mmol/kg dry weight, P < 0.001; see Fig. 4 C, compare first and third bars). To determine if ryanodine lowers the resting [Ca]ER level by reducing the basal rate of ER Ca2+ uptake, the initial rate at which [Ca2+]i rises after rapid application of the reversible SERCA inhibitor t-BuBHQ was measured before and after treatment with ryanodine (Fig. 4D and Fig. E). Before ryanodine, t-BuBHQ elicits a transient [Ca2+]i elevation that reflects Ca2+ release from an intracellular store; such transients are not observed after treatment with Tg, arguing that Tg and t-BuBHQ deplete the same store (not shown). The abrupt rise in [Ca2+]i that follows t-BuBHQ application indicates that, under resting conditions, ongoing Ca2+ uptake via SERCAs is balanced by passive Ca2+ release (Fig. 4 E, see diagrams). The initial rate of rise after SERCA inhibition provides a measure of the basal rate of release, as well as the rate of uptake that balances release under resting conditions. After treatment with ryanodine, t-BuBHQ also elicits a [Ca2+]i transient (Fig. 4 D), indicating that there is still a gradient favoring passive Ca2+ release, but the transient is smaller and shorter in duration than the control, as expected given the lower basal [Ca]ER. Nonetheless, the initial rate of rise is unchanged (Fig. 4D and Fig. E; 5/5 cells), indicating that ryanodine does not alter the resting rate of Ca2+ uptake or release. Since ryanodine lowers resting [Ca]ER (and presumably the free Ca concentration within the ER, [Ca2+]ER) without altering the basal release rate, it must increase the resting ER Ca2+ permeability, defined as (release rate)/([Ca2+]i − [Ca2+]ER). This is the opposite of the high concentration effect of ryanodine described previously (Jones et al. 1979) but is precisely the result expected if, in addition to preventing Ca2+-dependent RyR channel activation, ryanodine increases basal channel open probability.
These observations lead to the fourth important conclusion of this study. Since the ER normally accumulates Ca2+ when [Ca2+]i is elevated during weak depolarization, and inhibition of a ryanodine-sensitive CICR pathway increases the rate of ER Ca accumulation, activation of this pathway must normally attenuate Ca2+ accumulation by the ER. This is an interesting mechanism, since it would involve Ca2+-induced Ca2+ release at the level of a population of RyRs, providing a [Ca2+]i-sensitive pathway for passive Ca2+ release from the ER that accelerates evoked [Ca2+]i elevations, but in a capacity that downregulates ER Ca2+ accumulation.
Simulations Based on a Model of CICR Operating in a Low Gain Mode
To determine if activation of a CICR pathway could, in principle, attenuate ER Ca accumulation and accelerate depolarization-evoked [Ca2+]i responses, simulations were performed based on a model of Ca2+ regulation (Friel 1995) that includes two compartments representing the cytoplasm and ER containing Ca2+ at concentrations ci and cER, respectively. This model assumes that Ca2+ is distributed uniformly within each compartment, an approximation that becomes increasingly valid as the rate of stimulated Ca2+ entry becomes slow enough that the rate of Ca2+ transport between compartments is slow compared with diffusion within compartments. Also, mitochondria are not explicitly included to facilitate analysis of the impact of a CICR pathway on net Ca2+ transport by an intracellular pool. Although this approximation leads to results that agree with the experiment only when [Ca2+]i is low and mitochondrial Ca2+ transport is weak, the conclusions reached below regarding the [Ca2+] dependence of net ER Ca2+ transport are expected to apply generally (see discussion).
In the model, Ca2+ extrusion across the plasma membrane is represented by an experimentally determined rate equation (Colegrove et al. 2000b; see ), Ca2+ uptake by the store is controlled by a saturable pump, and the rate of passive Ca2+ release is the product of the total Ca2+ permeability of the store (PER) and a driving force (ci − cER). The permeability is the sum of a constant basal component (Pbasal) and a [Ca2+]i-sensitive component (with maximal value Pmax,RyR) that is intended to represent the macroscopic permeability conferred upon the store by a population of channels (e.g., RyR's) whose open probabilities increase with [Ca2+]i but do not have explicit time dependence. The parameter values defining the uptake and release pathways are estimates based on our observations in sympathetic neurons (unpublished data) but the general conclusions that follow hold over a range of parameter values.
Fig. 5 A illustrates responses to Ca2+ entry simulated under four different conditions for comparison with experiments described above. Under control conditions (Cont), Ca2+ entry (Fig. 5 A, first panel) leads to a rise in ci (second panel) that causes Ca2+ accumulation by the store, increasing cER from a high basal level (third panel). After suppressing Ca2+ uptake to represent the effects of Tg, Ca2+ accumulation is abolished and ci rises more rapidly, as observed experimentally. After blocking Ca2+-induced increases in permeability and raising the basal permeability of the store to model the effects of ryanodine, Ca2+ entry leads to a slower ci rise but a more robust increase in cER from a lower basal level, which is also in agreement with experiments described above. Finally, after increasing the strength and ci sensitivity of the release pathway to model the effects of caffeine (Fig. 5 A, bottom panel), ci rises more rapidly in response to the same stimulus, and cER declines, consistent with the observed effects of caffeine on depolarization-induced [Ca2+]i responses. Thus, the simple model accounts for the main observations in this study and illustrates how weak activation of a CICR pathway, operating in parallel with a Ca2+ uptake system, could accelerate depolarization-evoked [Ca2+]i elevations by reducing the rate at which the store accumulates Ca2+.
Fig. 5 B shows how the dynamics of ci and cER change as the stimulus strength is increased when the Ca2+ uptake and release pathways are described as in the control case in column A. Whereas weak stimuli cause Ca2+ accumulation (third panel, curve 1), increasing the stimulus strength leads to progressively weaker accumulation (Fig. 5 B second and third curves), until the balance between uptake and release tips in favor of net Ca2+ release (Fig. 5 B, curves 4–5). The accompanying study demonstrates such a transition in sympathetic neurons. Fig. 5 C illustrates a similar transition that results from increasing the maximal ci-sensitive permeability of the store (Pmax,RyR) in the case where the stimulus is fixed and the ci dependence of the permeability is steep. When Pmax,RyR is small (Fig. 5 C, bottom panel), the store is a Ca2+-regulated buffer (Fig. 5 C, second and third panels), but if this parameter is increased sufficiently, the same stimulus triggers net Ca2+ release (e.g., see curve 12). In the discussion, we will show how such quantitative properties of the CICR pathway are expected to contribute to qualitative properties of cellular Ca2+ regulation.
Returning to the main goal of the present study, we propose that small [Ca2+]i elevations elicited by weak depolarization increase the rate of passive Ca2+ release via a Ca2+- and ryanodine-sensitive CICR pathway, but because release is slower than uptake, the overall effect is to reduce the rate of ER Ca2+ accumulation. In terms of the ideas presented earlier, such a low gain mode of CICR would reduce the outward flux JΣ during depolarization and shift the total cytosolic Ca2+ flux during stimulation (JICa + JΣ) toward more negative values, leading to a faster rise in [Ca2+]i than would be expected without CICR. In this mode, activation of CICR accelerates the rise in [Ca2+]i elicited by Ca2+ entry, but does so by reducing the strength of ER Ca2+ buffering.
DISCUSSION
Our results show that small [Ca2+]i elevations evoked by weak depolarization lead to Ca2+ accumulation by the ER, and that Ca2+ accumulation becomes stronger after inhibiting CICR with ryanodine. The companion article (see Hongpaisan et al. 2001, in this issue) shows that as stimulus-evoked [Ca2+]i elevations become larger, ER Ca2+ accumulation becomes progressively weaker, and that at high [Ca2+]i, the ER becomes a Ca2+ source. Our results suggest a simple explanation: progressive activation of a [Ca2+]i-sensitive CICR pathway that operates in parallel with SERCA pumps to regulate net ER Ca2+ transport.
Comparison with Previous Studies
Studies in many neuronal cell types have identified a Ca2+ store that expresses functional RyRs and can be discharged by caffeine (Kuba 1994; Usachev and Thayer 1999). This and the companion article (see Hongpaisan et al. 2001, in this issue) provide direct confirmation that this Ca2+ store is the ER, contributing to the already large body of evidence that this organelle is important in cellular Ca2+ regulation (Meldolesi and Pozzan 1998). Although contributions from caffeine-modified RyRs to depolarization-evoked [Ca2+]i signals have been clear, it has not been obvious how RyRs participate in calcium signaling in the absence of CICR modifiers. In sympathetic neurons, the observation that ryanodine slows depolarization-evoked [Ca2+]i elevations raised the possibility that activation of a CICR pathway amplifies the effect of Ca2+ entry on [Ca2+]i (Friel and Tsien 1992a). This was supported by work of Hua et al. 1993 showing that depolarization-induced [Ca2+]i elevations increase supralinearly with Ca2+ load and exhibit a form of paired-pulse facilitation. Similar observations have been made in other cells (Shmigol et al. 1995; Llano et al. 1994). Although these results are consistent with Ca2+-induced net Ca2+ release from an intracellular Ca2+ store, they are also consistent with [Ca2+]i-dependent attenuation of intracellular Ca2+ buffering or sequestration. One approach to distinguishing between these possibilities is based on the use of CICR inhibitors like ryanodine. However, our results show that specific inhibition of CICR is expected to slow depolarization-evoked [Ca2+]i responses irrespective of the direction of net ER Ca2+ transport. Therefore, additional information is required. Several studies have provided examples where Ca2+ entry triggers net CICR (Cohen et al. 1997; Alonso et al. 1999; Emptage et al. 1999; Sandler and Barbara 1999). However, to our knowledge, the present study is the first to show that activation of a Ca2+- and ryanodine-sensitive Ca2+ release process during periods of Ca2+ entry accelerates [Ca2+]i responses by attenuating ER Ca accumulation.
Impact of CICR on Net ER Ca2+ Transport
At each instant in time during stimulation, the rate of net ER Ca2+ transport should depend on the relative rates of Ca2+ uptake via SERCA pumps and passive Ca2+ release via RyRs, d-myo-inositol 1,4,5-trisphosphate (InsP3) receptors (Pfaffinger et al. 1988), and possibly other uncharacterized Ca2+ transport pathways. The finding that weak depolarization leads to ER Ca accumulation indicates that small [Ca2+]i elevations stimulate Ca2+ uptake more strongly than release. Stimulated Ca2+ uptake is expected since SERCA activity increases with [Ca2+]i (Lytton et al. 1992), but how would the rate of Ca2+ release be expected to change in response to a rise in [Ca2+]i? This rate should depend on the driving force for passive Ca2+ movement between the ER and cytoplasm (∼[Ca2+]i − [Ca2+]ER) and the Ca2+ permeability of the ER (PER). If PER were constant, a rapid rise in [Ca2+]i (rapid enough so that [Ca2+]ER does not change very much) would reduce the driving force and lower the rate of release. If PER increased weakly with [Ca2+]i (e.g., as a result of [Ca2+]i-dependent RyR activation), the effect of reduced driving force on release rate would be partially overcome; if PER increased more steeply with [Ca2+]i, the rate of release could rise. Our results are consistent with a Ca2+-induced increase in the rate of passive Ca2+ release that is smaller than the stimulated increase in uptake rate. In this case, Ca2+ accumulation would occur as [Ca2+]i rises but at a reduced rate because of RyR activation that, in effect, short circuits the uptake process and permits [Ca2+]i to rise more rapidly during periods of Ca2+ entry. Inhibition of the permeability increase (e.g., with ryanodine) would prevent the increase in release rate, augment the imbalance between uptake and release, and strengthen Ca accumulation, causing [Ca2+]i to rise more slowly during stimulation.
Effects of Ryanodine on Net ER Ca2+ Transport
Slowing of depolarization-evoked [Ca2+]i responses by ryanodine has been interpreted to mean that Ca2+ entry normally triggers net Ca2+ release. This conclusion is based on the assumption, previously untested, that ryanodine renders the ER so leaky that active Ca2+ accumulation becomes impossible. However, if ryanodine only modestly increases ER Ca2+ permeability, its effects on evoked [Ca2+]i responses are consistent with either triggered net Ca2+ release or attenuated Ca2+ accumulation. We found that ryanodine (1 μM): abolishes caffeine responsiveness; reduces basal [Ca]ER (and presumably [Ca2+]ER, although this was not measured directly); and enhances depolarization-induced ER Ca accumulation. Each effect follows from known actions of ryanodine on RyRs (see materials and methods). Inhibition of caffeine responsiveness is expected based on reduced RyR sensitivity to Ca2+, and a reduction in basal [Ca]ER follows from increased channel open probability. The finding that ryanodine does not alter the basal rate of Ca2+ release argues that once RyRs are modified, [Ca2+]ER falls until a new steady-state level is reached where Ca2+ release once again balances Ca2+ uptake. In principle, either action of ryanodine could contribute to inhibition of caffeine-induced Ca2+ release. However, since net Ca2+ release can be stimulated by SERCA inhibition after ryanodine treatment, loss of caffeine responsiveness cannot be accounted for entirely by depletion of the ER, and is more likely a result of reduced RyR sensitivity to Ca2+. We propose that Ca2+ accumulation by the ER is enhanced in ryanodine-treated cells because as [Ca2+]i rises during stimulation, the rate of passive Ca2+ release falls, in contrast to the rise that occurs in untreated cells. This would increase the imbalance between ER Ca2+ uptake and release rates, favoring stronger Ca accumulation.
Clearly, other factors could contribute to our observations. For example, after ryanodine treatment, the reduction in basal [Ca]ER could cause the ER to become a less saturated, and therefore more powerful, Ca2+ buffer. Although our data do not address this point directly, it should be mentioned that this explanation cannot account for net Ca2+ release at high [Ca2+]i. In contrast, graded activation of a CICR pathway provides a parsimonious explanation for attenuated Ca accumulation at low [Ca2+]i, net Ca2+ release at high [Ca2+]i (Hongpaisan et al. 2001), and stronger ER Ca2+ buffering after ryanodine treatment, without making any assumptions about the properties of intraluminal Ca2+ buffers. Furthermore, simulations in Fig. 5 show that this idea is reasonable and can account for our general observations.
The findings described in this study are consistent with a single ER Ca2+ pool expressing multiple transport pathways (SERCAs, RyRs, InsP3s, etc.), but it must be asked if they can also be explained by structurally distinct ryanodine-sensitive and -insensitive pools that are both sensitive to Tg and, respectively, release and take up net Ca2+ during depolarization. Although the existence of such pools cannot be excluded, our measurements do not detect them (see companion article, Hongpaisan et al. 2001, in this issue). Moreover, the observation that ryanodine does not change the basal rate of Ca2+ uptake (Fig. 4D and Fig. E) severely limits such multipool models. If the ER did consist of two (or more) distinct pools, the basal rate of Ca2+ uptake would be the sum of the rates of uptake by the individual pools. If ryanodine specifically eliminated contributions from the ryanodine-sensitive pool by rendering it so leaky that Ca2+ becomes passively distributed between the ER and cytoplasm, then it would also reduce the initial rate at which [Ca2+]i rises after exposure to t-BuBHQ, but no such change was observed. Insensitivity of the initial rate to ryanodine would require that the resting rate of Ca2+ uptake by the ryanodine-sensitive pool be undetectably low, which is difficult to reconcile with the observation that caffeine-induced [Ca2+]i transients are larger if they are preceded by brief depolarizations that raise [Ca2+]i to levels comparable to those shown in this study to stimulate ER Ca2+ accumulation (unpublished data). The single-pool model, on the other hand, provides a simple explanation of the results. A similar conclusion was reached by Khodakhah and Armstrong 1997 from their studies of cerebellar Purkinje neurons.
Qualitative Properties of CICR: Dual Regulation of Cytoplasmic and ER Calcium Levels
Our results indicate that CICR modulates ER Ca2+ accumulation during weak stimulation, but they do not preclude net Ca2+ release via CICR at higher [Ca2+]i. Indeed, as illustrated in Fig. 5 B, theory suggests that under certain conditions, larger stimulus-evoked [Ca2+]i elevations can lead to a transition from regulated Ca2+ buffering to triggered net Ca2+ release, and the model provides a framework for evaluating the conditions under which such a transition can occur. Since mitochondria are not included in the model, simulations can only be compared directly to results obtained under conditions where mitochondrial Ca2+ transport is weak. However, Ca2+ transport by mitochondria is not expected to influence the [Ca2+]i dependence of net ER Ca2+ transport, per se, but rather the range over which [Ca2+]i varies during stimulation (see companion article, Hongpaisan et al. 2001, in this issue). Therefore, analysis of the case without mitochondria should provide insight into the properties of ER Ca2+ transport even when mitochondrial Ca2+ transport is appreciable.
According to the model, ci and cER dynamics depend on the total cytosolic Ca2+ flux (Ji) and the net ER Ca2+ flux (JER), respectively, which in turn display the combined [Ca2+] dependencies of the underlying transporters. Fig. 6 (A and B) shows how these fluxes depend on ci and cER when JICa = 0 in a particular case where the Ca2+ permeability of the store increases steeply with ci (Hill coefficient 3) and becomes large when ci is high because the ci-sensitive permeability (Pmax,RyR) is large (Fig. 6 E, left). Slices from the surfaces in Fig. 6 (A and B, dashed lines) show how Ji and JER change with ci at constant cER (Fig. 6C and Fig. D, left), in particular, giving the instantaneous rates at which ci and cER would change if ci were raised rapidly by a very brief stimulus without perturbing cER. These curves are analogous to the momentary current-voltage relations described by Jack et al. 1983. As with membrane potential dynamics, ci and cER dynamics depend on the stimulus protocol: two stimuli that raise ci to the same steady-state level but at different rates could have very different effects on ci and cER dynamics owing to differences in cER that arise during stimulation. Nevertheless, at each instant in time, the net fluxes Ji and JER would be defined by the magnitudes of ci, cER and JICa at that time.
Figure 6.
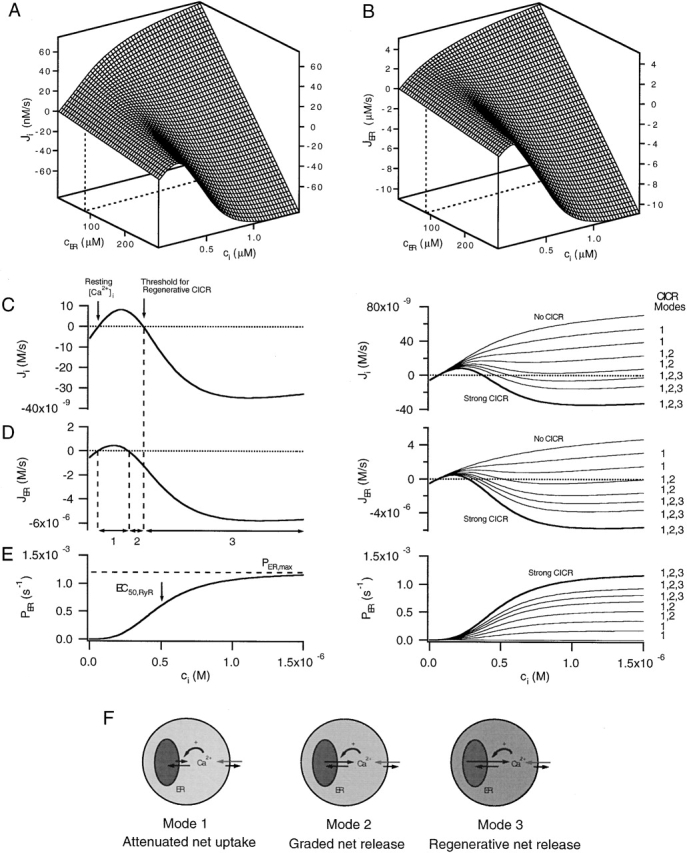
Three modes of net CICR. (A and B) Surfaces showing how the net cytoplasmic and ER Ca2+ fluxes, Ji and JER, depend on ci and cER in the case JICa= 0 according to the model described in the . Slices from these surfaces at constant cER (dashed lines) illustrate the nonlinear ci dependencies of the underlying Ca2+ fluxes, whereas curves at constant ci illustrate the linear cER dependence of the fluxes. Parameters describing the ci-sensitive permeability are as follows: Pmax,RyR = 1.2 × 10−3 s−1, EC50,RyR = 500 nM, nRyR = 3. Basal cER = 90.8 μM. (C and D, left) Slices from the surfaces in A and B at constant cER. These flux/ci relations illustrate the basis for three qualitatively distinct modes of net CICR: Mode 1 (Ji > 0, JER > 0), Mode 2 (Ji > 0, JER < 0), and Mode 3 (Ji < 0, JER < 0). The ranges of ci in which these modes occur are indicated in D (bottom). (E, left) ci dependence of the Ca2+ permeability, PER. (C and D, right) Slices from different Ji and JER surfaces with different values of Pmax,RyR show that as this parameter is increased from zero (no CICR) to a large value (strong CICR) additional modes of net CICR become available. Bold traces represent the same curves shown at left but on a different ordinate scale. In each case, cER was fixed at its resting value, which depended on Pmax,RyR. Values of Pmax,RyR were (10−4 s−1) as follows: 0, 1.8, 3.6, 5.4, 7.2, 8.4, 9.6, and 12. (F) Relationship between the rates of Ca2+ uptake, release, and extrusion in each CICR mode. Relative free Ca concentrations are represented by shading (darker = higher [Ca2+]). In Mode 1 (low ci), uptake is faster than release, whereas in Mode 2 (intermediate ci), release is faster than uptake, causing net release but at a slower rate than extrusion. In Mode 3 CICR (high ci), net release is faster than extrusion, leading to positive feedback and a regenerative rise in ci. Surface in A describes the total Ca2+ flux representing uptake, release, and extrusion (dark arrow on the plasma membrane in F) in the absence of stimulated Ca2+ entry. During stimulated Ca2+ entry (light arrows on the plasma membrane), the surface in A would be displaced downward by an amount JICa, which depends on time.
In the case illustrated, Ji first increases with ci and crosses the zero net flux axis at a (stable) steady-state value (Fig. 6 C, left arrow) that is well below the Ca2+ concentration (EC50,RyR) where the ci-sensitive permeability is half maximally activated (Fig. 6 E, left). However, when ci is higher, the permeability of the store is larger, causing Ji to turn downward, cross the zero flux axis with negative slope at an (unstable) steady-state (panel C, right arrow), and then become negative. There are three ranges of ci in which Ji and JER show distinct ci-dependencies, revealing three modes of CICR (Fig. 6 D, arrows 1–3; Friel 1998). When ci is above the resting level, but low compared with EC50,RyR, Ji and JER are both positive (Mode 1 CICR). JER is positive because passive Ca2+ release is slower than Ca2+ uptake, causing the store to act as a buffer. In this case, activation of the ci-sensitive permeability during stimulation lowers the rate of Ca2+ accumulation by the store and accelerates evoked increases in ci. Over the intermediate ci range, Ji is positive and JER is negative (Mode 2 CICR). JER becomes negative when passive Ca2+ release is faster than uptake, leading to net release, which further increases the rate at which ci rises in response to stimulated Ca2+ entry, and causes cER to decline. Finally, at higher ci, both Ji and JER become inward fluxes (Mode 3 CICR). A brief stimulus that brings ci within this range would stimulate Ca2+ release at such a high rate that it overwhelms Ca2+ extrusion, leading to a regenerative rise in ci and fall in cER. The zero crossing in this case represents the threshold for regenerative net CICR (Fig. 6C, see right arrow). When ci is very high compared with EC50,RyR, Ji once again increases monotonically with ci, representing the case where the total Ca2+ permeability is high and essentially constant (not shown).
With fixed rate characteristics for Ca2+ extrusion and uptake, the quantitative properties of the ci-dependent permeability determine which modes of net CICR can be expressed. Fig. 6 (C and D, right) shows families of slices from Ji and JER surfaces like those in Fig. 6 (A and B) at constant cER but with different values of Pmax,RyR (Fig. 6 E, right). Pmax,RyR would depend on the number of Ca2+ release channels, their maximal open probability at high ci, and unitary Ca2+ permeability. When Pmax,RyR is small, only Mode 1 CICR is available, irrespective of the stimulus strength. This would describe a cell in which Ca2+ release channels are present at low density compared with SERCA pumps. With intermediate values of Pmax,RyR, both Modes 1 and 2 are available, but not Mode 3, which could represent a cell in which RyR density relative to SERCA pumps is somewhat higher, but plasma membrane Ca2+ extrusion systems are powerful. Finally, when Pmax,RyR is large, all three modes are available, which includes the possibility of regenerative Ca2+ release if ci is raised beyond the appropriate threshold. For example, this could represent a case in which RyRs are expressed at high density compared with SERCAs or are modified pharmacologically so that their maximal open probability is high. Cells having different levels of channel expression would be described by different values of Pmax,RyR, as would a given cell type before and after a pharmacological modification (e.g., by caffeine). Naturally occurring variations in the density or unitary properties of RyRs or SERCAs could contribute to variability of response properties observed in different populations of sympathetic neurons (Cseresnyes et al. 1999) or different types of cells. These general conclusions are not limited to RyR-mediated CICR and apply equally well to InsP3 receptor mediated CICR.
Which modes of CICR are expressed by sympathetic neurons? Results from the present study demonstrate Mode 1 CICR, and results in the companion article (see Hongpaisan et al. 2001, in this issue) show that when [Ca2+]i is raised to higher levels by stronger depolarizing stimuli, Mode 2 or 3 CICR can occur. Regenerative depolarization-induced [Ca2+]i elevations have been reported in sympathetic neurons in the absence of caffeine (Lipscombe et al. 1988; Hua et al. 1993), further pointing to the possibility of Mode 3 CICR. Regenerative [Ca2+]i increases can be elicited in the presence of caffeine in these and other cells (Friel and Tsien 1992b; Usachev and Thayer 1997), clearly demonstrating Mode 3 CICR under these conditions.
Implications for Calcium Signaling
Activation of a CICR pathway is expected to have direct effects on stimulus-induced changes in [Ca2+] within the cytoplasm and the ER, as well as indirect effects on other organelles that exchange Ca2+ with the cytoplasmic pool. Modulation of depolarization-evoked changes in [Ca2+]i by CICR may well contribute to the control of membrane excitability and neurotransmitter release (Narita et al. 1998, Narita et al. 2000; Llano et al. 2000) in a way that is sensitive to stimulus history. Our results indicate that weak and strong depolarizing stimuli, while evoking graded increases in [Ca2+]i, could have opposite effects on [Ca2+]ER, leading to qualitatively different patterns of activity among Ca2+-sensitive effectors within the cytosol and the ER (Corbett and Michalak 2000). Weak activation of a CICR pathway would provide a mechanism for increasing the impact of Ca2+ entry on [Ca2+]i without reducing [Ca]ER. In this regard, it is interesting to note that reducing [Ca]ER has been shown in some cells to be proapoptotic (Wei et al. 1998). Finally, it is expected that the expression levels of Ca2+ transporters, as well as their state of modulation, define which modes of CICR can occur. This may play a role in defining differences between cells from different tissues, species and developmental stages, as well as adaptive changes that occur after perturbations of Ca2+ delivery in disease (Dove et al. 2000).
A Note on Terminology
Our results underscore a basic ambiguity in the phrase “Ca2+-induced Ca2+ release” that arises because there is a distinction between Ca2+ transport via a CICR pathway, and net Ca2+ transport by the ER. CICR is usually used to refer to net Ca2+ release, but the results presented here indicate that activation of a Ca2+-sensitive release pathway can regulate ER Ca2+ accumulation. In both cases, activation of the CICR pathway accelerates depolarization-evoked [Ca2+]i elevations, but, in one case, [Ca]ER rises whereas in the other it falls. In principle, CICR could refer to either situation, describing passive release via a Ca2+-sensitive permeability without reference to the direction of net organellar Ca2+ movement, or Ca2+-induced net Ca2+ release. Since the second definition necessarily involves the relationship between a CICR pathway and other transport systems, we prefer the first definition, and use the phrase “net CICR” to refer to the second. According to this usage, CICR refers to a passive macroscopic Ca2+ flux whose impact on intraluminal Ca2+ levels is context-dependent.
Acknowledgments
The authors thank Drs. S.W. Jones, D. Kunze and J. Ma for their helpful comments on the manuscript.
This work was supported by a grant (No. NS-33514) from the NIH and by the NIH Intramural Research Program.
Description of the Model
The dynamics of the free Ca2+ concentration within the cytosol (ci) and the ER (cER) were represented by the following differential equations ( and ; see 7).
Figure 7.

Schematic of the model. (left) Model includes two compartments representing the cytoplasm and ER with Ca2+ concentrations ci and cER. The extracellular Ca2+ concentration is assumed to be constant. ci and cER change under the influence of the intercompartmental net Ca2+ fluxes Jpm and JER. (right) Components of the intercompartmental Ca2+ fluxes. Jpm is the sum of the rates of Ca2+ entry through voltage-gated Ca2+ channels (JICa) and Ca2+ extrusion (Jextru). JER is the sum of the rates of Ca2+ uptake (JSERCA) and passive Ca2+ release (JRelease). See for definitions of the fluxes and their ci- and cER dependencies and figure legends for parameter values.
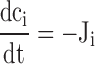 |
1 |
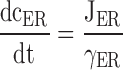 |
2 |
where
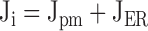 |
3 |
and the intercompartmental fluxes Jpm and JER depend on the relative rates of transport via different pathways as follows:
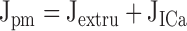 |
4 |
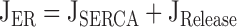 |
5 |
where
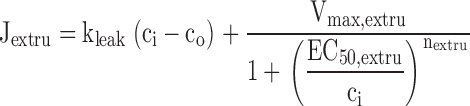 |
6 |
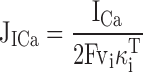 |
7 |
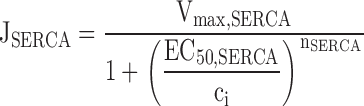 |
8 |
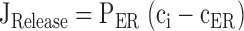 |
9 |
and
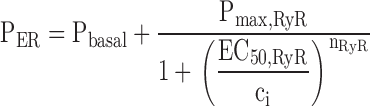 |
10 |
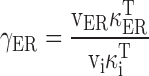 |
11 |
kleak, co, Vmax,extru, EC50,extru, nextru, F, Vmax,SERCA, EC50,SERCA, nSERCA, Pbasal, Pmax,RyR, EC50,RyR, nRyR, and γER are constants. co is the extracellular Ca2+ concentration and F is the Faraday constant.
describe the rate of total Ca2+ transport by the respective pathways divided by the cytoplasmic volume (vi) and a buffering factor that is the ratio of changes in total cytoplasmic Ca concentration that accompany small changes in free Ca concentration (κT i, Colegrove et al. 2000a,Colegrove et al. 2000b). Fluxes that raise ci are negative and fluxes that lower ci are positive. Jextru is the sum of plasma membrane pump and leak fluxes. PER describes the total Ca2+ permeability of the ER store and consists of a constant basal component (Pbasal) and a ci-dependent component that increases saturably with ci (half-maximal activation when ci = EC50,RyR) and approaches Pmax,RyR when is high ci (5, A–C, bottom). In the model, the ci dependence of PER is responsible for CICR. Estimation of parameters for is described in Colegrove et al. 2000b, measurement of κT iis described in materials and methods, and parameters for are estimates based on our unpublished data.
Footnotes
Abbreviations used in this paper: t-BuBHQ, 2,5-di-(t-butyl)-1,4-hydroquinone; EDX, energy-dispersive X-ray; InsP3, d-myo-inositol 1,4,5-trisphosphate; Ryr, ryanodine receptors; SERCA, sarco- and endoplasmic reticulum Ca ATPase; Tg, thapsigargin.
References
- Albrecht M.A., Friel D.D. Ryanodine-induced enhancement of Ca2+ sequestration by intracellular stores in sympathetic neurons Biophys. J. 72 1997. 298(Abstr.). [Google Scholar]
- Alonso M.T., Barrero M.J., Michelena P., Carnicero E., Cuchillo I., Garcia A.G., Garcia-Sancho J., Montero M., Alvarez J. Ca2+-induced Ca2+ release in chromaffin cells seen from inside the ER with targeted aequorin. J. Cell Biol. 1999;144:214–254. doi: 10.1083/jcb.144.2.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babcock D.F., Hille B. Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol. 1998;8:398–404. doi: 10.1016/s0959-4388(98)80067-6. [DOI] [PubMed] [Google Scholar]
- Berridge M.J. Neuronal calcium signaling. Neuron. 1998;21:13–26. doi: 10.1016/s0896-6273(00)80510-3. [DOI] [PubMed] [Google Scholar]
- Berridge M.J., Cheek T.R., Bennett D.L., Bootman M.D. Ryanodine Receptors and intracellular calcium signaling. In: Sorrentino V., editor. Ryanodine receptors. CRC Press; Boca Raton: 1995. pp. 119–153. [Google Scholar]
- Boyce W.E., DiPrima R.C. Elementary Differential Equations 1969. John Wiley and Sons, Inc; New York: pp. 353–357 [Google Scholar]
- Chen S.R.W., Li P., Zhang L. Sensitization of the Ca2+ release channel (ryanodine receptor) to Ca2+ activation by ryanodine Biophys. J. 80 2001. 384(Abstr.). [Google Scholar]
- Cohen A.S., Moore K.A., Bangalore R., Jafri M.S., Weinreich D., Kao J.P.Y. Ca2+-induced Ca2+ release mediates Ca2+ transients evoked by single action potentials in rabbit vagal afferent neurones. J. Physiol. 1997;499:315–328. doi: 10.1113/jphysiol.1997.sp021929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove S.L., Albrecht M.A., Friel D.D. Dissection of mitochondrial Ca2+ uptake and release fluxes after depolarization-evoked [Ca2+]i elevations in sympathetic neurons J. Gen. Physiol. 115 2000. 351 370a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colegrove S.L., Albrecht M.A., Friel D.D. Quantitative analysis of mitochondrial Ca2+ uptake and release pathways in sympathetic neuronsreconstruction of the recovery after depolarization-evoked [Ca2+]i elevations J. Gen. Physiol. 115 2000. 371 388b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corbett E.F., Michalak M. Calcium, a signaling molecule in the endoplasmic reticulum? Trends Biochem. Sci. 2000;25:307–311. doi: 10.1016/s0968-0004(00)01588-7. [DOI] [PubMed] [Google Scholar]
- Coronado R., Morrissette J., Sukhareva M., Vaughan D.M. Structure and function of ryanodine receptors. Am. J. Physiol. 1994;266:C1485–C1504. doi: 10.1152/ajpcell.1994.266.6.C1485. [DOI] [PubMed] [Google Scholar]
- Cseresnyes Z., Bustamante A.I., Schneider M.F. Caffeine-induced [Ca2+]i oscillations in neurones of frog sympathetic ganglia. J. Physiol. 1999;514:83–99. doi: 10.1111/j.1469-7793.1999.083af.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dove L.S., Nahm S.S., Murchison D., Abbott L.C., Griffith W.H. Altered calcium homeostasis in cerebellar Purkinje cells of leaner mutant mice. J. Neurophysiol. 2000;84:513–524. doi: 10.1152/jn.2000.84.1.513. [DOI] [PubMed] [Google Scholar]
- Emptage N., Bliss T.V., Fine A. Single synaptic events evoke NMDA receptor-mediated release of calcium from internal stores in hippocampal dendritic spines. Neuron. 1999;22:115–124. doi: 10.1016/s0896-6273(00)80683-2. [DOI] [PubMed] [Google Scholar]
- Friel D.D. [Ca2+]i oscillations in sympathetic neuronsan experimental test of a theoretical model. Biophys. J. 1995;68:1752–1766. doi: 10.1016/S0006-3495(95)80352-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel D.D., Tsien R.W. A caffeine- and ryanodine-sensitive Ca2+ store in bullfrog sympathetic neurones modulates effects of Ca2+ entry on [Ca2+]i J. Physiol. 450 1992. 217 246a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friel D.D., Tsien R.W. Phase-dependent contributions from Ca2+ entry and Ca2+ release to caffeine-induced [Ca2+]i oscillations in bullfrog sympathetic neurons Neuron. 8 1992. 1109 1125b [DOI] [PubMed] [Google Scholar]
- Friel D.D. Three modes of Ca2+ induced Ca2+ release in neurons. In: Holcombe M., Paton R., editors. Information Processing in Cells and Tissues. Plenum Press; New York: 1998. pp. 47–56. [Google Scholar]
- Hongpaisan J., Pivovarova N.B., Colgrove S.L., Friel D.D., Andrews S.B. Interactions between mitochondria and endoplasmic reticulum modulate Ca2+ signaling in sympathetic neurons Soc. Neurosci. 25 1999. 1249(Abstr.) [Google Scholar]
- Hongpaisan J., Pivovarova N.B., Colgrove S.L., Leapman R.D., Friel D.D., Andrews S.B. Multiple modes of calcium-induced calcium release in sympathetic neurons IIA [Ca2+]i- and location-dependent transition from ER Ca accumulation to net Ca release. J. Gen. Physiol. 2001;118:101–112. doi: 10.1085/jgp.118.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hua S.Y., Nohmi M., Kuba K. Characteristics of Ca2+ release induced by Ca2+ influx in cultured bullfrog sympathetic neurons. J. Physiol. 1993;464:245–272. doi: 10.1113/jphysiol.1993.sp019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jack, J.J.B., D. Noble, and R.W. Tsien. 1983. Electrical current flow in excitable cells. Clarendon Press, Oxford.
- Jones L.R., Besch H.R., Jr., Sutko J.L., Willerson J.T. Ryanodine-induced stimulation of net Ca++ uptake by cardiac sarcoplasmic reticulum vesicles. J. Pharmacol. Exp. Ther. 1979;209:48–55. [PubMed] [Google Scholar]
- Khodakhah K., Armstrong C.M. Inositol trisphosphate and ryanodine receptors share a common functional Ca2+ pool in cerebellar Purkinje neurons. Biophys. J. 1997;73:3349–3357. doi: 10.1016/S0006-3495(97)78359-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuba K. Ca2+-induced Ca2+ release in neurons. Jap. J. Physiol. 1994;44:613–650. doi: 10.2170/jjphysiol.44.613. [DOI] [PubMed] [Google Scholar]
- Lipscombe D., Madison D.V., Poenie M., Reuter H., Tsien R.W., Tsien R.Y. Imaging of cytosolic Ca2+ transients arising from Ca2+ stores and Ca2+ channels in sympathetic neurons. Neuron. 1988;1:355–365. doi: 10.1016/0896-6273(88)90185-7. [DOI] [PubMed] [Google Scholar]
- Llano I., DiPolo R., Marty A. Calcium-induced calcium release in cerebellar Purkinje neurones. Neuron. 1994;12:663–673. doi: 10.1016/0896-6273(94)90221-6. [DOI] [PubMed] [Google Scholar]
- Llano I., Gonzalez J., Caputo C., Lai F.A., Blayney L.M., Tan Y.P., Marty A. Presynaptic calcium stores underlie large-amplitude miniature IPSCs and spontaneous calcium transients. Nat. Neurosci. 2000;3:1256–1265. doi: 10.1038/81781. [DOI] [PubMed] [Google Scholar]
- Lytton J., Westlin M., Burk S.E., Shull G.E., MacLennan D.H. Functional comparisons between isoforms of the sarcoplasmic or endoplasmic reticulum family of calcium pumps. J. Biol. Chem. 1992;267:14483–14489. [PubMed] [Google Scholar]
- Meldolesi J., Pozzan T. The endoplasmic reticulum Ca2+ storea view from the lumen. Trends Biochem. Sci. 1998;23:10–14. doi: 10.1016/s0968-0004(97)01143-2. [DOI] [PubMed] [Google Scholar]
- Muschol M., Dasgupta B.R., Salzberg B.M. Caffeine interaction with fluorescent calcium indicator dyes. Biophys. J. 1999;77:577–586. doi: 10.1016/S0006-3495(99)76914-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K., Akita T., Osanai M., Shirasaki T., Kijima H., Kuba K. A Ca2+-induced Ca2+ release mechanism involved in asynchronous exocytosis at frog motor nerve terminals. J. Gen. Physiol. 1998;112:593–609. doi: 10.1085/jgp.112.5.593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narita K., Akita T., Hachisuka J., Huang S.-M., Ochi K., Kuba K. Functional coupling of Ca2+ channels to ryanodine receptors at presynaptic terminals. J. Gen. Physiol. 2000;115:519–532. doi: 10.1085/jgp.115.4.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neher E., Augustine G.J. Calcium gradients and buffers in bovine chromaffin cells. J. Physiol. 1992;450:273–301. doi: 10.1113/jphysiol.1992.sp019127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng Y.Y. Ryanodine-sensitive component of calcium transients evoked by nerve firing at presynaptic nerve terminals. J. Neurosci. 1996;12:6703–6712. doi: 10.1523/JNEUROSCI.16-21-06703.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfaffinger P.J., Leibowitz M.D., Subers E.M., Nathanson N.M, Almers W., Hille B. Agonists that suppress M-current elicit phosphoinositide turnover and Ca2+ transients, but these events do not explain M-current suppression. Neuron. 1988;1:477–484. doi: 10.1016/0896-6273(88)90178-x. [DOI] [PubMed] [Google Scholar]
- Pivovarova N.B., Hongpaisan J., Andrews S.B., Friel D.D. Depolarization-induced mitochondrial Ca accumulation in sympathetic neuronsSpatial and temporal characteristics. J. Neurosci. 1999;19:6372–6384. doi: 10.1523/JNEUROSCI.19-15-06372.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzo-Miller L.D., Pivovarova N.B., Leapman R.D., Buchanan R.A., Reese T.S., Andrews S.B. Activity-dependent calcium sequestration in dendrites of hippocampal neurons in brain slices. J. Neurosci. 1997;17:8729–8738. doi: 10.1523/JNEUROSCI.17-22-08729.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pozzan T., Rizzuto R., Volpe P., Meldolesi J. Molecular and cellular physiology of intracellular calcium stores. Physiol. Rev. 1994;74:595–636. doi: 10.1152/physrev.1994.74.3.595. [DOI] [PubMed] [Google Scholar]
- Rousseau E., Smith J.S., Meissner G. Ryanodine modifies conductance and gating behavior of single Ca2+ release channel. Am. J. Physiol. 1987;253:C364–C368. doi: 10.1152/ajpcell.1987.253.3.C364. [DOI] [PubMed] [Google Scholar]
- Rousseau E., LaDine J., Liu Q.Y., Meissner G. Activation of the Ca2+ release channel of skeletal muscle sarcoplasmic reticulum by caffeine and related compounds. Arch. Biochem. Biophys. 1988;267:75–86. doi: 10.1016/0003-9861(88)90010-0. [DOI] [PubMed] [Google Scholar]
- Sandler V.M., Barbara J.G. Calcium-induced calcium release contributes to action potential evoked calcium transients in hippocampal CA1 pyramidal neurons. J. Neurosci. 1999;19:4325–4336. doi: 10.1523/JNEUROSCI.19-11-04325.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shmigol A., Verkhratsky A., Isenberg G. Calcium-induced calcium release in rat sensory neurons. J. Physiol. 1995;489:627–636. doi: 10.1113/jphysiol.1995.sp021078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutko J.L., Airey J.A., Welch W., Ruest L. The pharmacology of ryanodine and related compounds. Pharmacol. Revs. 1997;49:53–98. [PubMed] [Google Scholar]
- Thayer S.A., Hirning L.D., Miller R.J. The role of caffeine-sensitive calcium stores in the regulation of intracellular free calcium concentration in rat sympathetic neurons in vitro. Mol. Pharmacol. 1988;34:664–673. [PubMed] [Google Scholar]
- Toescu E.C. Intraneuronal Ca2+ stores act mainly as a Ca2+ sink; in cerebellar granule cells. Neuroreport. 1998;9:1227–1231. doi: 10.1097/00001756-199804200-00049. [DOI] [PubMed] [Google Scholar]
- Usachev Y., Thayer S.A. All-or-none Ca2+ release from intracellular stores triggers by Ca2+ influx through voltage gates Ca2+ channels in rat sensory neurons. J. Neurosci. 1997;17:7404–7414. doi: 10.1523/JNEUROSCI.17-19-07404.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Usachev Y., Thayer S.A. Controlling the urge for a Ca2+ surgeall-or-none Ca2+ release in neurons. Bioessays. 1999;21:743–750. doi: 10.1002/(SICI)1521-1878(199909)21:9<743::AID-BIES5>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Verkhratsky A., Shmigol A. Calcium-induced calcium release in neurons. Cell Calcium. 1996;19:1–14. doi: 10.1016/s0143-4160(96)90009-3. [DOI] [PubMed] [Google Scholar]
- Wei H., Wenlin W., Bredesen D.E., Perry D.C. Bcl-2 protects against apoptosis in neuronal cell line caused by thapsigargin-induced depletion of intracellular calcium stores. J. Neurochem. 1998;70:2305–2314. doi: 10.1046/j.1471-4159.1998.70062305.x. [DOI] [PubMed] [Google Scholar]
- Zucchi R., Roncha-Testoni S. The sarcoplasmic reticulum Ca2+ release channel/ryanodine receptormodulation by endogenous effectors, drugs and disease states. Physiol. Rev. 1997;49:1–51. [PubMed] [Google Scholar]