

Abstract
In airway epithelia, purinergic receptor (P2Y2-R) stimulation of intracellular calcium (Ca2+ i)–regulated ion transport is restricted to the membrane domain ipsilateral to receptor activation, implying compartmentalization of Ca2+ i signaling. Because mitochondria can spatially restrict cellular Ca2+ i signals, immunocytochemical, electron microscopic, and fluorescent studies of mitochondria localization were performed in human airway epithelia. Although concentrated at the apical domain, mitochondria were found distributed at both the apical and the basolateral poles and in close association with the endoplasmic reticulum. The role of mitochondria in locally restricting P2Y2-R–induced Ca2+ i signals was investigated by measuring changes in mitochondrial Ca2+ (Ca2+ m) in human airway epithelial monolayers. P2Y2-R activation induced Ca2+ m accumulation in mitochondria confined to the domain ipsilateral to P2Y2-R stimulation, which was blocked by mitochondrial uncoupling with 1 μM CCCP and 2.5 μg/ml oligomycin. The role of mitochondria in restricting the cellular cross-talk between basolateral P2Y2-R–dependent Ca2+ i mobilization and apical membrane Ca2+-activated Cl− secretion was investigated in studies simultaneously measuring Ca2+ i and Cl− secretion in cystic fibrosis human airway epithelial monolayers. Activation of basolateral P2Y2-Rs produced similar increases in Ca2+ i in monolayers without and with pretreatment with uncouplers, whereas Ca2+ i-activated Cl− secretion was only efficiently triggered in mitochondria-uncoupled conditions. We conclude that (a) mitochondria function as a Ca2+ i-buffering system in airway epithelia, compartmentalizing Ca2+ i-dependent functions to the membrane ipsilateral to receptor stimulation; and (b) the mitochondria provide structural barriers that protect the airway epithelia against nonspecific activation of Ca2+ i-modulated functions associated with Ca2+ i signals emanating from the apical or the basolateral membrane domains.
Keywords: calcium signaling, mitochondria, endoplasmic reticulum, chloride secretion, purinergic receptors
INTRODUCTION
Epithelial cells are exposed to distinct physical and chemical stimuli in their mucosal and serosal environments. Epithelia can adapt to these different environments by confining functional responses selectively to their apical or basolateral epithelial membranes. In airway epithelia, membrane-specific intracellular calcium (Ca2+ i)-dependent responses can be demonstrated by activation of apical or basolateral purinergic receptors (P2Y2-Rs) coupled to phospholipase C stimulation and inositol 1,4,5-trisphosphate (IP3) formation (Paradiso et al., 1995; Ribeiro et al., 2001). Ca2+ i signals resulting from activation of P2Y2-Rs are restricted to the membrane domain ipsilateral to receptor activation (Paradiso et al., 1995).
The confinement of Ca2+ i signals to the apical or the basolateral cellular poles in airway epithelia has functional correlates. For example, apical P2Y2-R activation results in Ca2+ i mobilization that efficiently couples to apical Cl− secretion through Ca2+-activated Cl− channels (CaCC) in monolayers of cystic fibrosis (CF) human airway epithelia (Paradiso et al., 2001). In contrast, an effective coupling between P2Y2-R activation and CaCC-mediated Cl− secretion is not present after activation of basolateral P2Y2-Rs in CF airway epithelia (Clarke and Boucher, 1992; Paradiso et al., 1995, 2001). These data have led to the speculation that Ca2+ i mobilized by apical P2Y2-R activation locally activates CaCC, whereas Ca2+ i signals triggered by basolateral P2Y2-R activation do not transit the cell to activate CaCC in the apical membrane.
A number of studies suggest that mitochondria, if properly organized, play a role in restricting Ca2+ i permeation within polarized cells by buffering Ca2+ i signals generated by activation of plasma membrane receptors (Rizzuto et al., 1994; Simpson and Russell, 1996; Babcock et al., 1997; Landolfi et al., 1998; Boitier et al., 1999; Hajnoczky et al., 1999; Tinel et al., 1999). Agonist-induced ER Ca2+ store release can be buffered by mitochondrial Ca2+ uptake through a Ca2+ uniporter whose activity is dependent on the mitochondrial membrane potential (Gunter et al., 2000). After the return of cytoplasmic Ca2+ toward basal levels, Ca2+ can exit the mitochondria through three possible mechanisms that have different kinetics and inhibitor susceptibilities: A Na+-dependent efflux (3 Na+ to 1 Ca2+ or 2 Na+ to 1 Ca2+), a Na+-independent efflux (2 H+ for 1 Ca2+), and, in some cases, through the mitochondrial permeability transition (MPT) pore (Gunter et al., 2000). As evidence of the role for mitochondria in Ca2+ i signaling, Tinel et al. (1999) have shown that mitochondria localized at the apical pole of polarized pancreatic acinar cells limited Ca2+ i diffusion from the apical to the basolateral membrane. After mitochondrial inhibition, this mitochondrial “barrier” was lost and apical Ca2+ i signals were able to spread to the basolateral pole (Tinel et al., 1999).
The Tinel et al. (1999) studies suggest that segregation of Ca2+ i signals in human airway epithelia may result from the spatial distribution of ER Ca2+ stores and, possibly, mitochondria. Therefore, we investigated the role mitochondria play in compartmentalizing P2Y2-R–dependent Ca2+ i signals to the membrane ipsilateral to receptor activation. For these studies, analyses of mitochondria distribution and mitochondrial Ca2+ uptake upon P2Y2-R activation were performed in human airway epithelia. We also simultaneously measured ΔCa2+ i and anion (Cl−) secretion to test the functional role of mitochondria in preventing Ca2+ i signals generated at one pole of the cell from reaching the opposite pole.
MATERIALS AND METHODS
Tissues
Human bronchial airway epithelia were freshly isolated at the time of transplantation and obtained from the University of North Carolina Cystic Fibrosis Center Tissue Culture Core under the auspices of protocols approved by the Institutional Committee on the Protection of the Rights of Human Subjects.
Cell Culture
Human bronchial epithelial cells from main stem or lobar bronchi, harvested from excess tissues from excised CF and recipient lungs, were obtained from the University of North Carolina Cystic Fibrosis Center Tissue Culture Core at the time of lung transplantation. Disaggregated airway epithelial cells were seeded on 1-cm2 Transwell Col (T-Col) filters (pore diameter = 0.45 μm) at a density of 0.25 × 106/cm2 in Ham's F12-based medium supplemented with 10 μg/ml insulin, 5 μg/ml transferrin, 1 μM hydrocortisone, 30 nM triiodothyronine, 25 ng/ml epidermal growth factor, and 3.75 μg/ml endothelial cell growth substance. All cell preparations were maintained at an air–liquid surface interface, and polarized monolayer cultures were studied 6–11 d later.
Confocal Immunofluorescence Microscopy of Mitochondrial Distribution in Airway Epithelia
Bronchial epithelia from transplant lungs were fixed in 4% paraformaldehyde, embedded in paraffin blocks, 10-μm sections were obtained and deparaffinized, and immunocytochemistry performed according to a previous method (Ribeiro et al., 1997). Sections were washed with PBS (120 mM NaCl, 2.6 mM KCl, 8.1 mM Na2HPO4, 1.5 KH2PO4, pH 7.4; all incubations and rinses were performed with PBS), permeabilized with 1% Triton X-100 for 10 min at 25°C, rinsed three times, blocked overnight with 3% bovine serum albumin at 4°C, and rinsed three times. To stain mitochondria, sections were incubated with a mouse monoclonal anti-mitochondrial heat shock protein 70 antibody (Affinity Bioreagents) at 1:50 dilution for 60 min at 37°C, washed three times, incubated for 30 min at 25°C with a Texas red–labeled affinity-purified goat anti–mouse antibody (1:200 dilution; Jackson ImmunoResearch Laboratories), and rinsed three times. Mitochondria were localized by laser confocal microscopy (TCS 4D; PL APO 63×/1.20 mm water lens; Leica). As a control for the mitochondria immunostain, the primary antibody was omitted and only the secondary antibody was used. No stain was observed under these conditions.
Fluorescent Labeling of Mitochondria with MitoTracker
Primary culture monolayers of airway epithelia were bilaterally incubated with F12 medium containing 100 nM of MitoTracker Red CMX Ros (Molecular Probes) for 30 min at 37°C. Cultures were subsequently washed in a HEPES-buffered saline solution and mitochondrial distribution visualized by laser confocal microscopy (SP2-AOBS; PL APO 63×/1.20 mm water lens; Leica) in the XZ-scanning mode.
Electron Microscopy (EM) Studies of Mitochondrial Distribution in Airway Epithelia
Bronchial airway epithelia from transplant lungs were fixed in 2% gluteraldehyde + 2% paraformaldehyde + 0.25% tannic acid, postfixed in 1% OsO4, and processed for EM as described previously (Robinson and Gray, 1996). For EM studies of the physical interaction between mitochondria and ER, monolayers of primary cultures were fixed and processed for EM in the same manner as freshly excised bronchial epithelia.
Confocal Microscopic Studies of Mitochondrial Calcium (Ca2+ m)
For confocal Ca2+ m measurements, monolayers were loaded with rhod2 according to a modification of a previous method (Trollinger et al., 1997) by incubation with 5 μM rhod2/AM at 4°C for 18 h in a HEPES-buffered saline solution, followed by incubation with 5 μM rhod2/AM in F12 medium at 37°C for 1 h. Ca2+ m mobilization (changes in rhod2 fluorescence) was studied by laser confocal microscopy (TCS 4D; PL APO 63×/1.20 mm water lens; Leica) on the XY or XZ scanning mode. The fluorescence intensity of rhod-2 from XY or XZ confocal scans was measured with the MetaMorph software (Universal Imaging). Regions of interest were designated for the apical or the basolateral domains, depending on the protocol, and the same region was quantified at each time point. The same acquisition parameters (e.g., laser power, contrast, brightness, and pinhole value) were used throughout the time course. The fluorescence intensity values (in arbitrary units) from the designated regions were expressed as percentage of the fluorescence intensity from baseline (t = 0) in every experiment.
Studies with Mitochondrial Uncouplers
To uncouple the mitochondria, the mitochondrial membrane potential was dissipated by exposing the airway epithelia to 1 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) in conjunction with 2.5 μg/ml oligomycin (to block the activity of the F0F1 ATP synthase) before or after P2Y2-R activation.
Perfusion Chamber and Bioelectric Measurements
For simultaneous measurements of Cl− secretion (Ieq) and Ca2+ i, CF culture monolayers on T-Col filters were mounted in a miniature Ussing chamber over an objective of a microscope coupled to a microfluorimeter as described previously (Paradiso et al., 2001). For passing current, two circular Ag/AgCl electrodes were placed in the two half-chambers to generate a uniform density of current through the preparation. For measurements of transepithelial electrical potential difference (Vt), polyethylene bridges containing 2 M KCl in 3% agar were positioned in the two half-chambers and connected to calomel electrodes. Briefly, Vt was measured by a voltage-clamp/pulse generator (model VCC600; Physiologic Instruments) and recorded on a two-channel recorder (Linsesis model L200S). To calculate changes in Cl− secretory current (ΔIeq), a defined 1-s current pulse was delivered across the monolayer every 10 s. The airway epithelia were converted from their native Na+ absorptive state to a Cl− secretory state by exposing the monolayers to a basolateral medium of Krebs bicarbonate Ringer solution (KBR; in mM, 125 NaCl, 2.5 K2HPO4, 1.3 CaCl2, 1.3 MgCl2, 25 NaHCO3 and 5 d-glucose) equilibrated with 5% CO2/95% O2, and to a apical medium of KBR containing 0 Na+/low Cl− (N-methyl-D-glucamine substituting for Na+) as reported previously (Paradiso et al., 2001). Polarized monolayers of CF airway epithelia were loaded with Fura-2/AM (5 μM at 37°C for 25 min), as described earlier (Paradiso et al., 2001), before being mounted in a miniature Ussing chamber over an objective (ZEISS LD Achroplan ×40, NA 0.6; working distance 1.8 mm) of a ZEISS Axiovert 35 microscope.
Statistical Analyses
Data in bar graphs represent the mean ± SEM from at least three experiments from individual donors (tissue codes). Where appropriate, data were analyzed by unpaired t test or two-way analysis of variance (ANOVA) with the GraphPad InStat software and statistical significance was defined with P < 0.05.
RESULTS
Mitochondrial Distribution in Polarized Human Airway Epithelia
The immunocytochemical localization of mitochondrial heat shock protein 70 in freshly isolated bronchial airway epithelia demonstrated that mitochondria are present throughout the epithelial cells, but distributed predominantly toward the apical domain (Fig. 1 A). This localization was confirmed in studies of freshly isolated human bronchial airway epithelia subjected to transmission electron microscopic analysis (Fig. 1 B). The micrograph reveals a concentration of mitochondria, i.e., a “barrier”, at the apical cellular pole, although, to a lesser extent, mitochondria were also localized at the basolateral domain around and below the nucleus. Fig. 1 C illustrates that the mitochondrial distribution observed in Fig. 1, A and B, was reproduced in primary culture monolayers of human bronchial airway epithelia labeled with the mitochondrial fluorescent dye MitoTracker Red CMX Ros.
Figure 1.

Mitochondrial distribution in airway epithelia. (A) Immunostaining of the mitochondrial resident protein, mitochondrial heat shock protein 70, in freshly excised human bronchial airway epithelia. Bar, 10 μm. (B) Transmission electron micrograph displaying mitochondrial distribution in freshly excised human bronchial airway epithelia. Arrows indicate mitochondria. Magnification, 4,400×. (C) The mitochondrial distribution in human bronchial airway epithelia in primary culture (monolayer) labeled with MitoTracker Red CMX Ros. Bar, 10 μm. Immunostain is representative of five tissue codes; micrograph is representative of three tissue codes and mitotracker staining is representative of four tissue codes.
P2Y2-R Activation Induces Mitochondrial Ca2+ Uptake in the Cellular Pole Ipsilateral to Receptor Stimulation in Human Airway Epithelia
We next addressed whether the mitochondria are involved in compartmentalization of Ca2+ i signals in airway epithelia. If mitochondria restrict Ca2+ i movements via an uptake mechanism, changes in mitochondrial Ca2+ (Ca2+ m) should be detectable after P2Y2-R activation. To measure Ca2+ m, mitochondria were loaded with the fluorescent Ca2+ indicator rhod-2, and Ca2+ m uptake was studied by laser confocal–scanning microscopy. Fig. 2 A illustrates a time series of XY confocal images from the apical domain of CF human airway epithelial monolayers loaded with rhod-2. Mucosal application of UTP (100 μM), which activates apical P2Y2-Rs and triggers a rise in Ca2+ i (Clarke and Boucher, 1992; Paradiso et al., 2001), rapidly induced large elevations of Ca2+ m (Fig. 2 A, top). To test whether the changes in rhod-2 fluorescence resulted from P2Y2-R activation-dependent Ca2+ m uptake, the same monolayers were subsequently treated with 1 μM carbonyl cyanide m-chlorophenylhydrazone (CCCP) and 2.5 μg/ml oligomycin to depolarize the mitochondria and abolish the driving force for Ca2+ m uptake (Tinel et al., 1999). Mitochondrial uncoupling abolished the UTP-induced rise in mitochondrial rhod-2 fluorescence (Fig. 2 A, bottom), in agreement with previous studies (Hajnoczky et al., 2000). Fig. 2 B summarizes the time series studies illustrated in Fig. 2 A. Conversely, mitochondrial uncoupling before mucosal application of UTP prevented a subsequent P2Y2-R activation-induced Ca2+ m uptake (unpublished data). These studies strongly suggest that Ca2+ i, released from IP3-sensitive ER Ca2+ stores after P2Y2-R stimulation, accumulates in functioning mitochondria in airway epithelia.
Figure 2.
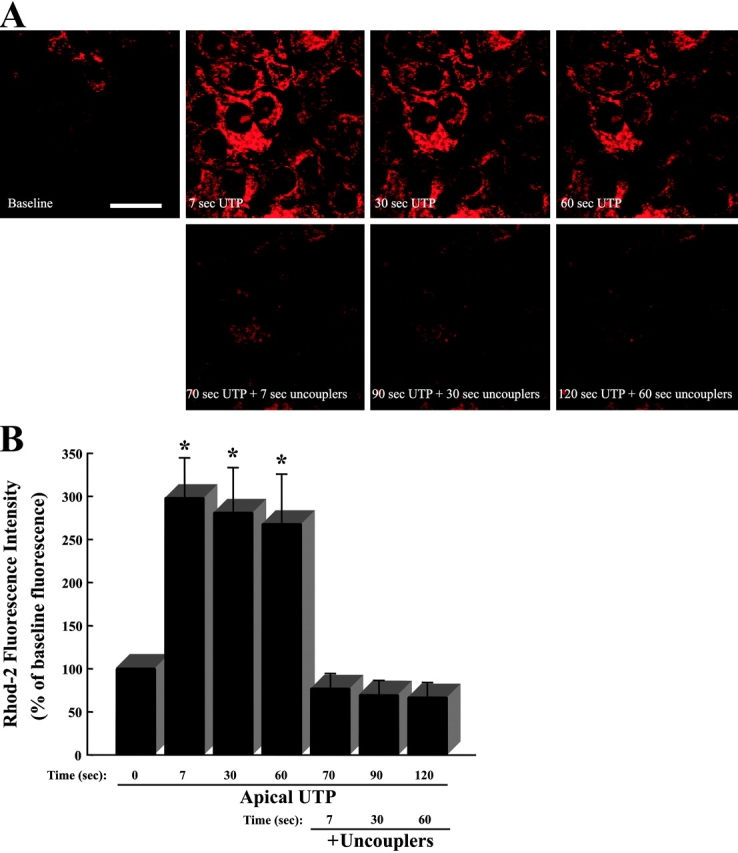
Mitochondrial calcium (Ca2+ m) uptake elicited by apical P2Y2-R activation is inhibited by mitochondrial uncouplers. Representative time series of XY confocal scans from the apical epithelial domain. (A, top) Time course for Ca2+ m uptake (visualized as increases in rhod-2 fluorescence) after addition of 100 μM mucosal UTP to a polarized primary culture monolayer of CF human bronchial airway epithelia. (A, bottom) Effect of subsequent mitochondrial uncoupling with 1 μM CCCP and 2.5 μg/ml oligomycin on Ca2+ m accumulation induced by apical P2Y2-R activation. Bar, 10 μm. (B) Summary of the time series studies depicted in A. Data are expressed as a percentage of baseline fluorescence and represent the mean of four experiments ± SEM. *, P < 0.05, 100 μM mucosal UTP-induced fluorescence vs. baseline (t = 0) fluorescence.
Since mitochondrial Ca2+ uptake after P2Y2-R activation may require an intimate association between mitochondria and IP3-sensitive ER sites (Simpson and Russell, 1996; Boitier et al., 1999; Hajnoczky et al., 1999; Khodorov et al., 1999; Tinel et al., 1999; Duchen, 2000), the spatial distribution of these organelles was studied in electron micrographs from primary cultures of human airway epithelia. Fig. 3 illustrates that the mitochondrial and the ER networks are intimately associated in airway epithelia, similar to previous findings (Rizzuto et al., 1993, 1998; Landolfi et al., 1998; Tinel et al., 1999). These data suggest that the association of mitochondria and ER may provide an efficient system for the mitochondrial role in restricting the spread of Ca2+ i signals resulting from P2Y2-R activation in airway epithelia.
Figure 3.
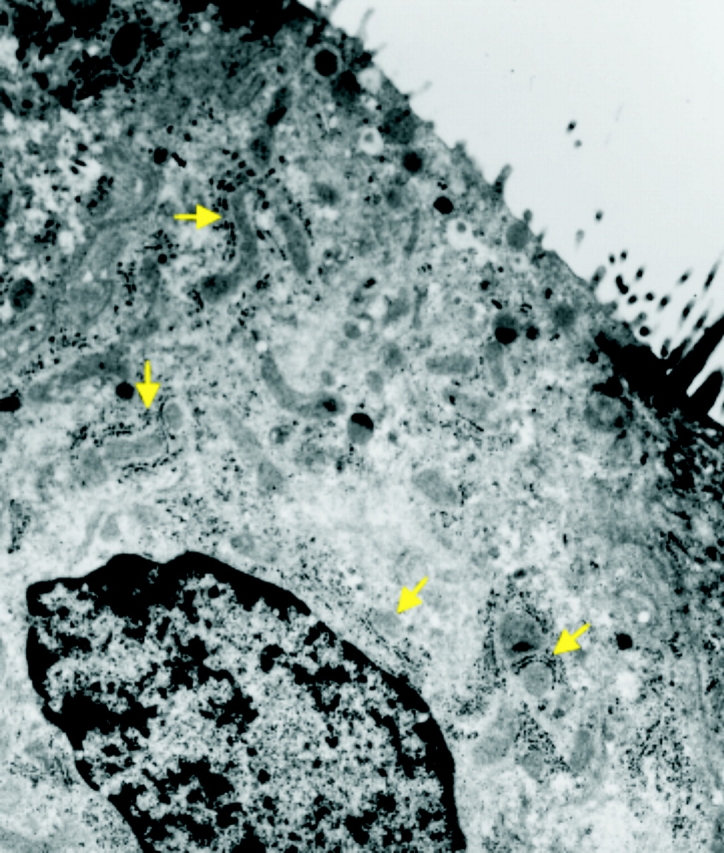
Mitochondria and ER are closely associated in human airway epithelia. Representative electron micrograph from a CF primary culture monolayer illustrating, as denoted by the arrows, that mitochondria and ER (depicted as rough ER membranes decorated with ribosomes) are intimately associated. Magnification, 20,000×. Micrograph is representative of four tissue codes.
To address the Ca2+ m uptake under conditions where Ca2+ i signals are of lower magnitude compared with those elicited by P2Y2-R activation, additional studies were performed with the ER Ca2+-ATPase inhibitor thapsigargin (TG). Since the kinetics of ER Ca2+ release are slower in TG-treated cells, Ca2+ i levels do not rise as quickly and do not reach the same magnitude compared with UTP-dependent Ca2+ i signals resulting from IP3-mediated ER Ca2+ release (Paradiso et al., 1995). Fig. 4 A depicts a time series of XY confocal images from the apical domain of CF human airway epithelial monolayers loaded with rhod-2. Mucosal application of TG (1 μM) induced Ca2+ m uptake, which was inhibited by mitochondrial uncouplers, but exhibited slower kinetics and lower magnitude when compared with the Ca2+ m uptake elicited by P2Y2-R activation (Fig. 2). Fig. 4 B summarizes the time series studies exemplified in Fig. 4 A. These findings suggest that, in airway epithelia, the magnitude of Ca2+ m uptake is a function of the magnitude of the Ca2+ signal that reaches the mitochondria after ER Ca2+ store depletion.
Figure 4.
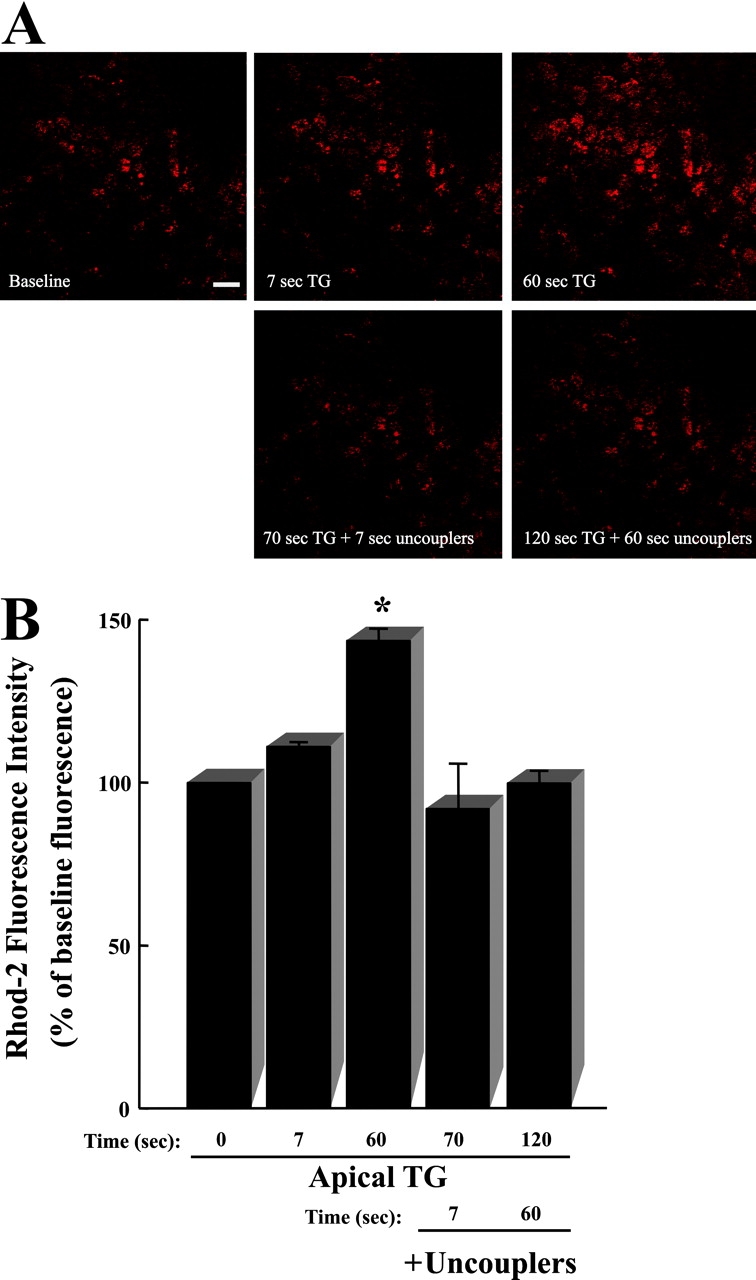
Mitochondrial calcium (Ca2+ m) uptake resulting from TG-dependent inhibition of ER Ca2+-ATPase activity in airway epithelia. Representative time series of XY confocal scans from the apical epithelial domain. (A, top) Time course for Ca2+ m uptake (visualized as increases in rhod-2 fluorescence) after addition of 1 μM mucosal TG to a polarized primary culture monolayer of CF human bronchial airway epithelia. (A, bottom) Effect of subsequent mitochondrial uncoupling with 1 μM CCCP and 2.5 μg/ml oligomycin on Ca2+ m accumulation induced by TG. Bar, 10 μm. (B) Summary of the time series studies illustrated in A. Data are expressed as a percentage of baseline fluorescence and represent the mean of 3 experiments ± SEM. *, P < 0.05, 1 μM mucosal TG-induced fluorescence vs. baseline (t = 0) fluorescence.
The role of mitochondria in the compartmentalization of Ca2+ i signals was further studied in rhod2-loaded CF monolayers with XZ confocal scans to simultaneously visualize the apical and the basolateral domains. Apical P2Y2-R activation induced a rapid and sustained apical Ca2+ m accumulation (Fig. 5 A). The rise in Ca2+ m was restricted to the mitochondria localized at the apical pole of the cells, with no changes in Ca2+ m detected at the contralateral, basolateral domain. The compiled data from these studies are depicted in Fig. 5 B.
Figure 5.

Mitochondrial calcium (Ca2+ m) uptake, as a read-out of localized Ca2+ i signals resulting from apical P2Y2-R activation, is restricted to the apical domain in airway epithelia. (A) Representative time course for apical P2Y2-R activation-promoted Ca2+ m uptake, as measured by increases in rhod-2 fluorescence, in a polarized primary culture monolayer of CF human airway epithelia. Bar, 10 μm. (B) Summary of the time series studies illustrated in A. Data are expressed as a percentage of baseline rhod-2 fluorescence and represent the mean of four experiments ± SEM.
Conversely, basolateral P2Y2-R activation promoted Ca2+ m accumulation in the mitochondria localized at the basolateral domain, without affecting Ca2+ m in the mitochondria localized at the apical cellular pole (Fig. 6 A). The average data from these studies are shown in Fig. 6 B. Collectively, these findings suggest that apically or basolaterally distributed mitochondria exert functional barriers against the spread of Ca2+ i signals toward the domain contralateral to the membrane of P2Y2-R activation.
Figure 6.

Mitochondrial calcium (Ca2+ m) uptake, as a read-out of localized Ca2+ i signals resulting from basolateral P2Y2-R activation, is restricted to the basolateral domain in airway epithelia. (A) Representative time course for basolateral P2Y2-R activation-promoted Ca2+ m uptake, as measured by increases in rhod-2 fluorescence, in a polarized primary culture monolayer of CF human airway epithelia. Bar, 10 μm. (B) Summary of the time series studies depicted in A. Data are expressed as a percentage of baseline rhod-2 fluorescence and represent the mean of three experiments ± SEM.
Basolateral UTP-dependent Ca2+ i Mobilization and Apical Cl− Secretion under Coupled and Uncoupled Mitochondrial Conditions in Human Airway Epithelia
We next addressed whether mitochondria play a functional role in restricting the spatial range of P2Y2-R-promoted Ca2+ i mobilization by using Ca2+-dependent Cl− secretion as a read-out of transcellular Ca2+ i permeation. These studies were performed in monolayers of polarized CF human airway epithelia which, due to the absence of functional CFTR, utilize CaCC as the sole pathway for apical Cl− secretion (Paradiso et al., 2001). We simultaneously measured Ca2+ i mobilization and CaCC-mediated Cl− secretion (equivalent current; Ieq) in a miniature Ussing chamber that permits the independent perfusion of the serosal and the mucosal compartments.
Fig. 7 A depicts representative simultaneous recordings of Ca2+ i levels and Vt (transepithelial electrical potential difference) from CF monolayers with intact mitochondrial function. Basolateral addition of forskolin (10 μM) failed to induce any changes in Vt, consistent with the absence of cAMP-induced Cl− secretion through CFTR in CF cultures. The subsequent maximal activation of basolateral P2Y2-Rs by addition of 100 μM UTP (Paradiso et al., 2001) elicited Ca2+ i mobilization. However, this response was only coupled to small changes in Vt as reported previously (Clarke and Boucher, 1992; Paradiso et al., 1995, 2001).
Figure 7.
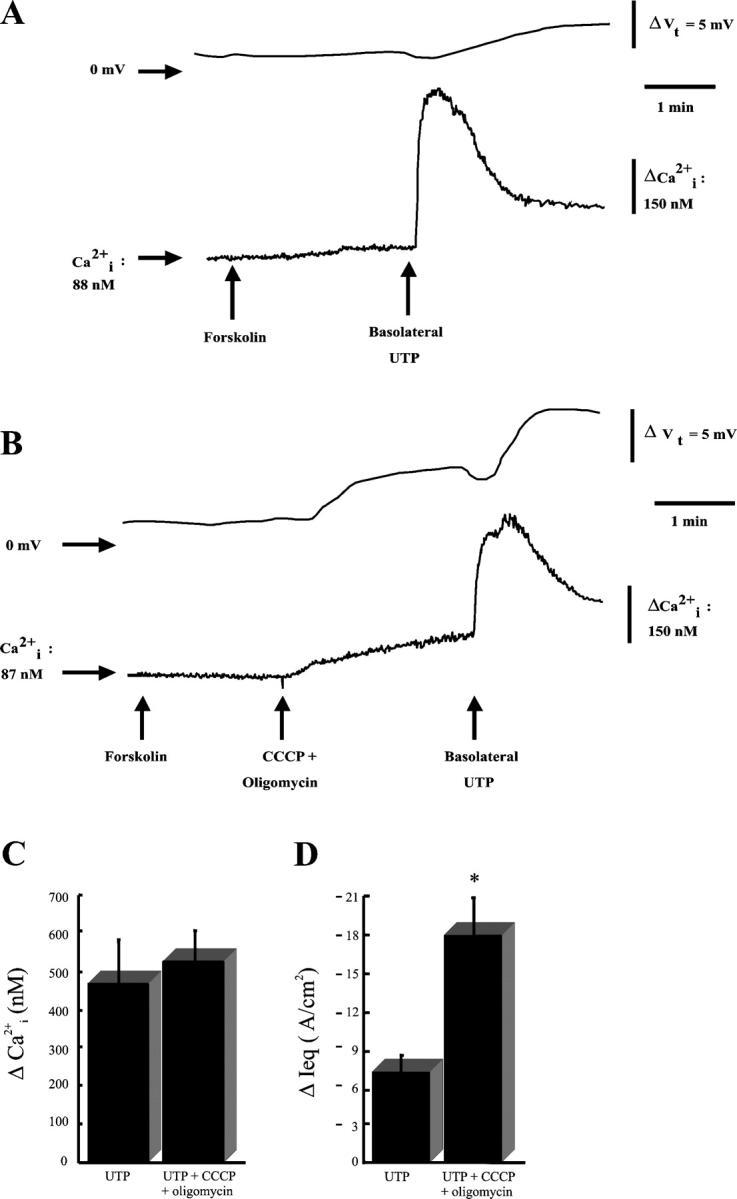
Basolateral P2Y2 receptor activation–dependent intracellular calcium (Ca2+ i) mobilization and apical Cl− secretion in CF human airway epithelia. (A) Simultaneous measurements of Ca2+ i and transepithelial electrical potential difference (Vt) in a CF monolayer exposed to luminal Na+ free/low Cl− KBR and basolateral KBR. 10 μM forskolin and 100 μM basolateral UTP were added at times denoted by arrows. Representative Vt and Ca2+ i tracings from 13 experiments. (B) Similar protocol as in A, but with addition of the mitochondrial uncouplers 1 μM CCCP + 2.5 μg/ml oligomycin at time depicted by arrow. Representative Vt and Ca2+ i tracings from 24 experiments. (C) Average changes in Ca2+ i (peak-baseline value) after basolateral UTP or basolateral UTP + uncouplers. n = 11 and n = 24 for UTP and UTP + CCCP + oligomycin, respectively. (D) Average changes in Ieq (calculated from the Vt values) from the experiments compiled in C. n = 13 and n = 24 for UTP and UTP + CCCP + oligomycin, respectively. *, P < 0.0005, UTP vs. UTP + CCCP + oligomycin.
Studies were then performed after pretreatment with mitochondrial uncouplers to inhibit mitochondrial function. Fig. 7 B illustrates representative simultaneous recordings of Ca2+ i mobilization and Vt from CF monolayers subjected to the same protocol described in Fig. 7 A except that, after basolateral forskolin addition, the mitochondria were uncoupled by bilateral treatment with 1 μM CCCP and 2.5 μg/ml oligomycin. Before UTP, CCCP and oligomycin promoted a small Ca2+ i rise, suggesting that mitochondrial uncoupling released Ca2+ from the mitochondria (Rizzuto et al., 1994; Simpson and Russell, 1996; Babcock et al., 1997; Hajnoczky et al., 1999). More importantly, this Ca2+ i mobilization correlated with a small increase in Vt, suggesting that this small Ca2+ i rise was effective in stimulating apical CaCC activity under mitochondria-uncoupled conditions. Since most mitochondria are distributed at the apical pole of airway epithelial cells (Fig. 1), these findings suggest that the close proximity of mitochondria to CaCC sites may account for the moderately efficient stimulation of apical Cl− secretion after Ca2+ m release. After mitochondrial inhibition, basolateral P2Y2-R activation with 100 μM UTP increased Ca2+ i to the same degree as to mitochondria-coupled cultures (Fig. 7 B). However, under mitochondria-uncoupled conditions, basolateral P2Y2-R activation much more efficiently triggered CaCC-mediated Cl− secretion. Fig. 7, C and D, depict the summary ΔCa2+ i (peak – baseline Ca2+ i levels) and ΔIeq (derived from the Vt values) data, respectively, from the experiments shown in Fig. 7, A and B.
DISCUSSION
Polarized airway epithelial cells express specific Ca2+ i-dependent functions, e.g., Ca2+ i-regulated ion channels, confined to the apical or the basolateral domains (Paradiso et al., 1995). Although both apical or basolateral P2Y2-R activation couple to PLC stimulation–dependent Ca2+ i mobilization, Ca2+ i-mediated channel regulation at the apical or the basolateral membrane can only be elicited by ipsilateral receptor activation (Clarke and Boucher, 1992; Paradiso et al., 1995, 2001). Therefore, P2Y2-R activation can be used as a tool to study mechanisms by which Ca2+ i-dependent responses are compartmentalized within airway epithelia.
This study demonstrates that mitochondria efficiently function as a Ca2+ i-buffering system in airway epithelia. The studies with mitochondria-compartmentalized rhod-2 revealed that Ca2+ m accumulated upon apical (Figs. 2 and 5) or basolateral (Fig. 6) P2Y2-R activation selectively in the mitochondria localized at the pole ipsilateral to receptor stimulation. Therefore, by virtue of their Ca2+ i-buffering activity, mitochondria appear functionally capable of restricting the distance that Ca2+ signals generated by P2Y2-R activation can travel across the airway epithelia.
This notion was tested functionally in experiments that demonstrated that, under mitochondria-coupled conditions (Fig. 7 A), basolateral P2Y2-R activation did not efficiently couple to Ca2+ i-regulated apical Cl− secretion, despite a robust Ca2+ i mobilization. However, after mitochondrial uncoupling (Fig. 7 B), basolateral P2Y2-R activation increased Ca2+ i to the same level compared with cultures with coupled mitochondria, but promoted efficient Ca2+ i-dependent apical secretion (Fig. 7, C and D).
The present data offer a mechanism to account for previous observations of membrane-restricted Ca2+ i signaling events in airway epithelia (Clarke and Boucher, 1992; Paradiso et al., 1995, 2001). Specifically, the mitochondria localized at the apical or the basolateral poles serve as barriers to prevent global cell Ca2+ i signaling upon plasma membrane receptor activation. Moreover, the basolateral mitochondrial Ca2+ i buffering activity provides a mechanism that explains the failure to couple basolateral P2Y2-R activation with CaCC-mediated apical anion secretion in CF airway epithelia (Clarke and Boucher, 1992; Paradiso et al., 1995, 2001).
Our findings are consistent with a previous study addressing the mitochondrial participation in Ca2+ i signaling in polarized pancreatic acinar cells. In this study, the mitochondria were shown to localize around the apical pole, the site of IP3-sensitive Ca2+ stores, to limit Ca2+ i diffusion from the apical to the basolateral domain (Tinel et al., 1999). After mitochondrial inhibition, this mitochondrial barrier was lost and Ca2+ i signals could spread toward the basolateral pole (Tinel et al., 1999).
The present observation of a close association between mitochondria and ER in airway epithelia (Fig. 3) is consistent with reports describing a role for mitochondria in Ca2+ i homeostasis in other systems. For example, mitochondria have been shown to localize in close proximity to ER Ca2+-releasing sites (Rizzuto et al., 1993, 1998; Landolfi et al., 1998; Tinel et al., 1999), where microdomains of high Ca2+ i resulting from IP3-dependent Ca2+ release trigger fast mitochondrial uptake of large amounts of Ca2+. The physical interaction between mitochondria and IP3-dependent Ca2+-releasing sites in the ER, together with the rapid mitochondrial Ca2+ uptake, not only allow for the fast modulation of mitochondrial metabolism, but may also limit the amplitude as well as the spatiotemporal aspects of agonist-generated Ca2+ i signals (Simpson and Russell, 1996; Boitier et al., 1999; Hajnoczky et al., 1999; Khodorov et al., 1999; Tinel et al., 1999; Duchen, 2000).
In conclusion, our findings have important implications for airway epithelial biology. A model illustrating the role of the apical and the basolateral mitochondrial barriers in confining Ca2+ i signals in human airway epithelia is depicted in Fig. 8 . The strategic apical polarization of the mitochondria and their association with ER Ca2+ stores provides for an optimal Ca2+ m uptake when Ca2+ i levels rise as a result of autocrine/paracrine regulation of apical P2Y2-Rs by, e.g., apical nucleotide release. The large mitochondrial barrier at the apical cellular pole, by acting as a buffer for Ca2+ i signals resulting from luminal airway stress (e.g., cough-induced shear stress), may provide a functional barrier between the apical domain and the rest of the cell to prevent global Ca2+ waves, thereby protecting the nuclear and basolateral cytoplasmic compartments from Ca2+ i-modulated functions in response to luminal stress. Conversely, the Ca2+ i-buffering activity of the basolateral mitochondria serves to restrict Ca2+ i signals generated by serosal agonists to the basolateral domain of airway epithelia, resulting in efficient Ca2+ i-dependent regulation of basolateral K+ channels, but not apical CaCC.
Figure 8.
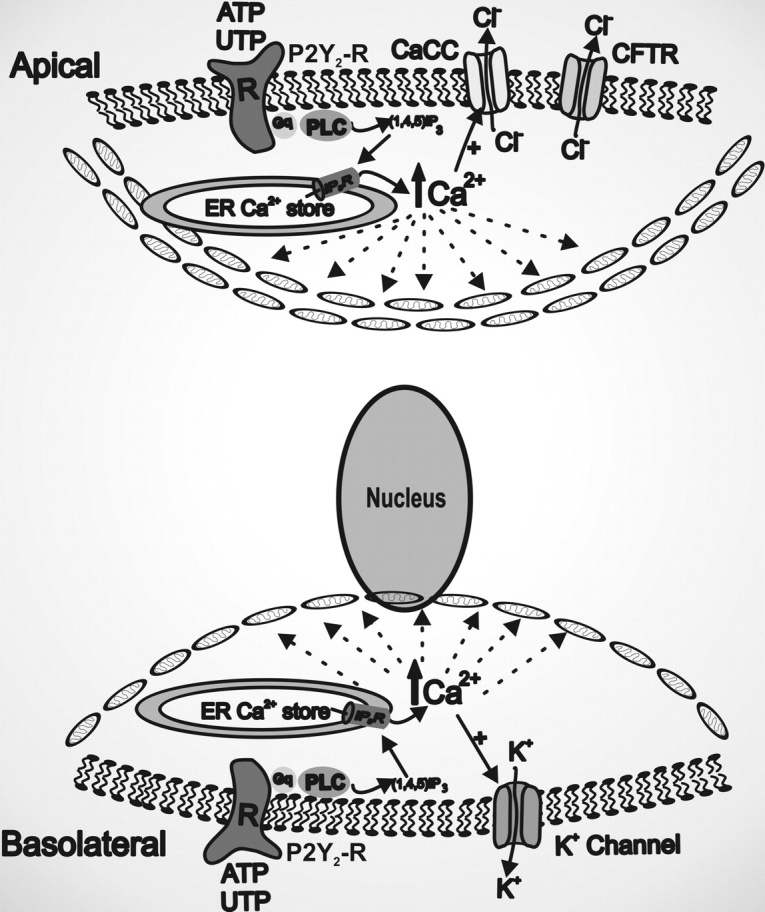
The apical and the basolateral mitochondrial barriers restrict Ca2+ i-dependent functions to the membrane domain ipsilateral to receptor activation in human airway epithelia. The mitochondria distributed toward the apical or the basolateral domains provide structural barriers between the apical and basolateral poles to global Ca2+ waves, thereby protecting the airway epithelia against nonspecific regulation of Ca2+ i-modulated functions associated with the domain contralateral to the membrane of receptor activation.
The mitochondrial compartmentalization of Ca2+ i signals resulting from basolateral P2Y2-R activation has profound consequences for CF as compared with normal airway epithelia. In normal airway epithelia, CFTR-mediated Cl− secretion can be triggered by basolateral P2Y2-R activation coupled to induction of two pathways (Paradiso et al., 2001): (a) membrane hyperpolarization due to Ca2+ i-activated basolateral K+ channels, which increases the driving force for CFTR-mediated Cl− secretion and (b) activation of a Ca2+ i-independent protein kinase C, which, directly or indirectly, activates CFTR at the apical membrane. On the other hand, in CF airway epithelia lacking functional CFTR, basolateral P2Y2-R activation does not induce Cl− secretion since the only Cl− secretory pathway available at the apical membrane, CaCC (Paradiso et al., 2001), cannot be activated by the resultant Ca2+ i mobilization by virtue of the Ca2+ i-buffering activity of basolateral mitochondria.
Acknowledgments
We are indebted to the UNC CF Center Tissue Core and Drs. Scott Randell and James Yankaskas for supplying cells and tissue blocks, Kim Burns for performing electron microscopy, Tracy Eldred for providing sections from native bronchial epithelia, and Lisa Brown for editorial assistance. We also thank Dr. John Lemasters for scientific advice.
This work has been supported by the Cystic Fibrosis Foundation through grants to C.M.P. Ribeiro (CFF #RIBEIR00Z0 and CFF #RIBEIR00G0) and by The National Institutes of Health through grants to R.C. Boucher (HL34322 and HL60280).
Lawrence G. Palmer served as editor.
Abbreviations used in this paper: CaCC, Ca2+-activated Cl− channels; P2Y2-Rs, purinergic receptors; TG, thapsigargin.
References
- Babcock, D.F., J. Herrington, P.C. Goodwin, Y.B. Park, and B. Hille. 1997. Mitochondrial participation in the intracellular Ca2+ network. J. Cell Biol. 136:833–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boitier, E., R. Rea, and M.R. Duchen. 1999. Mitochondria exert a negative feedback on the propagation of intracellular Ca2+ waves in rat cortical astrocytes. J. Cell Biol. 145:795–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke, L.L., and R.C. Boucher. 1992. Chloride secretory response to extracellular ATP in normal and cystic fibrosis nasal epithelia. Am. J. Physiol. 263:C348–C356. [DOI] [PubMed] [Google Scholar]
- Duchen, M.R. 2000. Mitochondria and Ca2+ in cell physiology and pathophysiology. Cell Calcium. 28:339–348. [DOI] [PubMed] [Google Scholar]
- Gunter, T.E., L. Buntinas, G. Sparagna, R. Eliseev, and K. Gunter. 2000. Mitochondrial calcium transport: mechanisms and functions. Cell Calcium. 28:285–296. [DOI] [PubMed] [Google Scholar]
- Hajnoczky, G., G. Csordas, M. Madesh, and P. Pacher. 2000. Control of apoptosis by IP3 and ryanodine receptor driven calcium signals. Cell Calcium. 28:349–363. [DOI] [PubMed] [Google Scholar]
- Hajnoczky, G., R. Hager, and A.P. Thomas. 1999. Mitochondria suppress local feedback activation of inositol 1,4, 5-trisphosphate receptors by Ca2+. J. Biol. Chem. 274:14157–14162. [DOI] [PubMed] [Google Scholar]
- Khodorov, B., V. Pinelis, T. Storozhevykh, A. Yuravichus, and L. Khaspekhov. 1999. Blockade of mitochondrial Ca2+ uptake by mitochondrial inhibitors amplifies the glutamate-induced calcium response in cultured cerebellar granule cells. FEBS Lett. 458:162–166. [DOI] [PubMed] [Google Scholar]
- Landolfi, B., S. Curci, L. Debellis, T. Pozzan, and A.M. Hofer. 1998. Ca2+ homeostasis in the agonist-sensitive internal store: functional interactions between mitochondria and the ER measured In situ in intact cells. J. Cell Biol. 142:1235–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradiso, A.M., S.J. Mason, E.R. Lazarowski, and R.C. Boucher. 1995. Membrane-restricted regulation of Ca2+ release and influx in polarized epithelia. Nature. 377:643–646. [DOI] [PubMed] [Google Scholar]
- Paradiso, A.M., C.M.P. Ribeiro, and R.C. Boucher. 2001. Polarized signaling via purinoceptors in normal and cystic fibrosis airway epithelia. J. Gen. Physiol. 117:53–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro, C.M., J. Reece, and J.W. Putney, Jr. 1997. Role of the cytoskeleton in calcium signaling in NIH 3T3 cells. An intact cytoskeleton is required for agonist-induced [Ca2+]i signaling, but not for capacitative calcium entry. J. Biol. Chem. 272:26555–26561. [DOI] [PubMed] [Google Scholar]
- Ribeiro, C.M.P., A.M. Paradiso, E. Lazarowski, and R.C. Boucher. 2001. P2Y2 receptors and Ca2+-dependent Cl− secretion in normal and cystic fibrosis human airway epithelia. Cilia, Mucus and Mucociliary Interactions. M. Salathe, editor. Marcel Dekker, Inc., New York. 303–314.
- Rizzuto, R., C. Bastianutto, M. Brini, M. Murgia, and T. Pozzan. 1994. Mitochondrial Ca2+ homeostasis in intact cells. J. Cell Biol. 126:1183–1194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rizzuto, R., M. Brini, M. Murgia, and T. Pozzan. 1993. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science. 262:744–747. [DOI] [PubMed] [Google Scholar]
- Rizzuto, R., P. Pinton, W. Carrington, F.S. Fay, K.E. Fogarty, L.M. Lifshitz, R.A. Tuft, and T. Pozzan. 1998. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science. 280:1763–1766. [DOI] [PubMed] [Google Scholar]
- Robinson, G., and T. Gray. 1996. Electron microscopy 2: practical procedures. Theory and Practice of Histological Techniques. J.D. Bancroft and A. Stevens, editors. Churchill Livingstone, Inc., New York. 585-627.
- Simpson, P.B., and J.T. Russell. 1996. Mitochondria support inositol 1,4,5-trisphosphate-mediated Ca2+ waves in cultured oligodendrocytes. J. Biol. Chem. 271:33493–33501. [DOI] [PubMed] [Google Scholar]
- Tinel, H., J.M. Cancela, H. Mogami, J.V. Gerasimenko, P.V. Gerasimenko, A.V. Tepikin, and O.H. Petersen. 1999. Active mitochondria surrounding the pancreatic acinar granule region prevent spreading of inositol trisphosphate-evoked local cytosolic Ca2+ signals. EMBO J. 18:4999–5008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trollinger, D.R., W.E. Cascio, and J.J. Lemasters. 1997. Selective loading of Rhod 2 into mitochondria shows mitochondrial Ca2+ transients during the contractile cycle in adult rabbit cardiac myocytes. Biochem. Biophys. Res. Commun. 236:738–742. [DOI] [PubMed] [Google Scholar]