

Abstract
Calcium binding and signaling orchestrate a wide variety of essential cellular functions, many of which employ the EF-hand Ca2+ binding motif. The ion binding parameters of this motif are controlled, in part, by the structure of its Ca2+ binding loop, termed the EF-loop. The EF-loops of different proteins are carefully specialized, or fine-tuned, to yield optimized Ca2+ binding parameters for their unique cellular roles. The present study uses a structurally homologous Ca2+ binding loop, that of the Escherichia coli galactose binding protein, as a model for the EF-loop in studies examining the contribution of the third loop position to intramolecular tuning. 10 different side chains are compared at the third position of the model EF-loop with respect to their effects on protein stability, sugar binding, and metal binding equilibria and kinetics. Substitution of an acidic Asp side chain for the native Asn is found to generate a 6,000-fold increase in the ion selectivity for trivalent over divalent cations, providing strong support for the electrostatic repulsion model of divalent cation charge selectivity. Replacement of Asn by neutral side chains differing in size and shape each alter the ionic size selectivity in a similar manner, supporting a model in which large-ion size selectivity is controlled by complex interactions between multiple side chains rather than by the dimensions of a single coordinating side chain. Finally, the pattern of perturbations generated by side chain substitutions helps to explain the prevalence of Asn and Asp at the third position of natural EF-loops and provides further evidence supporting the unique kinetic tuning role of the gateway side chain at the ninth EF-loop position.
Keywords: calcium signaling, calmodulin, troponin C, metal binding site, ion channels
introduction
Numerous proteins use the ubiquitous EF-hand Ca2+ binding motif to coordinate diverse cellular functions ranging from muscle control and neuronal signaling to cell cycle control (for reviews see Ikura, 1996; Bootman and Berridge, 1995; Clapham, 1995; Ghosh and Greenberg, 1995). Such widespread use arises, in part, from the tunable architecture of the motif, which has enabled its Ca2+ binding parameters to be specialized by evolution for different biological roles. Recent studies have begun to define general molecular mechanisms used by the motif to tune Ca2+ binding affinity, selectivity, and kinetics over ranges of 103-fold or more (reviewed by Linse and Forsén, 1995; Falke et al., 1994). For example, several laboratories have shown that the Ca2+ parameters of the motif can be modulated by isolated substitutions altering one or more coordinating side chains (Drake et al., 1996; Henzl et al., 1996; DaSilva et al., 1995; Procyshyn and Reid, 1994; Renner et al., 1993; Maune et al., 1992; Negele et al., 1992; Babu et al., 1992; Palmisano et al., 1990; MacManus et al., 1989), while other investigators have demonstrated that regions outside the coordination site can also play an important role in intramolecular tuning (Trigo-Gonzalez et al., 1993; Monera et al., 1992; Linse et al., 1991; Reid et al., 1981). The present study investigates the tuning contribution of a specific coordinating side chain, namely that provided by the third position of the EF-loop. Ultimately, the molecular principles elucidated by studies of Ca2+ binding sites of known structure are likely to have relevance for other important classes of sites sharing the same general Ca2+ coordination chemistry, including the essential sites of Ca2+ channels and pumps (Chen et al., 1996; Carafoli et al., 1996).
The canonical EF-hand motif is an independent folding domain containing two coupled Ca2+ binding sites, each exhibiting a helix-loop-helix architecture where the semi-conserved nine-residue loop, termed the “EF-loop,” provides five of the six coordinating residues (at loop positions 1,3,5,7,9) as illustrated in Fig. 1 A. The sixth coordinating residue lies on the trailing helix, three residues beyond the COOH terminus of the EF-loop (Kretsinger and Nockolds, 1973; reviewed by Linse and Forsén, 1995; Falke et al., 1994; Kawasaki and Kretsinger, 1994; Skelton et al., 1994; Marsden et al., 1990; and Strynadka and James, 1989). All of the 45 known EF-hand crystal structures display a pentagonal bipyramidyl coordination geometry in which the seven coordinating oxygens are arranged around the bound Ca2+ ion (Falke et al., 1994; Kawasaki and Kretsinger, 1994; Strynadka and James, 1989). The side chain of EF-loop position 9 lies at an axial coordinating position where it modulates the major pathway for ion binding and release and has therefore been termed the gateway side chain (Drake and Falke, 1996; Renner et al., 1993). Positions 3, 5, and 7 provide three of the five planar coordinating oxygens while the remaining two planar oxygens are donated by the side chain carboxylate of a conserved glutamate lying outside to the EF-loop, which serves as the only bidentate ligand in the site. This bidentate carboxylate appears to play an important role in Ca2+ versus Mg2+ selectivity, and in the triggering of Ca2+-induced conformational changes (DaSilva et al., 1995; Declercq et al., 1991; Gagne et al., 1997). Finally, below the planar pentagon of coordinating oxygens lies the buried axial oxygen provided by EF-loop position 1.
Figure 1.


(A) The model EF-loop of GBP, including a comparison to a canonical EF-loop. Shown in stereo are the superimposed nine-residue Ca2+ binding loop backbones of GBP (solid bonds ; Vyas et al., 1987) and EF-hand site IV of human calmodulin (fine bonds; Chattopadhyaya et al., 1992). Also depicted are the coordinating side chains for each site, and, for the GBP site, the coordinating oxygens (small spheres) and bound Ca2+ ion (large sphere). The external glutamates of the two sites (n), which provide bidentate coordination in each site, originate from different sequence positions but display the same spatial location. In the GBP site, as in some canonical EF-hand sites (reviewed by Drake and Falke, 1996), the side chain at EF-loop position nine is a Gln or Glu residue that provides direct Ca2+ coordination at the solvent-facing axial position. By contrast, many canonical sites, including site IV of calmodulin, exhibit coordination by a solvent oxygen (not shown) at this position. Overall, the observed differences between the GBP loop and CaM loop IV fall within the range of differences observed between canonical EF-hand loops. (B) Crystal structures illustrating the three most common types of Ca2+ coordination by the third loop position. Shown are CPK representations of the bound Ca2+ ion (center), the surrounding seven oxygens forming the pentagonal bipyramidal coordination array (medium gray), and the position 3 side chain (carbons are light gray, nitrogen is dark gray). Position 3 coordination by Asn is illustrated by the galactose binding protein (GBP ; Vyas et al., 1987); Asp is exemplified by site I of parvalbumin (Parv; Kumar et al., 1990), and Ser is represented by site III of sarcoplasmic Ca2+ binding protein (Sarc ; Cook et al., 1993). (In the Sarc structure, the water molecule providing coordination at the upper axial position is not present in the crystallographic coordinates.)
In natural EF-loops, the planar position 3 is one of the variable positions exhibiting a range of different side chains as expected for a role in EF-hand tuning (Linse and Forsén, 1995; Falke et al., 1994; Strynadka and James, 1989), but a systematic study of the effects of various side chains at position 3 on the ion binding parameters of a given site has not yet been carried out. Table I summarizes an analysis of the current EF-hand database (Kawasaki and Kretsinger, 1994) indicating that evolution has placed at least 17 different side chains at the third EF-loop position, although only six (Asp, Asn, Glu, Gly, Thr, and Ser) are present there at frequencies of 2% or greater in the 1,073 canonical EF-hand sequences. The same six side chains are most prevalent in both major sub-groups of the canonical sequences: (a) the “classical” sub-group of 658 sequences sharing primary structure elements known to promote high affinity Ca2+ binding, and (b) the remaining 415 “nonclassical” sequences lacking one or more of these elements (Table I; reviewed by Kretsinger, 1996; Kawasaki and Kretsinger, 1994; Falke et al., 1994). In the classical sub-group the two most common residues, Asp and Asn, greatly predominate over the other side chains, together accounting for over 97% of the third position residues (Table I). The next most prevalent side chain, Ser, is found in <1% of the classical sites. The reasons for this conservation are unclear, as are the types of tuning which may be provided by different side chains at position 3. Crystallographic structures have been obtained for sites possessing three different position 3 side chains: Asn, Asp and Ser, as illustrated in Fig. 1 B.
Table I.
Amino Acids Observed at Position 3 of Canonical EF-Loops
| Amino acid | Full EF-hand database* | Classical EF-hand motif‡ | Kretsinger consensus§ | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Asp | 596 | 55.5% | 482 | 73.3% | 417 | 80.3% | ||||||
| Asn | 182 | 17.0% | 161 | 24.5% | 102 | 19.7% | ||||||
| Glu | 96 | 8.9% | 2 | 0.3% | ||||||||
| Gly | 45 | 4.2% | 2 | 0.3% | ||||||||
| Thr | 30 | 2.8% | 3 | 0.5% | ||||||||
| Ser | 21 | 2.0% | 6 | 0.9% | ||||||||
| Met | 19 | 1.8% | ||||||||||
| Cys | 16 | 1.5% | ||||||||||
| Ala | 14 | 1.3% | ||||||||||
| Leu | 11 | 1.0% | ||||||||||
| Lys | 10 | 0.9% | 1 | 0.1% | ||||||||
| Gln | 9 | 0.8% | ||||||||||
| Ile | 7 | 0.6% | ||||||||||
| Arg | 6 | 0.6% | 1 | 0.1% | ||||||||
| Pro | 5 | 0.5% | ||||||||||
| Val | 5 | 0.5% | ||||||||||
| Tyr | 1 | 0.1% | ||||||||||
| Phe | ||||||||||||
| His | ||||||||||||
| Trp | ||||||||||||
| Total | 1,073 | 658 | 519 | |||||||||
Database courtesy of Dr. R.H. Kretsinger (Kawasaki and Kretsinger, 1994).
Defined by the previously described consensus algorithim (Prosite algorithm; Falke et al., 1994) except that position 3 is allowed to vary freely.
Defined by the Kretsinger consensus algorithm for functional EF-hands (Linse and Forsen, 1995).
Several previous studies have substituted nonclassical residues (Ala, Lys, and Glu) for Asp or Asn at the third EF-loop position of various sites (Dotson and Putkey, 1993; Linse and Chazin, 1995; Sage et al., 1995; Babu et al., 1992). All of these substitutions either eliminated or dramatically reduced Ca2+ binding or function, thereby demonstrating the importance of the third EF-loop position to Ca2+ coordination and activity. On the other hand, studies exchanging Asp for Asn at the third EF-loop positions of three different EF-hand proteins have increased, decreased, or had little effect on Ca2+ affinity (Henzl et al., 1996; Waltersson et al., 1993; Babu et al., 1992), suggesting that such substitutions are context dependent.
The present study systematically compares substitutions at the third EF-loop position of the E. coli galactose binding protein (GBP).1 The GBP site, which serves to stabilize the folded protein in the proteolytic environment of the periplasmic space, exists as a single independent site that lacks the multi-site cooperativity of most EF-hand proteins (Falke et al., 1994, 1991; Snyder et al., 1990; Vyas et al., 1987). Other differences from the EF-hand motif include a Ca2+ binding loop that bridges an α-helix and a β-strand rather than the canonical pair of α-helices. In addition, the bidentate glutamate of the coordinating array is provided by a sequence position 63 residues, rather than 3 residues, beyond the loop COOH terminus. Such an arrangement allows Ca2+ binding to provide maximal protein stabilization by bridging distal regions of the primary structure. Despite the fact that the GBP site is not an EF-hand site, its ion binding parameters fall within the range exhibited by canonical EF-hands, and its ion binding cavity utilizes the same pentagonal bipyramid coordination scheme as the canonical sites. Moreover, the structure of the nine-residue GBP Ca2+ binding loop is strikingly homologous to that of the canonical EF-loop, which shares the same backbone conformation and places the coordinating side chains in the same spatial location (Snyder et al., 1990; Vyas et al., 1987). The close correspondence between the GBP Ca2+ binding loop and a typical EF-loop is illustrated in Fig. 1 A, which presents an overlay of the GBP loop (Vyas et al., 1987) on that of site IV from human calmodulin (Chattopadhyaya et al., 1992).
The GBP Ca2+ binding loop is useful as a model system in which to probe the ion binding characteristics of canonical EF-loops, due both to its EF-loop-like structure and its unique experimental advantages. The simplicity provided by the single-site nature of the GBP system facilitates quantitative studies of metal ion binding to an isolated loop, thereby eliminating the complexities encountered in cooperative, multi-site systems. Moreover, GBP contains five tryptophans including one, Trp127, located within 5 Å of the bound metal ion. The indole ring of Trp127 can be used to excite bound Tb3+ ion in a sensitive fluorescence energy transfer assay for site occupancy. Upon excitation, the indole transfers its energy to the nearby Tb3+ ion, which re-emits the energy via phosphorescence (Snyder et al., 1990). Previous studies have shown that Tb3+ is a functional replacement for Ca2+ in EF-hand proteins (Horrocks and Albin, 1984; Brittain et al., 1976; Moews and Kretsinger, 1975) because Tb3+ and Ca2+ both possess spherical, filled outer electronic subshells and are similar in size (the effective ionic radii of Tb3+ and Ca2+ are 0.98 and 1.06 Å, respectively). The efficiency of Trp-to-Tb3+ energy transfer decreases rapidly with distance (Stryer and Haugland, 1967; Horrocks, 1993). As a result, only the Tb3+ that is actually bound in the site is capable of accepting the energy transfer and emitting a signal.
In GBP, the native residue at the third position of the Ca2+ binding loop is Asn. Here we replace Asn with other side chains in order to compare the six most prevalent position 3 residues (Asp, Asn, Gln, Gly, Thr, and Ser) as well as four found less frequently in natural sequences (Glu, Val, Cys, and Ala; Table I). These substitutions generate a wide range of side chain sizes, charges, and hydrophobicities at the third position. The effects of the substitutions on metal binding affinity, selectivity, and kinetics indicate that (a) incorporation of a side chain carboxylate can dramatically shift the charge selectivity of the site toward trivalent cations, as predicted by the electrostatic repulsion model of charge selectivity. In addition, the results (b) yield further information regarding the mechanism of size selectivity, (c) suggest a model for the observed side chain preferences at position 3 of canonical EF-loops, and (d) provide further support for the unique kinetic tuning role played by the gateway residue at the ninth loop position.
materials and methods
Protein Engineering, Purification, and Characterization
GBP substitutions were engineered via a standard Kunkel-based mutagenesis protocol (Falke et al., 1991), and protein was purified and characterized as previously described (Drake and Falke, 1996). Protein expression was screened by growth in standard media containing 1 mM each of added MgCl2, CaCl2, and SrCl2 followed by expression analysis on SDS-PAGE. Mutants were deemed nonexpressers if they yielded expression levels <5% that of wild type. Protein masses were measured by a Sciex API-III triple-quadropole electrospray mass spectrometer to confirm that each protein contained only the desired single point mutation. Thermal melting curves and galactose binding assays were used to characterize the protein stability and functionality, respectively. Detailed descriptions of these procedures are published elsewhere (Drake and Falke, 1996).
Ion Affinities and Dissociation Rates
The affinities of wild-type and mutant proteins for Tb3+, as well as the dissociation rates of bound Tb3+ were measured using the previously described Tb3+ fluorescence energy transfer assay (Drake and Falke, 1996; Drake et al., 1996). Only the N136D substitution possessed a sufficiently high Tb3+ affinity to require measurement by the Indo-1 competition assay (Drake and Falke, 1996). The binding affinities of other metal ions were determined by competitive displacement of the bound Tb3+ fluorescence, which yielded an apparent dissociation constant (K Dapp) that was corrected for the Tb3+ competition to yield the true dissociation constant (K D) (Drake et al., 1996). Tb3+ dissociation rates were measured by monitoring the time course of Tb3+ release following rapid mixing with excess EDTA (Drake and Falke, 1996). For technical reasons, it is not possible to measure the Ca2+ dissociation rate of the GBP site (Drake and Falke, 1996). Steady state and rapid mixing fluorescence experiments were carried out using an SLM 48000S fluorescence spectrometer equipped with a stopped-flow module.
Molecular Graphics
Molecular images were displayed using the Insight II program (Biosym, v. 2.3.5) running on a Silicon Graphics Personal Iris 4D/35. Structural coordinates were retrieved from the Brookhaven Protein Data Bank (PDB). The relevant PDB identification codes are GBP = 2gbp, sarcoplasmic Ca2+ binding protein = 2scp, parvalbumin = 4cpv.
results
Protein Characterization
Table II presents the 10 residues compared in the present study of the GBP Ca2+ binding loop, all located at position 3 of this model EF-loop. The 10 residues include the native Asn and nine engineered side chains. Four of the engineered side chains (Glu, Cys, Ala, Gly) were found to block protein expression, most likely because the resulting mutant proteins were not stably folded which leads to degradation in the proteolytic environment of the periplasm. Similar inhibition of expression has been previously seen for several disallowed substitutions at the ninth position of the model EF-loop (Drake and Falke, 1996). The remaining five engineered side chains yielded proteins that were successfully overexpressed and purified. The molecular masses of these five mutant proteins were confirmed by electrospray mass spectrometry (Table II).
Table II.
Substitutions at Position 3 of the GBP Ca2+ Binding Loop: Characterization of Mutant Proteins
| Site | Residue | Side chain volume* | Expression | Protein Mass | Tm | K D(Gal) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Predicted | Measured | |||||||||||||
| (Å3) | (D) | (±4 D) | (°C) | (μM) | ||||||||||
| N136Q | Gln | 66 | + | 33,383 | 33,381 | 50.4 | 0.2 ± 0.1 | |||||||
| N136E | Glu | 61 | − | — | — | — | — | |||||||
| N136V | Val | 57 | + | 33,353 | 33,353 | 52.8 | 0.14 ± 0.08 | |||||||
| WT(N136) | Asn | 48 | + | 33,368 | 33,370 | 58.5 | 0.5 ± 0.1 | |||||||
| N136T | Thr | 45 | + | 33,355 | 33,354 | 53.2 | 0.8 ± 0.4 | |||||||
| N136D | Asp | 43 | + | 33,369 | 33,368 | 55.2 | 0.8 ± 0.2 | |||||||
| N136C | Cys | 38 | − | — | — | — | — | |||||||
| N136S | Ser | 25 | + | 33,341 | 33,340 | 53.4 | 0.17 ± 0.02 | |||||||
| N136A | Ala | 19 | − | — | — | — | — | |||||||
| N136G | Gly | 0 | − | — | — | — | — | |||||||
Sugar binding assays demonstrated that all five of the isolated GBP mutants retain near wild-type affinity for D-galactose. Three of the five mutants exhibit slightly higher D-galactose affinities than the wild-type protein, while the affinities of the remaining two mutants are reduced less than twofold as summarized in Table II. Such native activities imply that each of the isolated mutants retain a native conformation. Substitutions to coordinating positions of the Ca2+ binding site, which serves to stabilize GBP structure, are expected to decrease the melting temperature (Tm) and, as shown in Table II, all five of the isolated mutations decreased Tm approximately 4 ± 1°C except Gln, which decreased Tm by 8 ± 1°C. Similar stability losses were observed for substitutions at the gateway coordinating position (Drake and Falke, 1996). Despite these minor effects on stability, each of the expressed proteins retains a Tm exceeding 50°C and is stable and functional under the relevant experimental conditions.
Effects of Substitutions on Metal Binding Equilibria
To probe the effects of substitutions on the ion binding affinity and selectivity of the model EF-hand motif, equilibrium dissociation constants (K D) were measured for 24 spherical cations from groups Ia (Li+, Na+, K+, Rb+); IIa (Mg2+, Ca2+, Sr2+, Ba2+); IIIa (Sc3+, Y3+, La3+); and the lanthanides (Ce3+, Pr3+, Nd3+, Sm3+, Eu3+, Gd3+, Tb3+, Dy3+, Ho3+, Er3+, Tm3+, Yb3+, Lu3+). These spherical cations, which all possess filled outer electronic subshells, together provide a range of ionic radii and charges with which to probe the size and electrostatic features of the coordination site. For a given wild type or engineered site, the Tb3+ competition assay was used to measure the dissociation constant (K D) of each spherical cation as summarized in Table III, enabling a plot of binding free energy versus ionic radius for cations of identical charge. Figs. 2 and 3 display the resulting size selectivity profiles for divalent and trivalent cations, respectively. Monovalent cations yielded no detectable binding to any of the sites.
Table III.
Substitutions at Position 3 of the GBP Ca2+ Binding Loop: Effects on Metal Binding and Dissociation*
| Site | Sidechain | k off(Tb3+) | K D(Tb3+) | K D(Ca2+) | ||||
|---|---|---|---|---|---|---|---|---|
| (s−1) | (μM) | (μM) | ||||||
| N136D | Asp | 8.8 ± 0.6 × 10−5 | 0.03 ± 0.02 | >600 | ||||
| WT(N136) | Asn | 6.41 ± 0.03 × 10−3 | 3.0 ± 0.9 | 1.6 ± 0.4 | ||||
| N136S | Ser | 8.46 ± 0.09 × 10−2 | 0.7 ± 0.1 | 3.7 ± 0.5 | ||||
| N136T | Thr | 9.28 ± 0.06 × 10−2 | 0.6 ± 0.1 | 13.3 ± 0.9 | ||||
| N136V | Val | 9.4 ± 0.2 × 10−2 | 0.5 ± 0.3 | 10.3 ± 0.4 | ||||
| N136Q | Gln | 2.21 ± 0.04 × 100 | 3.0 ± 0.7 | 26 ± 3 |
Errors are standard deviation for n ≥ 7.
Figure 2.

Binding free energy as a function of divalent cation size: effect of side chain substitutions at position 3 of the GBP Ca2+ binding loop. Plotted against effective ionic radius are binding free energies for divalent cations of group IIa. Each panel shows data for the native site (open circles, fine curve) and, excepting the first panel, for a site possessing an engineered side chain at the third loop position (filled circles, bold curve). The binding free energies were calculated as ΔGo = RT ln(K D), while effective ionic radii are those of Shannon (1976) for sevenfold coordination. Group IIa cations used were, in order of increasing radius: Mg2+, Ca2+, Sr2+, Ba2+. Dashed curves indicate regions of the free energy profile which were incompletely determined by the available radii. Lower limits (up arrows) indicate ions which yielded less than 50% displacement of Tb3+ at their maximum attainable concentrations in the Tb3+ competition assay (see materials and methods). An error bar is indicated for the largest error in each data set, calculated as RT ln(K D ± 1 standard deviation). All measurements were at 25°C with 2.5 μM protein, 100 mM KCl, and 10 mM PIPES, pH 6.0.
Figure 3.

Binding free energy as a function of trivalent cation size: effect of side chain substitutions at position 3 of the GBP Ca2+ binding loop. Legend as for Fig. 2, except that the indicated binding free energies are for trivalent cations from group IIIa (triangles) and the lanthanides (circles). In order of increasing radius, these were: Sc3+, Lu3+, Yb3+, Tm3+, Er3+, Ho3+, Y3+, Dy3+, Tb3+, Gd3+, Eu3+, Sm3+, Nd3+, Pr3+, Ce3+, and La3+.
The effects of substitutions on the ionic charge selectivity of the site can be divided into two classes. The acidic Asp substitution dramatically shifted the ionic charge selectivity toward trivalent cations by decreasing the Ca2+ affinity at least 360-fold, while increasing the Tb3+ affinity 100-fold (Table III). The latter Tb3+ affinity enhancement nearly matches the 70-fold slowing of the Tb3+ dissociation kinetics caused by this same substitution (see below), indicating that most of the affinity enhancement is due to a slower Tb3+ off-rate. By contrast, the neutral Gln, Val, Thr, and Ser substitutions had only small effects on charge selectivity. These neutral substitutions either left the Tb3+ affinity unchanged or increased it by no more than 6-fold, while decreasing the Ca2+ affinity from a minimum of 2.3-fold (Ser) up to a maximum of 16-fold (Gln). Thus, the charge selectivity of the model EF-loop is highly sensitive to the incorporation of a new acidic coordinating side chain but is relatively unperturbed by the substitution of one neutral side chain for another.
In general, the position 3 substitutions caused remarkably similar changes in the ionic size selectivity, as illustrated by the size selectivity profiles of Figs. 2 and 3. For divalent cations, the size selectivity profiles retained an optimal radius near that of Ca2+, although the size selectivity was slightly weakened due to decreased Ca2+ affinity or, in some instances, enhanced affinity for the larger Ba2+ ion. The one exception was the acidic Asp substitution, which indiscriminately eliminated the high affinity binding of divalent cations. For trivalent cations, each of the position 3 substitutions generated a new local minimum in the size selectivity profile, where the new optimal size is increased in radius by as much as 0.1 Å. There was also a general loss of trivalent ion size selectivity, as evidenced by the flattened shapes of the deformed free energy wells (Figs. 2 and 3). Overall, the observed effects of position 3 substitutions on ionic charge and size selectivity are quite similar to those previously observed for substitutions at the ninth EF-loop position (Drake et al., 1996).
Effects of Substitutions on Metal Dissociation Rates
All of the position 3 substitutions significantly altered the rate of Tb3+ dissociation, as seen in Table III. The observed effects can again be divided into two classes: substitution of the native Asn with the acidic side chain Asp generated 70-fold slower Tb3+ dissociation, consistent with the increased Tb3+ affinity observed for this mutant (Table III). In contrast, substitution with a neutral side chain (Gln, Val, Thr, Ser) yielded faster Tb3+ dissociation rates. Of the neutral side chain substitutions, Val, Thr, and Ser speeded the dissociation by similar factors of approximately 14-fold, while the Gln substitution had a much larger effect, yielding 340-fold more rapid dissociation. Interestingly, the Val, Thr, and Ser substitutions increase the Tb3+ affinity while speeding Tb3+ dissociation kinetics and weakening the trivalent ion size selectivity (Table III, Fig. 3). One explanation for this counterintuitive result is that the Val, Thr, and Ser substitutions may enhance the conformational flexibility of the coordination site, thereby allowing (a) better accommodation of differently sized ions, (b) more rapid rearrangement of the site during ion binding and dissociation, and (c) an improved ability to adopt an optimal coordination structure for the Tb3+ ion.
discussion
Implications for Ionic Charge Selectivity
The substitution of an acidic residue for a neutral residue by the N136D modification confirms the predictions of the electrostatic repulsion model of ionic charge selectivity in Ca2+ binding sites (Drake et al., 1996; Falke et al., 1994; Falke et al., 1991). This model proposes that excess electrostatic repulsion between coordinating oxygens will destabilize the closed conformation of the occupied site, in which the coordinating oxygens are in closer proximity than in the unoccupied site (Falke et al., 1994), unless the occupied site contains a cation of sufficient positive charge. The model thereby explains the charge discrimination universally observed for known EF-hand sites, which exclude monovalent cations but bind multivalent spherical cations as required by their cellular functions in high background concentrations of Na+ and K+ (Linse and Forsén, 1995; Falke et al., 1994). The same general principle of charge selectivity is exhibited by organic chelators of the EDTA family, which also provide multiple, negatively charged oxygens for chelation, yielding exclusion of monovalent cations while divalent and trivalent cations are bound with high affinities (Falke et al., 1994; Christensen et al., 1975).
The electrostatic repulsion model predicts that the excess negative charge density triggered by the substitution of too many acidic side chains into the coordinating array will require additional positive charge to stabilize the occupied site, thereby shifting the charge selectivity toward higher cation charges. Substitution of an acidic side chain at the ninth EF-loop position (Q142E) was previously observed to yield a 3,000-fold shift toward trivalent ion selectivity (Drake et al., 1996). Here, the analogous N136D substitution decreases the optimal divalent affinity at least 380-fold and increases the optimal trivalent affinity 16-fold, yielding an overall 6,000-fold shift toward trivalent selectivity. Significantly, such behavior is not observed for any neutral side chain substitutions at either the third or the ninth positions (Table III and Drake et al., 1996). An alternative model, suggesting that the N136D substitution modulates the stability of the apo state rather than that of the metal-occupied state, is inconsistent with the effects of the substitution on the relative affinities of different metal ions. Since the apo state is the same for different ions, modulation of its stability should have little or no effect on the ionic selectivity. Instead, it is the stability of the metal-occupied state that is altered by the N136D substitution, which is observed to enhance the stability of the trivalent cation–occupied site and to destabilize the divalent cation–occupied site.
It follows that the electrostatic repulsion model successfully predicts the behavior of acidic substitutions at both the third and ninth positions of the model EF-loop. Such simple behavior is not likely to be universal, however, since the degree of electrostatic repulsion and charge selectivity will strongly depend on the electrostatic context of the coordination site and the surrounding protein. For example, incorporation of an additional negative charge should have a larger effect at a position adjacent to a highly charged oxygen. This natural context dependence explains the observation that canonical EF-hand sites possess variable numbers of coordinating acidic side chains, ranging from 3 to 5 (Marsden et al., 1990). Even more striking evidence for electrostatic context effects has been provided by the interchange of Asn and Asp side chains at the third position of canonical EF-loops. In some cases Asp increases Ca2+ affinity relative to Asn, while in other cases it decreases affinity or has little effect (Henzl et al., 1996; Waltersson et al., 1993; Babu et al., 1992).
The electrostatic repulsion model can explain the apparently contradictory effects of acidic substitutions on different proteins. For a given protein context, there is an optimal level of negative charge density which yields the highest affinity binding of a divalent cation. This optimal charge density is sufficient to provide adequate electrostatic attraction for the bound ion but not too great to prevent the coordinating oxygens from packing around the ion in close proximity to one another. Thus, if a given site possesses less than the optimal negative charge density, then addition of an acidic residue may increase Ca2+ affinity. In contrast, if a site already contains negative charge density at a level meeting or exceeding the optimal level for Ca2+ binding, then additional negative charge will prevent effective close-packing of the coordinating oxygens and will thereby reduce the Ca2+ affinity. In the latter cases the affinity for divalent cations will decrease while that for trivalent cations will generally increase (as observed for the N136D and Q142E substitutions in GBP). Another context-dependent factor is the effect of a given acidic substitution on the effective negative charge density within the coordination site, which can vary since the protonation state of the substituted side chain carboxyl will depend on the local electrostatic and hydrogen bonding environment. In EDTA, for example, where there are four symmetrical carboxyl moieties, the pKa of the final deprotonation event is shifted from 2.0 to over 10.0 due to the proximity of the deprotonating carboxylate to three negative charges. Thus, one cannot assume that the multiple carboxyls in a protein metal binding site are fully deprotonated. Moreover, the electric field of a given carboxylate may be channeled by the dielectric environment into solvent (Honig and Nichols, 1995), an effect which is highly context dependent. In short, environment-specific effects are a built-in feature of the electrostatic repulsion model.
Implications for Ionic Size Selectivity
Certain EF-hand sites exhibit a remarkable ability to discriminate between multivalent cations of different size. The spherical cations of groups IIa, IIIa, and the lanthanides have enabled quantitation of the striking size selectivity of the model EF-loop in GBP, particularly in the large ion limit which is best defined by the available spherical metal ions. Examination of the size selectivity profile for the binding of spherical divalent cations to the native model site (Fig. 2 A) reveals, for example, that the binding free energy of Ca2+ is significantly more favorable than that of the larger Ba2+ ion (ΔΔGB o = −5.8 kcal mol−1), despite the much larger free energy cost of dehydrating the smaller Ca2+ ion (ΔΔGW o = +61 kcal mol−1; Falke et al., 1994; Marcus, 1985). As shown in the Appendix, the binding free energy includes the energetic cost of metal ion dehydration; thus, the stability of bound Ca2+ must exceed that of bound Ba2+ by over 66 kcal mol−1. The ionic radii of these two cations differ by only 0.36 Å, making the Ca2+ binding site of GBP, to our knowledge, the most size-specific ligand binding site yet characterized in nature. The molecular mechanisms providing such dramatic size selectivity are likely to be relevant for the EF-hand and other Ca2+ binding motifs, including those of Ca2+-specific channels which exhibit qualitatively similar (though less extreme) size selectivities (Park and MacKinnon, 1995; Hess et al., 1986).
Altering the size and shape of the coordinating side chain at position 3 causes moderate changes to the ionic size selectivity of the model EF-loop, thereby shedding light on the mechanisms underlying size discrimination as illustrated by the size selectivity profiles of Figs. 2 and 3. The neutral side chain substitutions (Gln, Val, Thr, and Ser) yield divalent ion size selectivity profiles resembling that of the native protein, except that selectivity is slightly but significantly weakened by decreased affinity for Ca2+, or by increased affinity for the larger Ba2+ ion. Each of the neutral side chain substitutions also weakens the size selectivity for trivalent ions while generating a new local minimum in the selectivity profile, indicating that the optimal metal ion radius is increased by as much as 0.1 Å.
Notably, neutral side chains substituted at position 3 of the model loop all change the ionic size selectivity in similar ways, despite the different sizes, shapes, and chemical properties of these side chains. Furthermore, diverse neutral side chain substitutions at loop position 9 yielded nearly the same effects observed for substitutions at position 3 (Drake et al., 1996). Together, the available evidence indicates that it is difficult or impossible to generate predictable changes in large ion size selectivity by simply engineering the size or shape of coordinating side chains. Instead, the results are consistent with a model in which the metal binding loop provides a constrained coordination “cage” to disfavor the binding of large cations, where the constraints are provided by complex interactions between multiple loop residues. Recent studies have indicated that the side chains of the coordinating loop, rather than a rigid loop backbone, provide the major size constraints (Drake et al., 1997). In this model, replacement of any critical side chain disrupts the coordination cage and weakens its size selectivity, thereby explaining the similar changes in large ion size discrimination observed for a range of dissimilar substitutions at two different coordinating positions of the model EF-loop. Moreover, different proteins could possess highly specialized coordination cages, thereby explaining the range of size selectivities described for canonical EF-hand sites (Chao et al., 1984; Williams et al., 1984; Wang et al., 1984; Corson et al., 1983). Such a complex, multi-residue tuning mechanism can be contrasted with the simpler mechanism that excludes the smaller Mg2+ ion, which appears to involve the conserved, bidentate side chain carboxylate located on the COOH-terminal helix of canonical EF-hands (DaSilva et al., 1995; Declercq et al., 1991) as well as the greater dehydration free energy of the solvated Mg2+ ion (Sussman and Weinstein, 1989).
The Consensus Residues at the Third EF-loop Position
The present findings for the model EF-loop provide clues which help explain the prevalence of specific side chains at the third position of canonical EF-loops. The size and shape of the native Asn side chain is clearly well suited for use at the third position of the model EF-loop, since smaller and larger side chains were found to significantly decrease the Ca2+ affinity, yielding either (a) a higher dissociation constant for Ca2+ binding (Gln, Thr, Val, Ser substitutions) or (b) a significant drop in GBP expression, most likely due to a catastrophic loss of Ca2+ binding (Glu, Cys, Ala, Gly substitutions). Not surprisingly, Asn is commonly used as a coordinating side chain at the third position of canonical EF-loops, as is Asp which has a similar size and shape. One of these two side chains is found at the third EF-loop position in over 97% of classical EF-hand sites (Table I). Asn and Asp are proposed to be the preferred side chains simply because they provide the ideal spacing between the EF-loop backbone and the coordinating oxygen. For a given site, the choice between Asn or Asp for a given site is probably determined by the electrostatic context (see above).
Deviations from the size and shape of Asn are observed to perturb the metal binding parameters of the model EF-loop. Among the neutral side chains retaining expression, the most dramatic perturbation was observed when the native Asn was substituted with the larger Gln, which caused the greatest increase in the metal dissociation rate and the largest measurable loss of Ca2+ affinity, as well as the greatest decrease in Tm yet measured for a GBP substitution. Together, these deleterious effects suggest that the longer Gln side chain generates a substantial change in the conformation or dynamics of the EF-loop. In contrast, the smallest perturbation was caused by the Ser substitution, which is more easily accommodated due to its smaller size and thus follows Asp and Asn in frequency of usage in the classical subset of EF-hand sites (Table I). Modeling of Ser into the third position of the model EF-loop reveals that the interatomic distance between Ca2+ and the side chain oxygen must be significantly increased by the mutation unless the backbone structure of the loop changes by ∼0.7 Å, thereby accounting for the observed perturbations. In Sarc site III, which displays direct Ca2+ coordination by Ser at the third EF-loop position (Fig. 1 B), the rearrangement of the loop backbone appears to be facilitated by the presence of an adjacent Gly residue at the fourth loop position. The GBP loop, however, lacks Gly at the fourth position, as do nearly half of the canonical EF-loops (Falke et al., 1994).
At first glance, the perturbations of Ca2+ binding and dissociation caused by the Val substitution are surprisingly small, considering the larger size of this side chain compared with Asn, and its lack of suitable coordinating atoms. It seems likely that coordination at this position is replaced by a solvent oxygen.
Implications for the Gateway Model of Kinetic Tuning
Finally, a comparison of substitutions at the third (present work) and ninth (Drake and Falke, 1996) positions of the model EF-loop provides further support for the gateway model of kinetic tuning, which proposes that the side chain at the ninth loop position serves as a gate to control the rates of metal ion binding and release of a given EF-hand site (Drake and Falke, 1996; Renner et al., 1993). Neutral side chain substitutions decreasing the size of the gateway residue in the model EF-loop were observed to increase the Tb3+ dissociation rate from 100- to 590-fold, while changing the equilibrium Tb3+ and Ca2+ affinities less than 4-fold. By contrast, the present study shows that neutral substitutions at the third loop position generally increase the Tb3+ dissociation rate by a smaller factor of 14-fold, while decreasing the equilibrium Ca2+ affinity nearly 10-fold. (The larger effects observed for the Gln substitution at the third loop position appear to be aberrant, stemming from the unique perturbations generated by this substitution.)
Fig. 4 illustrates positions 3 and 9 in the coordination structure and their locations relative to solvent, which are the same as those observed in canonical EF-hand sites (Falke et al., 1994). Substitutions at the ninth position of the model EF-loop were previously found to simply modulate the height of the transition state barrier for metal ion binding and release, thereby yielding nearly pure kinetic tuning without altering affinity (Drake and Falke, 1996). Substitutions at the third loop position do not tune the metal dissociation rate over as large of a range and do not provide pure kinetic tuning since they significantly weaken the Ca2+ binding affinity (present findings). As a result, the model EF-loop appears to use its ninth loop position for kinetic tuning, with comparatively little contribution from the third position. Overall, the available evidence is consistent with the proposal that ion binding and release normally occurs via the axial position guarded by the gateway side chain at loop position 9. Such a model accounts for the unique suitability of the ninth loop position as the locus of kinetic tuning by a gateway mechanism. Substitutions at the third loop position may generate an alternative, nonnatural association-dissociation pathway, but dissociation via this pathway is slower and more strongly coupled to affinity than that of the normal gateway route.
Figure 4.

Schematic structure illustrating alternate Ca2+ dissociation pathways in the galactose binding protein. Essentially the same picture can be proposed for Ca2+ dissociation from EF-hand sites (Drake and Falke, 1996). The gateway model hypothesizes that the preferred route to solvent is the axial pathway controlled by the gateway side chain at the ninth position of the Ca2+ binding loop; dissociation via this pathway yields upward movement of the bound metal ion toward solvent. The alternative dissociation pathway, leading to the right past the side chain at loop position 3, provides slower dissociation and is thus less important.
As predicted by the gateway hypothesis developed for the model EF-loop of GBP, classical EF-hand sites use side chains at their ninth EF-loop positions whose identities are strongly correlated with their Ca2+ dissociation kinetics and functions (Drake and Falke, 1996). Thus, although certain features of EF-hand Ca2+ binding sites are highly context dependent, other features appear to be general. The identification of such general features requires that hypotheses developed by studies of a given site be tested in several EF-hand proteins. Studies of models generated for the GBP site are currently underway in site II of cardiac troponin C, the Ca2+-triggered activation site of heart muscle contraction (manuscript in preparation).
Acknowledgments
The authors thank Cory L. Miller for expert technical assistance, Drs. Andrea Hazard, Eric Nalefski and Olve Peersen for helpful discussions.
appendix
A general reaction scheme that describes the above experiments considers the binding of metals Ma and Mb to proteins P and P′, respectively:
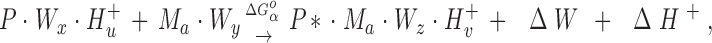 |
1 |
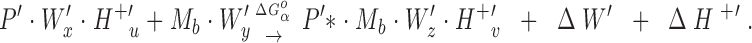 |
2 |
Here the reactants are the hydrated and protonated apo-protein (P · Wx · H+ u or P′ · W′x · H+′u) and the hydrated metal ion (Ma · Wy or Mb · W′y), while the products are the metal-occupied protein (P* · Ma · Wz · H+ v or P′* · Mb · W′z · H+′v) and the freed water molecules (ΔW or ΔW′) and protons (ΔH+ or ΔH+′). The asterisk indicates possible changes to the protein conformation or dynamics upon metal binding.
The difference in the binding free energies of these two reactions, ΔGa o − ΔGb o = ΔΔGB o, can be expressed in terms of the free energies of formation for the relevant reactants and products, as given by:
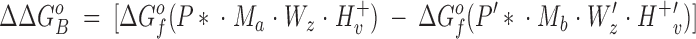 |
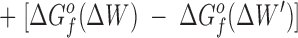 |
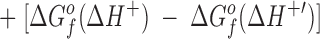 |
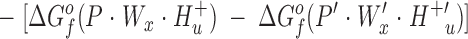 |
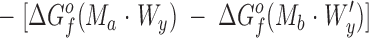 |
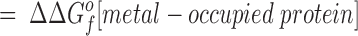 |
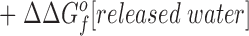 |
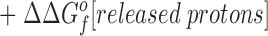 |
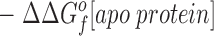 |
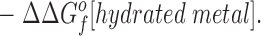 |
3 |
This relationship simplifies for the two specific comparisons made in the present work. (a) One type of comparison considers the binding of different metal ions to the same wild-type or engineered site, in which case the apo protein is identical (ΔΔGf o[apo protein] = 0), yielding:
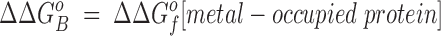 |
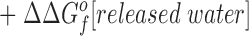 |
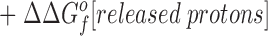 |
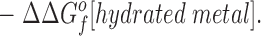 |
4 |
Eq. 4 indicates that when two metal ions release similar numbers of water molecules and protons, the overall ΔΔGB o is dominated by the stability differences between the two metal ions, both in solution and the protein complex. (b) The other type of comparison considers the binding of the same metal ion to different sites, in which case the hydrated metal term drops out (ΔΔGf o[hydrated metal] = 0) to give:
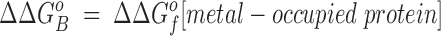 |
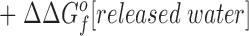 |
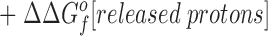 |
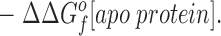 |
5 |
Eq. 5 shows that when two sites release similar numbers of water molecules and protons upon the binding of the same metal, the overall ΔΔGB o is now dominated by the stability differences between the apo and metal-occupied states of the two proteins.
Footnotes
Support provided by National Institutes of Health grant GM48203 (to J.J. Falke).
Abbreviations used in this paper: GBP, galactose binding protein; PDB, Brookhaven Protein Data Bank.
references
- Babu A, Su H, Ryu Y, Gulati J. Determination of residue specificity in the EF-hand of troponin C for Ca2+coordination, by genetic engineering. J Biol Chem. 1992;267:15469–15474. [PubMed] [Google Scholar]
- Bootman MD, Berridge MJ. The elemental principles of calcium signaling. Cell. 1995;83:675–678. doi: 10.1016/0092-8674(95)90179-5. [DOI] [PubMed] [Google Scholar]
- Brittain HG, Richardson FS, Martin RB. Terbium emission as a probe of calcium binding sites in proteins. J Am Chem Soc. 1976;98:8255–8260. doi: 10.1021/ja00441a060. [DOI] [PubMed] [Google Scholar]
- Carafoli E, Garciamartin E, Guerini D. The plasma-membrane calcium pump: Recent developments and future perspectives. Experientia. 1996;52:1091–1100. doi: 10.1007/BF01952107. [DOI] [PubMed] [Google Scholar]
- Chao SH, Suzuki Y, Zysk JR, Cheung WY. Activation of calmodulin by various meta cations as a function of ionic radius. Mol Pharmacol. 1984;26:75–82. [PubMed] [Google Scholar]
- Chattopadhyaya R, Meador WE, Means AR, Quiocho FA. Calmodulin structure refined at 1.7 Å resolution. J Mol Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- Chen X-H, Bezprozvanny I, Tsien RW. Molecular basis of proton block of L-type Ca2+channels. J Gen Physiol. 1996;108:363–374. doi: 10.1085/jgp.108.5.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen, J.J., D.J. Eatough, and R.M. Izatt. 1975. Handbook of Metal Ligand Heats and Related Thermodynamic Quantities. 2nd ed. Marcel Dekker, Inc., New York. 495 pp.
- Clapham DE. Calcium signaling. Cell. 1995;80:259–268. doi: 10.1016/0092-8674(95)90408-5. [DOI] [PubMed] [Google Scholar]
- Cook WJ, Jeffrey LC, Cox JA. Structure of a sarcoplasmic calcium-binding protein from amphioxus revined at 2.4 Å resolution. J Mol Biol. 1993;229:461–471. doi: 10.1006/jmbi.1993.1046. [DOI] [PubMed] [Google Scholar]
- Corson DC, Williams TC, Sykes BD. Calcium binding proteins: optical stopped-flow and proton NMR studies of the binding of lanthanide metal ions to parvalbumin. Biochemistry. 1983;22:5882–5889. doi: 10.1021/bi00294a030. [DOI] [PubMed] [Google Scholar]
- Creighton, T.E. 1993. Proteins: Structures and Molecular Properties. W.H. Freeman & Co., New York. pp. 4.
- DaSilva ACR, Kendrick-Jones J, Reinach FC. Determinants of ion-specificity on EF-hand sites: conversion of the Ca2+/Mg2+ site of smooth-muscle myosin regulatory light-chain into a Ca2+-specific site. J Biol Chem. 1995;270:6773–6778. doi: 10.1074/jbc.270.12.6773. [DOI] [PubMed] [Google Scholar]
- Declercq J-P, Tinant B, Parello J, Rambaud J. Ionic interactions with parvalbumins. Crystal structure determination of pike 4.10 parvalbumin in four different ionic environments. J Mol Biol. 1991;220:1017–1039. doi: 10.1016/0022-2836(91)90369-h. [DOI] [PubMed] [Google Scholar]
- Dotson DG, Putkey JA. Differential recover of Ca2+-binding activity in mutated EF-hands of cardiac troponin C. J Biol Chem. 1993;268:24067–24073. [PubMed] [Google Scholar]
- Drake SK, Falke JJ. Kinetic tuning of the EF-hand calcium binding motif: the gateway residue independently adjusts (i) barrier height and (ii) equilibrium. Biochemistry. 1996;35:1753–1760. doi: 10.1021/bi952335c. [DOI] [PubMed] [Google Scholar]
- Drake SK, Lee KL, Falke JJ. Tuning the equilibrium ion affinity and selectivity of the EF-hand calcium binding motif: substitutions at the gateway position. Biochemistry. 1996;35:6697–6705. doi: 10.1021/bi952430l. [DOI] [PubMed] [Google Scholar]
- Drake, S.K., M.A. Zimmer, and J.J. Falke. 1997. Optimizing the metal binding parameters of an EF-hand-like calcium chelation loop: coordinating side chains play a more important tuning role than chelation loop flexibility. Biochemistry. In press. [DOI] [PubMed]
- Falke JJ, Drake SK, Hazard AL, Peersen OB. Molecular tuning of ion binding to calcium signaling proteins. Q Rev Biophys. 1994;27:219–290. doi: 10.1017/s0033583500003012. [DOI] [PubMed] [Google Scholar]
- Falke JJ, Snyder EE, Thatcher KC, Voertler CS. Quantitating and engineering the ion specificity of an EF-hand-like calcium binding site. Biochemistry. 1991;30:8690–8697. doi: 10.1021/bi00099a029. [DOI] [PubMed] [Google Scholar]
- Gagne, S.M., M.X. Li, and B.D. Sykes. 1997. Mechanism of direct coupling between binding and calcium-induced structural change. Biochemistry. 36:4386–4392. [DOI] [PubMed]
- Ghosh A, Greenberg ME. Calcium signaling in neurons: molecular mechanisms and cellular consequences. Science (Wash DC) 1995;268:239–247. doi: 10.1126/science.7716515. [DOI] [PubMed] [Google Scholar]
- Henzl MT, Hapak RC, Goodpasture EA. Introduction of a fifth carboxylate ligand heightens the affinity of the oncomodulin CD and EF sites for Ca2+ . Biochemistry. 1996;35:5856–5869. doi: 10.1021/bi952184d. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, Tsien RW. Calcium channel selectivity for divalent and monovalent cations. J Gen Physiol. 1986;88:293–319. doi: 10.1085/jgp.88.3.293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science (Wash DC) 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Horrocks WD. Luminescence spectroscopy. Methods Enzymol. 1993;226:495–538. doi: 10.1016/0076-6879(93)26023-3. [DOI] [PubMed] [Google Scholar]
- Horrocks WD, Albin M. Lanthanide ion luminescence in coordination chemistry and biochemistry. Prog Inorg Chem. 1984;31:1–104. [Google Scholar]
- Ikura M. Calcium-binding and conformational response in EF-hand proteins. Trends Biochem Sci. 1996;21:14–17. [PubMed] [Google Scholar]
- Kawasaki H, Kretsinger RH. Calcium binding proteins. 1. EF-hands. Protein Profiles. 1994;1:343–517. [PubMed] [Google Scholar]
- Kretsinger RH. EF-hands reach out. Nature Struct Biol. 1996;3:12–15. doi: 10.1038/nsb0196-12. [DOI] [PubMed] [Google Scholar]
- Kretsinger RH, Nockolds CE. Carp muscle calcium-binding protein. J Biol Chem. 1973;248:3313–3326. [PubMed] [Google Scholar]
- Kumar VD, Lee L, Edwards BFP. Refined crystal structure of calcium-liganded carp parvalbumin 4.25 at 1.5 Å resolution. Biochemistry. 1990;29:1404–1412. doi: 10.1021/bi00458a010. [DOI] [PubMed] [Google Scholar]
- Linse S, Chazin WJ. Quantitative measurements of the cooperativity in an EF-hand protein with sequential calcium binding. Prot Sci. 1995;4:1038–1044. doi: 10.1002/pro.5560040602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linse S, Forsén S. Determinants that govern high-affinity calcium binding. Adv Second Messenger Phosphoprotein Res. 1995;30:89–151. doi: 10.1016/s1040-7952(05)80005-9. [DOI] [PubMed] [Google Scholar]
- Linse S, Johansson C, Brodin P, Grundström T, Drakenberg T, Forsén S. Electrostatic contributions to the binding of Ca2+ in calbindin D9K . Biochemistry. 1991;30:154–152. doi: 10.1021/bi00215a023. [DOI] [PubMed] [Google Scholar]
- MacManus JP, Hutnik CM, Sykes BD, Szabo AG, Williams TC, Banville D. Characterization and site-specific mutagenesis of oncomodulin. J Biol Chem. 1989;264:3470–3477. [PubMed] [Google Scholar]
- Marcus, Y. 1985. Ion Solvation. John Wiley, and Sons Limited, Chichester, UK. pp. 164.
- Marsden BJ, Shaw GS, Sykes BD. Calcium binding proteins. Elucidating the contributions to calcium affinity from an analysis of species variants and peptide fragments. Biochem Cell Biol. 1990;68:587–601. doi: 10.1139/o90-084. [DOI] [PubMed] [Google Scholar]
- Maune JF, Klee CB, Beckingham K. Ca2+ binding and conformational change in two series of point mutations to the individual Ca2+binding sites of calmodulin. J Biol Chem. 1992;267:5286–5295. [PubMed] [Google Scholar]
- Moews PC, Kretsinger RH. Terbium replacement of calcium in carp muscle parvalbumin: an X-ray crystallographic study. J Mol Biol. 1975;91:229–232. doi: 10.1016/0022-2836(75)90162-x. [DOI] [PubMed] [Google Scholar]
- Monera OD, Shaw GS, Zhu B-Y, Sykes BD, Kay CM, Hodges R S. Role of interchain helical hydrophobic interactions in Ca2+affinity, formation and stability of a two-site domain in troponin C. Prot Sci. 1992;1:945–955. doi: 10.1002/pro.5560010713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negele JC, Dotson DG, Liu W, Sweeney HL, Putkey JA. Mutation of the high-affinity Ca2+binding sites in cardiac troponin C. J Biol Chem. 1992;267:825–831. [PubMed] [Google Scholar]
- Palmisano WA, Treviño CL, Henzl MT. Site-specific replacement of amino acid residues within the CD binding loop of rat oncomodulin. J Biol Chem. 1990;265:14450–14456. [PubMed] [Google Scholar]
- Park C-S, MacKinnon R. Divalent cation selectivity in a cyclic nucleotide-gated ion channel. Biochemistry. 1995;34:13328–13333. doi: 10.1021/bi00041a008. [DOI] [PubMed] [Google Scholar]
- Procyshyn RM, Reid RE. A structure-activity study of calcium affinity and selectivity using a synthetic peptide model of the helix-loop-helix calcium-binding motif. J Biol Chem. 1994;269:1641–1647. [PubMed] [Google Scholar]
- Reid RE, Gariépy J, Saund AK, Hodges RS. Calcium-induced protein folding. Structure-affinity relationships in synthetic analogs of the helix-loop-helix calcium binding unit. J Biol Chem. 1981;256:2742–2751. [PubMed] [Google Scholar]
- Renner M, Danielson MA, Falke JJ. Kinetic control of Ca2+signaling: tuning the ion dissociation rates of EF-hand sites. Proc Natl Acad Sci USA. 1993;90:6493–6497. doi: 10.1073/pnas.90.14.6493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sage EH, Bassuk JA, Yost JC, Folkman MJ, Lane TF. Inhibition of endothelial cell proliferation by SPARC is mediated by a Ca2+-binding EF-hand sequence. J Cell Biochem. 1995;57:127–140. doi: 10.1002/jcb.240570113. [DOI] [PubMed] [Google Scholar]
- Shannon RD. Revised effective ionic radii. Acta Crystallogr. 1976;A32:751–767. [Google Scholar]
- Skelton NJ, Körde-Akke M, Forsén S, Chazin WJ. Signal transduction versus buffering activity in Ca2+binding proteins. Nature Struct Biol. 1994;1:239–245. doi: 10.1038/nsb0494-239. [DOI] [PubMed] [Google Scholar]
- Snyder EE, Buoscio BW, Falke JJ. Calcium-site specificity: effect of size and charge on metal ion binding to an EF-hand-like site. Biochemistry. 1990;29:3937–3943. doi: 10.1021/bi00468a021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stryer L, Haugland RP. Energy transfer: a spectroscopic ruler. Proc Natl Acad Sci USA. 1967;58:719–726. doi: 10.1073/pnas.58.2.719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strynadka NCJ, James MNG. Crystal structures of the helix-loop-helix calcium-binding proteins. Annu Rev Biochem. 1989;58:951–998. doi: 10.1146/annurev.bi.58.070189.004511. [DOI] [PubMed] [Google Scholar]
- Sussman F, Weinstein H. The ion selectivity of calcium binding proteins: the cyclo(-L-Pro-Gly-)3peptide as a model. Proc Natl Acad Sci USA. 1989;86:7880–7884. doi: 10.1073/pnas.86.20.7880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trigo-Gonzalez G, Awang G, Racher K, Neden K, Borgford T. Helix variants of troponin C with tailored calcium affinities. Biochemistry. 1993;32:9826–9831. doi: 10.1021/bi00088a038. [DOI] [PubMed] [Google Scholar]
- Vyas NK, Vyas MN, Quiocho FA. A novel calcium binding site in the galactose-binding protein of bacterial transport and chemotaxis. Nature (Lond) 1987;327:635–638. doi: 10.1038/327635a0. [DOI] [PubMed] [Google Scholar]
- Waltersson Y, Linse S, Brodin P, Grundström T. Mutational effects on the cooperativity of Ca2+binding to calmodulin. Biochemistry. 1993;32:7866–7871. doi: 10.1021/bi00082a005. [DOI] [PubMed] [Google Scholar]
- Wang C-LA, Leavis PC, Gergely J. Kinetic studies show that Ca2+ and Tb3+ have different binding preferences toward the four Ca2+binding sites of calmodulin. Biochemistry. 1984;23:6410–6415. doi: 10.1021/bi00321a020. [DOI] [PubMed] [Google Scholar]
- Williams TC, Corson DC, Sykes BD. Calcium binding proteins: calcium-lanthanide exchange in carp parvalbumin. J Am Chem Soc. 1984;106:5698–5702. [Google Scholar]