

Abstract
CNG channels in vivo are heteromers of homologous α and β subunits that each contain a six-transmembrane segment domain and a COOH-terminal cytoplasmic cyclic nucleotide binding domain (BD). In heterologous expression systems, heteromeric αβ channels activate with greater sensitivity to ligand than do homomeric α channels; however, ligand-gating of channels containing only β subunit BDs has never been studied because β subunits cannot form functional homomeric CNG channels. To characterize directly the contribution of the β subunit BD to ligand-gating, we constructed a chimeric subunit, X-β, whose BD sequence was that of the β subunit CNG5 from rat, but whose sequence outside the BD was derived from α subunits. For comparison, we constructed another chimera, X-α, whose sequence outside the BD was identical to that of X-β, but whose BD sequence was that of the α subunit CNG2 from catfish. When expressed in Xenopus oocytes, X-β and X-α each formed functional homomeric channels activated by both cAMP and cGMP. This is the first demonstration that the β subunit BD can couple ligand binding to activation in the absence of α subunit BD residues. Notably, both agonists activate X-β more effectively than X-α (higher opening efficacy and lower K1/2). The BD is believed to comprise two functionally distinct subdomains: (1) the roll subdomain (β-roll and flanking A- and B-helices) and (2) the C-helix subdomain. Opening efficacy was previously believed to be controlled primarily by the C-helix, but when we made additional chimeras by exchanging the subdomains between X-β and X-α, we found that both subdomains contain significant determinants of efficacy and agonist selectivity. In particular, only channels containing the roll subdomain of the β subunit had high efficacy. Thermodynamic linkage analysis shows that interaction between the two subdomains accounts for a significant portion of their contribution to activation energetics.
Keywords: cAMP, cGMP, ligand-gated channels, allosteric activation, thermodynamic linkage
INTRODUCTION
CNG channels are important for visual (Fesenko et al. 1985; Yau 1994) and olfactory (Nakamura and Gold 1987) signal transduction. They conduct cations nonselectively upon activation in response to direct binding of cytoplasmic cAMP or cGMP (for review see Zagotta and Siegelbaum 1996). In vertebrates, six distinct homologous CNG channel genes have been identified (called CNG1–CNG6 in the nomenclature proposed by Biel et al. 1999). Tissue-specific combinatorial expression of paralogous subunits is likely to prove a widespread strategy for customizing the dynamic range of the CNG channel response to the tissue's specific physiological requirements. Thus, it is important to understand how sequence variation among isoforms gives rise to functional differences.
In heterologous expression systems, some isoforms can form functional homomeric channels, whereas other isoforms cannot; the former are termed α subunits, and the latter are termed β subunits (Chen et al. 1994). All known orthologues of CNG1, 2, and 3 are α subunits, and all known orthologues of CNG4, 5, and 6 are β subunits; thus, the strictly operational designation probably accurately reflects differences in the in vivo function between the two groups. Channels formed from coexpressed α and β subunits differ in their activation and permeation properties from α subunit homomers. Thus, β subunits act as modulatory subunits, presumably coassembling with α subunits in heteromers. It is most likely that such αβ heteromers form in vivo, because functional properties of native channels are reproduced more closely in heterologous systems by coexpressing the relevant tissue-specific combination of α and β paralogues than by expressing only α homomers (Chen et al. 1993; Bradley et al. 1994; Liman and Buck 1994; Körschen et al. 1995; Sautter et al. 1998; Bönigk et al. 1999; Komatsu et al. 1999; Gerstner et al. 2000). The defect that prevents β subunits from forming functional CNG channels without α subunits remains unknown.
Despite their marked functional difference, both α and β subunits contain the conserved structural motifs considered essential to CNG channel function, arranged with the same modular organization. All subunits contain a transmembrane domain homologous to that of the voltage-gated potassium channel family (Jan and Jan 1992), with six putative membrane-spanning segments S1–S6 and a reentrant “P-loop” between S5 and S6. The P-loop controls the conductance and pharmacology of the aqueous pore (Goulding et al. 1993; Kramer et al. 1994; Root and MacKinnon 1994) as it does in potassium channels (MacKinnon 1995). Like the voltage-gated potassium channels, CNG channels formed from α subunits are tetrameric (Gordon and Zagotta 1995a; Liu et al. 1996), and αβ heteromers are thought to be also tetrameric with two α and two β subunits (Shapiro and Zagotta 1998; Shammat and Gordon 1999; He et al. 2000). At the cytoplasmic COOH-terminal end of S6, all CNG channel subunits contain a highly conserved “C-linker” region of ∼80 residues, followed by a cyclic nucleotide binding domain (BD) homologous to those of cAMP-dependent protein kinases and the catabolite activator protein (CAP) of Escherichia coli (Kaupp et al. 1989; Shabb and Corbin 1992). The BDs of both α and β subunits in αβ heteromers can be labeled by photoreactive agonists (Brown et al. 1995), and the BD is a natural focus for studies of CNG channel activation properties. Unfortunately, since the cyclic nucleotide activation properties of β subunits cannot be studied in the absence of α subunits, it has not been possible to make a direct comparison of the functional consequences of sequence differences between the α and β subunit BDs.
Structure–function studies of the BD in α homomers, guided by homology modeling, suggest that the BD itself has a modular organization, with two structural subdomains that are also functionally distinct. The homology models (Kumar and Weber 1992; Varnum et al. 1995; Scott et al. 1996) were based on the known 3-D structures of the cAMP-liganded BDs in CAP (Weber and Steitz 1987) and PKA (Su et al. 1995). In these structures, the BD contains a roll subdomain and a C-helix subdomain separated by a proline residue: the roll subdomain consists of a “β-roll” of four pairs of antiparallel β-strands (β1 through β8) flanked by two short α-helices (A- and B-helix), and the C-helix subdomain consists of a single, long α-helix. The cyclic nucleotide molecule sits between these two subdomains, with the cyclic phosphate moiety contacting the β-roll and the purine moiety contacting the C-helix. The C-helix, and in particular residue 604 (see materials and methods for numbering convention), serves as the major determinant of cGMP selectivity in certain α subunit BDs (Goulding et al. 1994; Varnum et al. 1995); similarly, mutations in the β subunit BD at position 604 influence the selectivity of αβ heteromers (Pagès et al. 2000; Shapiro and Zagotta 2000; He and Karpen 2001). A current model for C-helix function is that the purine ring's interaction with the C-helix is stronger when the channel is open than when the channel is closed, and as a consequence, this interaction contributes “activation coupling energy” to preferential stabilization of the open state (increasing open probability). In contrast, the cyclic phosphate interaction with the roll subdomain is state-independent (Tibbs et al. 1998) and so contributes binding energy to strengthen the affinity of the BD for ligand without contributing coupling energy.
In this light, the previous observations that the β subunit BD contributes to αβ heteromer activation leave open the question of how much activation coupling energy and/or binding energy can be derived from the β subunit BD itself. Interactions between BDs of neighboring subunits have been proposed to contribute to activation coupling energy in α homomers (Liu et al. 1998; Paoletti et al. 1999), and CAP forms a homodimer in which interaction between subunits is essential for activation (Cheng et al. 1995); thus, an attractive hypothesis is that the β subunit BD must interact with the BD of a neighboring α subunit to contribute significant coupling energy to channel activation. This would predict that a channel containing only β subunit BDs and no α subunit BDs would be incompetent or inefficient in coupling ligand binding to opening, or might even fail to bind agonist at all. In this report, we provide the first direct evidence that the β subunit BD can bind ligand and efficiently couple binding to channel opening, without assistance from α subunit BD residues. Our approach was to construct a chimeric CNG channel subunit composed of α subunit sequence, in which the α subunit BD sequence has been replaced with the BD sequence from a β subunit —namely rat CNG5 (rCNG5), the first olfactory β subunit to be cloned (Bradley et al. 1994; Liman and Buck 1994). This chimera (called X-β) forms functional homomeric channels that are efficiently activated by cyclic nucleotide. Our result definitively rules out the possibility of a BD defect, and allows us to conclude that the β subunit rCNG5 is deficient only in sequence regions outside the BD. We compared X-β with other similar chimeras that contain α subunit BDs but that are identical to X-β in all sequence outside the BD. This provided the first direct functional comparison of BDs from α and β subunits as they operate in homomeric channels, and had several unanticipated findings. The rCNG5 BD in X-β turns out to be more effective in ligand-gating than some α subunit BDs, such as that from the olfactory α subunit CNG2 from catfish (fCNG2; Goulding et al. 1992). We demonstrate that the differences in activation properties between the chimeras containing rCNG5 and fCNG2 BDs are determined by both the C-helix and roll subdomain sequences, and that the roll subdomain from the fCNG2 BD imparts an unusual new form of agonist selectivity, namely agonist-selective desensitization. Finally, we demonstrate that the roll and C-helix subdomains interact strongly with each other in determining preferential open state stabilization.
MATERIALS AND METHODS
Molecular Subcloning
All CNG channel subunits and chimeras were subcloned into the oocyte expression vector pGEM-HE (Liman et al. 1992). Chimeras were constructed from the α subunit CNG1 from bovine rod photoreceptors (bCNG1; Kaupp et al. 1989), α subunit CNG2 from catfish olfactory neurons (fCNG2; Goulding et al. 1992), and β subunit CNG5 from rat olfactory neurons (rCNG5; Bradley et al. 1994; Liman and Buck 1994). (Note: CNG5 also has been called OCNC2, OCNCβ, olfactory β, or CNCα4.) The chimeric α subunit ROON-S2 was previously described (Tibbs et al. 1997) and consists of bCNG1 in which bCNG1 N91-S240 (i.e., “N-S2” region; Goulding et al. 1994) is replaced by fCNG2 E89-R215, and bCNG1 A344-A378 (i.e., P-loop) is replaced by fCNG2 S314-F348. The BD sequences are defined as bCNG1 L485-A614, and the homologous sequences are defined as fCNG2 L455-G584 and rCNG5 L356-A485. These three 130-residue BD sequences can be aligned with no gaps. Since most extant literature deals with bCNG1, for the sake of uniformity in the text an individual BD residue is referred to by its corresponding position number in bCNG1 regardless of the residue's identity or its position number in a particular subunit chimera. The C-helix sequence is defined as bCNG1 D588-A614, and corresponding sequences in fCNG2 and rCNG5. X-β is ROON-S2 in which the bCNG1 BD is replaced by the rCNG5 BD. X-α is ROON-S2 in which the bCNG1 BD is replaced by the fCNG2 BD. The subscripted symbols αR and αC denote the α subunit fCNG2 roll (R) and C-helix (C) subdomains, respectively, and the symbols βR and βC denote β subunit rCNG5 roll and C-helix subdomains, respectively. Hence, X-αR/βC has a BD that contains the roll subdomain (R) from the α subunit fCNG2, and the C-helix (C) from the β subunit rCNG5; specifically, it is X-α in which the fCNG2 C-helix is replaced by the rCNG5 C-helix. Similarly, X-βR/αC is X-β in which the rCNG5 (β subunit) C-helix is replaced by the fCNG2 (α subunit) C-helix. Chimeras were constructed using PCR techniques as described previously (Goulding et al. 1993; Paoletti et al. 1999); all four X-chimeras were dideoxy sequenced in their entirety.
Patch-clamp Recording of Macroscopic Currents
Xenopus oocytes were prepared and injected with 0.25–25 ng RNA transcribed from linearized cDNA as previously described (Paoletti et al. 1999). Inside-out patches were obtained from oocytes 1–5 d after injection, using electrodes of resistance 1–5 MΩ. Divalent-free solution (Liu et al. 1998) was identical in both pipette and bath, and contained the following (in mM): 67 KCl, 30 NaCl, 10 HEPES, 10 EGTA, 1 EDTA, pH 7.2 with KOH. For Ni2+ potentiation experiments, the pipette solution was unchanged, whereas the bath (intracellular) solution contained 89 mM KCl, with 5 or 10 μM NiCl2 (Sigma-Aldrich) included and EGTA and EDTA omitted. Na-cGMP or Na-cAMP (Sigma-Aldrich) were included as needed in the bath solution by iso-osmolar replacement of NaCl. Patches were washed continuously with bath solution by a gravity perfusion system. Data was acquired with an Axopatch 200A amplifier (Axon Instruments), recorded via a VR10B digital data recorder (Instrutech) to VHS tape in a VCR (model VC-A206; Sharp), filtered at 4 kHz (eight-pole Bessel, No. 900; Frequency Devices) and digitized at 1 kHz with either a DigiData 1200 interface and pClamp 6.0, or a Digidata 1320A interface and pClamp 8.0 (Axon Instruments, Inc.).
Macroscopic current in the presence of agonist was measured after steady-state current level was reached at a holding voltage of −80 mV; currents from two to four repetitions of a voltage step protocol (+100 to −100 mV in 40-mV steps) were averaged, and corrected by subtraction of capacitance transients and leak currents obtained by running the same protocol in agonist-free solution. Dose–response curves were measured from currents at −100 mV; rectification was calculated from the ratio of currents at +60 and −60 mV. To verify time-stationarity of the dose–response curve, measurements at selected saturating and subsaturating agonist concentrations were repeated before and after dose–response data collection. The criterion for stationarity was that the duplicate measurements deviate by ≤15% from each other. Spontaneous changes in maximal current amplitudes and in K1/2 were observed (”runup” and/or “rundown”) and the time required to reach stationarity was variable, often >10 min after patch excision. This is significantly longer than the ∼5-min times reported previously for spontaneous dephosphorylation at Y498 of bCNG1 (Molokanova et al. 1999), and may be due to the presence of an additional unknown phosphorylation site.
Potentiation of channel activation by intracellular Ni2+ in saturating cAMP concentration was measured as follows. Macroscopic currents in 30 mM cAMP plus 5–10 μM Ni2+ (Imax,cAMP+Ni) were measured at −80 mV. These measurements were bracketed by measurements of currents in 30 mM cAMP without Ni2+, which were averaged to give Imax,cAMP. The Ni2+ concentration was saturating since 5 and 10 μM Ni2+ produced identical potentiation.
Fitting of Dose–Response Curves
For chimeras X-β and X-βR/αC, complete macroscopic current dose–response curves (≥4 concentrations distributed from saturating level to below K1/2) were collected from individual patches. For each curve, response current I at a given agonist concentration, [A], was fitted with the Hill equation, I = Imax/(1 + (K1/2/[A])h), where K1/2 is the concentration of agonist A eliciting half-maximal activation, h is the Hill coefficient, and Imax is the maximal current amplitude. Data were weighted by 1/SD during fitting (SigmaPlot). The K1/2 selectivity ratio (K1/2,cGMP/K1/2,cAMP) was evaluated for each patch; a value >1 for this ratio corresponds to cAMP selectivity.
For the low efficacy chimeras, X-α and X-αR/βC, stationarity was often not reached until very long times (20–40 min) after patch excision, and complete macroscopic dose–response curves could not always be collected before degradation of patch quality. Therefore, complete and partial datasets for each chimera (all sets checked for stationarity) were composited as follows. For each patch, currents were normalized by dividing by the current measured in the same patch in 3 mM cGMP. Normalized data for all patches of a given chimera were then pooled and averaged to give a mean relative current, Irel, for each agonist concentration [A]. This composite dataset was then fitted (weighted by 1/SD) with the Hill equation, Irel = Imax,rel/(1 + (K1/2/[A])h), where K1/2 and h are the usual Hill parameters and Imax,rel is the maximal relative current amplitude. The K1/2 selectivity ratio was calculated as K1/2,cGMP/K1/2,cAMP. To plot data in terms of open probability for illustrative purposes (see Fig. 5 and Fig. 6), the composite dataset and fitted Hill equation for both cAMP and cGMP activation were multiplied by the factor Pmax,cAMP/Imax,rel,cAMP, where Pmax,cAMP is the mean efficacy in cAMP determined from single-channel recordings (see next section).
Figure 5.
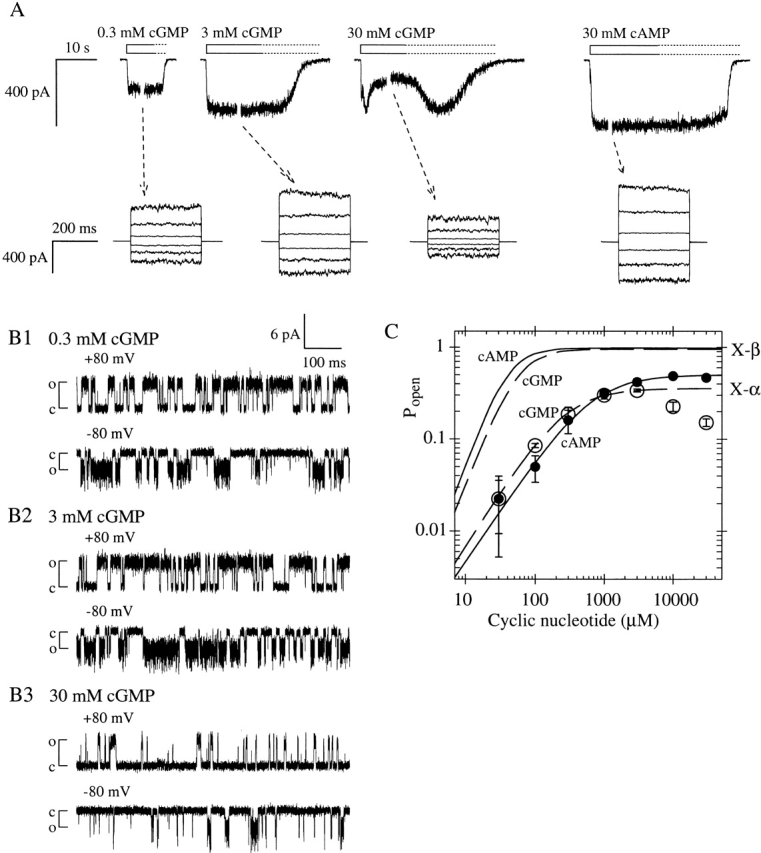
Chimera X-α is less efficiently activated by cGMP than is X-β, and exhibits desensitization at high cGMP concentrations. (A, top traces) Macroscopic current traces from homomeric X-α channels (different patch than that shown in Fig. 4 A), elicited by indicated concentrations of agonist. Channels desensitize in 30 mM cGMP and recover to the activation level of 3 mM cGMP during the slow washout of agonist (dotted portion of open bars). (bottom traces) Currents during voltage steps at indicated steady-state cGMP concentrations (tested as in Fig. 1). (B1–B3) Excerpts of single X-α channel recordings at indicated potentials and steady-state cGMP concentrations (same patch as in Fig. 4, B1–B4). Popen values from excerpt in 3 mM cGMP are 0.676 at +80 mV and 0.462 at −80 mV; values from excerpt in 30 mM cGMP (collected at steady-state after onset of desensitization) are 0.131 at +80 mV and 0.125 at −80 mV. (C) Mean dose–response data for X-α activation by cAMP (closed circles) and cGMP (open circles) at −100 mV, compiled from six patches expressing macroscopic currents; graph is normalized with Pmax,cAMP = 0.49, the mean from single-channel measurements (materials and methods). Lines show Hill equation fits for cAMP (solid) and cGMP (dashed; excluding data >3 mM); see Table for values of K1/2 and h. Using fitted values for Imax, the ratio Imax,cAMP/Imax,cGMP is 1.405 ± 0.034. For comparison, X-β dose–response curves for cAMP (solid line) and cGMP (dashed line), taken from Fig. 1C and normalized as in Fig. 4 C, are shown.
Figure 6.
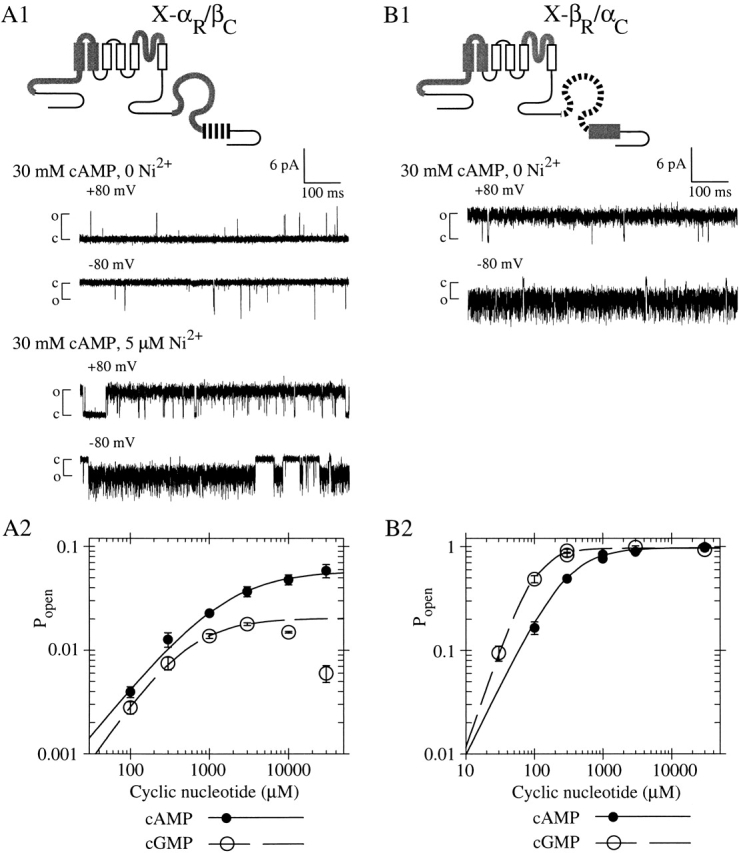
Chimera X-αR/βC is poorly activated by cyclic nucleotide, whereas the complementary chimera X-βR/αC is efficiently activated by cyclic nucleotide. (A1) X-αR/βC contains the roll subdomain from fCNG2 and the C-helix from rCNG5. Single-channel recordings were made at indicated potentials with a steady-state concentration of 30 mM cAMP, with or without 5 μM Ni2+ present. (top traces) Pmax is low in absence of Ni2+ (Popen = 0.004 at +80 mV and 0.012 at −80 mV). (bottom traces) Channel opening is potentiated by Ni2+, showing only one channel is present (Popen = 0.874 at +80 mV and 0.878 at −80 mV). (A2) Mean dose–response data for X-αR/βC activation by cAMP (closed circles) and cGMP (open circles) at −100 mV, compiled from four patches expressing macroscopic currents (materials and methods); graph is normalized using Pmax,cAMP = 0.056, the mean from single-channel measurements. Lines show Hill equation fits with parameters (±SEM) as follows: for cAMP (solid), K1/2 = 1,570 ± 370 μM, h = 0.932 ± 0.065; for cGMP (dashed, excluding data >3 mM), K1/2 = 511 ± 37 μM, h = 1.120 ± 0.044; Imax,cAMP/Imax,cGMP = 2.85 ± 0.30. (B1) X-βR/αC contains the roll subdomain from rCNG5 and the C-helix from fCNG2. Single-channel recording excerpt (no Ni2+ present) shows high Pmax in 30 mM cAMP (Popen = 0.9878 at +80 mV and 0.9896 at −80 mV). (B2) Dose–response data for X-βR/αC activation by cAMP (solid circles) and cGMP (open circles) at −100 mV, from a patch with maximal current ∼400 pA; graph is normalized using Pmax,cAMP = 0.954, the mean from single-channel measurements. Lines show Hill equation fits with parameters (±SE) as follows: for cAMP (solid), K1/2 = 288 ± 20 μM, h = 1.37 ± 0.12; for cGMP (dashed), K1/2 = 95.8 ± 9.2 μM, h = 1.95 ± 0.22; Imax,cAMP/Imax,cGMP = 1.0078 ± 0.030.
Single-channel Recording and Analysis
Solutions and instrumentation for recording single-channel currents in inside-out patches were identical to those used in macroscopic current experiments; pipettes were coated with Sylgard 184 (Corning). Currents were recorded from either 10–150-s stretches of data at −80 mV or 800-ms pulses at +80 mV, and were digitized at 20 kHz. As reported for previous observations of intact fCNG2 (Goulding et al. 1992), we sometimes observed slow (1–5 s timescale) slight fluctuations in open probability or, infrequently, periods of complete inactivity lasting 0.5–5 s; however, we did not observe sudden switching between long-lived (5–10 s) modes with very low Popen and very high Popen.
Continuous single-channel records were visually inspected and manually baseline-corrected (Clampfit 8.0). All-points amplitude histograms were compiled for each record and each was fitted (custom routine in LabScript; Origin 5.0) with a sum of Gaussian peaks representing a single closed state and either a single open state (at +80 mV) or multiple open states (at −80 mV). In the fit, the Gaussian means, amplitudes, and SDs were free parameters; amplitudes were constrained to be positive and SDs constrained to the range 0.05–1 pA. Goodness of fit was judged by satisfaction of three criteria: (1) visual inspection, (2) χ2 decreased by a factor of <10−7 with successive Levenburg-Marquardt iterations, and (3) total fit residuals were less than the area of the least-populated Gaussian component. To determine open state conductances, the fitted open state Gaussian means were corrected by subtraction of the fitted closed state Gaussian mean.
The closed probability Pclosed was calculated as the area of the closed state Gaussian divided by the area of the summed Gaussians. For some records with Pclosed < 0.01, the closed state Gaussian mean was fixed at zero to ensure a good fit. For one X-β recording, the closed state Gaussian area was less than the total fit residuals when other goodness of fit criteria were satisfied, so an upper limit for Pclosed was calculated by dividing the total fit residuals by the area of the summed Gaussians (0.0002). For records with large Pclosed, where the histogram was dominated by a single closed state peak, only the histogram data on the side of the peak away from the open state was fitted to a single Gaussian; Pclosed was calculated as the area of the Gaussian divided by the total histogram area. Finally, open probability was calculated as Popen = 1 − Pclosed.
Pmax was determined as Popen at −80 mV in concentrations of agonist as follows, with range and mean ± SD of data record lengths given in parentheses: for X-β, 3 mM cAMP (14–59 s, mean 39 ± 18 s) or 3 mM cGMP (30–146 s, mean 62 ± 48 s); for X-α, 10–30 mM cAMP (15–56 s, mean 29 ± 13) or 3 mM cGMP (17–55 s, mean 30 ± 11 s); for X-αR/βC, 30 mM cAMP (23–111 s, mean 53 ± 28 s); and for X-βR/αC, 30 mM cAMP (26–66 s, mean 54 ± 16 s) or 30 mM cGMP (32–98 s, mean 60 ± 26 s). Where applications of the noted agonist concentration occurred more than once over the course of a patch experiment, only the recording from the later application was included.
Unless otherwise noted, all experimental uncertainties reported are ±SD, with n being the number of patches, and significance was assessed by t test (population comparisons by unpaired t test).
RESULTS
Construction of a Chimeric CNG Channel Subunit Containing the BD of a β Subunit
We sought to examine the unknown functionality of the BD of the β subunit CNG5; to date, the only functionally characterized CNG5 subunit is that from rat (rCNG5). This subunit was the first modulatory subunit found in CNG channels of olfactory epithelium (Bradley et al. 1994; Liman and Buck 1994); it also exists in vomeronasal neurons (Berghard et al. 1996) and hippocampus (Bradley et al. 1997). Our strategy was to incorporate the rCNG5 BD sequence into a chimeric channel subunit that contained α-subunit sequence outside the BD. We also anticipated comparing this chimera with other similar chimeras, incorporating the BDs from a variety of channels into the same non-BD sequence; differences in function among all of these chimeras must directly reflect differences in their BD sequences. This projected comparative study would require both single channel and macroscopic current measurements on all chimeras, but we anticipated that some BDs might function very poorly in ligand-gating and thereby produce chimeras with low activity. Thus, we chose the non-BD sequence of our chimeras to impart a large single-channel conductance and a channel opening transition that was intrinsically energetically favorable.
The non-BD sequence in our chimeras is that of a previously characterized chimeric α subunit, ROON-S2 (Tibbs et al. 1997, Tibbs et al. 1998). ROON-S2 consists of the full-length sequence of the α subunit of bovine rods (bCNG1; Kaupp et al. 1989) in which selected sequences have been replaced by corresponding sequences from the α subunit of catfish olfactory neurons (fCNG2; Goulding et al. 1992). Replacement of the P-loop imparts a larger single-channel conductance, but does not affect channel activation (Goulding et al. 1993). Replacement of the “N-S2” region (comprising the portion of the transmembrane domain NH2-terminal to S3 together with the immediately adjacent portion of the cytoplasmic NH2 terminus) imparts a more favorable energy of opening in the absence of agonists and a parallel enhancement of channel activation for any agonist (Goulding et al. 1994). In ROON-S2 as well as in other previously characterized chimeras (Goulding et al. 1994), the replacement of bCNG1 N-S2 with fCNG2 N-S2 leaves unchanged the agonist selectivity derived from the BD, which suggests strongly that the canonical ligand-gating mechanism is also unchanged. ROON-S2 can be expressed as functional homomers in oocytes either at the single-channel level or at very high levels producing large macroscopic currents. Thus the non-BD sequence of ROON-S2 provides a suitable platform for testing function of a variety of BD sequences. Our new chimera, X-β, is identical to ROON-S2 from the NH2 terminus to the end of the C-linker (denoted by the X- prefix) but contains the BD of rCNG5 (see Fig. 1 A).
Figure 1.
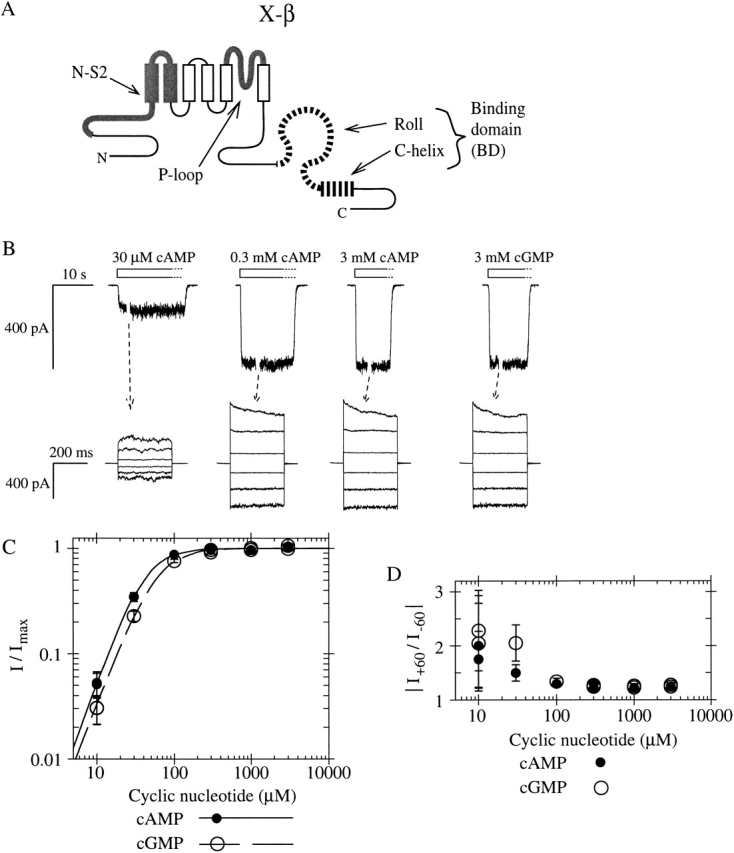
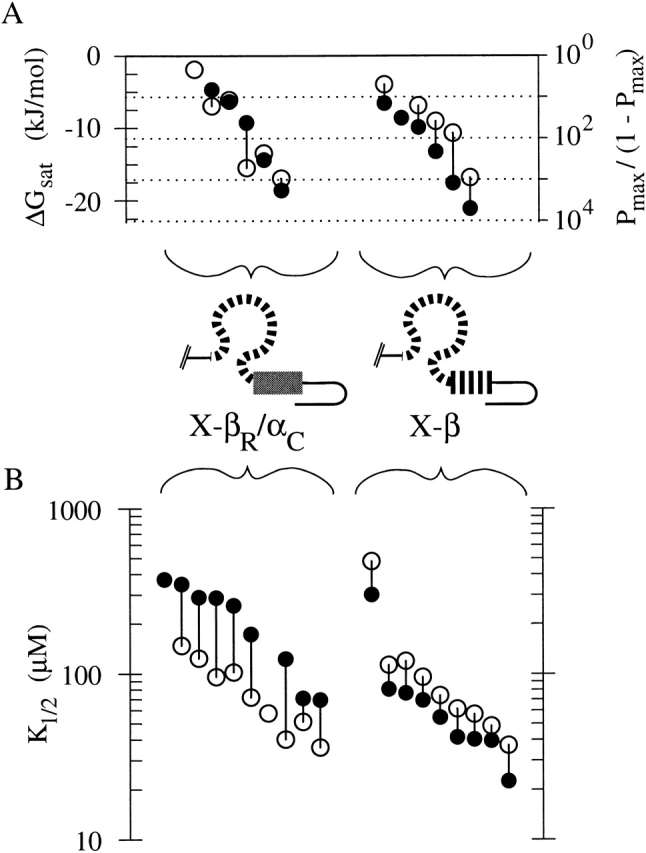
A chimeric CNG channel subunit containing the BD from a modulatory (β) subunit forms a functional homomeric CNG channel. (A) Schematic of CNG channel subunit sequence showing six transmembrane segments (vertical rectangles) and a cytoplasmic COOH-terminal binding domain with two parts: the roll subdomain (a β-roll flanked by A- and B-helices, shown as an omega-shaped loop) followed by the C-helix (horizontal rectangle). Chimera X-β consists of α subunit bCNG1 sequence (thin black lines) with N-S2 region and P-loop replaced by those of the α subunit fCNG2 (thick gray), and the BD replaced by that of the β subunit rCNG5 (striped). (B, top traces) Macroscopic currents from X-β homomeric channels in an inside-out patch, elicited in response to perfused cyclic nucleotide (open bars). Washout of agonist takes several seconds (dotted outline of bars). Holding voltage is −80 mV. Gap in each current trace during perfusion represents an arbitrary interval during which the voltage-step protocol was performed. (bottom traces) Currents elicited in steady-state concentrations of cyclic nucleotide (as indicated in top traces) were measured during voltage pulses from 0 mV to potentials between +100 and −100 mV in intervals of 40 mV. Traces are averages of duplicate trials and are leak subtracted. (C) Relative currents from the patch shown in B at −100 mV in response to cAMP (closed circles) or cGMP (open circles). Lines show fits of the Hill equation with parameters (± SE) as follows: for cAMP (solid line), K1/2 = 40.3 ± 2.8 μM, h = 2.08 ± 0.16; for cGMP (dashed line), K1/2 = 57.1 ± 4.0 μM, h = 1.94 ± 0.13; Imax,cAMP/Imax,cGMP = 0.996 ± 0.021. (D) Outward rectification in the same experiments shown in C, measured by the ratio of currents at +60 and −60 mV for different agonist concentrations.
When RNA for X-β alone is injected into oocytes, large currents are elicited in inside-out patches in response to cyclic nucleotide (Fig. 1 B). Both cAMP and cGMP are full agonists for activating X-β, and the dose–response curves (Fig. 1 C) exhibit steep Hill coefficients, indicating that multiple agonist molecules contribute to channel activation. This result stands in marked contrast with the failure of intact rCNG5 to express functional homomeric channels, and shows that the BD of rCNG5 is competent to bind cyclic nucleotide and to engage the coupling machinery that promotes channel opening.
We next characterized more closely the gating properties of X-β. Maximal currents (Imax) elicited by saturating concentrations of cAMP and cGMP are similar. However, at low agonist concentrations, the channel is clearly more sensitive to cAMP than to cGMP, i.e., the K1/2 for cAMP is lower than that for cGMP (Fig. 1 C). This is consistent with the observation that heteromers of rCNG5 with rat CNG2 (rCNG2) exhibit enhanced cAMP activation compared with homomers of rCNG2 (Bradley et al. 1994; Liman and Buck 1994). An empirical measure of the agonist selectivity of X-β is the ratio K1/2,cGMP/K1/2,cAMP. Since absolute K1/2 values of CNG channels typically vary from patch to patch (Ruiz et al. 1999), it is more reliable to evaluate the K1/2 ratio for each patch individually than to take one ratio of the mean values of K1/2,cGMP and K1/2,cAMP. In every macroscopic current patch of X-β channels, the K1/2 ratio was >1 (indicating cAMP selectivity); the mean of the individual ratios was 1.45 ± 0.13 (n = 9), which indicates the cAMP selectivity is statistically significant (P < 0.0001). It has also been reported that coexpression of rCNG5 with rCNG2 leads to a marked increase in outward rectification compared with rCNG2 homomers, and to a slow (seconds timescale) desensitization in response to application of millimolar concentrations of agonist (Bradley et al. 1994; Liman and Buck 1994). In contrast, over a range of agonist concentrations, X-β currents exhibit only weak outward rectification (Fig. 1 D) typical of α subunits containing the NH2-terminal region of CNG2 (Goulding et al. 1992; Möttig et al. 2000), and exhibit no slow desensitization (up to 30 mM agonist; unpublished data). This shows that rCNG5 sequence outside the BD is not only sufficient for marked rectification and desensitization (Shapiro and Zagotta 2000), but is also absolutely necessary.
Single Channels of X-β Display High Efficacy and Normal Conductance
Single-channel recordings of X-β show that at 3 mM (saturating) cAMP or cGMP, the efficacy of opening (or maximum open probability, Pmax) is very high (Fig. 2A Fig. 3 and A4). Channel closings are rare and short-lived. For cAMP, Pmax = 0.980 ± 0.025 (n = 6), and for cGMP, Pmax = 0.947 ± 0.068 (n = 5). Open probability has a strongly nonlinear relationship with the thermodynamic parameter (free energy) associated with the closed-open conformational change; when Popen is close to 1, a small variation in Popen represents a significant energetic difference in activation. To measure efficacy in thermodynamic terms, we used Pmax values to calculate the free energy of opening with saturating agonist concentrations (ΔGsat), according to a simple model with a single open and closed state
Figure 2.

Single-channel recordings of homomeric X-β channels show high efficacy of opening with either cAMP or cGMP. (A1–A4) Typical 750-ms excerpts of recordings from one single-channel patch at +80 and −80 mV, with steady-state concentration of agonist as indicated; closed and open state current levels are marked by c and o, respectively.
Figure 3.

X-β has normal single-channel conductance. (A1–A4) All-points current amplitude histograms compiled from the excerpts in Fig. 2 (A1–A4 respectively). Insets expand the feet of histograms in A1 (30 μM cAMP, very low Popen) and A3 (3 mM cAMP, very high Popen). With the exception of the histogram in A1 at −80 mV (materials and methods), the complete histograms (connected dots) were fitted with a sum (solid line) of Gaussian peaks (dashed lines). Arrows mark current means for multiple open states at −80 mV: O0 (unprotonated), and O+ and O++ (single and doubly protonated, respectively). Popen values from the excerpts are as follows: in 30 μM cAMP, 0.0745 at +80 mV and 0.015 at −80 mV; in 300 μM cAMP, 0.943 at +80 mV and 0.9299 at −80 mV; in 3 mM cAMP, 0.9929 at +80 mV and 0.9905 at −80 mV; and in 3 mM cGMP, 0.9523 at +80 mV and 0.9671 at −80 mV.
where AnC is the fraction of channels in the closed state with all n ligand binding sites occupied and AnO is the fraction of fully liganded channels in the open state. According to this simple Fig. 1, ΔGsat ≡ −RT ln([AnO]/[AnC]) = −RT ln(Pmax/(1 − Pmax)).
Scheme S1.

CNG channels composed of wild-type subunits are known to exhibit variation in activation properties at the single-channel level (Ruiz et al. 1999). Single X-β channels also had efficacies that varied from patch to patch over a ∼15-kJ/mol range, with mean values of ΔGsat,cAMP = −12.8 ± 5.6 kJ/mol (n = 6) and ΔGsat,cGMP = −9.5 ± 4.8 kJ/mol (n = 5). To evaluate agonist selectivity in the presence of patch-to-patch variation, we compared ΔGsat for cAMP and cGMP for each individual patch. The “efficacy selectivity” value, ΔGsel ≡ ΔGsat,cAMP − ΔGsat,cGMP, was negative in every patch, with a mean (−4.2 ± 1.7 kJ/mol, n = 5), which is significantly less than zero (P < 0.02). The negative value indicates that X-β is selective for cAMP over cGMP in terms of efficacy, just as for the K1/2 selectivity ratio.
Single-channel currents of X-β exhibit three open state conductances at −80 mV, but only one open state conductance at +80 mV (seen in all-points histograms, Fig. 3). These conductances match those previously described and attributed to external proton block in fCNG2, and in chimeric channels containing the fCNG2 P-loop, including ROON-S2 (Goulding et al. 1992; Root and MacKinnon 1994; Liu et al. 1996; Tibbs et al. 1998). The largest of the three conductances at −80 mV is ∼75 pS and corresponds to an unblocked state (O0). The two subconductances are attributed to block by one proton (O+, ∼50 pS) or two protons (O++, ∼25 pS; Root and MacKinnon 1994), and account for ∼95% of the channel open time, giving a mean conductance of ∼42 pS at −80 mV. Relief of proton block at positive voltages (giving a mean conductance ∼56 pS at +80 mV) accounts for the outward rectification seen in macroscopic currents at saturating agonist concentrations when Popen is ∼1 (Fig. 1 C). Preservation of the conductance properties of ROON-S2 in the new chimera X-β indicates that the substitution of the BD did not dramatically affect the structure of the open aqueous pore. This makes it unlikely that the activity of X-β in the oocytes depends on coassembly with an endogenous subunit, and also makes it unlikely that the transmembrane moiety of an X-β homomer has a structure significantly different than that of homomers of ROON-S2 or intact fCNG2. In summary, X-β shows all the essential functional properties of an intact α subunit: it assembles into homomeric channels that are activated by cyclic nucleotide in a selective fashion, and which exhibit conductance properties that match those predicted from the transmembrane domain sequence.
The BD of the β Subunit Produces More Efficient Activation than the BD from an α Subunit
The high Pmax and steep Hill coefficient of X-β indicate highly effective activation by ligands. This clearly rules out one plausible explanation for failure of the β subunit to form functional homomers, namely that the BD is incapable of strong coupling of ligand binding to channel opening. We were now in a position to make a comparative study asking, how does the performance of the rCNG5 BD (in the context of X-β) compare with that of BDs from closely related α subunit homologues? The BD of bCNG1 is present in the previously characterized ROON-S2; for further comparison we constructed a new chimera, X-α, identical to X-β and to ROON-S2 up to the end of the C-linker, but containing the BD of fCNG2, another α subunit.
X-α is activated to similar extents by both cAMP and cGMP, like X-β. However, X-α exhibits an unusual desensitization at high concentrations of cGMP (to be discussed below) so we first focus on cAMP activation. X-α preserves the very weak rectification (Fig. 4 A, voltage step traces) and single-channel conductance profile at +80 and −80 mV (Fig. 4 B; unpublished all-points histograms) that are characteristic of the X-based chimeras, confirming that these properties are determined by the invariant sequence outside the BD. However, single-channel recordings of X-α at 30 mM cAMP (Fig. 4 B4) show lower opening efficacy (mean Pmax,cAMP of 0.49 ± 0.30, n = 9) than seen for X-β (Pmax,cAMP = 0.980 ± 0.025, n = 6). There is a large variation in Pmax of X-α from patch to patch, which reflects the fact that when the open and closed states have similar stability (Popen near 0.5), small differences in the energetics of the opening reaction are manifest as numerically large changes in Popen. Despite this variation, the range of Pmax values for X-α (0.1150–0.9549) is clearly distinct from the range observed for X-β (range 0.9342–0.9998). Furthermore, when the Pmax values are expressed as free energies (mean ΔGsat,cAMP for X-α is −0.1 ± 4.1 kJ/mol; n = 9) the patch-to-patch variation (range of ∼13 kJ/mol) turns out to be similar in magnitude to that found for X-β, and is thus probably derived from a similar structural basis. The mean ΔGsat,cAMP for X-α is more positive (i.e., less favorable) than that of X-β by ∼13 kJ/mol (P < 0.0002). An additional observation comes from the cAMP dose–response curve for macroscopic X-α currents: X-α has a higher K1/2 and a lower Hill coefficient than X-β (Fig. 4 C). Therefore, in terms of both sensitivity and efficacy, cAMP is a poorer agonist for X-α than for X-β.
Figure 4.
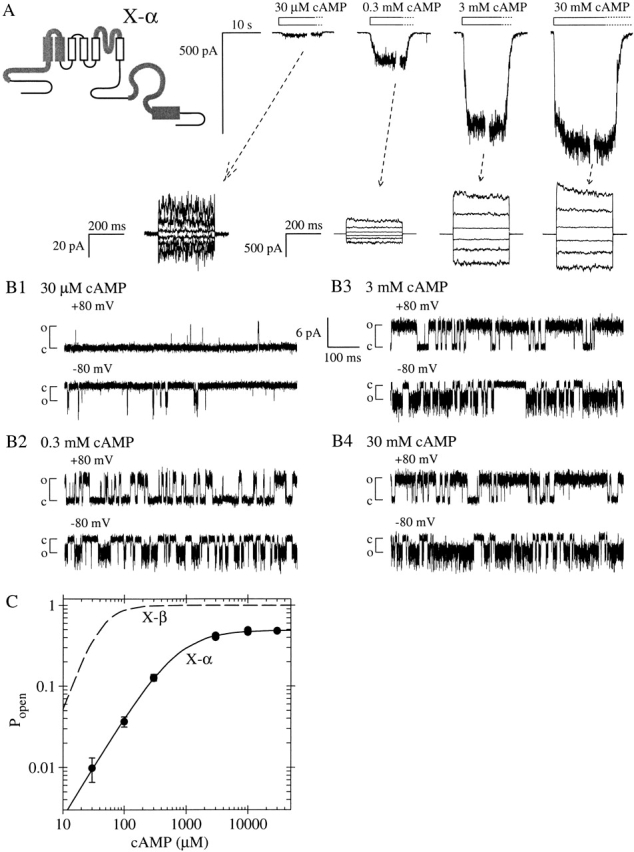
Chimeric CNG channel subunit X-α containing BD from the α subunit fCNG2 is less efficiently activated by cAMP than is X-β. (A) Schematic shows sequence of X-α is identical to that of X-β except that the BD sequence is that of fCNG2. (top traces) Macroscopic current traces from homomeric X-α channels elicited by the indicated concentrations of cAMP. (bottom traces) Currents during voltage steps at indicated steady-state cAMP concentrations (tested as in Fig. 1). (B1–B4) Excerpts of single X-α channel recordings at indicated potentials and steady-state cAMP concentrations (collected as in Fig. 2). Popen values from the excerpt in B4 (30 mM cAMP) are 0.778 at +80 mV and 0.646 at −80 mV. (C) Dose–response data (closed circles) for X-α activation by cAMP at −100 mV, from the patch shown in A. Solid line shows Hill equation fit with K1/2 = 717 ± 47 μM, h = 1.241 ± 0.051, and the graph is normalized with Pmax,cAMP at cAMP set to be 0.49, the mean from single-channel measurements. For comparison, the dashed line shows cAMP dose–response curve for X-β, taken from Fig. 1 C and normalized using the Pmax,cAMP of 0.98 derived from single-channel measurements.
X-α activation by cGMP at low concentrations resembles activation by similar concentrations of cAMP, but upon exposure to cGMP concentrations of 10 mM or higher the elicited macroscopic current decays in amplitude to a steady-state level that is lower than that elicited by 3 mM cGMP (Fig. 5 A; compare current traces in 3 mM cGMP and 30 mM cGMP). This indicates that the channels enter a desensitized state; entry to and exit from the desensitized state occur faster than the rate at which solutions are exchanged by the perfusion system (within a few seconds). This phenomenon is also found in homomeric channels composed of intact fCNG2 subunits (Goulding et al. 1992) but until now has not been examined closely. Desensitization of X-α is not relieved by positive voltage and does not change the extent of outward rectification, which remains slight, as seen for X-β (Fig. 5 A, voltage step traces). Note that there is no evidence for this desensitization in X-α when cAMP is the agonist (up to 30 mM), nor in X-β when either cAMP or cGMP is the agonist (up to 30 mM). Single-channel recordings show that desensitization does not result from an altered single-channel conductance (see 30 mM cGMP trace, Fig. 5 B3; unpublished all-points histograms) but rather from a decrease in open probability. Thus, the dose–response curve of steady-state Popen versus cGMP concentration shows a maximum at ∼3 mM, with a decrease in Popen at a higher agonist concentration (Fig. 5 C). K1/2,cGMP was determined only for the rising phase of the dose–response curve, using data collected at cGMP concentrations ≤3 mM. Similarly, the maximum open probability (Pmax,cGMP) was measured at only 3 mM in single-channel recordings, with a mean of 0.25 ± 0.28 (n = 9). When these Pmax measurements are converted to free energies, the mean ΔGsat,cGMP is +4.0 ± 4.8 kJ/mol (n = 9). Although we retain the “max” and “sat” subscripts in the efficacy symbols, we emphasize that the Pmax,cGMP values and ΔGsat,cGMP values do not measure opening energetics for X-α channels with binding sites saturated by ligand, but are empirical measures of the highest observable open probability on the cGMP dose–response curve.
Because of cGMP-dependent desensitization, comparisons of cAMP and cGMP activation using conventional parameters like Pmax (or ΔGsat) and K1/2 cannot be interpreted mechanistically for X-α as they can for X-β. Notably, for X-α the parameters ΔGsat,cGMP and ΔGsat,cAMP characterize channels at different degrees of binding site saturation, so they cannot be directly compared or used to calculate the efficacy selectivity, ΔGsel, for quantitative comparison with the ΔGsel of X-β. Nonetheless, comparisons of Pmax or of K1/2 do provide empirical descriptions of the agonist selectivity of X-α. When cAMP and cGMP activation are compared at a given high (millimolar) concentration, desensitization is marked and Popen is much higher in cAMP than in cGMP; thus X-α channels can be described as selective for cAMP. Specifically, Pmax,cAMP (measured at saturating cAMP concentrations) was greater than Pmax,cGMP (measured at 3 mM, the peak of the dose–response curve) in every single-channel patch for which both these measurements were made; the mean ratio Pmax,cAMP/Pmax,cGMP was 2.3 ± 1.7 (n = 6). The ratio of open probabilities in cAMP and cGMP is even greater than this factor when cGMP concentrations higher than 3 mM are considered. In contrast, when agonist concentration is decreased below millimolar levels, desensitization is not apparent, and X-α exhibits a reversal of selectivity (crossing of dose–response curves) so that cGMP produces a higher Popen than does cAMP. This cGMP selectivity is reflected empirically in the low K1/2 selectivity ratio (K1/2,cGMP/K1/2,cAMP = 0.407 ± 0.026; Fig. 5 C).
Table summarizes and compares activation parameters for the two new chimeras as well as for ROON-S2. Each chimera combines a different BD sequence with an invariant non-BD sequence (”X-”), and each was tested as a homomeric channel, like a conventional α subunit. The functional differences among the chimeras, hence, can be attributed wholly to differences in how their respective BDs interact with agonist and/or with the invariant non-BD regions. One way to compare BDs is to examine how efficiently a given agonist like cAMP activates each X-chimera: of the three chimeras, X-β (containing the BD of the β subunit rCNG5) shows the most efficient cAMP activation (the lowest K1/2 and highest Pmax), and X-α shows the least efficient activation. Thus, not only can the BD of rCNG5 participate in cAMP activation of a homomeric channel, but it also can do so with higher efficiency than the BDs of the α subunits bCNG1 and fCNG2. Note that any ranking of BDs by activation efficiency will depend on which agonist is considered: the chimera most efficiently activated by cGMP is not X-β but ROON-S2 (with the BD of bCNG1).
Table 1.
Activation Properties of CNG Channel Subunit Chimeras Differing only in BD Sequences
| Chimera | X-β | X-α | ROON-S2 | |
|---|---|---|---|---|
| BD source | rCNG5 | fCNG2 | bCNG1 | |
| Macroscopic current | ||||
| Hill equation parameters | K1/2,cAMP | 81 ± 85 (9) | 643 ± 29 | 432 ± 99 (4) |
| K1/2,cGMP | 120 ± 140 (9) | 261 ± 12 | 1.8 ± 1.0 (10) | |
| hcAMP | 2.14 ± 0.26 (9) | 1.123 ± 0.070 | 1.31 ± 0.34 (4) | |
| hcGMP | 2.07 ± 0.27 (9) | 1.210 ± 0.046 | 1.89 ± 0.38 (10) | |
| K1/2 selectivity ratio | K1/2,cGMP/K1/2,cAMP | 1.45 ± 0.13 (9) | 0.407 ± 0.026 | 0.0042 ± 0.0025 |
| Single channel | Pmax,cAMP | 0.980 ± 0.025 (6) | 0.49 ± 0.30 (9) | 0.98 |
| Pmax,cGMP | 0.947 ± 0.068 (5) | 0.25 ± 0.28 (9) | 0.9987 ± 0.0021 (5) | |
| ΔGsat,cAMP | −12.8 ± 5.6 (6) | −0.1 ± 4.1 (9) | −9.6 | |
| ΔGsat,cGMP | −9.5 ± 4.8 (5) | +4.0 ± 4.8 (9) | −18.7 ± 3.6 (5) | |
| Efficacy selectivity | ΔGsel | −4.2 ± 1.7 (5) | n.d. | +9.1 |
Except where otherwise noted, all entries are means ± SD of values evaluated in individual patches (number of patches is given in parentheses). Selectivity ratio above unity or efficacy selectivity below zero corresponds to selectivity for cAMP over cGMP. Units are μM for K1/2 values and kJ/mol for ΔG values.
Another way to compare the three BDs is to examine the selectivity for one agonist over another in each X-chimera. The chimera X-β shows for the first time that in a homomeric channel, the rCNG5 BD imparts selectivity for cAMP over cGMP, as measured by both efficacy and K1/2. This agrees with observations that a hallmark modulatory effect of assembly of intact rCNG5 in αβ channels is an enhancement of cAMP activation compared with that found in α subunit homomers (Bradley et al. 1994; Liman and Buck 1994; Finn et al. 1998). In contrast, the bCNG1 BD imparts to ROON-S2 a strong selectivity for cGMP, just as it does in intact bCNG1 homomers (Kaupp et al. 1989; Altenhofen et al. 1991; Goulding et al. 1994). The cGMP-dependent desensitization observed in X-α mimics that found in intact fCNG2 homomers (Goulding et al. 1992). Therefore, in general, the BDs in the X-chimeras show selectivities that reflect those of the intact subunits from which they are derived.
The very existence of agonist selectivity in the X-chimeras implies that the interaction of agonist with the BD is structurally specific. Previous studies (Varnum et al. 1995; Scott and Tanaka 1998; Sunderman and Zagotta 1999; Pagès et al. 2000; Shapiro and Zagotta 2000; He and Karpen 2001) found that agonist selectivity in both α homomers and αβ heteromers is strongly influenced by the amino acid at position 604 in the C-helix subdomain (numbering is based on bCNG1; see materials and methods). At this position, aspartate (found in bCNG1) and methionine (found in rCNG5) imparted cGMP- and cAMP selectivity, respectively, to bCNG1 homomers (Varnum et al. 1995; Sunderman and Zagotta 1999). That finding is borne out in the current study, since ROON-S2 (with aspartate at position 604) is cGMP-selective and X-β (with methionine) is cAMP-selective. The fCNG2 BD in X-α has a glutamine at position 604; this amino acid was previously found to impart weak cGMP selectivity to bCNG1 homomers (Varnum et al. 1995; Sunderman and Zagotta 1999). Although evaluation of selectivity in X-α is complex because of desensitization (see previous page), this channel is indeed selective for cGMP at low agonist concentrations where desensitization is less likely to be a confounding factor. Therefore, the agonist selectivity in all three chimeras is correlated with the amino acid identity at position 604 as previously characterized. This correlation suggests strongly that the structures of the BDs in the X-chimeras closely resemble those found in previously studied intact α subunits, and more generally, the mechanism of ligand-gating in the X-chimeras reflects the canonical mechanism of wild-type CNG channels.
The putative C-helix subdomain, which contains residue 604, has been assigned a key role in some models of how the BD functions during ligand-gating of CNG channels. Previous studies suggested that coupling energy for channel activation depends largely on a contact between the C-helix and the ligand which is stronger in the open than in the closed state of the channel (Goulding et al. 1994; Varnum et al. 1995; Sunderman and Zagotta 1999). It has moreover been proposed that the C-helix undergoes a large rigid-body rotation during channel activation (Matulef et al. 1999). In contrast the roll subdomain has been proposed to make state-independent contacts that contribute ligand binding energy but not coupling energy toward activation, so that the BD has a “functional polarity” (Tibbs et al. 1998). The two BD sequences of fCNG2 and rCNG5 diverge far less in the roll subdomain (89% similarity) than in the C-helix (64% similarity). This fact, along with the correlation between the identity of residue 604 and agonist selectivity in our BD chimeras, suggested to us that sequence variation in the C-helix might also be the primary determinant of the absolute differences in agonist efficacy between X-α and X-β.
High Activation Efficiency of X-β Compared with X-α Cannot Be Attributed Solely to C-helix Residues
We constructed two new chimeras to ask whether differences in absolute activation efficacy (ΔGsat) and in sensitivity (K1/2) between X-α and X-β were wholly due to differences in their C-helix sequences or if other elements, such as the roll subdomain, also contribute. The first chimera, X-αR/βC, has a BD comprising the roll subdomain (denoted by “R”) from the α subunit fCNG2 and the C-helix (denoted by “C”) from the β subunit rCNG5. The second chimera, X-βR/αC, is complementary in organization, with a BD comprising the roll subdomain from the β subunit and the C-helix from the α subunit.
Because of the prominence of the C-helix in previous models, we expected that activation of X-αR/βC would be similar to X-β, or at least significantly better than X-α, but this is far from the case. We find (to our surprise) that the C-helix of rCNG5 contains sequence elements with a markedly negative influence on channel activation. X-αR/βC exhibits cGMP-dependent desensitization similar to that of X-α, so cAMP activation will be discussed first. Single-channel recordings for X-αR/βC show very low Popen even at high cAMP concentrations (Fig. 6 A1). The mean Pmax,cAMP is 0.056 ± 0.091 (n = 6), which is ninefold lower than that of X-α. In thermodynamic terms, the mean ΔGsat,cAMP is +9.2 ± 3.6 kJ/mol (n = 6). Note that the Pmax values for X-αR/βC have a SD that is larger than the mean (the range of values is 0.0047–0.27), whereas the SD of ΔGsat,cAMP values is smaller than the mean. This is consistent with the highly nonlinear relationship between ΔGsat and Pmax since Pmax is very low for this chimera. For example, a relatively small variation of 2 kJ/mol in ΔGsat corresponds to a variation in Pmax which is very small (∼1%) if Pmax ≈ 0.99 as for X-β, but large (∼50%) if Pmax ≈ 0.5 as for X-α, and enormous (∼100%) if Pmax ≈ 0.05 as for X-αR/βC. The variation in efficacies of single X-αR/βC channels is similar in energetic terms (ΔGsat,cAMP has a spread of ∼11 kJ/mol) to those found for X-β and X-α, whereas the means of the three chimeras are widely separated. Specifically, the mean ΔGsat,cAMP for X-αR/βC is more positive (i.e., less favorable) than that of X-α by ∼9 kJ/mol (P < 0.0002).
Because a patch that appears to contain a single channel may actually contain more than one if Pmax is very low, the “single-channel” experiments provide only an upper limit for the true Pmax of X-αR/βC. In the patch shown in Fig. 6 A1, Pmax in saturating cAMP was potentiated to a value close to 1 by application of the allosteric modulator Ni2+ (Ildefonse et al. 1992; Gordon and Zagotta 1995b), confirming that a single channel was present. To obtain an independent estimate of Pmax that does not depend on knowing the number of channels in the patch, we measured the extent of potentiation of macroscopic X-αR/βC currents by Ni2+ in the presence of saturating concentrations of cAMP (Varnum et al. 1995). The ratio of the current elicited without Ni2+ to the current elicited with Ni2+ (Imax,cAMP/Imax,cAMP+Ni) provides an estimate for Pmax,cAMP; this estimate, like that from single-channel experiments, is an upper limit since Ni2+ may not increase Pmax to 1. For X-αR/βC, this ratio is 0.071 ± 0.010 (n = 8), similar to Pmax,cAMP from single-channel recordings. In contrast, X-α currents gave a ratio Imax,cAMP/Imax,cAMP+Ni = 0.38 ± 0.17 (n = 8), which is consistent with the Pmax values from single X-α channel recordings. Thus, two different approaches show that the cAMP activation of X-αR/βC has a lower efficacy, not higher efficacy, compared with that of X-α. An additional indicator that X-αR/βC activates less efficiently than X-α is that the K1/2,cAMP of X-αR/βC (Fig. 6 A2) is higher than that of X-α (Fig. 4 C and Table ). Therefore, replacing the fCNG2 C-helix sequence in X-α with the rCNG5 C-helix sequence has a deleterious effect on both the efficacy and sensitivity of cAMP activation.
The activation of X-αR/βC by cGMP is even less efficient than its activation by cAMP. The cGMP dose–response curve from macroscopic current experiments is biphasic (like that of X-α) with desensitization above 3 mM, so that at high agonist concentrations X-αR/βC exhibits markedly larger currents in cAMP than in cGMP (Fig. 6 A2). The maximal cAMP-activated current (Imax,cAMP, measured at saturating cAMP concentrations) is greater than the maximal current for cGMP activation (Imax,cGMP, measured at 3 mM, the peak of the dose–response curve) by a factor of 2.85 ± 0.30 (n = 4). The cAMP selectivity is not limited to the concentration regime where cGMP-dependent desensitization is apparent: even at the lowest tested agonist concentrations, cAMP elicits larger X-αR/βC currents than does cGMP. The Pmax in 3 mM cGMP was not measured in single X-αR/βC channel recordings; however, assuming the ratio Imax,cAMP /Imax,cGMP in macroscopic currents measures the true ratio of Pmax,cAMP /Pmax,cGMP for each single-channel patch, we calculate that Pmax,cGMP values would have a range from 0.0017 to 0.10, and a mean of 0.020; these Pmax,cGMP values for X-αR/βC are ∼10-fold lower than those for X-α, indicating that like cAMP activation, cGMP activation is made less efficient by the replacement of the fCNG2 C-helix with the rCNG5 C-helix.
We were wary that the unexpectedly poor activation of X-αR/βC might indicate merely that its chimeric BD was grossly misfolded. However, several lines of evidence argue that the BD of X-αR/βC is similar to intact BDs in structural organization. First, cAMP activates homomers of intact bCNG1 with low efficacy, so low efficacy is not in itself an absolute indicator of BD misfolding. Second, single channels of X-αR/βC can indeed achieve high levels of cyclic nucleotide-dependent activation when potentiated by Ni2+. This reagent acts through binding to the C-linker region and so is not expected to influence the folding of the BD structure directly (Gordon and Zagotta 1995b). Third, X-αR/βC mimics X-α (and intact fCNG2) in exhibiting desensitization in high concentrations of cGMP but not cAMP, implying that these subunits have BDs with similar structures. Fourth, the selectivity for cAMP over cGMP of X-αR/βC is strong over all agonist concentrations, as predicted from the presence of M604 in the rCNG5 C-helix. It is unlikely that a misfolded BD could preserve the specific interaction of ligand with the C-helix residue that is believed to impart this selectivity (Varnum et al. 1995). All evidence supports the conclusion that X-αR/βC has a BD whose structure closely resembles that of BDs in intact subunits, but that at some functionally important position(s) in the BD sequence, X-αR/βC has an amino acid that decreases the efficiency of ligand-gating.
Another simple interpretation of the poor efficacy of activation of chimera X-αR/βC might be that the rCNG5 C-helix sequence is not inherently unfavorable for activation compared with the fCNG2 C-helix sequence, but rather that mixed pairing of subdomains from α and β subunits would always have a deleterious effect on activation energetics. This interpretation cannot be correct, however, because X-βR/αC, which has a mixed pair of subdomains complementary to X-αR/βC, shows a very high efficacy of activation. In single-channel recordings (Fig. 6 B1), X-βR/αC has a high efficacy for both agonists (Pmax,cAMP = 0.954 ± 0.056 and ΔGsat,cAMP = −10.6 ± 5.8 kJ/mol, n = 5; Pmax,cGMP = 0.92 ± 0.12 and ΔGsat,cGMP = −10.1 ± 6.0 kJ/mol, n = 6). In particular, the mean ΔGsat,cAMP efficacy of X-βR/αC is significantly more favorable than that of X-α (which has an intact fCNG2 BD) by ∼10 kJ/mol (P < 0.002). Thus mixing subdomains from α and β subunits cannot be predicted to have a positive or negative effect on activation energetics as a general rule. To summarize the results from the group of four chimeras, both X-α and X-αR/βC show relatively low efficacy of activation with either cAMP or cGMP, whereas both X-β and X-βR/αC show relatively high efficacy of activation with either agonist. Consequently the C-helix cannot be reckoned the sole major BD determinant of the absolute extent of activation in CNG channels, since efficacy in the four chimeras does not correlate with the identity of the C-helix subdomain. Rather, the efficacy correlates with the identity of the roll subdomain: low efficacy derives from the fCNG2 sequence and high efficacy from the rCNG5 sequence.
Moreover, the C-helix is also not the sole determinant of the relative extent of activation by different agonists, i.e., agonist selectivity. X-α and X-βR/αC both contain the same C-helix sequence (from fCNG2), but X-βR/αC fails to show the cGMP-dependent desensitization seen in X-α. This means that the roll subdomain of the α subunit (fCNG2) is responsible for desensitization, an unusual but nonetheless agonist-specific determinant of open probability. Therefore, the roll subdomain sequence can contain residues that exert large effects on agonist selectivity as well as absolute open probability at high agonist concentrations.
Although the above results show that the C-helix is not the sole determinant of agonist selectivity, at the same time, they do confirm that the C-helix does indeed contribute important determinants of selectivity. Thus, X-β and X-βR/αC differ only in their C-helix sequences, and show a different selectivity. In terms of efficacy, X-β exhibits strong cAMP selectivity (see ΔGsel in Table , and Fig. 7 A), whereas X-βR/αC does not select among the two agonists, with a very small ΔGsel value (+1.1 ± 3.2 kJ, n = 5; ΔGsel values for the two chimeras are significantly different, P < 0.02). The difference between the chimeras is even more pronounced at low (submillimolar) agonist concentration, where X-β is selective for cAMP, but X-βR/αC is selective for cGMP (see dose–response curves, Fig. 1 C and Fig. 6 B2). The ratio K1/2,cGMP/K1/2,cAMP was tested in individual macroscopic current patches of X-βR/αC (as was done for X-β) and was <1 in every case, with a mean of 0.420 ± 0.095, n = 8 (indicating significant cGMP selectivity, P < 0.002). This is significantly different (P < 0.0001) from the K1/2 ratio of X-β (1.45 ± 0.13, n = 9; Fig. 7 B). In general, at submillimolar agonist concentrations, the chimeras containing the rCNG5 C-helix (X-β and X-αR/βC) are cAMP-selective and the chimeras containing the fCNG2 C-helix (X-α and X-βR/αC) are cGMP-selective. These selectivities are consistent with the respective identities of residue 604 in the two chimeras.
Figure 7.
Chimeras X-βR/αC and X-β exhibit different agonist selectivity. (A) Points plot the efficacies of the two chimeras obtained from single-channel patches from distinct oocytes. Each vertical line connects a solid and an open point plotting data for cAMP and cGMP, respectively, obtained from an individual patch. For some patches, efficacy was only measured for one agonist so only one point is plotted without a vertical line. Efficacies are plotted as ΔGsat (left axis) or equivalently as Pmax/(1 − Pmax) on a logarithmic scale (right axis). (B) Points plot the K1/2 values of the two chimeras obtained from macroscopic current patches from distinct oocytes, with cAMP and cGMP data represented as in A.
Efficacy Determinants Inside and Outside the C-helix Do Not Act Independently
The above results show that differences in activation properties of X-α and X-β are derived from elements in both the C-helix and roll subdomains. Do these two regions act independently or do they interact during activation of the channel? If these subdomains act independently of one another, then their energetic contributions to channel activation will be invariant when quantified separately and will be strictly additive. This independence hypothesis can be tested by thermodynamic linkage analysis of the four chimeras we have studied here (Fig. 8; see materials and methods), which form corners of a linkage cycle (Wyman 1964; Weber 1975); the chimeras represent all combinations of the roll and C-helix subdomains from fCNG2 and rCNG5. Thus, if the C-helix acts independently of the roll subdomain, we would predict that the effects on activation of exchanging the α subunit C-helix for the β subunit C-helix should be the same whether we measure the difference in activation between X-α and X-αR/βC or whether we measure the difference in activation between X-βR/αC and X-β (see Fig. 8 B, double arrows). (The same principles have been applied to point mutations in “double mutant cycle” analysis [Carter et al. 1984; Scott and Tanaka 1998].)
Figure 8.

Analysis of cAMP activation data for chimeras using a thermodynamic linkage cycle. (A) Efficacies of cAMP activation for all single-channel patches in this study are plotted using the same axes as in Fig. 6 A; each solid point plots data obtained from an individual patch from a distinct oocyte. (B) BDs of four chimeras arranged at the corners of the linkage cycle. Wide arrows represent replacements of fCNG2 C-helix by rCNG5 C-helix; dashed arrows represent replacements of fCNG2 roll subdomain by rCNG5 subdomain. For each replacement, ΔΔGsat,cAMP (defined as the change in ΔGsat,cAMP that results from performing that replacement) is shown, in units of kJ/mol. The independence hypothesis fails because parallel sides of the cycle have different values of ΔΔGsat,cAMP.
We find that the two subdomains do not act independently, as shown in Fig. 8 and Table , which summarize activation efficacy (ΔGsat) and other activation parameters for the corners of the linkage cycle. We consider only cAMP efficacy because its analysis is not complicated by desensitization. Converting X-α into X-αR/βC (Fig. 8 B, double arrow on left side of cycle) leads to a decrease in efficacy (the difference in ΔGsat,cAMP is ΔΔGsat,cAMP = +9.3 kJ/mol, and is statistically significant, P < 0.0002). In this sequence substitution, the C-helix of the β subunit appears to have a deleterious effect on activation. (Allowing the possibility that the Pmax,cAMP of X-αR/βC may be overestimated by single-channel recordings and Ni2+ potentiation experiments, the deleterious effect of the rCNG5 C-helix may be even greater than we report.) However, making the same C-helix replacement in the conversion of X-βR/αC into X-β (Fig. 8 B, double arrow on right side of cycle) has little effect on efficacy (ΔΔGsat,cAMP = −2.2 kJ/mol, which is not a significant change in ΔGsat,cAMP, P < 0.6), as if the deleterious effect of the rCNG5 C-helix has been mitigated. This is inconsistent with the independence hypothesis; i.e., the contributions made by the C-helix and roll subdomains toward cAMP activation are not independent but are energetically coupled. This implies that subdomain interaction contributes to preferential open state stabilization in at least one of the four chimeras.
Table 2.
Summarized Activation Parameters for Chimeras in Thermodynamic Linkage Relationship
| Chimera | X-α | X-αR/βC | X-βR/αC | X-β | |
|---|---|---|---|---|---|
| cAMP | ΔGsat | −0.1 ± 4.1 (9) | +9.2 ± 3.6 (8) | −10.6 ± 5.8 (5) | −12.8 ± 5.6 (6) |
| K1/2 | 643 ± 29 | 1,570 ± 370 | 222 ± 116 (8) | 81 ± 85 (9) | |
| cGMP | Desensitization | Yes | Yes | No | No |
| ΔGsat | +4.0 ± 4.8 (9) | n.d. | −10.1 ± 6.0 (6) | −9.5 ± 4.8 (5) | |
| K1/2 | 261 ± 12 | 511 ± 37 | 81 ± 39 (8) | 120 ± 140 (9) | |
| Selectivity | |||||
| Efficacy | ΔGsel | n.d. | n.d. | +1.1 ± 3.2 (5) | −4.2 ± 1.7 (5) |
| K1/2 | K1/2,cGMP/K1/2,cAMP | 0.407 ± 0.026 | 0.325 ± 0.080 | 0.420 ± 0.095 (8) | 1.45 ± 0.13 (9) |
Except where otherwise noted, all entries are means ± SD of values evaluated in individual patches (number of patches is given in parentheses). Units are μM for K1/2 values and kJ/mol for ΔG values.
To restate our analysis: when the α subunit BD is replaced with the β subunit BD (converting X-α to X-β), the absolute opening efficacy for cAMP activation ( ΔGsat,cAMP) is strongly enhanced (ΔΔGsat,cAMP between X-α and X-β is negative). Both subdomains contain sequence elements that influence efficacy, because replacements of each of the major subdomains alone also change ΔGsat,cAMP. However, a simple summation of the efficacy changes (ΔΔGsat,cAMP) produced by individual subdomain replacements cannot account for all of the ΔΔGsat,cAMP between X-α and X-β. The discrepancy (the portion of the ΔΔGsat,cAMP between X-α and X-β that remains not accounted for) can be termed “interaction coupling energy,” because it can exist only if one or more of the four possible subdomain pairs (αR/αC, αR/βC, βR/αC, or βR/βC) contains an interaction between subdomains. The subdomain interaction responsible for interaction coupling energy must change in strength during the channel opening transition, and if it occurs in all four subdomain pairs, then it cannot be identical in all pairs.
Note that interaction coupling energy is distinct from the “activation coupling energy” previously discussed. The activation coupling energy is a property of an individual chimera, and represents the portion of ΔGsat,cAMP that is contributed by interactions of the BD with the agonist molecule. The interaction coupling energy is a property of the set of four subdomain pairs (roll + C-helix) formed from two given BD sequences (e.g., fCNG2 and rCNG5). It represents the contribution of subdomain interactions toward the variation in ΔGsat,cAMP among the four subdomain pairs. Quantitative determination of interaction coupling energy for cAMP activation (ΔGint,cAMP) is done simply by taking the difference between parallel sides of the linkage cycle, (−2.2) − (+9.3) = (−22.0) − (−10.5) = −11.5 kJ/mol. Under the sign convention used here (Weber 1975), the negative sign of ΔGint,cAMP indicates that the most favorable contribution to cAMP activation from the interaction occurs when both subdomains derive from the same isoform rather than from different isoforms.
DISCUSSION
The β Subunit BD Is Functional
The CNG channel subunit rCNG5 is classed as a modulatory or β subunit because it contains some as yet unidentified “defects” in its structure that prevent assembly of homomeric channels that can be activated by cyclic nucleotide. Interaction with an α subunit neutralizes the defects, so heteromeric αβ channels are fully functional. Paradoxically, the defects coexist in the β subunit with elements that contribute favorably to cyclic nucleotide activation, so that αβ heteromeric channels activate with greater sensitivity to cyclic nucleotide than do α homomers. This study provides strong evidence that the residues responsible for failure of the β subunit to form functional homomers lie outside the cyclic nucleotide binding domain. This conclusion is based on our demonstration, for the first time, that the BD of the β subunit rCNG5 participates in coupling of ligand binding to channel opening in a homomeric CNG channel that does not contain residues from α subunit BDs. If interactions between BDs are important for ligand-gating as has been proposed, these interactions can occur between rCNG5 BDs in the same manner as they occur between α subunit BDs. Possibly the failure to form functional β homomers might be due to a deleterious interaction (or the lack of a required interaction) between the β subunit BD and β subunit non-BD regions; however, the “defective” residues in this partnership must be those of the β subunit non-BD regions rather than those of the β subunit BD, since in our work the α subunit non-BD regions can interact properly with the β subunit BD to form functional homomers.
We went further with a comparative study of a series of chimeras containing different BD sequences from either α or β subunits. All the chimeras shared an identical non-BD sequence so functional differences must be derived from BD sequence polymorphism. We found that when cAMP is used as an agonist, the chimera (X-β) containing the BD of the β subunit rCNG5 is more efficiently activated than are two other chimeras containing BDs from the α subunits bCNG1 or fCNG2. When cGMP is the agonist, the activation of the β subunit BD chimera also far surpasses that of the fCNG2 BD chimera, although the rCNG5 BD chimera is activated less well than the chimera containing the BD of bCNG1. Thus, the β subunit BD certainly contains no defect that precludes ligand-gating, and rather contains structural elements that are highly favorable to channel activation in comparison to BDs from some α subunits.
The functional capability of the β subunit BD was addressed in a previous study (Shapiro and Zagotta 2000) using chimeras derived from the α subunit rCNG2 and the same β subunit rCNG5 that we used. In that study, a chimeric CNG channel subunit, β-CNBRα, was constructed, consisting of the β subunit with its BD sequence replaced by that of the α subunit (a composition complementary to our chimera X-β). It was found that β-CNBRα failed to form functional homomeric channels but did coassemble with α subunits to form functional heteromeric channels. This showed that the β subunit sequence outside of the BD contains defects that prevent formation of functional homomeric channels, but did not argue for or against the presence of an additional defect in the β subunit BD. Another chimera from that study was composed of the α subunit rCNG2 in which the BD sequence was replaced with that of the β subunit rCNG5. Even though this (unnamed) chimera had a similar design to that of X-β in our study, it failed to form functional channels, either alone or as heteromers with α subunits, which might be taken to suggest that the β subunit BD was indeed defective in some way. The activity of X-β in our study definitively rules out a BD defect; an explanation for the previous study's negative result might be a suppression of channel expression due to some combination of rCNG2 and rCNG5 residues that conflicted with polypeptide folding. An ongoing search for deficient regions of the β subunit can, after our study, be limited to sequence outside of the BD.
Both Subdomains of the BD Determine Efficacy
Another finding of this study concerns the individual contributions of two putative subdomains of the β subunit BD, namely the roll subdomain (β-roll with flanking A- and B-helices), and the C-helix. We replaced each subdomain in the fCNG2 BD with the respective subdomain of rCNG5, and found that both swaps had significant effects on activation properties. Although the importance of the C-helix in controlling selectivity and efficacy is well studied, a comparably large influence of the roll subdomain has not previously been reported for CNG channels. Recent evidence suggests that important control elements for activation do exist in the roll subdomains of HCN (pacemaker) channels (Wainger et al. 2001) and hERG channels (Cui et al. 2000, Cui et al. 2001).
Our analysis of chimeric subunits, like that of any mutagenesis study, depends on the assumption that all of our chimeras resemble intact CNG channel subunits in structure and in activation mechanism. We verified that multiple functional properties of our chimeras replicate phenomena observed in intact CNG channels. Most obviously, the chimeras exhibit cyclic nucleotide activation with C-helix–dependent agonist selectivity; in addition, the chimeras replicate the P-loop–dependent single-channel conductance behavior and (in the case of X-α and X-αR/βC) the BD-dependent desensitization in high cGMP concentration that are found in the intact CNG channels from which the chimeras are derived. X-α and X-αR/βC exhibit relatively low efficacy in cAMP as do intact homomers of bCNG1 and fCNG2 (Altenhofen et al. 1991; Goulding et al. 1992), and this efficacy is increased by the binding of Ni2+ to the C-linker of bCNG1 (Gordon and Zagotta 1995b) that is present in all the X-chimeras. (Ni2+ also potentiates X-β and X-βR/αC, increasing the Popen observed at subsaturating cyclic nucleotide concentrations; unpublished data). Although no amount of observation could prove beyond doubt that all aspects of normal CNG channel structure and mechanism are intact in the X-chimeras, there is no aspect of X-chimera function that is not also found in intact CNG channels. Thus, the assumption that X-chimeras resemble intact CNG channels seems reasonable.
In our comparison of the fCNG2 and rCNG5 BDs, the phenotypes of high and low efficacy were correlated with the presence of the roll subdomain of rCNG5 and fCNG2, respectively, whereas the efficacy was not correlated with the identity of the C-helix. Swapping the roll subdomain sequence has a surprisingly large influence on activation (as much as 22 kJ/mol change in ΔGsat). Earlier studies of residues in the roll subdomain typically showed only small efficacy effects: data in the literature (expressed on a free energy basis under the minimal two-state model) show changes in ΔGsat,cAMP of ∼4 kJ/mol for the mutation T560A (Varnum et al. 1995), ∼1 kJ/mol for F533Y (Scott and Tanaka 1998), or ∼2 kJ/mol for the covalent modification of C505 by thimerosal (Matulef et al. 1999). The most prominent effects of roll subdomain mutations were in K1/2 values. Indeed, the highly conserved residue R559 was mutated to a variety of amino acids with >1,000-fold shifts in K1/2 but no observed effects on efficacy (Tibbs et al. 1998).
These previous results were generalized in the concept of “functional polarity” in the agonist binding site (Tibbs et al. 1998), whereby the sequence in the β-roll determined the strength of binding of the cyclic phosphate and ribose moieties of the agonist, but the preferential stabilization of the open state was wholly determined by interaction of the C-helix with the purine base moiety of the agonist. In this model, modifying the roll subdomain sequence would change the channel–ligand interactions in the closed and open states to a similar extent, and thus have no effect on open probability of a fully liganded channel. This notion has been expressed both explicitly (Tibbs et al. 1998) and implicitly in models of the lobster-claw variety that picture the β-roll as an invariant ligand-binding site and the C-helix as a dynamic element that contacts the ligand only when the channel opens (Matulef et al. 1999; Sunderman and Zagotta 1999). Our findings do not contradict the notion that the subdomains may have different degrees of mobility during channel opening, and functional polarity is certainly a valid description for some specific pairs of residues (such as R559/D604) in the two subdomains. However, our findings provide a counterexample against functional polarity, and thus show that functional polarity is not an accurate general description of relative contributions of the complete subdomains to channel activation energetics.
Both Subdomains of the BD Determine Selectivity
Identifying molecular determinants of agonist selectivity has been a dominant goal of structure–function studies on CNG channels ever since the first analyses of the BD of CNG channels (Altenhofen et al. 1991; Goulding et al. 1994). A notable success was identification of D604 in the bCNG1 C-helix as a strong determinant of cGMP selectivity (Varnum et al. 1995). Because the β subunit rCNG5 contains methionine rather than aspartate at the analogous C-helix position, and because αβ heteromers containing rCNG5 show enhanced activation by cAMP relative to α subunit homomers, we were interested in characterizing the agonist selectivity of homomeric channels containing BD residues from the β subunit. Our findings are consistent with the predicted contribution to selectivity of methionine at position 604 of the rCNG5 C-helix. X-β was selectively activated by cAMP, and replacing the C-helix of rCNG5 with that of fCNG2 (and, thus, replacing the methionine at position 604 with a glutamine) removed cAMP selectivity. Oddly, although the resultant chimera (X-βR/αC) was cGMP-selective at low concentrations (as predicted from the glutamine at position 604), it showed no significant efficacy selectivity (ΔGsel) at high concentrations. This illustrates that there is no uniquely correct way to characterize agonist selectivity. K1/2 is commonly interpreted as measuring sensitivity (i.e., the extent to which the channel responds to low concentrations of agonist), but in general K1/2 will be partially determined by ligand interactions with the BD that do not contribute activation coupling energy, and thus do not influence the open probability. Hence, the BD of X-βR/αC forms stronger interactions with cGMP than with cAMP, but the higher binding energy with cGMP contributes equally to open and closed state stability.
Chimeras containing the roll subdomain of the fCNG2 α subunit (X-α and X-αR/βC) exhibit an unusual form of selectivity at high agonist concentration, namely desensitization by cGMP but not by cAMP. This phenomenon is also seen in intact fCNG2 homomers (Goulding et al. 1992) and is thus not an artifact arising from the X-chimera construction. No other form of desensitization has ever been reported in wild-type α homomeric CNG channels. Some point mutations of a glutamate residue in the pore-forming P-loop were found to impart desensitization to bCNG1 homomers (Bucossi et al. 1996), but that desensitization differed from that seen in this study in several ways: it occurred to a similar extent at all tested concentrations of cGMP (10–1,000 μM), and the single-channel conductance of desensitizing channels differed from that of nondesensitizing channels, which is consistent with a change in the structure of the ion permeation pathway; moreover, recovery from desensitization was slow (tens of seconds). By contrast, in our experiments, desensitization is most pronounced at concentrations above 3 mM cGMP, and it does not involve a change in single-channel conductance, which is consistent with a change in channel gating without a change in permeation properties; moreover, recovery from desensitization is relatively fast (seconds time scale). Another form of desensitization, which occurs when cAMP or cGMP is present at concentrations of 100 μM or greater, and which has slow recovery, has been reported in αβ heteromeric CNG channels (Liman and Buck 1994), but that phenomenon depends on β subunit residues outside the BD (Shapiro and Zagotta 2000), as confirmed by the lack of desensitization in X-β. Desensitization in our chimeras arises from some unidentified element in the fCNG2 roll subdomain. An interesting question for future work is whether the sequence positions in the fCNG2 roll subdomain responsible for cGMP-dependent desensitization are identical to those positions in the rCNG5 roll subdomain responsible for high cAMP efficacy.
A minimal kinetic mechanism for our desensitizing channels consists of an activating process (transition from closed nondesensitizing state to open state) and a desensitizing process (transition from open state to closed desensitized state). The steady-state Popen values probably underestimate the efficiency of the activating process, and quantitative delineation of the two processes will require analysis of opening and closing kinetics. Nonetheless, we can deduce that the two processes must be associated with distinct ligand-binding events because the curves of steady-state Popen versus [cGMP] are biphasic. (Pmax,cGMP and K1/2,cGMP provide empirical descriptors only of the rising phase of the dose–response curves.) For the ligand-binding event associated with the desensitizing process, BDs containing the fCNG2 roll subdomain clearly distinguish between cyclic nucleotides, because binding of cGMP but not cAMP causes a decrease in open probability. Hence, the roll subdomain of fCNG2 influences agonist selectivity in the desensitizing process. We cannot conclude whether the roll subdomain also exerts agonist-selective effects on the activating process, but we note that the difference between “closed nondesensitizing” and “closed desensitized” states might be nothing more than the number of cGMP molecules bound to the channel.
We conclude that cAMP selectivity in channel opening can arise not only from the C-helix of the β subunit rCNG5 as previously predicted, but also from the roll subdomain of the α subunit fCNG2. Moreover, the latter subdomain confers a unique form of agonist selectivity: a cGMP-dependent decrease in channel open probability (desensitization). These two forms of selectivity are not mutually exclusive and, indeed, appear to be of similar importance in determining the agonist selectivity of a given BD sequence. Hence, either the fCNG2 roll subdomain or the rCNG5 C-helix subdomain is sufficient to make Popen higher in cAMP than in cGMP over some range of agonist concentration; the chimera X-αR/βC that combines both elements produces larger currents in cAMP than in cGMP at every tested concentration. Residues controlling selectivity for the activating and desensitizing processes may indeed turn out to be segregated in the C-helix and roll subdomains, respectively. Thus, some form of functional polarity may apply to agonist selectivity.
Strong Interaction between BD Subdomains Determines Preferential Open State Stabilization
An important and striking finding is that the molecular interactions contributed by the rCNG5 C-helix have widely different effects on activation efficacy, depending on the sequence of the roll subdomain. This nonadditivity of the effects of subdomain replacement indicates the subdomains are energetically coupled, which implies that an interaction between them partially determines the relative stability of the channel's open state. These interactions may be direct through immediate contact, or indirect through another structural element, such as the agonist molecule or a non-BD region. (Note all non-BD regions are conserved in the chimeras of this study.) The interaction in question depends on some residues that are not conserved between the fCNG2 and rCNG5 BD sequences (if all residues pertinent to the interaction were conserved in our four chimeras, then nonadditivity would not have been observed). It remains to be seen if nonadditivity effects can be found for other pairs of BD sequences.
Our analysis applies to a group of four chimeras representing all combinations of the roll and C-helix subdomains of fCNG2 and rCNG5, so it does not assign the subdomain interaction to a specific subdomain pair. We cannot distinguish, for example, whether the extremely low efficacy of X-αR/βC reflects the absence of a favorable interaction or the presence of an unfavorable interaction between the fCNG2 roll and rCNG5 C-helix subdomains. However, our result makes the key point that among the four chimeras of this study, energetic coupling through subdomain interaction is necessary to account for the variation in efficacy. This is significant: subdomain interaction (direct or indirect) can determine efficacy only if the relative orientation and distance between subdomains are subject to a rigid constraint at some stage of the channel opening transition. The interaction itself may exist only in particular BD sequences, but if the BDs in all four chimeras have structures that closely resemble the BD structure in intact CNG channels (as is suggested by numerous lines of evidence), then it is likely that the structural constraint that is prerequisite for the subdomain interaction will be a general feature of normal BD structure.
Nonadditivity of subdomain replacement effects is demonstrable on the basis of qualitative differences in opening efficacies among the four subdomain-pair chimeras, and is therefore essentially model-independent. However, adopting a minimal model with a single open and closed state enables estimation of the strength of the subdomain interaction from thermodynamic linkage analysis of ΔGsat,cAMP. We have not validated the minimal model with kinetic analysis of our chimeras, but the use of more complex models would change only the quantitative estimates and not our essential conclusions. We stress that the interaction coupling energy ( ΔGint,cAMP) calculated from linkage analysis represents the energy of those particular subdomain interactions that cause preferential stabilization of the open state, and not simply the energy of all subdomain interactions.
Our calculated ΔGint,cAMP of −11.5 kJ/mol is large enough to represent multiple van der Waals contacts. Interaction coupling energies of this magnitude have not previously been reported either for interactions between the two BD subdomains or between the BD and non-BD regions. A previous point mutation study (Scott and Tanaka 1998) found coupling between residues F533, K596 and D604 within the BD of bCNG1, and these residues were also found to influence the action of Ni2+ on the C-linker, but all the interaction coupling energies were <3 kJ/mol, markedly smaller than ours. This difference in magnitude is reasonable because our study examines the combined effects of all sequence differences between two BDs rather than the effects of single point mutations. Another interaction has been identified, between the NH2- and COOH-terminal regions (Gordon et al. 1997; Varnum and Zagotta 1997), but it was found that the bCNG1 and rCNG2 COOH-terminal regions formed identical interactions with the rCNG2 NH2-terminal region (presumably because the COOH-terminal residues involved in the interaction are conserved between bCNG1 and rCNG2). If indeed energetic coupling of the BD with the NH2-terminal region is a determinant of efficacy, the magnitude of its contribution remains unknown. Our study is the first to show that the total interaction coupling energy from BD subdomain interactions can be several-fold larger than kT; indeed, the interaction energy is comparable in magnitude to the entire ΔΔGsat,cAMP between some chimeras, such as that between X-α and X-β. Therefore, subdomain interaction can be a major determinant of the functional differences between CNG channel subunits.
Simultaneous interaction of the two subdomains with ligand is an intuitive premise of models for CNG channel activation. Our measurement of significant subdomain interaction coupling energy places constraints on the construction of such models. For instance, one simple model posits state-dependent subdomain interactions with the purine ring in the cyclic nucleotide as a mechanistic basis for switching the channel between the open and closed state (Varnum et al. 1995). When the channel is closed, the purine ring makes contacts with the roll rather than the C-helix; during channel opening, the purine rotates around the N-glycosidic bond, breaking its contacts with the roll to form a highly favorable interaction with the C-helix. (The cyclic phosphate and ribose are thought to make interactions with the roll subdomain that are the same in the open and closed channel.) This “rotamer” model in its simplest form posits no other roll–C-helix interactions save that taking place through the ligand. However, this would predict that changes in the strength of the roll–purine contact should be independent of changes in the strength of the C-helix–purine contact, because the purine does not contact the two subdomains simultaneously, and the connection between the purine and the ribose is not rigid. Thus, nonadditivity could be explained if the rotamer model were modified to include either (1) a direct state-dependent roll–C-helix interaction at some other locus, or (2) a purine–roll interaction in the open state (i.e., simultaneous interaction of the C-helix and roll with different halves of the purine ring during the open state).
Roll residues interacting with the purine ring likely include the previously studied position 533 (Scott and Tanaka 1998), which is phenylalanine in fCNG2 and tyrosine in rCNG5. However, interaction coupling energy ≥10 kJ/mol is generally too large to be derived from a single pair of residues, except in the case of oppositely charged side chains <4 Å apart (Schreiber and Fersht 1995). We feel it is most likely that the large interaction coupling energy found in our study indicates the presence of novel determinants of activation that were not previously identified; future work will aim at pinpointing such residues. A parallel avenue of study is the continued exploration of the functional properties of BDs from other CNG channel subunits, whether of the conventional (α) or modulatory (β) type. This will eventually allow compilation of a roster of BD residues that can be used in different combinations to tune the efficiency of ligand-gating in CNG channels.
Acknowledgments
We thank Emily R. Liman for the gift of the rCNG5 clone, John Riley and Huan Yao for technical assistance, and Gareth Tibbs and members of the Siegelbaum lab for comments.
This work was partially supported by grant NS36658 from the National Institutes of Health (to S.A. Siegelbaum); E.C. Young is a Research Associate, D.M. Sciubba is a Medical Student Research Fellow, and S.A. Siegelbaum is an Investigator of the Howard Hughes Medical Institute.
Footnotes
Abbreviations used in this paper: bCNG1, bovine CNG1; BD, binding domain; CAP, catabolite activator protein; fCNG2, catfish CNG2; rCNG2 and 5, rat CNG2 and rat CNG5, respectively.
References
- Altenhofen W., Ludwig J., Eismann E., Kraus W., Bönigk W., Kaupp U.B. Control of ligand specificity in cyclic nucleotide-gated channels from rod photoreceptors and olfactory epithelium. Proc. Natl. Acad. Sci. USA. 1991;88:9868–9872. doi: 10.1073/pnas.88.21.9868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berghard A., Buck L.B., Liman E.R. Evidence for distinct signaling mechanisms in two mammalian olfactory sense organs. Proc. Natl. Acad. Sci. USA. 1996;93:2365–2369. doi: 10.1073/pnas.93.6.2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Biel M., Zong X., Ludwig A., Sautter A., Hofmann F. Structure and function of cyclic nucleotide-gated channels. Rev. Physiol. Biochem. Pharmacol. 1999;135:151–171. doi: 10.1007/BFb0033672. [DOI] [PubMed] [Google Scholar]
- Bönigk W., Bradley J., Müller F., Sesti F., Boekhoff I., Ronnett G.V., Kaupp U.B., Frings S. The native rat olfactory cyclic nucleotide-gated channel is composed of three distinct subunits. J. Neurosci. 1999;19:5332–5347. doi: 10.1523/JNEUROSCI.19-13-05332.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J., Li J., Davidson N., Lester H.A., Zinn K. Heteromeric olfactory cyclic nucleotide-gated channelsa subunit that confers increased sensitivity to cAMP. Proc. Natl. Acad. Sci. USA. 1994;91:8890–8894. doi: 10.1073/pnas.91.19.8890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J., Zhang Y., Bakin R., Lester H.A., Ronnett G.V., Zinn K. Functional expression of the heteromeric “olfactory” cyclic nucleotide-gated channel in the hippocampusa potential effector of synaptic plasticity in brain neurons. J. Neurosci. 1997;17:1993–2005. doi: 10.1523/JNEUROSCI.17-06-01993.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown R.L., Gramling R., Bert R.J., Karpen J.W. Cyclic GMP contact points within the 63-kDa subunit and a 240-kDa associated protein of retinal rod cGMP-activated channels. Biochemistry. 1995;34:8365–8370. doi: 10.1021/bi00026a018. [DOI] [PubMed] [Google Scholar]
- Bucossi G., Eismann E., Sesti F., Nizzari M., Seri M., Kaupp U.B., Torre V. Time-dependent current decline in cyclic GMP-gated bovine channels caused by point mutations in the pore region expressed in Xenopus oocytes. J. Physiol. 1996;493:409–418. doi: 10.1113/jphysiol.1996.sp021392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter P.J., Winter G., Wilkinson A.J., Fersht A.R. The use of double mutants to detect structural changes in the active site of the tyrosyl-tRNA synthetase (Bacillus stearothermophilus) Cell. 1984;38:835–840. doi: 10.1016/0092-8674(84)90278-2. [DOI] [PubMed] [Google Scholar]
- Chen T.-Y., Peng Y.-W., Dhallan R.S., Ahamed B., Reed R.R., Yau K.-W. A new subunit of the cyclic nucleotide-gated cation channel in retinal rods. Nature. 1993;362:764–767. doi: 10.1038/362764a0. [DOI] [PubMed] [Google Scholar]
- Chen T.-Y., Illing M., Molday L.L., Hsu Y.-T., Yau K.-W., Molday R.S. Subunit 2 (or β) of retinal rod cGMP-gated cation channel is a component of the 240-kDa channel-associated protein and mediates Ca2+-calmodulin modulation. Proc. Natl. Acad. Sci. USA. 1994;91:11757–11761. doi: 10.1073/pnas.91.24.11757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng X., Kovac L., Lee J.C. Probing the mechanism of CRP activation by site-directed mutagenesisthe role of serine 128 in the allosteric pathway of cAMP receptor protein activation. Biochemistry. 1995;34:10816–10826. doi: 10.1021/bi00034a014. [DOI] [PubMed] [Google Scholar]
- Cui J., Melman Y., Palma E., Fishman G.I., McDonald T.V. Cyclic AMP regulates the HERG K+ channel by dual pathways. Curr. Biol. 2000;10:671–674. doi: 10.1016/s0960-9822(00)00516-9. [DOI] [PubMed] [Google Scholar]
- Cui J., Kagan A., Qin D., Mathew J., Melman Y.F., McDonald T.V. Analysis of the cyclic nucleotide binding domain of the HERG potassium channel and interactions with KCNE2. J. Biol. Chem. 2001;276:17244–17251. doi: 10.1074/jbc.M010904200. [DOI] [PubMed] [Google Scholar]
- Fesenko E.E., Kolesnikov S.S., Lyubarsky A.L. Induction by cyclic GMP of cationic conductance in plasma membrane of retinal rod outer segment. Nature. 1985;313:310–313. doi: 10.1038/313310a0. [DOI] [PubMed] [Google Scholar]
- Finn J.T., Krautwurst D., Schroeder J.E., Chen T.-Y., Reed R.R., Yau K.-W. Functional co-assembly among subunits of cyclic-nucleotide-activated, nonselective cation channels, and across species from nematode to human. Biophys. J. 1998;74:1333–1345. doi: 10.1016/S0006-3495(98)77846-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstner A., Zong X., Hofmann F., Biel M. Molecular cloning and functional characterization of a new modulatory cyclic nucleotide-gated channel subunit from mouse retina. J. Neurosci. 2000;20:1324–1332. doi: 10.1523/JNEUROSCI.20-04-01324.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S.E., Zagotta W.N. Subunit interactions in coordination of Ni2+ in cyclic nucleotide-gated channels Proc. Natl. Acad. Sci. USA. 92 1995. 10222 10226a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon S.E., Zagotta W.N. A histidine residue associated with the gate of the cyclic nucleotide-activated channels in rod photoreceptors Neuron. 14 1995. 177 183b [DOI] [PubMed] [Google Scholar]
- Gordon S.E., Varnum M.D., Zagotta W.N. Direct interaction between amino- and carboxyl-terminal domains of cyclic nucleotide-gated channels. Neuron. 1997;19:431–441. doi: 10.1016/s0896-6273(00)80951-4. [DOI] [PubMed] [Google Scholar]
- Goulding E.H., Ngai J., Kramer R.H., Colicos S., Axel R., Siegelbaum S.A., Chess A. Molecular cloning and single-channel properties of the cyclic nucleotide-gated channel from catfish olfactory neurons. Neuron. 1992;8:45–58. doi: 10.1016/0896-6273(92)90107-o. [DOI] [PubMed] [Google Scholar]
- Goulding E.H., Tibbs G.R., Liu D., Siegelbaum S.A. Role of H5 domain in determining pore diameter and ion permeation through cyclic nucleotide-gated channels. Nature. 1993;364:61–64. doi: 10.1038/364061a0. [DOI] [PubMed] [Google Scholar]
- Goulding E.H., Tibbs G.R., Siegelbaum S.A. Molecular mechanism of cyclic-nucleotide-gated channel activation. Nature. 1994;372:369–374. doi: 10.1038/372369a0. [DOI] [PubMed] [Google Scholar]
- He Y., Karpen J.W. Probing the interactions between cAMP and cGMP in cyclic nucleotide-gated channels using covalently tethered ligands. Biochemistry. 2001;40:286–295. doi: 10.1021/bi002014n. [DOI] [PubMed] [Google Scholar]
- He Y., Ruiz M., Karpen J.W. Constraining the subunit order of rod cyclic nucleotide-gated channels reveals a diagonal arrangement of like subunits. Proc. Natl. Acad. Sci. USA. 2000;97:895–900. doi: 10.1073/pnas.97.2.895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ildefonse M., Crouzy S., Bennett N. Gating of retinal rod cation channel by different nucleotidescomparative study of unitary currents. J. Membr. Biol. 1992;130:91–104. doi: 10.1007/BF00233741. [DOI] [PubMed] [Google Scholar]
- Jan L.Y., Jan Y.N. Tracing the roots of ion channels. Cell. 1992;69:715–718. doi: 10.1016/0092-8674(92)90280-p. [DOI] [PubMed] [Google Scholar]
- Kaupp U.B., Niidome T., Tanabe T., Terada S., Bönigk W., Stühmer W., Cook N.J., Kangawa K., Matsuo H., Hirose T. Primary structure and functional expression from complementary DNA of the rod photoreceptor cyclic GMP-gated channel. Nature. 1989;342:762–766. doi: 10.1038/342762a0. [DOI] [PubMed] [Google Scholar]
- Komatsu H., Jin Y.H., L'Etoile N., Mori I., Bargmann C.I., Akaike N., Ohshima Y. Functional reconstitution of a heteromeric cyclic nucleotide-gated channel of Caenorhabditis elegans in cultured cells. Brain Res. 1999;821:160–168. doi: 10.1016/s0006-8993(99)01111-7. [DOI] [PubMed] [Google Scholar]
- Körschen H.G., Illing M., Seifert R., Sesti F., Williams A., Gotzes S., Colville C., Müller F., Dose A., Godde M. A 240 kDa protein represents the complete β subunit of the cyclic nucleotide-gated channel from rod photoreceptor. Neuron. 1995;15:627–636. doi: 10.1016/0896-6273(95)90151-5. [DOI] [PubMed] [Google Scholar]
- Kramer R.H., Goulding E., Siegelbaum S.A. Potassium channel inactivation peptide blocks cyclic nucleotide-gated channels by binding to the conserved pore domain. Neuron. 1994;12:655–662. doi: 10.1016/0896-6273(94)90220-8. [DOI] [PubMed] [Google Scholar]
- Kumar V.D., Weber I.T. Molecular model of the cyclic GMP-binding domain of the cyclic GMP-gated ion channel. Biochemistry. 1992;31:4643–4649. doi: 10.1021/bi00134a015. [DOI] [PubMed] [Google Scholar]
- Liman E.R., Tytgat J., Hess P. Subunit stoichiometry of a mammalian K+ channel determined by construction of multimeric cDNAs. Neuron. 1992;9:861–871. doi: 10.1016/0896-6273(92)90239-a. [DOI] [PubMed] [Google Scholar]
- Liman E.R., Buck L.B. A second subunit of the olfactory cyclic nucleotide-gated channel confers high sensitivity to cAMP. Neuron. 1994;13:611–621. doi: 10.1016/0896-6273(94)90029-9. [DOI] [PubMed] [Google Scholar]
- Liu D.T., Tibbs G.R., Siegelbaum S.A. Subunit stoichiometry of cyclic nucleotide-gated channels and effects of subunit order on channel function. Neuron. 1996;16:983–990. doi: 10.1016/s0896-6273(00)80121-x. [DOI] [PubMed] [Google Scholar]
- Liu D.T., Tibbs G.R., Paoletti P., Siegelbaum S.A. Constraining ligand-binding site stoichiometry suggests that a cyclic nucleotide-gated channel is composed of two functional dimers. Neuron. 1998;21:235–248. doi: 10.1016/s0896-6273(00)80530-9. [DOI] [PubMed] [Google Scholar]
- MacKinnon R. Pore loopsan emerging theme in ion channel structure. Neuron. 1995;14:889–892. doi: 10.1016/0896-6273(95)90327-5. [DOI] [PubMed] [Google Scholar]
- Matulef K., Flynn G.E., Zagotta W.N. Molecular rearrangements in the ligand-binding domain of cyclic nucleotide-gated channels. Neuron. 1999;24:443–452. doi: 10.1016/s0896-6273(00)80857-0. [DOI] [PubMed] [Google Scholar]
- Molokanova E., Maddox F., Luetje C.W., Kram R.H. Activity-dependent modulation of rod photoreceptor cyclic nucleotide-gated channels mediated by phosphorylation of a specific tyrosine residue. J. Neurosci. 1999;19:4786–4795. doi: 10.1523/JNEUROSCI.19-12-04786.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Möttig H., Zimmer T., Koopmann R., Benndorf K. Structural elements involved in the voltage-dependent gating of cyclic nucleotide gated channels Biophys. J. 78 2000. 352A(Abstr [Google Scholar]
- Nakamura T., Gold G.H. A cyclic nucleotide-gated conductance in olfactory receptor cilia. Nature. 1987;325:442–444. doi: 10.1038/325442a0. [DOI] [PubMed] [Google Scholar]
- Pagès F., Ildefonse M., Ragno M., Crouzy S., Bennett N. Coexpression of α and β subunits of the rod cyclic GMP-gated channel restores native sensitivity to cyclic AMProle of D604/N1201. Biophys. J. 2000;78:1227–1239. doi: 10.1016/S0006-3495(00)76680-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paoletti P., Young E.C., Siegelbaum S.A. C-linker of cyclic nucleotide-gated channels controls coupling of ligand binding to channel gating. J. Gen. Physiol. 1999;113:17–33. doi: 10.1085/jgp.113.1.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Root M.J., MacKinnon R. Two identical noninteracting sites in an ion channel revealed by proton transfer. Science. 1994;265:1852–1856. doi: 10.1126/science.7522344. [DOI] [PubMed] [Google Scholar]
- Ruiz M., Brown R.L., He Y., Haley T.L., Karpen J.W. The single-channel dose–response relation is consistently steep for rod cyclic nucleotide-gated channelsimplications for the interpretation of macroscopic dose–response relations. Biochemistry. 1999;38:10642–10648. doi: 10.1021/bi990532w. [DOI] [PubMed] [Google Scholar]
- Sautter A., Zong X., Hofmann F., Biel M. An isoform of the rod photoreceptor cyclic nucleotide-gated channel β subunit expressed in olfactory neurons. Proc. Natl. Acad. Sci. USA. 1998;95:4696–4701. doi: 10.1073/pnas.95.8.4696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schreiber G., Fersht A.R. Energetics of protein-protein interactionsanalysis of the barnase-barstar interface by single mutations and double mutant cycles. J. Mol. Biol. 1995;248:478–486. doi: 10.1016/s0022-2836(95)80064-6. [DOI] [PubMed] [Google Scholar]
- Scott S.-P., Tanaka J.C. Three residues predicted by molecular modeling to interact with the purine moiety alter ligand binding and channel gating in cyclic nucleotide-gated channels. Biochemistry. 1998;37:17239–17252. doi: 10.1021/bi981185d. [DOI] [PubMed] [Google Scholar]
- Scott S.-P., Harrison R.W., Weber I.T., Tanaka J.C. Predicted ligand interactions of 3′, 5′-cyclic nucleotide-gated channel binding sitescomparison of retina and olfactory binding site models. Prot. Eng. 1996;9:333–344. doi: 10.1093/protein/9.4.333. [DOI] [PubMed] [Google Scholar]
- Shabb J.B., Corbin J.D. Cyclic nucleotide-binding domains in proteins having diverse functions. J. Biol Chem. 1992;267:5723–5726. [PubMed] [Google Scholar]
- Shammat I.M., Gordon S.E. Stoichiometry and arrangement of subunits in rod cyclic nucleotide-gated channels. Neuron. 1999;23:809–819. doi: 10.1016/s0896-6273(01)80038-6. [DOI] [PubMed] [Google Scholar]
- Shapiro M.S., Zagotta W.N. Stoichiometry and arrangement of heteromeric olfactory cyclic nucleotide-gated ion channels. Proc. Natl. Acad. Sci. USA. 1998;95:14546–14551. doi: 10.1073/pnas.95.24.14546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro M.S., Zagotta W.N. Structural basis for ligand selectivity of heteromeric olfactory cyclic nucleotide-gated channels. Biophys. J. 2000;78:2307–2320. doi: 10.1016/S0006-3495(00)76777-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su Y., Dostmann W.R., Herberg F.W., Durick K., Xuong N.H., Ten Eyck L., Taylor S.S., Varughese K.I. Regulatory subunit of protein kinase Astructure of deletion mutant with cAMP binding domains. Science. 1995;269:807–813. doi: 10.1126/science.7638597. [DOI] [PubMed] [Google Scholar]
- Sunderman E.R., Zagotta W.N. Sequence of events underlying the allosteric transition of rod cyclic nucleotide-gated channels. J. Gen. Physiol. 1999;113:621–640. doi: 10.1085/jgp.113.5.621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tibbs G.R., Goulding E.H., Siegelbaum S.A. Allosteric activation and tuning of ligand efficacy in cyclic-nucleotide-gated channels. Nature. 1997;386:612–615. doi: 10.1038/386612a0. [DOI] [PubMed] [Google Scholar]
- Tibbs G.R., Liu D.T., Leypold B.G., Siegelbaum S.A. A state-independent interaction between ligand and a conserved arginine residue in cyclic nucleotide-gated channels reveals a functional polarity of the cyclic nucleotide binding site. J. Biol Chem. 1998;273:4497–4505. doi: 10.1074/jbc.273.8.4497. [DOI] [PubMed] [Google Scholar]
- Varnum M.D., Zagotta W.N. Interdomain interactions underlying activation of cyclic nucleotide-gated channels. Science. 1997;278:110–113. doi: 10.1126/science.278.5335.110. [DOI] [PubMed] [Google Scholar]
- Varnum M.D., Black K.D., Zagotta W.N. Molecular mechanism for ligand discrimination of cyclic nucleotide-gated channels. Neuron. 1995;15:619–625. doi: 10.1016/0896-6273(95)90150-7. [DOI] [PubMed] [Google Scholar]
- Wainger B.J., DeGennaro M., Santoro B., Siegelbaum S.A., Tibbs G.R. Molecular mechanism of cAMP modulation of HCN pacemaker channels. Nature. 2001;411:805–810. doi: 10.1038/35081088. [DOI] [PubMed] [Google Scholar]
- Weber G. Energetics of ligand binding to proteins. Adv. Protein Chem. 1975;29:1–83. doi: 10.1016/s0065-3233(08)60410-6. [DOI] [PubMed] [Google Scholar]
- Weber I.T., Steitz T.A. Structure of a complex of catabolite gene activator protein and cyclic AMP refined at 2.5 Å resolution. J. Mol. Biol. 1987;198:311–326. doi: 10.1016/0022-2836(87)90315-9. [DOI] [PubMed] [Google Scholar]
- Wyman J. Linked functions and reciprocal effects in hemoglobina second look. Adv. Protein Chem. 1964;19:223–290. doi: 10.1016/s0065-3233(08)60190-4. [DOI] [PubMed] [Google Scholar]
- Yau K.-W. Cyclic nucleotide-gated channelsan expanding new family of ion channels. Proc. Natl. Acad. Sci. USA. 1994;91:3481–3483. doi: 10.1073/pnas.91.9.3481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta W.N., Siegelbaum S.A. Structure and function of cyclic nucleotide-gated channels. Annu. Rev. Neurosci. 1996;19:235–263. doi: 10.1146/annurev.ne.19.030196.001315. [DOI] [PubMed] [Google Scholar]