

Abstract
The permeation pathway of the Na channel is formed by asymmetric loops (P segments) contributed by each of the four domains of the protein. In contrast to the analogous region of K channels, previously we (Yamagishi, T., M. Janecki, E. Marban, and G. Tomaselli. 1997. Biophys. J. 73:195–204) have shown that the P segments do not span the selectivity region, that is, they are accessible only from the extracellular surface. The portion of the P-segment NH2-terminal to the selectivity region is referred to as SS1. To explore further the topology and functional role of the SS1 region, 40 amino acids NH2-terminal to the selectivity ring (10 in each of the P segments) of the rat skeletal muscle Na channel were substituted by cysteine and expressed in tsA-201 cells. Selected mutants in each domain could be blocked with high affinity by externally applied Cd2+ and were resistant to tetrodotoxin as compared with the wild-type channel. None of the externally applied sulfhydryl-specific methanethiosulfonate reagents modified the current through any of the mutant channels. Both R395C and R750C altered ionic selectivity, producing significant increases in K+ and NH4 + currents. The pattern of side chain accessibility is consistent with a pore helix like that observed in the crystal structure of the bacterial K channel, KcsA. Structure prediction of the Na channel using the program PHDhtm suggests an α helix in the SS1 region of each domain channel. We conclude that each of the P segments undergoes a hairpin turn in the permeation pathway, such that amino acids on both sides of the putative selectivity filter line the outer mouth of the pore. Evolutionary conservation of the pore helix motif from bacterial K channels to mammalian Na channels identifies this structure as a critical feature in the architecture of ion selective pores.
Keywords: pore helix, cysteine mutagenesis, tetrodotoxin, methanethiosulfonates, cadmium
INTRODUCTION
In voltage-gated ion channels the pore-lining (P) segments in each of four internally homologous domains (Na and Ca channels) or subunits (K channels) have been proposed to form hairpins that partially traverse the membrane and together create an ion-selective pore (Stuhmer 1991; Catterall 1995; Marban et al. 1998). Based on mutagenesis data and homology with a ring of glutamic acid residues in the Ca channel, the sequence-aligned residues in the Na channel pore D400, E755, K1237, and A1529, (DEKA; see Fig. 1) form at least part of the selectivity filter (Heinemann et al. 1992). The external mouth of the pore has been extensively studied by site-directed mutagenesis. Mutations introduced on the COOH-terminal side of the DEKA ring have been electrophysiologically characterized using toxin or metal cation blockers as structural probes (Backx et al. 1992; Satin et al. 1992; Dudley et al. 1995; Favre et al. 1995, Favre et al. 1996; Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Chen et al. 1997; Li et al. 1997, Li et al. 2000a,Li et al. 2000b; Tsushima et al. 1997a,Tsushima et al. 1997b; Yamagishi et al. 1997; Chang et al. 1998; Penzotti et al. 1998; Sunami et al. 2000). High affinity Cd2+ block and modification by extracellular methanethiosulfonate (MTS) reagents of cysteine substitution mutants on the COOH-terminal side of the DEKA ring suggests that this part of the pore is composed of random coils rather than periodic structures such as α helices or β strands (Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Tsushima et al. 1997b; Yamagishi et al. 1997). The topology of the amino acid residues immediately NH2-terminal to the DEKA ring (Yamagishi et al. 1997) is distinct from that in voltage-dependent K channels (MacKinnon and Yellen 1990; Yellen et al. 1991). Cysteine mutants on the NH2-terminal side of the DEKA ring are not accessible from the internal mouth of the channel; in fact, two substitutions immediately NH2-terminal of the DEKA residues (F1236C and T1528C) are accessible from the external surface (Yamagishi et al. 1997). It appears that the P segments of the Na channel do not traverse the membrane as they do in K channels (MacKinnon and Yellen 1990; Yellen et al. 1991).
Figure 1.

Single-letter amino acid alignment of the residues in the P segments of the Na channel with KcsA. The bars over the KcsA sequence identify the turret and pore helix with the selectivity signature sequence enclosed in the box. Y62 is the first residue of the pore helix in KcsA, the underlined residues have their side chains exposed to the channel pore based on the crystal structure (Doyle et al. 1998). The residues in bold in all of the sequences are those predicted to be helical by PHDhtm. The italicized residues in the dShaker sequence were not categorized as helical by PHDhtm but exhibited a probability of >0.4 of being helical by the algorithm. The dotted box identifies the residues mutated in the μ1 channel in this study. The hH1 sequence is shown for comparison. The underlined residues indicate that the cysteine mutant at this position is accessible (i.e., altered Cd2+ and/or TTX block sensitivity) from the extracellular pore. The jagged underlined residues have been shown previously to be accessible from the outside of the channel. The DEKA selectivity filter of the Na channel is enclosed in a solid box. The numbering in parentheses, is according to the μ1 sequence.
As might be predicted by the sequence divergence of the P segments in each domain of the Na channel, the contribution of each of the segments to the formation of the pore is not equivalent (Chiamvimonvat et al. 1996). Amino acids in each of the P segments contribute differently to the permeation phenotype of the channel; residues in the DEKA ring importantly influence conductance (DEK) and ion selectivity (K; Heinemann et al. 1994; Chiamvimonvat et al. 1996; Favre et al. 1996; Perez-Garcia et al. 1997; Li et al. 2000b). Other residues in the domain IV P segment (G1530, W1531, and D1532) are major determinants of monovalent cation selectivity (Chiamvimonvat et al. 1996; Tsushima et al. 1997b).
The crystal structure of the pore of a bacterial K channel (KcsA) exhibits several features predicted to be conserved across the superfamily of voltage-gated ion channels (Doyle et al. 1998). A pore helix is present immediately NH2-terminal to the selectivity filter of KcsA. The pore helix is a critical determinant of the structural integrity of the pore and ensures the proper alignment of the selectivity filter residues. However, Na channels must be structurally distinct from K channels based on the limited amino acid homology and different ion selectivity. It is uncertain which, if any, of the structural motifs in KcsA have been retained in the Na channel pore. Structural modeling suggests that the P segments of the Na (Hsu et al. 1999; Lipkind and Fozzard 2000) and Ca channels (Hsu et al. 1999) retain the α helix-turn-coil motif described in the KcsA structure (Doyle et al. 1998). We sought to test experimentally the structure predictions of the P segments of the Na channel using a combination of cysteine mutagenesis, electrophysiological recording, and site-specific modification. Cysteine substitutions were made at 40 positions NH2-terminal to the DEKA residues in the μ1-skeletal muscle Na channel and expressed in tsA-201 cells. Cd2+ and tetrodotoxin (TTX) block or MTS modification gauged the external accessibility of these mutants, and monovalent cation selectivity was examined by ionic substitution. The experimental data are incorporated into a revised structural model of the Na channel P segments.
MATERIALS AND METHODS
Mutagenesis and Channel Expression
Mutagenesis of the expression plasmid containing the rat skeletal muscle (μ1) α subunit of the Na channel was performed as described previously (Yamagishi et al. 1997). The wild-type μl Na channel α subunit cDNA (Trimmer et al. 1989) was cloned into the mammalian expression vector pGW1H for both mutagenesis and expression. The rat brain β1 subunit (Isom et al. 1992) was cloned into pCMV-5 (Phillipson et al. 1993). A plasmid expressing the green fluorescent protein (pGreenLantern; GIBCO BRL) was used as a transfection reporter. Cysteine substitutions were introduced into the μ1 cDNA by oligonucleotide-directed mutagenesis (Perez-Garcia et al. 1996) or by PCR with overlapping mutagenic primers (QuickchangeTM site-directed mutagenesis kit; Stratagene). All mutations were performed in duplicate and confirmed by DNA sequencing the region of the mutation. TsA-201 cells, a transformed human embryonic kidney cell line (HEK 293) stably expressing the SV40 T-antigen, were used for channel expression. The culture and transfection conditions were as described previously (Yamagishi et al. 1997). Transfection was performed by the calcium phosphate precipitation method for 6–12 h with a combination of 2.5 μg of the wild-type or mutant α subunit, 1 μg of the β1 subunit, and 0.3 μg of the green fluorescent protein cDNA-containing plasmids per 35-mm dish.
Electrophysiology and Data Analysis
Transfected cells were identified by epifluorescence microscopy and membrane currents were recorded with the whole-cell configuration of patch clamp (Hamill et al. 1981) 24–72 h after transfection. Cells were transferred to the stage of an inverted microscope and superfused with external solution at a rate of 1–2 ml/min. All experiments were performed at room temperature (22–23°C). Patch electrodes were pulled from borosilicate glass and had 2–5-MΩ tip resistances. Currents were recorded using a patch-clamp amplifier (model Axopatch 200A; Axon Instruments) interfaced to a personal computer. Cell capacitance was calculated by integrating the area under an uncompensated capacity transient elicited by a 20-mV hyperpolarizing test pulse from a holding potential of −80 mV. Series resistance was then compensated as much as possible without ringing, typically 70–90%. Given the average series resistance of our electrodes, the maximal uncompensated voltage error was <|6 mV| for the largest currents studied. Whole-cell currents were recorded in a bath solution containing (in mM): 140 NaCl, 5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, and 10 glucose, pH 7.4. The pipette solution was (in mM): 35 NaCl, 105 CsF, 1 MgCl2, 10 HEPES, and 1 EGTA, pH 7.2. Dose–response curves for block of the mutant channels by extracellular Cd2+ were determined by adding the chloride salt of the blocker to the bath at concentrations between 1 μM and 5 mM, and TTX was added at concentrations between 1 nM and 10 μM, depending on the sensitivity of the particular mutant. Full current-voltage relationships were determined at each Cd2+ or TTX concentration. The half-blocking concentration (IC50) was determined by a least-squares fit (Levenberg-Marquardt algorithm; Microcal Origin) of the data to the function: I/Io = 1/{1 + ([blocker]/IC50)n}, where I and Io are the currents in the presence and absence of blocker, respectively. In nearly all cases, block by Cd2+ and TTX was reversible, when the current amplitude did not return to >90% of control channel run down could not be excluded and the experiments were not included in the analysis.
Susceptibilities to extracellular methanethiosulfonate ethylammonium (MTSEA), methanethiosulfonate ethyltrimethylammonium (MTSET), methanethiosulfonate ethylsulfonate (MTSES; Toronto Research Chemicals) modification was determined by bath application of saturating concentrations of each reagent (2.5 mM MTSEA, 1 mM MTSET, and 5 mM MTSES). MTS modification of the current was verified by the irreversibility of the change in the current amplitude. In no case was there an irreversible change in current amplitude after exposure to MTS reagents.
Single-channel recording was performed in those mutant channels that exhibited sensitivity to block by Cd2+ at the whole-cell level. The cell-attached configuration of the patch clamp was used (Hamill et al. 1981). The currents were sampled at 10 kHz and low-pass filtered at 2 kHz. The pipette solution contained (in mM): 140 NaCl, 10 HEPES, pH 7.4, and 400 μM Cd2+. The bath contained (in mM): 140 KCl, 1 BaCl2, 10 HEPES, pH 7.4, and 20 μM fenvalerate (DuPont Inc.) to facilitate the analysis of open-channel blockade by Cd2+ (Yamagishi et al. 1997). Well-resolved single-channel opening events were determined using a half-height criterion (Colquhoun and Sigworth 1983). Amplitude histograms were fitted to the sum of Gaussians using a nonlinear least-squares method. The voltage dependence of Cd2+ blockade was used to estimate the fractional electrical distance to the substituted cysteine residue as described previously (Yamagishi et al. 1997). The average current during long openings in the presence of Cd2+ was used to quantify the blocked unitary current amplitude.
Pooled data are presented as means ± SD with at least three determinations of the IC50 for block by Cd2+ and TTX with the exceptions noted as follows. The wild-type IC50 for Cd2+ block was 2.5 ± 0.2 mM. The mutant channels S387C, W388C, L393C, F742C, H743C, S744C, F749C, G1224C, G1516C, and C1521A exhibited no difference in the IC50 compared with the wild-type channel in a single determination that was confirmed by the absence of 50% block of the peak current by 0.5 mM Cd2+ in at least two additional cells. The wild-type IC50 for TTX block was 17 ± 5 nM. The mutant channels S387C, W388C, A389C, F390C, L391C, F742C, H743C, S744C, S1229C, L1230C, Q1232C, G1516C, N1517C, S1518C, C1521A, F1523C, E1524C, and I1525C exhibited no difference in the IC50 compared with the wild-type channel in a single determination that was confirmed by the greater 50% block of the peak current by 50 nM Cd2+ in at least two additional cells. Statistical comparisons were made using an analysis of variance (ANOVA) with P < 0.05 considered to be significant.
Molecular Modeling
Each homologous domain of the Na channel was evaluated by the program PHDhtm (http://www.embl-heidelberg.de/predictprotein), which is a neural network system that predicts the locations of transmembrane helices in integral membrane proteins (Rost et al. 1995). This method uses evolutionary information as input to the network system and detects transmembrane helices with an accuracy of 95%. PHDhtm predictions were first validated with the known structure of the KcsA channel with its default parameters (Hsu et al. 1999): three helical segments were assigned by the program, and they corresponded to TM1, the pore helix, and TM2 of the KcsA. Several inward-rectifying (hROMK1 or hKir1.1, or hGIRK1 or hKir3.1) and voltage-gated K channels (hKv1.1, dShaker) were also tested with PHDhtm: the program assigned a helical segment adjacent to the NH2-terminal of the selectivity sequences in each sequence. The sodium channels μ1 and hH1 were then evaluated by input of the entire sequence, and the sequence of each homologous domain using the default setting of PHDhtm. Molecular models were generated using the Insight/Discover suite of programs (Molecular Simulation Inc.). The steepest descents and conjugate gradients were used for energy minimization.
RESULTS
Modeling Predicts Pore Helices in the Na Channel
We tested the SS1 region of the Na channel P segments for α helical content using PHDhtm, a neural network based program that detects transmembrane helices with 95% accuracy (Rost et al. 1995). We first validated the predictive power of the PHDhtm by testing the program on several proteins of known structure including KcsA (Doyle et al. 1998). The program accurately predicted the transmembrane and pore helices of KcsA (Doyle et al. 1998). PHDhtm predicts α helices involving the italicized residues in the KcsA pore and the P segments of each Na channel domain (Fig. 1). Based on the crystal structure, the second of the two vicinal tryptophans (W68), E71 and T72 in KcsA, are oriented with their side chains accessible to the channel pore (Doyle et al. 1998). PHDhtm predicted helices in sequence-aligned regions of the inwardly rectifying channels hROMK1 (Kir1.1) and hGIRK1 (Kir3.1), the voltage-dependent K channels hKv1.1 and Drosophila Shaker (Fig. 1), and the α1C subunit of the Ca channel (unpublished data). Several motifs in the putative helical regions of the P segments of the Na channel are conserved from the KcsA pore helix: a threonine residue one to two positions NH2-terminal to the selectivity filter (except domain II), and conserved aromatic and charged residues in the pore helix. We sought to experimentally test the hypothesis that the regions proximal to the selectivity filter in the Na channel are helical.
Accessibility Pattern to Cd 2+, MTS Reagents, and TTX
Fig. 1 shows the positions in the Na channel pore studied here. The rat μ1 channel P segment sequences are aligned with the bacterial inward-rectifier channel (KcsA). The residues that form the turret and pore helix of the KcsA channel are designated (Doyle et al. 1998). The amino acids underlined with a jagged line are known to be accessible from the external mouth of the channel by thiol-specific modifiers (Chiamvimonvat et al. 1996; Tsushima et al. 1997b; Yamagishi et al. 1997). 3 of the 40 mutations (G1226C, L1231C, and L1522C) did not yield functional channels. To evaluate the possibility that an internal disulfide bond had formed spontaneously to cripple these mutant channels (Benitah et al. 1997), cells were exposed to 1 mM DTT. The application of DTT did not reveal any current (unpublished data).
The permeation phenotype of each expressing mutant channel was characterized by determination of the IC50 for block by Cd2+ and TTX, accessibility to modification by MTS reagents, and peak current size with cationic substitutions. Fig. 2 shows the sensitivity to externally applied Cd2+ of wild-type and mutant Na channels. Eight mutants had enhanced sensitivity to block by Cd2+ compared with the wild-type channel: F390C and R395C in domain I; R750C, L746C, and F745C in domain II; Y1227C and L1228C in domain III; and I1519C in domain IV (Fig. 2 A). It is notable that the mutated residues in the same P segments that exhibit enhanced sensitivity to Cd2+ are either adjacent to one another or separated by three to four amino acids. The easy reversibility of Cd2+ block and the relatively low, micromolar affinity of sensitive channels suggests that single cysteine residues participate in the divalent cation block.
Figure 2.

The effect of external Cd2+ and TTX on the cysteine substitution mutants on the NH2-terminal side of putative selectivity filter in μ1. (A) Bar plot of the IC50s for Cd2+ block of each of the mutants that express current. The mutants R395C, R750C, L746C, F745C, Y1227C, L1228C, and I1519C have IC50s (mean ± SD) 999 ± 64, 201 ± 10, 813 ± 10, 123 ± 19, 650 ± 106, 292 ± 94, and 173 ± 19 μM (P < 0.05 versus WT, 1,955 ± 207 μM), respectively. (B) Bar plot of the IC50s for TTX of each of the mutants that express current. The mutants R395C, F745C, R750C, and L1228C have IC50s for TTX block of (mean ± SD) 112 ± 32, 1,660 ± 425, 4,431 ± 661, and 441 ± 268 nM (P < 0.05 versus WT, 13 ± 4 nM), respectively. In cases without standard error bars, full dose–response curves were obtained in individual cells, and the lack of deviation from wild-type was confirmed in several other experiments using single doses of Cd2+ (0.5 mM) and TTX (50 nM).
Mutations in the pore of the Na channel also alter sensitivity to block by guanidinium toxins. The toxin molecule is much larger than Cd2+, with more points of contact with amino acids in the channel pore. The IC50 for TTX of the wild-type μ1 channel expressed in tsA201 cells is 17 ± 0.5 nM, comparable to that in the oocyte expression system (Backx et al. 1992; Satin et al. 1992). Of the eight mutants with altered Cd2+ sensitivity, four significantly destabilized TTX block (R395C, F745C, R750C, and L1228C). No mutant channels with unchanged sensitivity to Cd2+ block exhibited altered TTX sensitivity (Fig. 2 B). The IC50s (mean ± SD) for TTX block of R395C, R750C, F745C, and L1228C were 112 ± 32, 4,431 ± 661, 1,660 ± 425, and 441 ± 270 nM, respectively.
MTS reagents selectively modify free cysteinyl side chains in an aqueous environment. External application of saturating concentrations of MTS reagents produced no significant effect on the current through any of the mutant channels; data from R395C are summarized in Fig. 3. To test the MTS accessibility from the inside of the channel, MTSEA or MTSET was applied to the cytoplasmic face of F394C, R395C, and L396C (domain I) and I751C and R750C (domain II). Internal MTS reagents had no effect on the mutant channels (unpublished data).
Figure 3.

No effect of MTS reagents on R395C. (A) Representative experiment with repeated external application of 1 mM Cd2+ and saturating concentrations of MTS reagents. None of the Cd2+-sensitive mutants exhibited current reduction or a change in the IC50 for Cd2+ block after MTS application.
The lack of effect of MTS reagents may result from an inability of the reagents to form a mixed thiol. Alternatively, the cysteinyl may be modified, but the adduct does not block the permeation pathway and, therefore, does not affect current amplitude. To distinguish between these possibilities, we measured sensitivity to Cd2+ block before and after application of MTS reagents. Fig. 3 shows representative experiments for R395C. Application of 1 mM Cd2+ blocks ∼60% of the current; the subsequent administration of MTSEA, MTSET, and MTSES does not alter the current amplitude nor does it modify the sensitivity to subsequent applications of Cd2+ (Benitah et al. 1996, Benitah et al. 1997). Similar results were observed with the other Cd2+-sensitive mutants (unpublished data).
Single-channel Analysis
The voltage dependence of blockade of the unitary current by external Cd2+ was used to determine the fractional electrical distance of divalent cation binding. The fractional electrical distance for Cd2+ block of cysteine mutants provides the relative location of the substituted thiol in the channel pore. Fig. 4 (top) shows unitary Na+ currents for R395C and R750C, Cd2+-sensitive mutants with robust whole-cell expression, in the presence and absence of Cd2+. In the absence of Cd2+, the single-channel conductances of R395C and R750C differed from the wild-type (50 ± 0.3 pS in 140 mM [Na+]o) by <30%. In the presence of Cd2+, block is rapid and, therefore, appears as a reduction in the single-channel current amplitude. The voltage dependence of block is evident from the change in single-channel current through the mutant channels in the presence and absence of Cd2+ over a wide voltage range (Fig. 4, middle). In each case, the reduction in unitary current is more prominent at negative voltages. By plotting the ratio of the blocked and unblocked unitary currents and assuming a single-site model for Cd2+ binding (Yamagishi et al. 1997), the fraction of the voltage field traversed by Cd2+ to reach its binding site was estimated to be 0.12 and 0.23 for R395C and R750C, respectively (Fig. 4, bottom).
Figure 4.

Single-channel Cd2+ block of R395C and R750C. (top) Representative single-channel currents after modification by 20 μM fenvalerate, elicited by voltage steps to −50 and −80 mV from a holding potential of −120 mV with 140 mM Na+ in the pipette. The single-channel current is reduced in the presence of 400 or 800 μM Cd2+ (middle) Single-channel current-voltage relationships for each of the mutant channels determined from three patches in the presence and absence of Cd2+. Cd2+ blocks each of the mutants in a voltage-dependent fashion. (bottom) A plot of the logarithm of the ratio of the blocked and unblocked single-channel current amplitude versus voltage gives the fractional electrical distance (δ) for Cd2+ binding.
Ion Selectivity of the Mutant Na Channels
Several residues in both the third (K1237) and fourth domain (G1530, W1531, and D1532) P segments of the Na channel influence ionic selectivity (Heinemann et al. 1992; Chiamvimonvat et al. 1996; Favre et al. 1996; Perez-Garcia et al. 1997; Tsushima et al. 1997b). We examined all mutants for changes in ion selectivity; only R395C and R750C altered the selectivity of the channel, and R395C altered selectivity most dramatically. We compared the whole-cell conductances in external solutions of different monovalent and divalent cation composition to the Na+ conductance of the mutants. Fig. 5 (A and B) shows representative R395C currents and current-voltage relationships. Fig. 5 C summarizes the ion-specific conductances compared with Na+. For comparison, the previously reported domain III DEKA mutant, K1237C, is shown (Chiamvimonvat et al. 1996; Perez-Garcia et al. 1997). R395C is more permeable to NH4 +, K+, and Ca2+ than the wild-type channel. R750C exhibits enhanced permeability to NH4 + and modest K+ permeability, but does not support divalent cation flux. The selectivity phenotype of F745C is similar to the wild-type.
Figure 5.
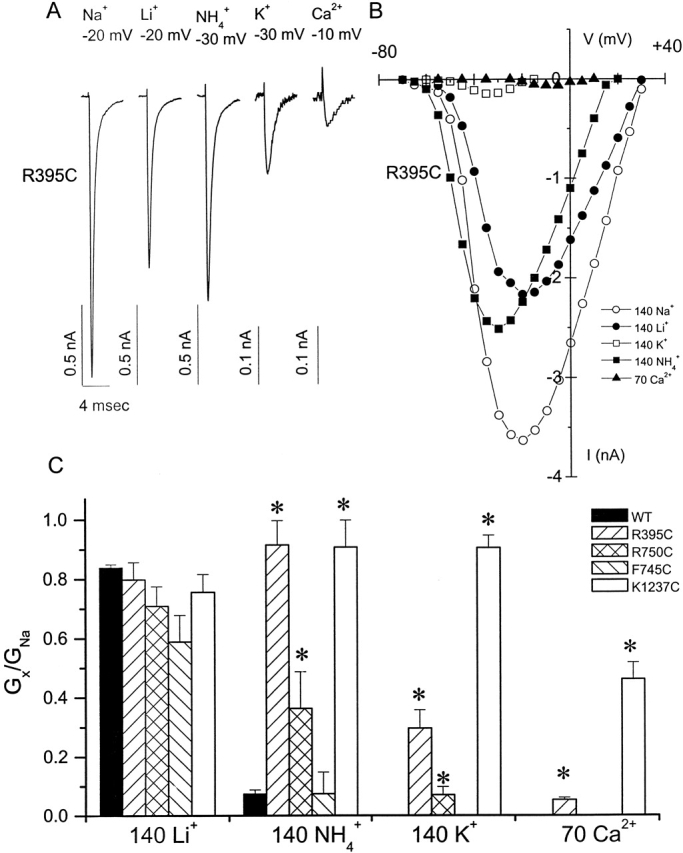
Selectivity of the cysteine mutants with altered Cd2+ and TTX affinities. Peak inward currents (A) and current-voltage relationship (B) through R395C in the presence of different cations. (C) Plot of the ratio of the whole-cell conductance in 140 mM Li+, 140 mM NH4 +, 140 mM K+, and 70 mM Ca2+ (Gx) compared with that in 140 mM Na+(GNa). The selectivity of the wild-type channel and the selectivity filter mutant K1237C are shown for comparison.
DISCUSSION
The P segments contain the major determinants of permeation in voltage-gated ion channels. In the Na channel, the DEKA ring is a central, but not the only element that mediates conductance and selectivity (Chiamvimonvat et al. 1996; Tsushima et al. 1997b). Amino acid residues on both the NH2- and COOH-terminal sides of the DEKA ring are accessible from the extracellular side of the channel pore, but no P segment residues have been demonstrated to contribute to the inner mouth of the Na channel (Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Tsushima et al. 1997b; Yamagishi et al. 1997). We have previously shown that cysteine mutants immediately NH2-terminal of the DEKA ring in domains III (F1236C and T1235C) and IV (S1528C) are accessible from the outer pore mouth. The data suggest that these P segments descend then ascend in the outer pore but, unlike the K channel, do not span the selectivity region (Yamagishi et al. 1997).
The role of the residues NH2-terminal to the DEKA ring (often referred to as the SS1 region) in the formation of the outer pore mouth is unknown. In the KcsA channel, the analogous region forms an α-helical structure referred to as the pore helix. Side chain accessibility to the aqueous phase of SS1 residues was gauged by a combination of cysteine substitution mutagenesis, and sensitivity to block or modification by hydrophilic, thiol-specific modifying reagents. The probes used provide complementary structural information. Alterations in external Cd2+ block sensitivity and MTS modification of the current imply side chain accessibility in the outer mouth of the pore. Modest changes in TTX block may be associated with global changes in pore structure with retained specific channel–toxin interaction sites. Dramatic changes in toxin affinity are more likely to result if a specific channel–toxin interaction is disrupted (Hidalgo and MacKinnon 1995). In this regard, it is interesting to note that only mutations in domain II in this region of the P segment produce dramatic (more than three orders of magnitude) changes in sensitivity to TTX block (Fig. 2 B).
In each domain, at least one substituted thiol side chain is accessible to the extracellular pore. In domain I, the side chains of both F390C and R395C (separated by four residues) are accessible. In domain II, F745C, L746C, and R750C exhibit side chain accessibility. In domain III, Y1227C and L1228C have enhanced sensitivity to Cd2+ block, whereas G1226C and L1231C have an indeterminate phenotype since they do not express functional channels. Similarly, in domain IV, the mutant I1519C exhibits side chain accessibility to Cd2+ and is three residues NH2-terminal from the nonfunctional mutant L1522C. In the context of previous mutagenesis data in adjacent regions of the P segments (Yamagishi et al. 1997), side chains that are accessible or indeterminate are observed every three to five positions NH2-terminal to the DEKA ring in domains II–IV (Fig. 1 and Fig. 6). This periodicity of side chain accessibility in the SS1 regions is consistent with α helices.
Figure 6.


Structure of Na channel SS1 region of the P segments. (top) Helical wheel representations of the residues mutated in this study. The residues in bold italics are accessible from the aqueous pore; those enclosed in a box do not express current and thus have an indeterminate phenotype. The cysteine mutants that alter Cd2+/ TTX block sensitivity or have an indeterminate phenotype are on the same side of the helix. (bottom) Molecular model of the putative pore helices in the Na channel pore, views from the top (left) and side (right) are shown. The domains are positioned to approximate the distances between selectivity filter residues. The pore helices are rendered in ribbon format, the DEKA ring in yellow stick format, and accessible (green) or nonfunctional (salmon) positions in ball-and-stick format.
Helical wheel representations of the SS1 regions demonstrate that the accessible or indeterminate side chains lie on the same face of the predicted α helices with the exception of G1226 (Fig. 6). The secondary structure of amino acid residues on the NH2-terminal side of DEKA are distinct from the random coil structure predicted by mutagenesis of amino acids on the COOH-terminal side of the ring (Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Tsushima et al. 1997b).
The detailed pattern of accessibility differs for each of the residues that alter channel interaction with the structural probes. The mutations R395C (domain I), F745C, R750C (domain II), and L1228C (domain III) increase the affinity for Cd2+ block and are resistant to TTX. However, external application of MTS reagents does not alter the current through these mutant channels. R395C and R750C, but not F745C or l1228C, alter cation selectivity of the channel (Fig. 5). R395 and R750 are at homologous positions by sequence alignment in domains I and II, respectively. It is possible that they form a charge ring in the external pore that alters permeation. However, R395C and R750C do not effect permeation by a simple electrostatic mechanism. Neutralization of positive charges in the external pore would be expected to increase channel conductance; neither mutant increases conductance compared with wild type, in fact, R750C is ∼30% smaller than R395C or wild type (Fig. 4). A simple electrostatic mechanism cannot explain the change in ion selectivity observed with these two mutations (Fig. 5). Furthermore, an electrostatic impediment to TTX binding is inconsistent with the data: a reduction in the net positive charge of the pore, all else being equal, should increase TTX sensitivity. Instead, these two charge-reducing mutations decrease the channel sensitivity to block by TTX, perhaps by changing the interaction of other toxin channel contact sites (Noda et al. 1989; Terlau et al. 1991; Lipkind and Fozzard 1994; Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Chen et al. 1997). It is possible that these residues are involved in an electrostatically mediated stabilization of the pore-forming salt bridges with the acidic filter residues in domains I (D400) and II (E755). Of course more global changes in the pore structure cannot be ruled out with certainty, but the relative preservation of Na+ selectivity (Fig. 5 C) argues against this possibility.
Another mutation that alters both Cd2+ and TTX sensitivity is replacement of aromatic phenylalanine in domain II (F745C). Both mutations in domain II that alter Cd2+ sensitivity (F745C and R750C) decrease TTX affinity by nearly two orders of magnitude. If F745 and R750 participate in the formation of a pore helix, the cysteine substitutions at these positions disrupt an important interaction between the channel and toxin. F745C produces no change in channel selectivity and is not modified by MTS reagents. Similarly, L1228C increases affinity for Cd2+ block and makes the channel resistant to TTX block. The magnitude of the destabilization of TTX block is similar to that exhibited by the domain I mutant R395C and less than that of the domain II mutants F745C and R750C. The remainder of the cysteine mutations, Y1227C (domain III) and I1519C (domain IV) increase Cd2+ affinity without affecting toxin block or MTS binding.
The IC50 for Cd2+ block of all of the mutants is not affected by prior addition of MTS reagents. This suggests that the cysteine mutants rather than being modified but not altering current are, instead, not modified by MTS reagents. These data imply that some parts of the P segments in the external pore are not accessible to modification by external MTS reagents, which require a reaction sphere of ∼6 A.
The pattern of side chain accessibility in the pore helix region has been studied in a number of K channels. As in the case of the Na channel, and as predicted by the KcsA structure, hydrophobic residues at the NH2-terminal side of the pore helix appear to have their side chains exposed using Ag+ (Lu and Miller 1995) or charybodotoxin (Ranganathan et al. 1996) as structural probes. In contrast, there is less experimental data to assign the location of the side chains of the residues at the COOH-terminal side of the helix. However, cysteine scanning mutagenesis followed by MTS modification generally has shown that the residues in this region of the K channel pore are inaccessible (Kurz et al. 1995; Pascual et al. 1995). Residues at or near the COOH-terminal side of the putative pore helix are involved in the determination of sensitivity to block by internal TEA (Yellen et al. 1991). The absence of an internally accessible site flanking the pore helices of the Na channel suggests a different orientation compared with K channel pore helices.
Summary
Our data demonstrate that the P segments on the NH2-terminal side of the DEKA residues of the Na channel participate in formation of the outer pore. The P segments do not span the selectivity filter; none of the cysteine substitutions are accessible from the internal mouth of the channel. The pattern of side chain accessibility is consistent with pore helices similar to that seen in the KcsA channel pore crystal structure and side chain accessibility in other K channels. Two of the mutations in domain II (F745C and R750C) that alter Cd2+ block dramatically reduce TTX sensitivity, and may be sites of direct toxin–channel interaction. The data are consistent with previous studies that demonstrate the importance of domains I and II in guanidinium toxin binding and block (Noda et al. 1989; Terlau et al. 1991; Backx et al. 1992; Satin et al. 1992; Lipkind and Fozzard 1994; Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Chen et al. 1997). Arginine residues in the first and second domains (R395 and R750) alter cation selectivity, and in conjunction with the DEKA ring and residues in domain IV, comprise the selectivity determinants of the Na channel.
Our data and previous work (Chiamvimonvat et al. 1996; Perez-Garcia et al. 1996; Chen et al. 1997; Tsushima et al. 1997a,Tsushima et al. 1997b; Yamagishi et al. 1997) provide experimental evidence to support the hypothesis that the SS1 regions of the Na channel form an α helix-turn-coil motif (Fig. 6, bottom). The precise location of the end of the helix and beginning of the turn are not defined; however, modeling predicts that 8–10 amino acids preceding the selectivity filter residue in each domain, have helical character. Overall, this region is hydrophobic, and helical packing is likely to be thermodynamically favorable for this water-accessible part of the protein. The preference for acidic residues to flank the NH2 terminus and basic residues the COOH terminus (Lipkind and Fozzard 2000) suggests the possibility that the NH2 termini of the α helices may extend beyond the region studied; however, in domains III and IV, this would mean the incorporation of one or more glycines into the helices.
The KcsA channel appears to be an evolutionary forerunner of the voltage-dependent family of K channels (MacKinnon et al. 1998). In turn, Na and Ca channels are likely to have evolved from voltage-dependent K channels by gene duplication (Strong et al. 1993). Such evolutionary relationships may explain the remarkable homologies of the mammalian Na channel pore with that of a bacterial K channel. It is likely that the pore helix is essential for the integrity of the transmembrane pore common to all ion channels (Doyle et al. 1998). However, it is the substantial differences in structural detail that mediate the unique functions of the Na channel pore.
Acknowledgments
We thank Ailsa Mendez-Fitzwilliam for construction of the mutants.
This work was supported by the National Institutes of Health (R01 HL50411 and R01 HL52376). R.A. Li is supported by the Heart and Stroke Foundation of Canada. E. Marbán holds the Michel Mirowski, M.D. Professorship of Cardiology of the Johns Hopkins University.
Footnotes
Abbreviations used in this paper: DEKA, Na channel pore resi-dues D400, E755, K1237, and A1529; MTS, methanethiosulfonate; MTSEA, methanethiosulfonate ethylammonium; MTSES, methanethiosulfonate ethylsulfonate; MTSET, methanethiosulfonate ethyltrimethylammonium; P, pore-lining; TTX, tetrodotoxin.
References
- Backx P.H., Yue D.T., Lawrence J.H., Marban E., Tomaselli G.F. Molecular localization of an ion-binding site within the pore of mammalian sodium channels. Science. 1992;257:248–251. doi: 10.1126/science.1321496. [DOI] [PubMed] [Google Scholar]
- Benitah J.P., Tomaselli G.F., Marban E. Adjacent pore-lining residues within sodium channels identified by paired cysteine mutagenesis. Proc. Natl. Acad. Sci. USA. 1996;93:7392–7396. doi: 10.1073/pnas.93.14.7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benitah J.P., Ranjan R., Yamagishi T., Janecki M., Tomaselli G.F., Marban E. Molecular motions within the pore of voltage-dependent sodium channels. Biophys. J. 1997;73:603–613. doi: 10.1016/S0006-3495(97)78096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall W.A. Structure and function of voltage-gated ion channels. Annu. Rev. Biochem. 1995;64:493–531. doi: 10.1146/annurev.bi.64.070195.002425. [DOI] [PubMed] [Google Scholar]
- Chang N.S., French R.J., Lipkind G.M., Fozzard H.A., Dudley S., Jr. Predominant interactions between mu-conotoxin Arg-13 and the skeletal muscle Na+ channel localized by mutant cycle analysis. Biochemistry. 1998;37:4407–4419. doi: 10.1021/bi9724927. [DOI] [PubMed] [Google Scholar]
- Chen S., Hartmann H., Kirsch G. Cysteine mapping in the ion selectivity and toxin binding region of the cardiac Na+ channel pore. J. Membr. Biol. 1997;155:11–25. doi: 10.1007/s002329900154. [DOI] [PubMed] [Google Scholar]
- Chiamvimonvat N., Perez-Garcia M.T., Ranjan R., Marban E., Tomaselli G.F. Depth asymmetries of the pore-lining segments of the Na+ channel revealed by cysteine mutagenesis. Neuron. 1996;16:1037–1047. doi: 10.1016/s0896-6273(00)80127-0. [DOI] [PubMed] [Google Scholar]
- Colquhoun D., Sigworth F.J. Fitting and statistical analysis of single-channel records. In: Sakmann B., Neher E., editors. Single-channel Recording. Plenum Press; New York: 1983. pp. 191–263. [Google Scholar]
- Doyle D.A., Cabral J.M., Pfuetzner R.A., Kuo A., Gulbis J.M., Cohen S.L., Chait B.T., Mackinnon R. The structure of the potassium channelmolecular basis of K+ conduction and selectivity. Science. 1998;280:69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- Dudley S., Jr., Todt H., Lipkind G., Fozzard H. A mu-conotoxin-insensitive Na+ channel mutantpossible localization of a binding site at the outer vestibule. Biophys. J. 1995;69:1657–1665. doi: 10.1016/S0006-3495(95)80045-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre I., Moczydlowski E., Schild L. Specificity for block by saxitoxin and divalent cations at a residue which determines sensitivity of sodium channel subtypes to guanidinium toxins. J. Gen. Physiol. 1995;106:203–229. doi: 10.1085/jgp.106.2.203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre I., Moczydlowski E., Schild L. On the structural basis for ionic selectivity among Na+, K+, and Ca2+ in the voltage-gated sodium channel. Biophys. J. 1996;71:3110–3125. doi: 10.1016/S0006-3495(96)79505-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording form cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Heinemann S., Terlau H., Stuhmer W., Imoto K., Numa S. Calcium channel characteristics conferred on the sodium channel by single mutations. Nature. 1992;356:441–443. doi: 10.1038/356441a0. [DOI] [PubMed] [Google Scholar]
- Heinemann S.H., Schlief T., Mori Y., Imoto K. Molecular pore structure of voltage-gated sodium and calcium channels. Braz. J. Med. Biol. Res. 1994;27:2781–2802. [PubMed] [Google Scholar]
- Hidalgo P., MacKinnon R. Revealing the architecture of a K+ channel pore through mutant cycles with a peptide inhibitor. Science. 1995;268:307–310. doi: 10.1126/science.7716527. [DOI] [PubMed] [Google Scholar]
- Hsu K., Amzel M., Tomaselli G.F., Marban E. Prediction of pore helices in sodium and calcium channels. Biophys. J. 1999;76:A82. [Google Scholar]
- Isom L.L., De Jongh K.S., Patton D.E., Reber B.F., Offord J., Charbonneau H., Walsh K., Goldin A.L., Catterall W.A. Primary structure and functional expression of the beta 1 subunit of the rat brain sodium channel. Science. 1992;256:839–842. doi: 10.1126/science.1375395. [DOI] [PubMed] [Google Scholar]
- Kurz L.L., Zuhlke R.D., Zhang H.J., Joho R.H. Side-chain accessibilities in the pore of a K+ channel probed by sulfhydryl-specific reagents after cysteine-scanning mutagenesis. Biophys. J. 1995;68:900–905. doi: 10.1016/S0006-3495(95)80266-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.A., Tsushima R.G., Kallen R.G., Backx P.H. Pore residues critical for mu-CTX binding to rat skeletal muscle Na+ channels revealed by cysteine mutagenesis. Biophys. J. 1997;73:1874–1884. doi: 10.1016/S0006-3495(97)78218-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R.A., Ennis I.L., Velez P., Tomaselli G.F., Marban E. Novel structural determinants of mu-conotoxin block in rat skeletal muscle Na+ channels J. Biol. Chem. 275 2000. 27551 27558a [DOI] [PubMed] [Google Scholar]
- Li R.A., Velez P., Chiamvimonvat N., Tomaselli G.F., Marban E. Charged residues between the selectivity filter and S6 segments contribute to the permeation phenotype of the sodium channel J. Gen. Physiol. 115 2000. 81 92b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind G., Fozzard H. A structural model of the tetrodotoxin and saxitoxin binding site of the Na+ channel. Biophys. J. 1994;66:1–13. doi: 10.1016/S0006-3495(94)80746-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipkind G.M., Fozzard H.A. KcsA crystal structure as framework for a molecular model of the Na(+) channel pore. Biochemistry. 2000;39:8161–8170. doi: 10.1021/bi000486w. [DOI] [PubMed] [Google Scholar]
- Lu Q., Miller C. Silver as a probe of pore-forming residues in a potassium channel. Science. 1995;268:304–307. doi: 10.1126/science.7716526. [DOI] [PubMed] [Google Scholar]
- MacKinnon R., Cohen S.L., Kuo A., Lee A., Chait B.T. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 1998;280:106–109. doi: 10.1126/science.280.5360.106. [DOI] [PubMed] [Google Scholar]
- MacKinnon R., Yellen G. Mutations affecting TEA blockade and ion permeation in voltage-activated K+ channels. Science. 1990;250:276–279. doi: 10.1126/science.2218530. [DOI] [PubMed] [Google Scholar]
- Marban E., Yamagishi T., Tomaselli G.F. Structure and function of voltage-gated sodium channel. J. Physiol. 1998;508:647–657. doi: 10.1111/j.1469-7793.1998.647bp.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda M., Suzuki H., Numa S., Stühmer W. A single point mutation confers tetrodotoxin and saxitoxin insensitivity on the sodium channel II. FEBS Lett. 1989;259:213–216. doi: 10.1016/0014-5793(89)81531-5. [DOI] [PubMed] [Google Scholar]
- Pascual J.M., Shieh C.C., Kirsch G.E., Brown A.M. K+ pore structure revealed by reporter cysteines at inner and outer surfaces. Neuron. 1995;14:1055–1063. doi: 10.1016/0896-6273(95)90344-5. [DOI] [PubMed] [Google Scholar]
- Penzotti J.L., Fozzard H.A., Lipkind G.M., Dudley S.C., Jr. Differences in saxitoxin and tetrodotoxin binding revealed by mutagenesis of the Na+ channel outer vestibule. Biophys. J. 1998;75:2647–2657. doi: 10.1016/S0006-3495(98)77710-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia M., Chiamvimonvat N., Ranjan R., Balser J.R., Tomaselli G.F., Marban E. Mechanisms of sodium/calcium selectivity in sodium channels probed by cysteine mutagenesis and sulfhydryl modification. Biophys. J. 1997;72:989–996. doi: 10.1016/S0006-3495(97)78751-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Garcia M.T., Chiamvimonvat N., Marban E., Tomaselli G.F. Structure of the sodium channel pore revealed by serial cysteine mutagenesis. Proc. Natl. Acad. Sci. USA. 1996;93:300–304. doi: 10.1073/pnas.93.1.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillipson L.H., Malayev A., Kuznetkov A., Chang C., Nelson D. Functional and biochemical characterization of the human potassium channel Kv1.5 with a transplanted carboxyl-terminal epitope in stable mammalian cell lines. Biochem. Biophys. Acta. 1993;1153:112–121. doi: 10.1016/0005-2736(93)90282-5. [DOI] [PubMed] [Google Scholar]
- Ranganathan R., Lewis J.H., MacKinnon R. Spatial localization of the K+ channel selectivity filter by mutant cycle-based structure analysis. Neuron. 1996;16:131–139. doi: 10.1016/s0896-6273(00)80030-6. [DOI] [PubMed] [Google Scholar]
- Rost B., Casadio R., Fariselli P., Sander C. Transmembrane helices predicted at 95% accuracy. Prot. Sci. 1995;4:521–533. doi: 10.1002/pro.5560040318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin J., Kyle J., Chen M., Bell P., Cribbs L., Fozzard H., Rogart R. A mutant of TTX-resistant cardiac sodium channels with TTX-sensitive properties. Science. 1992;256:1202–1205. doi: 10.1126/science.256.5060.1202. [DOI] [PubMed] [Google Scholar]
- Strong M., Chandy K.G., Gutman G.A. Molecular evolution of voltage-sensitive ion channel geneson the origins of electrical excitability. Mol. Biol. Evol. 1993;10:221–242. doi: 10.1093/oxfordjournals.molbev.a039986. [DOI] [PubMed] [Google Scholar]
- Stuhmer W. Structure–function studies of voltage-gated ion channels. Annu. Rev. Biophys. Biophys. Chem. 1991;20:65–78. doi: 10.1146/annurev.bb.20.060191.000433. [DOI] [PubMed] [Google Scholar]
- Sunami A., Glaaser I.W., Fozzard H.A. A critical residue for isoform difference in tetrodotoxin affinity is a molecular determinant of the external access path for local anesthetics in the cardiac sodium channel. Proc. Natl. Acad. Sci. USA. 2000;97:2326–2331. doi: 10.1073/pnas.030438797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terlau H., Heinemann S., Stühmer W., Pusch M., Conti F., Imoto K., Numa S. Mapping the site of block by tetrodotoxin and saxitoxin of sodium channel II. FEBS Lett. 1991;293:93–96. doi: 10.1016/0014-5793(91)81159-6. [DOI] [PubMed] [Google Scholar]
- Trimmer J.S., Cooperman S.S., Tomiko S.A., Zhou J.Y., Crean S.M., Boyle M.B., Kallen R.G., Sheng Z.H., Barchi R.L., Sigworth F.J. Primary structure and functional expression of a mammalian skeletal muscle sodium channel. Neuron. 1989;3:33–49. doi: 10.1016/0896-6273(89)90113-x. [DOI] [PubMed] [Google Scholar]
- Tsushima R., Li R., Backx P. P-loop flexibility in Na+ channel pores revealed by single- and double-cysteine replacements J. Gen. Physiol. 110 1997. 59 72a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsushima R., Li R., Backx P.H. Altered ionic selectivity of the sodium channel revealed by cysteine mutations within the pore J. Gen. Physiol. 109 1997. 463 475b [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamagishi T., Janecki M., Marban E., Tomaselli G. Topology of the P segments in the sodium channel pore revealed by cysteine mutagenesis. Biophys. J. 1997;73:195–204. doi: 10.1016/S0006-3495(97)78060-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellen G., Jurman M.E., Abramson T., MacKinnon R. Mutations affecting internal TEA blockade identify the probable pore-forming region of a K+ channel. Science. 1991;251:939–942. doi: 10.1126/science.2000494. [DOI] [PubMed] [Google Scholar]