

Abstract
KATP channels are a functional complex of sulphonylurea receptor (SUR1, SUR2) and inward rectifier K+ (Kir6.1, Kir6.2) channel subunits. We have studied the role of the putative pore forming subunit (Kir6.2) in regulation of rectification and gating of KATP channels generated by transfection of SUR1 and Kir6.2 cDNAs in COSm6 cells. In the absence of internal polyvalent cations, the current-voltage relationship is sigmoidal. Mg2+ or spermine4+ (spm) each induces a mild inward rectification. Mutation of the asparagine at position 160 in Kir6.2 to aspartate (N160D) or glutamate (N160E) increases the degree of rectification induced by Mg2+ or spermine4+, whereas wild-type rectification is still observed after mutation to other neutral residues (alanine–N160A, glutamine–N160Q). These results are consistent with this residue lining the pore of the channel and contributing to the binding of these cations, as demonstrated for the equivalent site in homomeric ROMK1 (Kir1.1) channels. Since Kir6.2 contains no consensus ATP binding site, whereas SUR1 does, inhibition by ATP has been assumed to depend on interactions with SUR1. However, we found that the [ATP] causing half-maximal inhibition of current (K i) was affected by mutation of N160. Channels formed from N160D or N160Q mutant subunits had lower apparent sensitivity to ATP (K i,N160D = 46.1 μM; K i,N160Q = 62.9 μM) than wild-type, N160E, or N160A channels (K i = 10.4, 17.7, 6.4 μM, respectively). This might suggest that ATP binding to the channel complex was altered, although examination of channel open probabilities indicates instead that the residue at position 160 alters the ATP-independent open probability, i.e., it controls the free energy of the open state, thereby affecting the “coupling” of ATP binding to channel inhibition. The results can be interpreted in terms of a kinetic scheme whereby the residue at Kir6.2 position 160 controls the rate constants governing transitions to and from the open state, without directly affecting ATP binding or unbinding transitions.
Keywords: sulphonylurea receptor, Kir6.2, ATP, gating, rectification
introduction
KATP channels are present in most, if not all, excitable tissues, and share the property of being inhibited by intracellular nucleotide triphosphates (Ashcroft, 1988). Structurally unique amongst K channels, these KATP channels are formed by coexpression of an ABC protein (SUR1, or SUR2; Aguilar-Bryan et al., 1995; Inagaki et al., 1996) and an inward rectifier K channel subunit (Kir6.1 or Kir6.2; Inagaki et al., 1995a , b ; Inagaki et al., 1996). Expression of Kir6.2 alone does not result in functional ion channels, suggesting an intimate and requisite interaction between these two subunits. The nature of the inhibitory action of ATP is unknown, but the recent expression of functional KATP channels from cloned subunits (Inagaki et al., 1995b ; 1996; Nichols et al., 1996) provides the necessary tools to begin to address this important question. Given the presence of nucleotide binding fold in SUR, and the homology between Kir6.2 and other Kir subunits that form homomeric inward rectifier channels (Nichols and Lopatin, 1997), it seems likely that nucleotide sensitivity of the channel resides in the SUR protein and that the ion channel is formed predominantly by the Kir6.2 subunit. We have recently demonstrated that at least part of the nucleotide sensitivity of KATP channels is conferred by the SUR subunit (Nichols et al., 1996; Gribble et al., 1997). The present study utilizes mutations in a putative pore lining residue in Kir6.2 to demonstrate that Kir6.2 does indeed form the ion-conducting pathway, since mutations at this residue cause changes in the rectification induced by Mg2+ ions or polyamines that can be predicted from the effects of similar mutations in homomeric inward rectifying K channels (Fakler et al., 1994; Ficker et al., 1994; Lopatin et al., 1994; Lu and MacKinnon, 1994; Stanfield et al., 1994; Wible et al., 1994). Significantly, we find that this same pore-lining residue controls the apparent sensitivity of the channel to inhibition by ATP. Analysis of channel open probability in the absence of ATP indicates that this effect results from an alteration of the stability of the open state of the channel, not from a modification in ATP binding affinity. These results have important implications for the interpretation of experiments assessing nucleotide sensitivity of KATP channels.
methods
Expression of Recombinant KATP Channels in COSm6 Cells
COSm6 cells were plated on glass coverslips at a density of 2.5 × 105 cells per well (30-mm six-well dishes) and cultured in Dulbecco's Modified Eagle Medium plus 10 mM glucose (DMEM-HG), supplemented with FCS (10%). The following day, pCMV-Kir6.2 (5 μg) and pECE-haSUR cDNA (5 μg) were cotransfected into the COSm6 cells with diethylaminoethyl-dextran (0.5 mg/ml). Cells were incubated for 2 min in HEPES-buffered salt solution containing DMSO (10%), and then for 4 h in DMEM-HG plus 2% FCS and chloroquine (100 μM), and then returned to DMEM-HG plus 10% FCS.
Generation of Kir6.2 Mutations
Mutant constructs were prepared by overlap extension at the junctions of the relevant domains by sequential PCR. Resulting PCR products were subcloned into pCMV vector and sequenced to verify the correct mutation before transfection.
86Rb+ Efflux Measurements
For 86Rb+ flux experiments, 86RbCl (1 μCi/ml) was added in fresh DMEM-HG containing FCS (10%) 24 h after transfection. Cells were incubated for 12–24 h before measurement of Rb-efflux. For efflux measurements cells were incubated for 30 min at 25°C in Krebs' Ringer solution, with or without metabolic inhibitors (2.5 μg/ml oligomycin plus 1 mM 2-deoxy-d-glucose), glibenclamide, or diazoxide. At selected time points, the solution was aspirated from the cells and replaced with fresh solution. The 86Rb+ in the aspirated solution was counted.
Patch-clamp Measurements
Patch-clamp experiments were made at room temperature in an oil-gate chamber which allowed the solution bathing the exposed surface of the isolated patch to be changed in less than 50 ms (Lederer and Nichols, 1989). Shards of glass were removed from the culture dishes and placed in the experimental chamber. Micropipettes were pulled from thin-walled glass (WPI Inc., New Haven, CT) on a horizontal puller (Sutter Instrument Co., Novato, CA), fire polished, and the tips coated with a 1:1 mixture of light mineral oil and Parafilm (American National Can Co., Greenwich, CT) to reduce capacitative currents. Electrode resistance was typically 0.5–1 MΩ when filled with K-INT solution (see below). Microelectrodes were “sealed” onto cells by applying light suction to the rear of the pipette. Inside-out patches were obtained by lifting the electrode and then passing the electrode tip through the oil-gate. Membrane patches were voltage-clamped with an Axopatch 1B patch-clamp (Axon Instruments, Foster City, CA). PClamp software and a Labmaster TL125 D/A converter (Axon Instruments) were used to generate voltage pulses. Data were normally filtered at 0.5–3 kHz; signals were digitized at 22 kHz (Neurocorder; Neurodata, New York, NY) and stored on video tape. Experiments were replayed onto a chart recorder or digitized into a microcomputer using Axotape software (Axon Instruments). The standard bath (intracellular) and pipette (extracellular) solution used throughout these experiments (K-INT) had the following composition: 140 mM KCl, 10 mM K-HEPES, 1 mM K-EGTA, with additions as described. The solution pH was 7.3. Calculations of free [Mg2+] were made with a program written by M. Kurzmak (Department of Biological Chemistry, University of Maryland, Baltimore, MD), which was based on the formulations of Fabiato and Fabiato (1979).
Data Analysis
Off-line analysis was performed using ClampFit and Microsoft Excel programs. Stationary fluctuation analysis of macroscopic currents (Neher and Stevens, 1977; Sigworth, 1980) was performed on short (<1 s) recordings of currents at +50 mV following a step to zero [ATP] (over which time mean channel activity run-down, or inactivation, was negligible) and in 5 mM [ATP]. Currents (50 pA < mean current < 1 nA, corresponding to ∼25–500 channels) were filtered at fc = 3 kHz and digitized at 10 kHz with 12-bit amplitude resolution. Mean channel current (I), and variance (σ2) in the absence of ATP were obtained by subtraction of the mean patch current and variance of patch current in 5 mM ATP (all channels closed) from mean current and variance estimated in zero ATP. Single channel current (i) was assumed to be constant at 3.75 pA, corresponding to single channel conductance of 75 pS (see Fig. 1). The mean open probability (PO) was then calculated by fitting the Eq. 1:
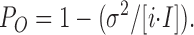 |
1 |
Figure 1.



Single channel currents from KATP channels generated by expression of SUR1 with Kir6.2 subunits. (A) Representative single channel currents recorded from an inside-out patch containing wild-type KATP channel expressed from SUR1 and Kir6.2 subunits at the indicated voltages. (B) Current-voltage relationship for wild-type KATP channels in inside-out membrane patches, together with currents from KATP channels expressed from SUR1 and mutant Kir6.2 subunits as indicated. (C) Single channel currents recorded from an inside-out patch containing a KATP channel expressed from SUR1 and N160D mutant Kir6.2 subunits (top) or N160A mutant Kir6.2 subunits (bottom) at −50 mV. (D) Single channel currents recorded from an inside-out patch containing KATP channels expressed from SUR1 and N160Q mutant Kir6.2 subunits at −50 mV. 5 mM ATP was present for the time indicated by the horizontal bar. The fully closed state (c) and the fully open (75 pS, o) state are indicated. Movement artifacts obscure the record during the switch of the solution.
Model simulations of steady-state [ATP] dose response relationships were generated using Microsoft Excel. Simulations of current relaxations were generated with an adaptive Runge-Kutta method using MathCad5.0+ (Mathsoft Inc.). Wherever possible, data are presented as mean ± SE (standard error). Microsoft Solver was used to fit data by a least-square algorithm.
results
Asparagine 160 in Kir6.2 Is a Critical Pore Lining Residue in KATP Channels
KATP channels in native tissues do not show strong inward rectification, and neither do exogenous KATP channels generated by coexpression of wild-type Kir6.2 and SUR1 subunits (Figs. 1 and 2). In the absence of Mg2+ or polyamines the current-voltage relationship through channels is essentially linear up to about +100 mV. Addition of 20–100 μM spermine, spermidine, or putrescine induces a very weak inward rectification, detectable above about +20 mV (Figs. 1 and 2). Work on cloned inward rectifiers has demonstrated that strong inward rectification is controlled by a pore lining residue in the M2 transmembrane domain (Stanfield et al., 1994; Lu and MacKinnon, 1994) and that the presence of a negative charge at this “rectification controller” position confers strong inward rectification by generation of a high affinity site for the voltage-dependent binding of cytoplasmic polyamines or Mg2+ (Fakler et al., 1994; Ficker et al., 1994; Lopatin et al., 1994). Kir6.2 subunits are homologous to Kir1.1 (ROMK1, Ho et al., 1993) channels in this region of the M2 segment—both subunits contain an asparagine (N160 in Kir6.2) at the “rectification controller” position. Mutation of this residue to a negatively charged aspartate (N160D) or glutamate (N160E) residue results in the expression of KATP channels that rectify strongly in the presence of cytoplasmic polyamines or Mg2+ ions (Figs. 2 and 3). As shown in Fig. 1, single channel conductance is unaltered, and, as shown in Fig. 2, channels are still sensitive to inhibition by high concentration (5 mM) of ATP. Substitution of a neutral glutamine (N160Q, Fig. 2) or alanine (N160A) results in channels that still do not rectify strongly in the presence of spermine. These results suggest that KATP channel pores are lined by Kir6.2 subunits in a directly analogous way to those formed by homologous combinations of Kir1 and Kir2 subfamily subunits.
Figure 2.
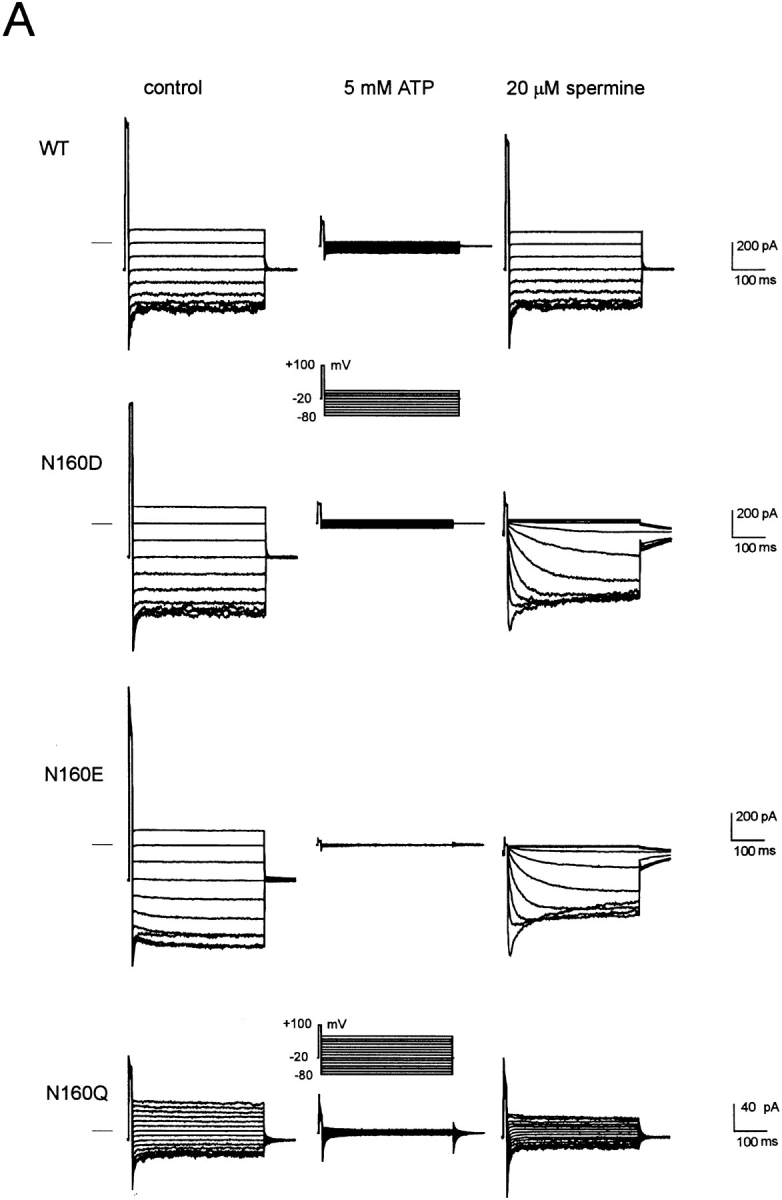

Substitution of negative charge at position 160 (D or E) induces strong inward rectification in the presence of spermine. (A) Representative currents recorded from inside-out membrane patches containing wild-type or mutant KATP channels, with mutations at position 160 in Kir6.2, as indicated. The membrane potential was stepped briefly from a holding potential of −20 to +100 mV and then to voltages between −80 and +10 mV (or +60 mV, N160Q). Currents were recorded in control (left), after inhibiting all KATP current with 5 mM [ATP] (middle), or 20 μM spermine (right). Residual linear conductance in 5 mM ATP is leak current in the patch. (B) Time constant of activation (τACT, spermine unblock) versus membrane potential (mV) for representative currents recorded from inside-out patches containing N160D or N160E mutant channels in the presence of 20 μM spermine.
Figure 3.

Mutation N160D in Kir6.2 induces strong rectification in the presence of Mg2+, as well as di- and trivalent polyamines. Representative currents recorded from inside-out membrane patches containing wild-type (WT, left) or N160D mutant (N160D, right) KATP channels in the absence (c) and presence of 1 mM Mg2+ (Mg), 100 μM putrescine (put), or 100 μM spermidine (spd). Quasi–steady-state current-voltage relationships were obtained by performing 1 s ramps from −100 to +100 mV.
N160D and N160E mutant channels show identical, very slow “activation” kinetics resulting from unblock of spermine (Fig. 2). It has previously been demonstrated that neutralization of the naturally occurring negative charge at this position (D172) in Kir2.1 is insufficient to completely remove polyamine sensitivity (Yang et al., 1995) and that a second region in the COOH terminus (Taglialatela et al., 1994), specifically involving E224 (Yang et al., 1995), is also a contributor to polyamine binding within the channel pore. It seems that within the Kir6.2 pore, a single negative charge at position 160 can introduce an extremely high affinity polyamine binding site, with very slow blocker off-rate (Fig. 2).
Rapid assessment of the blocking ability of other polyamines and Mg2+ was obtained for wild-type (WT) and N160D mutant channels using voltage ramps between −100 and +100 mV (Fig. 3). The slow kinetics of spermine unblock and the slow kinetics of channel deactivation at very negative voltages (see Fig. 2) give rise to different shaped current-voltage (I-V) relationships depending on the direction and speed (V/s−1) of the ramp. In Fig. 3, quasi–steady-state I-Vs were obtained using 1-s ramps. The mutation N160D increases sensitivity of the channels to each of the polyamines and to Mg2+. As shown in Fig. 1, the single channel conductance of each mutant channel is the same, and as shown in Fig. 4 B, the density of current through N160D, N160E, and N160Q mutant channels in patches was comparable to that through wild-type channels. However, although for the neutral substitution N160Q there was a similar rate of 86Rb+ efflux when cells were metabolically poisoned and channels maximally activated (Fig. 4 A), the efflux was reduced to about 40% of wild type for the N160D mutant channels. This reduction of macroscopic conductance of N160D channels can then be explained by suggesting that, at the resting potential of transfected cells (i.e., ∼EK), rectification of the N160D mutant channels is ∼60% complete due to intrinsic polyamine and Mg2+ levels (Shyng et al., 1996; Bianchi et al., 1996). In Rb+ efflux experiments, the flux through N160A mutant channels was also considerably lower than that through wild-type channels. However, the hydrophobic N160A substitution does not induce strong rectification. In this case, the observed density of channels formed by the N160A mutant subunits in excised patches was also considerably lower than that observed with wild type and the other mutant subunits (Fig. 4 B), suggesting channel formation is impaired by this mutation such that the reduced Rb+ efflux is explained by a lower number of channels. Substitution of positively charged arginine (N160R) or histidine (N160H) did not result in measurable KATP conductance in excised membrane patches, although 86Rb+ efflux was slightly increased above the untransfected cell level (Fig. 4 A).
Figure 4.

Expression levels of N160 mutant channels. (A) 86Rb+ efflux from untransfected COSm6 cells and cells expressing SUR1 and N160 mutant Kir6.2 subunits. Graphs show percent Rb+ released into the medium as a function of time in the presence of metabolic inhibitors for a typical experiment. (B) Estimated current density in patch-clamp experiments for wild-type and mutant channels. Bar graph shows mean current at −50 mV (in zero ATP and no added polyamines) ± SEM (n = 14–32 patches) measured over the first 5–10 s after isolation. Inactivation of N160Q channels during this time leads to underestimation of the peak N160Q current by ∼50% (*).
Apparent Nucleotide Sensitivity Is Altered by N160 Mutations
We have recently demonstrated that mutations in the second nucleotide binding fold of SUR1 abolish the channel activating effects of MgADP (Nichols et al., 1996). Mutations at N160 do not abolish the ability of MgADP to activate recombinant channels (not shown), but, unexpectedly, they affect the apparent sensitivity to inhibition by ATP itself. Fig. 5 shows the currents recorded from patches expressing wild-type and mutant channels in response to different [ATP] in the absence of Mg2+. In each case, the patches were exposed to zero ATP before and after the test solutions in order to assess the degree of run-down during the maneuver. Fig. 7 A shows [ATP]-response relationships averaged for all patches in which there was less than 20% run-down during exposure to test solutions. It is clear that certain mutations (N160D, N160Q, and N160E in particular) reduce the apparent sensitivity to inhibition by ATP (shifting the K i from 10.4 μM [N160, wild type] to 46.1, 62.9, and 17.7 μM, respectively). Conversely, the N160A mutation causes a slight shift in K i to lower [ATP] (6.4 μM).
Figure 5.

Mutations at position 160 in Kir6.2 alter [ATP] sensitivity of expressed channels. Representative currents recorded from inside-out membrane patches containing wild type or mutant KATP channels (as indicated) at −50 mV. Patches were exposed to differing [ATP] as indicated.
Figure 7.

N160 mutations shift apparent ATP sensitivity and [ATP]-independent channel open probability. (A) Steady-state dependence of membrane current (relative to current in zero ATP) on [ATP] for wild-type and mutant KATP channels (as indicated). Data points represent the mean ± SEM (n = 3–8 patches). The fitted lines correspond to least-squares fits of the Hill equation (relative current = 100/ {1+([ATP]/K i)H}, with H = 1.8, and K i = 10.4 μM (N160, wild type), 46.1 μM (N160D), 62.9 μM (N160Q), 17.7 μM (N160E), and 6.4 μM (N160A). (B) Relationship between estimated K i and PO(max) (see methods) for wild-type and mutant KATP channels. The curved line is a least squares fit of the empirical equation K i,ATP = 263 · PO(max) 11.
Mutation N160Q Induces a Prominent Inactivated State
Novel and complicating behavior was observed with N160Q mutant channels (Figs. 1 D and 4–6). Current through N160Q channels declines slowly in zero ATP. However, this current is transiently and repeatedly recovered after returning to ATP-containing solution (Fig. 6). This phenomenon is observed in the complete absence of Mg2+, indicating that it represents transition to a reversibly “inactivated” state in zero ATP and distinguishing it from the “run-down” that is observed for wild-type and mutant channels in zero ATP which can only be recovered by exposure to hydrolyzable ATP analogues (Findlay and Dunne, 1986; Ohno-Shosaku et al., 1987; Nichols and Lederer, 1991). The rate of this inactivation was generally well fit by a single exponential in any given patch, and the exponential did not change with time after patch excision (see Fig. 6 B and C). However, there was considerable variation of the time course between patches (see Fig. 6 B) from a time constant of about 1 s to about 18 s (see Fig. 6 C),1 consistent with this process being regulated by the cellular environment such that the rate of inactivation in any given patch is a reflection of the status of the cell from which it was isolated. While we have no immediate physical explanation for the inactivation process, it is curious that N160Q channels also showed prominent subconductance states. As shown in Fig. 1 D, when a patch is moved from high [ATP] to zero [ATP], an initial burst of activity rapidly declines to a steady level where the channel spends a lot of time in subconductance states, the predominant one being about half of the full open state. It is conceivable that the inactivation process actually represents a switch from the full conducting to the partially conducting state.
Figure 6.
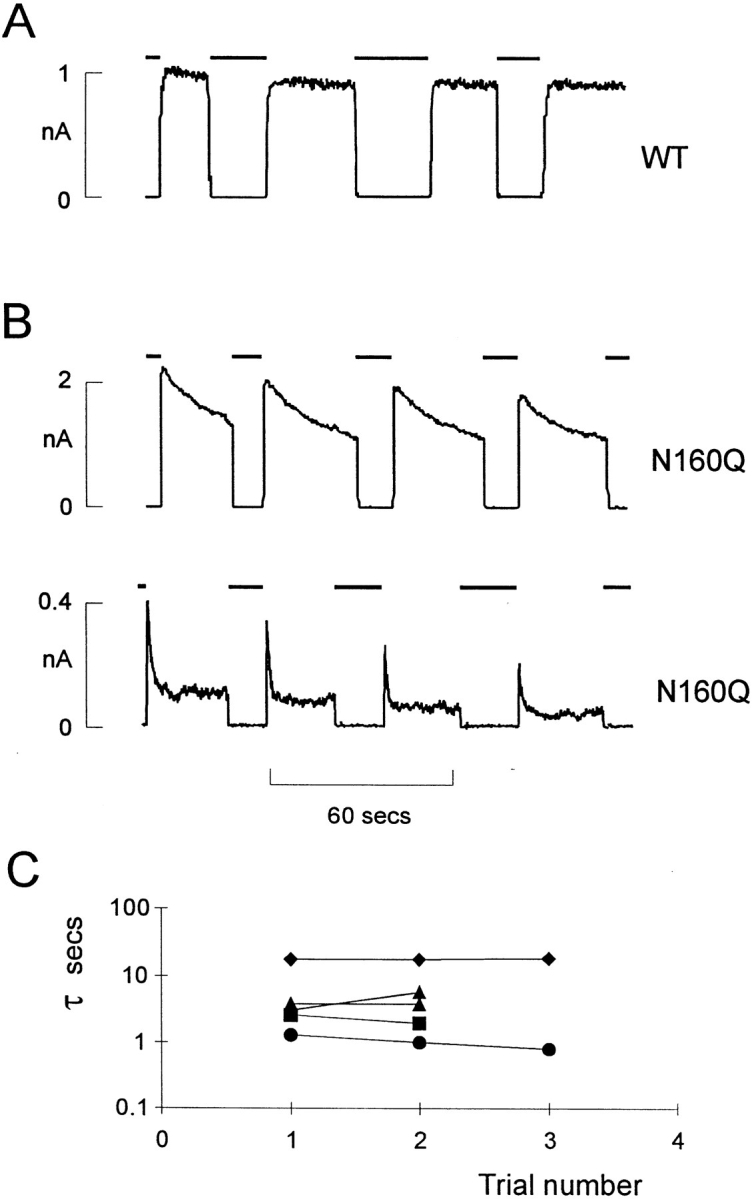
Inactivation of N160Q mutant channels. (A) Representative currents recorded from inside-out membrane patches containing wild-type (A) or N160Q mutant (B) KATP channels (as indicated) at −50 mV. Patches were repeatedly exposed to 5 mM [ATP] as indicated by the bars above the records. In B, examples of both slowly inactivating (top) and rapidly inactivating (bottom) N160Q mutant channels are shown. (C) Time constant (τ) of inactivation of individual N160Q patches versus trial number after removal from 5 mM ATP. The time constant of inactivation does not change appreciably with time after patch excision within any given patch.
N160 Mutations Affect the ATP-independent Open Probability
Are N160 mutations affecting nucleotide binding? Given the homology between Kir6.2 and other ATP-insensitive Kir channel subunits, the lack of reports of permeant ion effects on ATP inhibition, and the lack of consensus nucleotide binding sites on Kir6.2 subunits, it seems a reasonable hypothesis that ATP inhibition of channel activity results from ATP interaction with a site on another protein. Observation of the records in Fig. 1 provides a clue to an alternative explanation for the shifts in K i. N160D mutant channels have a noticeably higher open probability in the absence of ATP and polyamines than do wild-type or N160A channels. If the open state of the channel is stabilized, then the apparent ATP-sensitivity will be shifted for any coupled process, as is the case for cyclic nucleotide sensitivity of cyclic nucleotide–gated channels (Goulding et al., 1994; Gordon and Zagotta, 1995; Varnum et al., 1995). Given the variable density of expressed channels, very few patches contained only one or a few channels, as necessary to directly assess channel open probability. To obtain a quantitative estimate of the channel open probability, we therefore performed noise analysis of short sections of recordings from patches containing typically 10–200 channels (see methods). Samples of currents were recorded immediately after patch excision in zero ATP and after exposure to 5 or 10 mM ATP. As shown in Fig. 7 B, there is a correlation between channel open probability and apparent K i for ATP inhibition when compared between different mutant channels.
A Change in Open Probability Can Account for Shifts in Apparent ATP Sensitivity
We considered the consequences of such changes in open probability on apparent ATP sensitivity by examining a simplified version of a model for the native KATP channel (Nichols et al., 1991). This model and other kinetic models for the KATP channel (Qin et al., 1989) are based on detailed analysis of the kinetic response of endogenous KATP channels to changes of ATP concentration. They assume that multiple, sequential, ATP binding steps move the channel into closed states that are increasingly distant from the open state. In a comprehensive version of the model (Nichols et al., 1991), a total of four sequential binding steps were required to give full agreement with kinetic data. In the present simulations, we have simplified the simulations by reducing the number of ATP binding steps to two, which adequately predicts steady-state [ATP] dependence (Fig. 8). Previous analysis of single channel events (e.g., Qin et al., 1989) have suggested that within bursts there is no dependence of the open duration on [ATP], and hence in the present simulations we placed an additional closed state between the first ATP binding step and the open state (Fig. 8), such that open duration will be independent of [ATP]. The steady-state [ATP] dependence and peak open probability of the wild-type channel are well described by this model (Fig. 8), with the equilibrium constant (K CO) for the allosteric transition between the ATP unbound state (C) and the open state (O) = 5. The steady-state [ATP]-response curves and peak open probability of other mutations can be predicted solely by varying K CO (to values for each mutant of N160D, 80; N160E, 10; N160A, 5). The ability to reproduce the effects of mutations on both K i and peak PO by varying only K CO is consistent with the effect of mutations at position 160 being to alter the free energy of the open state relative to the free energy of closed states, with WT and N160A channel open states being the least stable and N160D being the most stable.
Figure 8.

The gating effects of Kir6.2 N160 mutants can be explained by changes in the stability of the open state. (A) Model scheme for simulation of KATP channel activity. Only two sequential ATP binding steps are necessary to approximate the steepness of steady-state dependence of channel activity on [ATP] (see below). The open state is ATP independent (see text). (B) The steady-state [ATP] dependence of wild-type channels is well described by the model in A, with equilibrium constants as follows: K A = 5 μM, K CO = 5, K OI = 0.1. The ATP dependence of N160D mutant channels is well described by the same model, but with K CO = 80. (C) Model predicted K i versus measured K i (from the graphs in Fig. 7 A). Model predicted K i is the [ATP] causing half-maximal inhibition of steady-state current for curves such as those shown in (B), with adjustments to the wild-type model as follows: wild type (WT), none; N160A, none; N160E, K CO = 10; N160D, K CO = 80; N160Q, K CO = 80, K OI = 1. (D) Modeled PO(max) using adjustments above, versus measured PO(max). Note that there is not agreement between measured and modelled PO for N160Q mutant channels since the significant occupancy of the I state reduces the modelled PO(max) to ∼0.5, although the slow transition rates between the I and O states would not be detectable by the method of experimental measurement of PO (see text).
By assigning specific values to individual rate constants (Fig. 9) we can simulate the time dependence of currents in response to step changes in [ATP]. The model qualitatively reproduces all of the essential behavior of patch currents, solely by changing K CO, corresponding to alterations in the free energy of the open state. As discussed above, the behavior of N160Q mutant channels differs qualitatively from other channels in exhibiting a marked inactivation following channel opening upon exposure to zero ATP (Fig. 6 B). This inactivation is clearly distinguishable from MgATP-dependent run-down (Ohno-Shosaku et al., 1987; Ashcroft, 1988; Kozlowski and Ashford, 1990; Takano et al., 1990; Nichols and Lederer, 1991; Tung and Kurachi, 1991; Furukawa et al., 1994; Hussain and Wareham, 1994; Furukawa et al., 1996) in being recoverable without exposure to Mg2+-containing solutions. We propose that this inactivation results again from alteration in the stability of ATP-independent states. In order to simulate the N160Q mutant channel behavior, we make the additional assumption that K CO for N160Q channels is the same as that for N160D channels, but that the K IO is now also reduced 10-fold from 1.0 to 0.1.
Figure 9.

The model simulates the kinetic behavior observed in excised membrane patches. (A, top) The simulated kinetic scheme. (Bottom) Table showing the model parameters for individual rate constants indicated in the kinetic scheme. Only the rate constants boxed and bold were altered to produce the necessary simulations. (B) Simulated changes in PO, in response to changes in [ATP].
discussion
Asparagine 160 of Kir6.2 Is a Critical Pore Lining Residue in KATP Channels
KATP channels in native tissues do not show strong inward rectification (Ashcroft, 1988; Nichols and Lederer, 1991), and neither do exogenous KATP channels generated by coexpression of Kir6.2 and SUR1 subunits (Figs. 1 and 2). Work on cloned inward rectifiers has demonstrated that strong inward rectification is controlled by a pore lining residue in the M2 transmembrane domain (Lu and MacKinnon, 1994; Stanfield et al., 1994) and that the presence of a negative charge at this “rectification controller” position confers strong inward rectification by generation of a high affinity site for the voltage-dependent binding of cytoplasmic polyamines or Mg2+ (Fakler et al., 1994; Ficker et al., 1994; Lopatin et al., 1994). Kir6.2 subunits are homologous to Kir1.1 (ROMK1, Ho et al., 1993) channels in this region of the M2 segment, both subunits contain an asparagine (N160 in Kir6.2) at the rectification controller position. We have introduced mutations at position 160 in order to determine whether this residue also lines the KATP channel pore and acts as a rectification controller in Kir6.2. Several mutations resulted in expression of functional KATP channels (Figs. 1–5). In each of the four expressed mutations, the single channel conductance of fully open channels was unaffected (Fig. 1), and channels were still sensitive to inhibition by ATP (Fig. 5). Substitution of a neutral glutamine (N160Q) or alanine (N160A) resulted in channels with unchanged rectification properties, but as shown in Fig. 2, mutation of this residue to a negatively charged aspartate (N160D) or glutamate (N160E) residue resulted in the expression of KATP channels that rectify strongly in the presence of cytoplasmic polyamines or Mg2+ ions. These results suggest that KATP channel pores are lined by Kir6.2 subunits in a directly analogous way to those that are formed by Kir subunits without an additional SUR-like subunit. Substitution of positively charged arginine (N160R) or histidine (N160H) did not result in measurable KATP conductance in excised membrane patches, but in Rb efflux experiments we did observe a slight increase in efflux rate compared to untransfected cells (Fig. 4 A), indicating that a small conductance was expressed. Lu and MacKinnon (1994) have previously demonstrated that the equivalent mutation in ROMK1 results in low level expression of permanently rectified channels. It is possible that such is the case here. If so, however, expression may be too low to be detected in patch experiments.
In ROMK1 channels, the mutation N171D (Lu and MacKinnon, 1994) increases sensitivity to block by spermine by ∼3 orders of magnitude. Mutation N160D in Kir6.2 increases spermine sensitivity ∼5 orders of magnitude and leads to a very tight binding of spermine (Fig. 2). The off-rate of spermine from N160D or N160E mutant channels is 10- to 100-fold slower than in other Kir channels containing a negative charge at the rectification controller position. This implies that at least one other, as yet unrecognized, residue must also contribute to stabilizing the binding of spermine in the channel. In all Kir channel subunits examined to date, introduction or removal of a negative charge at this position leads to a significant increase or decrease in the degree of rectification induced by Mg2+ or polyamines (Stanfield et al., 1994; Lu and MacKinnon, 1994; Wible et al., 1994; Lopatin et al., 1994; Ficker et al., 1994; Fakler et al., 1994; Yang et al., 1995), and Reuveny et al. (1996) have further shown that mutation of D172 to asparagine increases the selectivity of Kir2.1 (IRK1) channels to Rb+ compared to K+. Previous investigations have not examined the effects of such mutations on single channel current. The relatively large single channel conductance (∼75 pS, Fig. 1) facilitates such measurements in channels formed by Kir6.2 subunits. It is apparent that even though large changes in rectification result from N160 mutations, single channel conductance is unaltered. This result is consistent with earlier work on voltage-gated K+ channels, which demonstrated that the H5, or P-loop between the 5th and 6th transmembrane regions (S5 and S6, corresponding to M1 and M2 transmembrane regions in Kir subunits) was the major determinant of both K+ selectivity and single channel conductance (Hartmann et al., 1991; Heginbotham et al., 1992; Heginbotham and MacKinnon, 1992; MacKinnon and Yellen, 1990; Yellen et al., 1991; Yool and Schwartz, 1991). Thus, although position 160 in the Kir6.2 M2 segment contributes to the internal entrance to the pore, K+ passage through this region is apparently not the rate limiting step in permeation.
Pore Lining Residues Control Gating of the KATP Channel
Since the Kir6.2 subunit contains no consensus ATP-binding site, it is initially surprising that N160 mutations (that alter the pore structure) should alter the ATP sensitivity of channel activity (Figs. 4 and 5). It is clear that certain mutations (N160D, N160Q, and N160E in particular) reduce the apparent sensitivity to inhibition by ATP, shifting the K i (ATP concentration causing inhibition to half that in zero [ATP]) from 10.4 μM (N160, wild type) to 46.1, 62.9, and 17.7 μM, respectively. However, the correlation between apparent K i and peak open probability in the absence of nucleotides (Fig. 7) is consistent with the idea that the effect of N160 mutations is predominantly to alter the stability of the ATP-independent open state, and thus indirectly affect the efficacy of ATP inhibition, rather than altering ATP binding. Such an effect is thus analogous to those of mutations in cyclic nucleotide–activated ion channels that alter the activating efficacy, but not the binding affinity, of cyclic nucleotides for these channels (Goulding et al., 1994; Gordon and Zagotta, 1995; Varnum et al., 1995; Tibbs et al., 1997), although this coupling of open probability to apparent ligand sensitivity is a previously unconsidered notion with regards to KATP channels, wherein many studies have demonstrated apparent shifts in [ATP] sensitivity without considering the possibility that open probability might be the affected parameter (e.g., Cameron et al., 1988; Findlay and Faivre, 1991). N160D and N160E mutant effects are well explained by assuming that the mutations stabilize the open state, and as shown in Figs. 8 and 9, can be simulated by increasing the equilibrium constant (K CO) of the allosteric transition between the last closed and the open state.
The N160Q mutation was unique in uncovering a marked inactivation following channel opening after removal of ATP. This result can be explained by assuming that the mutation additionally stabilizes an inactivated state that is reached from the open state. The behavior can be simulated by an additional change in the K OI equilibrium constant, with K CO shifted to be approximately equal to that resulting from the N160D mutation (Fig. 8). The physical reality of such alterations is presumably alterations in the stability of the open pore that result from changes in the pore structure. Unfortunately, we are not yet able to construct a consistent picture of the effects of residue substitutions based on the residue side-chain structure. The stabilization of the open state by N160D and N160Q mutations might be consistent with a weak long range electrostatic interaction between N160 and another residue, such that this interaction is strengthened by substitution of a permanent negative charge (N160D) or by elongating the side chain (N160Q). The loss of electrostatic interaction by substitution of a hydrophobic residue (N160A) would lead to destabilization and decreased PO and hence K i, as is observed. The substitution of both an elongated side-chain and a permanent negative charge (N160E) might then be expected to have an even more significant effect. However, although by comparison to wild-type channels the trend towards higher K i and increased PO is continued, the N160E mutant phenotype is not quantitatively as strong as predicted. N160Q mutant channels demonstrate a state-dependent inactivation mechanism that is not seen with native KATP channels or with the other pore mutants examined in this study. Although we presently have no mechanistic explanation for this inactivation, it provides evidence for a change in the pore structure (or at least of some portion of the channel involving Q160 in the Kir6.2 pore) before ATP-gating, such that transition to the inactivated state cannot occur when the channel is in the ATP-bound state. Although there is a present bias towards the assumption that ATP-gating of the channel is essentially due to interaction with the SUR subunit (Inagaki et al., 1995a , 1996; Nichols and Lopatin, 1997), the lack of effect of SUR1 mutations on sensitivity to ATP inhibition (Nichols et al., 1996; Gribble et al., 1997) combined with this interaction between Kir6.2 pore structure and ATP gating might indicate that ATP inhibition is more intimately involved with the Kir6.2 subunit than supposed.
In conclusion, the present results establish that the KATP channel pore is formed from Kir6.2 subunits in an analogous way to the pores formed by other Kir subunits that do not require a SUR-like subunit for channel activity. The results demonstrate an intimate relationship between pore structure and nucleotide gating of the channel, and demonstrate how the stability of the open channel can be critical in determining apparent inhibitory affinity of gating nucleotides. The results have further implications for the interpretation of studies examining the ATP-sensitivity of KATP channels. The simulations demonstrate how it is possible to generate changes in the K i for nucleotide inhibition of current, without altering nucleotide binding itself. Thus, for instance, the difference in ATP sensitivity reported for KATP channels formed from SUR1 or SUR2 subunits (Inagaki et al., 1996) may not necessarily reflect differences in nucleotide binding to the two subunits. Indeed, it is apparent from published records comparing channels formed by coexpression of Kir6.2 with SUR1 and with SUR2 (Inagaki et al., 1996), that intra-burst open probability is higher when channels are formed with the latter sulfonylurea receptor subunit.
Acknowledgments
We are grateful to Dr. S. Seino for providing us with the Kir6.2 clone, and to Dr. Joe Bryan and Jack Clement IV (Baylor College of Medicine) for discussion during the course of this work and for providing us with the N160D mutant of Kir6.2.
This work was supported by grants HL451231 and HL54171 from the National Institutes of Health (C.G. Nichols) and an Established Investigatorship from the American Heart Association (C.G. Nichols).
Footnotes
Address correspondence and reprint requests to C.G. Nichols, Department of Cell Biology and Physiology, Washington University School of Medicine, 660 South Euclid Avenue, St. Louis, Missouri 63110. Fax: 314-362-7463; E-mail: cnichols@cellbio.wustl.edu
This slowly developing inactivation makes it difficult to obtain an accurate steady-state dose-response curve (since in slowly inactivating patches there can be considerable overlap of inactivation and run-down), and hence the dose-response curve for N160Q channels is bell-shaped in Fig. 4 B and not particularly well fit by an empirical Hill equation.
references
- Ashcroft FM. Adenosine 5′-triphosphate-sensitive potassium channels. Annu Rev Neurosci. 1988;11:97–118. doi: 10.1146/annurev.ne.11.030188.000525. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L, Nichols CG, Wechsler SW, Clement JP, IV, Boyd AE, III, Gonzalez G, Herrera-Sosa H, Nguy K, Bryan J, Nelson DA. Cloning of the β-cell high affinity sulfonylurea receptor: a regulator of insulin secretion. Science (Wash DC) 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Bianchi L, Roy ML, Taglialatela M, Lundgren DW, Brown AM, Ficker E. Regulation by spermine of native inward rectifier K+channels in RBL-1 cells. J Biol Chem. 1996;271:6114–6121. doi: 10.1074/jbc.271.11.6114. [DOI] [PubMed] [Google Scholar]
- Cameron JS, Kimura S, Jackson-Burns DA, Smith DB, Bassett AL. ATP-sensitive K+channels are altered in hypertrophied ventricular myocytes. Am J Physiol. 1988;255:H1254–H1258. doi: 10.1152/ajpheart.1988.255.5.H1254. [DOI] [PubMed] [Google Scholar]
- Fabiato A, Fabiato F. Calculator programs for computing the composition of the solutions containing multiple metals and ligands used for experiments in skinned muscle cells. J Physiol (Paris) 1979;75:463–505. [PubMed] [Google Scholar]
- Fakler B, Brandle U, Bond C, Glowatzki E, Konig C, Adelman JP, Zenner HP, Ruppersberg JP. A structural determinant of differential sensitivity of cloned inward rectifier K+channels to intracellular spermine. FEBS Lett. 1994;356:199–203. doi: 10.1016/0014-5793(94)01258-x. [DOI] [PubMed] [Google Scholar]
- Ficker E, Taglialatela M, Wible BA, Henley CM, Brown AM. Spermine and spermidine as gating molecules for inward rectifier K channels. Science (Wash DC) 1994;266:1068–1072. doi: 10.1126/science.7973666. [DOI] [PubMed] [Google Scholar]
- Findlay I, Dunne MJ. ATP maintains ATP-inhibited K+channels in an operational state. Pflüg Archiv. 1986;407:238–240. doi: 10.1007/BF00580683. [DOI] [PubMed] [Google Scholar]
- Findlay I, Faivre JF. ATP-sensitive K channels in heart muscle. Spare channels. FEBS Lett. 1991;279:95–97. doi: 10.1016/0014-5793(91)80259-6. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Virag L, Furukawa N, Sawanobori T, Hiraoka M. Mechanism for reactivation of the ATP-sensitive K+channel by MgATP complexes in guinea-pig ventricular myocytes. J Physiol (Lond) 1994;479:95–107. doi: 10.1113/jphysiol.1994.sp020280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furukawa T, Yamane T, Terai T, Katayama Y, Hiraoka M. Functional linkage of the cardiac ATP-sensitive K+channel to the actin cytoskeleton. Pflüg Archiv. 1996;431:504–512. doi: 10.1007/BF02191896. [DOI] [PubMed] [Google Scholar]
- Gordon SE, Zagotta WN. Localization of regions affecting an allosteric transition in cyclic nucleotide-activated channels. Neuron. 1995;14:857–864. doi: 10.1016/0896-6273(95)90229-5. [DOI] [PubMed] [Google Scholar]
- Goulding EH, Tibbs GR, Siegelbaum SA. Molecular mechanism of cyclic-nucleotide-gated channel activation. Nature (Lond) 1994;372:369–374. doi: 10.1038/372369a0. [DOI] [PubMed] [Google Scholar]
- Gribble FM, Tucker SJ, Ashcroft FM. The essential role of the Walker A motifs of SUR1 in K-ATP channel activation by Mg-ADP and diazoxide. EMBO (Eur Mol Biol Organ) J. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartmann HA, Kirsch GE, Drewe JA, Taglialatela M, Joho RH, Brown AM. Exchange of conduction pathways between two related K+channels. Science (Wash DC) 1991;251:942–944. doi: 10.1126/science.2000495. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, Abramson T, MacKinnon R. A functional connection between the pores of distantly related ion channels as revealed by mutant K+channels. Science (Wash DC) 1992;258:1152–1155. doi: 10.1126/science.1279807. [DOI] [PubMed] [Google Scholar]
- Heginbotham L, MacKinnon R. The aromatic binding site for tetraethylammonium ion on potassium channel. Neuron. 1992;8:483–491. doi: 10.1016/0896-6273(92)90276-j. [DOI] [PubMed] [Google Scholar]
- Ho K, Nichols CG, Lederer WJ, Lytton J, Vassilev PM, Kanazirska MV, Hebert SC. Cloning and expression of an inwardly rectifying ATP-regulated potassium channel. Nature (Lond) 1993;362:31–38. doi: 10.1038/362031a0. [DOI] [PubMed] [Google Scholar]
- Hussain M, Wareham AC. Rundown and reactivation of ATP-sensitive potassium channels (KATP) in mouse skeletal muscle. J Membr Biol. 1994;141:257–265. doi: 10.1007/BF00235135. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, IV, Wang CZ, Aguilar-Bryan L, Bryan J, Seino S. A family of sulfonylurea receptors determines the pharmacological properties of ATP-sensitive K+channels. Neuron. 1996;16:1011–1017. doi: 10.1016/s0896-6273(00)80124-5. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Gonoi T, Clement JP, Namba N, Inazawa L, Gonzalez G, Aguilar-Bryan L, Seino S, Bryan J. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science (Wash DC) 1995a;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- Inagaki N, Tsuura Y, Namba N, Masuda K, Gonoi T, Horie M, Seino Y, Mizuta M, Seino S. Cloning and functional characterization of a novel ATP-sensitive potassium channel ubiquitously expressed in rat tissues, including pancreatic islets, pituitary, skeletal muscle, and heart. J Biol Chem. 1995b;270:5691–5694. doi: 10.1074/jbc.270.11.5691. [DOI] [PubMed] [Google Scholar]
- Kozlowski RZ, Ashford ML. ATP-sensitive K(+)-channel run-down is Mg2+dependent. Proc R Soc Lond B Biol Sci. 1990;240:397–410. doi: 10.1098/rspb.1990.0044. [DOI] [PubMed] [Google Scholar]
- Lederer WJ, Nichols CG. Nucleotide modulation of the activity of rat heart KATPchannels in isolated membrane patches. J Physiol (Lond) 1989;419:193–211. doi: 10.1113/jphysiol.1989.sp017869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopatin AN, Makhina EN, Nichols CG. Potassium channel block by cytoplasmic polyamines as the mechanism of intrinsic rectification. Nature (Lond) 1994;372:366–369. doi: 10.1038/372366a0. [DOI] [PubMed] [Google Scholar]
- Lu Z, Mackinnon R. Electrostatic tuning of Mg2+ affinity in an inward rectifier K+channel. Nature (Lond) 1994;371:243–246. doi: 10.1038/371243a0. [DOI] [PubMed] [Google Scholar]
- MacKinnon R, Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+channels. Science (Wash DC) 1990;250:276–279. doi: 10.1126/science.2218530. [DOI] [PubMed] [Google Scholar]
- Neher E, Stevens CF. Conductance fluctuations and ionic pores in membranes. Ann Rev Biophys Bioeng. 1977;6:345–381. doi: 10.1146/annurev.bb.06.060177.002021. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ. ATP-sensitive potassium channels in the cardiovascular system. Am J Physiol. 1991;261:H1675–H1686. doi: 10.1152/ajpheart.1991.261.6.H1675. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Lederer WJ, Cannell MB. The ATP- dependence of KATPchannel kinetics in isolated membrane patches from rat ventricle. Biophys J. 1991;60:1164–1177. doi: 10.1016/S0006-3495(91)82152-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols CG, Lopatin AN. Inward rectifier potassium channels . Annu Rev Physiol. 1997;59:171–191. doi: 10.1146/annurev.physiol.59.1.171. [DOI] [PubMed] [Google Scholar]
- Nichols CG, Shyng S-L, Nestorowicz A, Glaser A, Clement JP, IV, Gonzales G, Aguilar-Bryan L, Permutt AM, Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science (Wash DC) 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Ohno-Shosaku T, Zunkler BJ, Trube G. Dual effects of ATP on K+currents of mouse pancreatic β-cells. Pflüg Archiv. 1987;408:133–138. doi: 10.1007/BF00581342. [DOI] [PubMed] [Google Scholar]
- Qin DY, Takano M, Noma A. Kinetics of ATP-sensitive K+channel revealed with oil-gate concentration jump method. Am J Physiol. 1989;257:H1624–H1633. doi: 10.1152/ajpheart.1989.257.5.H1624. [DOI] [PubMed] [Google Scholar]
- Reuveny E, Jan YN, Jan LY. Contributions of a negatively charged residue in the hydrophobic domain of the IRK1 inwardly rectifying K+channel to K(+)-selective permeation. Biophys J. 1996;70:754–761. doi: 10.1016/S0006-3495(96)79615-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng SL, Sha Q, Ferrigni T, Lopatin AN, Nichols CG. Depletion of intracellular polyamines relieves inward rectification of potassium channels. Proc Natl Acad Sci USA. 1996;93:12014–12019. doi: 10.1073/pnas.93.21.12014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth FJ. The variance of sodium current fluctuations at the node of Ranvier. J Physiol (Lond) 1980;307:97–129. doi: 10.1113/jphysiol.1980.sp013426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanfield PR, Davies NW, Shelton PA, Sutcliffe MJ, Khan IA, Brammar WJ, Conley EC. A single aspartate residue is involved in both intrinsic gating and blockage by Mg2+of the inward rectifier, IRK1. J Physiol (Lond) 1994;478:1–6. doi: 10.1113/jphysiol.1994.sp020225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taglialatela M, Wible BA, Caporoso R, Brown AM. Specification of the pore properties by the carboxyl terminus of inwardly rectifying K+channels. Science (Wash DC) 1994;264:844–847. doi: 10.1126/science.8171340. [DOI] [PubMed] [Google Scholar]
- Takano M, Qin DY, Noma A. ATP-dependent decay and recovery of K+channels in guinea pig cardiac myocytes. Am J Physiol. 1990;258:H45–H50. doi: 10.1152/ajpheart.1990.258.1.H45. [DOI] [PubMed] [Google Scholar]
- Tibbs GR, Goulding EH, Siegelbaum SA. Allosteric activation and tuning of ligand efficacy in cyclic-nucleotide-gated channels. Nature (Lond) 1997;386:612–615. doi: 10.1038/386612a0. [DOI] [PubMed] [Google Scholar]
- Tung RT, Kurachi Y. On the mechanism of nucleotide diphosphate activation of the ATP-sensitive K+channel in ventricular cell of guinea-pig. J Physiol (Lond) 1991;437:239–256. doi: 10.1113/jphysiol.1991.sp018593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnum MD, Black KD, Zagotta WN. Molecular mechanism for ligand discrimination of cyclic nucleotide-gated channels. Neuron. 1995;15:619–625. doi: 10.1016/0896-6273(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Wible BA, Taglialatela M, Ficker E, Brown AM. Gating of inwardly rectifying K+channels localized to a single negatively charged residue. Nature (Lond) 1994;371:246–249. doi: 10.1038/371246a0. [DOI] [PubMed] [Google Scholar]
- Yang J, Jan YN, Jan LY. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+channel. Neuron. 1995;14:1047–1054. doi: 10.1016/0896-6273(95)90343-7. [DOI] [PubMed] [Google Scholar]
- Yellen G, Jurman ME, Abramson T, MacKinnon R. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+channel. Science (Wash DC) 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]
- Yool AJ, Schwartz TL. Alteration of ionic selectivity of a K+channel by mutation in the H5 region. Nature (Lond) 1991;349:700–704. doi: 10.1038/349700a0. [DOI] [PubMed] [Google Scholar]