

Abstract
We characterized the functional and molecular properties of nicotinic acetylcholine receptors (AChRs) expressed by IMR-32, a human neuroblastoma cell line, and compared them to human α3 AChRs expressed in stably transfected human embryonic kidney (HEK) cells. IMR-32 cells, like neurons of autonomic ganglia, have been shown to express α3, α5, α7, β2, and β4 AChR subunits. From these subunits, several types of α3 AChRs as well as homomeric α7 AChRs could be formed. However, as we show, the properties of functional AChRs in these cells overwhelmingly reflect α3β4 AChRs. α7 AChR function was not detected, yet we estimate that there are 70% as many surface α7 AChRs in IMR-32 when compared with α3 AChRs. Agonist potencies (EC50 values) followed the rank order of 1,1-dimethyl-4-phenylpiperazinium (DMPP; 16±1 μM) > nicotine (Nic; 48 ± 7 μM) ≥ cytisine (Cyt; 57 ± 3 μM) = acetylcholine (ACh; 59 ± 6 μM). All agonists exhibited efficacies of at least 80% relative to ACh. The currents showed strong inward rectification and desensitized at a rate of 3 s−1 (300 μM ACh; −60 mV). Assays that used mAbs confirmed the predominance of α3- and β4-containing AChRs in IMR-32 cells. Although 18% of total α3 AChRs contained β2 subunits, no β2 subunit was detected on the cell surface. Chronic Nic incubation increased the amount of total, but not surface α3β2 AChRs in IMR-32 cells. Nic incubation and reduced culture temperature increased total and surface AChRs in α3β2 transfected HEK cells. Characterization of various α3 AChRs expressed in HEK cell lines revealed that the functional properties of the α3β4 cell line best matched those found for IMR-32 cells. The rank order of agonist potencies (EC50 values) for this line was DMPP (14 ± 1 μM) = Cyt (18 ± 1 μM) > Nic (56 ± 15 μM > ACh (79 ± 8 μM). The efficacies of both Cyt and DMPP were ∼80% when compared with ACh and the desensitization rate was 2 s−1. These data show that even with the potential to express several human nicotinic AChR subtypes, the functional properties of AChRs expressed by IMR-32 are completely attributable to α3β4 AChRs.
Keywords: nicotinic receptor, autonomic, patch clamp
INTRODUCTION
α3 acetylcholine receptors (AChRs) have been identified as the predominant neuronal AChR in ganglia of the autonomic nervous system (Sargent 1993; McGehee and Role 1995). Most ganglionic neurons express a repertoire of nicotinic subunits that typically includes α3, α5, α7, β2, and β4. Much evidence suggests that the α3 AChR is the main mediator of the nicotinic postsynaptic response in autonomic ganglia (Covernton et al. 1994; Mandelzys et al. 1994; Rust et al. 1994). Knockout mice that lack α3 subunits illustrate the critical role played by these AChRs in the peripheral nervous system. These animals have increased perinatal mortality and exhibit severe physiologic impairment in organs innervated by the autonomic nervous system (Xu et al. 1999a). α7 AChRs are expressed widely in ganglia, also. Selective inhibition of α3 AChRs failed to block synaptic transmission in chick ciliary ganglion showing that α7 AChRs are sufficient to sustain transmission in this preparation (Ullian et al. 1997). Knockout mice lacking α7 AChRs had normal nervous system development (Orr-Urtreger et al. 1997), but exhibited impaired hypotensive reflex tachycardia (Franceschini et al. 2000). Since the loss of α3 function in mice is lethal largely due to autonomic dysfunction, the inability of α7 AChRs to sustain adequate function brings into question its specialized role in the autonomic nervous system.
Knockout mice that lack both β2 and β4 subunits are similar in phenotype to the α3 knockout animals (Xu et al. 1999b). Mice that lacked only the β4 subunit survived but exhibited profound reduction in nicotine (Nic)-induced whole-cell currents in superior cervical ganglion. These animals maintained sufficient autonomic function, presumably, through β2 subunit redundancy or compensation. Mice lacking the β2 subunit had apparently normal organ function and normal whole-cell currents in superior cervical ganglion. Even after these elaborate genetic perturbations, many questions remain regarding nicotinic signaling within the autonomic nervous system. In particular, to what extent does α3β2, α3β4, or α7 AChRs normally contribute to ganglionic transmission, and what other roles do they have? A better understanding of the functional properties of these AChRs as well as how AChR expression might be regulated will help to clarify these issues. IMR-32 cells provide a useful model with which to study the subunit composition of human ganglionic AChRs as they express the characteristic complement of “ganglionic” AChR subunits. Additionally, we have developed a series of cell lines that express α3 AChRs formed from combinations of these ganglionic subunits. We determined and then compared the functional properties of recombinant AChRs with “native” AChRs that are expressed by IMR-32 cells. We use functional and pharmacological profiling to investigate further the subunit composition of human AChRs expressed by IMR-32 cells and support our conclusions with various antibody-based molecular techniques.
MATERIALS AND METHODS
Tissue Culture
Transfected human embryonic kidney (HEK) tsA201 cells were maintained in DME medium as described previously (Wang et al. 1998). Cytotoxic selection antibiotics were added to the media to ensure integrity of AChR subunit expression. 0.5 mg/ml Zeocin (Invitrogen) was used for α3 subunit expression, 0.6 mg/ml G-418 (Life Technologies) was used for β2 or β4 subunit selection, and 0.2 mg/ml hygromycin B (Boehringer Mannheim) was used for α5 subunit selection. IMR-32 neuroblastoma cells (Tumilowicz et al. 1970) were obtained from American Tissue Culture Collection and SH-SY5Y neuroblastoma cells (Ross et al. 1983) were provided by June Biedler and Barbara Spengler of Sloan Kettering Institute for Cancer Research. Both were maintained in media consisting of a 1:1 mixture of HAMs F-12 (Sigma-Aldrich) and Eagle's minimum essential medium (Sigma-Aldrich) with penicillin (100 U/ml) and streptomycin (100 μg/ml; Life Technologies) and 10% FBS (Hyclone).
Electrophysiology
At least 2 d before recording, HEK cells were plated onto glass coverslips coated with rat tail collagen (Type 1; Collaborative Biomedical Products), and the IMR-32 cells were plated onto glass coverslips coated with mouse laminin (Collaborative Biomedical Products). Currents were measured by standard patch-clamp techniques (Hamill et al. 1981). Agonist-containing solutions were applied to the cells by gravity fed fused glass tubing that was connected to multiple reservoirs mounted above the recording chamber. The recording solution contained the following (in mM): 150 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 5 HEPES, and was adjusted to pH 7.3 with NaOH. Electrodes (5–8 MΩ) were formed from borosilicate glass and were filled with a solution containing the following (in mM): 150 cesium gluconate, 10 Cs-EGTA, and 10 HEPES, adjusted to pH 7.2 with CsOH. Cell access resistances were typically 8–15 MΩ and were compensated (40–60%) when peak currents were in excess of 2 nA. Cells transfected with α3β2 or α3α5β2 AChRs were treated for 12 h with Nic (100 μM) followed by a minimum of 1-h wash with normal media before recording (Wang et al. 1998) or were incubated overnight at 29°C to increase the levels of functional AChRs. Currents were activated and recorded as described previously (Wang et al. 1998). Concentration–response curves were constructed and fitted to the following logistic equation in Origin (Ver. 4.1; Microcal Software, Inc.):
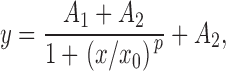 |
where y is the normalized response amplitude, A1 is the maximum amplitude asymptote of the fit, A2 is the minimum asymptote of the fit, x is the concentration of agonist, x0 is the EC50 value, and p is the steepness of the fitted curve. In some cases, the concentration–response relationship would peak and then decline with increasing concentrations of agonist. In the cases where the decline caused an obviously inferior fit, the reduced amplitude responses at higher concentrations were not included in the fit. Desensitization time constants were determined by fitting exponential equations to the data. Representative traces were constructed by opening data files in Axograph 3.55 (Axon Instruments) and exporting data to Canvas 5.0 (Deneba Software, Inc.).
Single-Channel Analysis
Single-channel currents were recorded and analyzed as described previously (Nelson and Lindstrom 1999). In brief, channel activity was recorded in outside-out configuration patches by isolating the patch in a stream of ACh (1–5 μM). Recordings were performed in ND-96 saline also containing 50 mM dextrose, as used previously to characterize single-channel properties for AChRs expressed by IMR-32 cells (Nelson and Lindstrom 1999). Data were sampled offline at 10 kHz (model Axoscope 2.0; Axon Instruments) and filtered at 3 kHz (model 902; 8-pole Bessel, −3 dB; Frequency Devices, Inc.) for analysis. All single-channel analysis and fitting were performed with pClamp 6.0.3 (Axon Instruments).
Production of mAb 337
mAb 337 (mouse IgG) was developed from a bacterially expressed fusion protein consisting of the large cytoplasmic loop located between the M3 and M4 transmembrane domains of the human β4 subunit (amino acids 305–419) coupled at the NH2 terminus to bacterial glutathione S-transferase (Wang et al. 1998). Mice were immunized with the fusion protein purified from bacterial inclusion bodies. The animals were boosted four times over several months while titers were monitored by test bleeds against human α3β4 AChRs (from transfected HEK cells) in immunoprecipitation assays. 5 d after a final boost with antigen, the animal with the highest titers was killed. Hybridomas for mAb production were formed by fusion of the animal's splenic lymphocytes with Sp2 myeloma cells according to a procedure adapted from (Lane et al. 1986). Wells containing hybridomas were then screened against human α3β4 AChRs by immunoprecipitation assays and positive wells were cloned. As we show here, in addition to recognizing the β4 subunit in its native confirmation, the antibody also recognizes the denatured subunit as well. Lack of cross-reactivity with human α3, α5, or β2 subunits was established using AChRs extracted from the α3α5β2 cell line with mAb 337-coated microwells using the procedure described below for radioimmune assays. All animals were handled in accordance with guidelines set forth by the Institutional Animal Care and Use Committee (IACUC) at the University of Pennsylvania under approved protocol on file with that office. IACUC operates under an institutional Animal Welfare Assurance (A3079-01) on file with the Office for Protection from Research Risks at the National Institutes of Health.
Radioimmune Assays (RIAs)
IMR-32 and SH-SY5Y cells were grown as described above until just before confluence in T-175 (175 cm2) tissue culture flasks (model Falcon 3112; Becton Dickinson) then detached with 5 mM EDTA in PBS, pelleted by centrifugation, and used immediately or frozen after removal of media. Fresh or frozen IMR-32 cells were suspended and triturated in a 2% Triton X-100 in buffer A (50 mM Na2HPO4-NaH2PO4, pH 7.5, 50 mM NaCl, 5 mM EDTA, 5 mM EGTA, 5 mM benzamidine, 15 mM iodoacetamide, and 2 mM phenylmethylsulfonyl fluoride at a 3:1–6:1 ratio (buffer volume/cell volume) and placed on a rotating mixer for 2–3 h at 4°C. Cellular debris was removed by centrifugation and supernatant extract was collected.
For solid phase RIA, AChRs solubilized in Triton were incubated in Immulon 4 microwells (Dynatech Laboratories) coated with mAb 210 (specific for α3 and α5 AChRs; Wang et al. 1996), mAb 295 (specific for β2 AChRs; Whiting and Lindstrom 1988), or mAb 337 (specific for β4 AChRs) overnight at 4°C. The labeling of bound AChRs was carried out by including a supersaturating concentration of 10 nM [3H]epibatidine during the incubation in the wells. Unbound material was removed from wells by washing three times with ice-cold 0.5% Triton in PBS. The remaining contents of the wells were stripped with 75 μl of 2.5% SDS containing 5% β-mercaptoethanol and then transferred to liquid scintillation tubes containing 3 ml of Optiphase “Hisafe” 3 scintillation fluid (Fisher Scientific). The amount of bound [3H]epibatidine was determined by liquid scintillation counting for 5 min using a liquid scintillation counter (model Wallac 1410; Perkin-Elmer Wallac). Background was determined using wells coated with BSA or goat anti–rat IgG (GART).
To measure the amount of α3-containing AChRs that could be labeled in intact cells by [3H]epibatidine, IMR-32 cells were plated in 35-mm tissue culture dishes and grown until confluent. At this time, the media was replaced with 0.5 ml of media that contained 2 nM [3H]epibatidine and returned to the tissue culture incubator. For background determinations, 300 μM Nic was added 45 min before [3H]epibatidine. After 2 h at 37°C, the medium was then removed and the cells quickly rinsed with PBS. The cells were removed from the dishes by trituration with 0.5 ml buffer A, and then lysed with an ultrasonic cell disrupter (model Sonifier 200; Branson Ultrasonics Corp.). Membrane fragments were collected on glass fiber filters (model GF/F; Whatman; 25 mm, treated with 0.3% polyethylenimine) followed by four rapid 1-ml washes with ice cold PBS. Filters were then transferred to scintillation tubes for counting of retained [3H]epibatidine.
For isolation and measurement of α7-containing AChRs, microwells were coated with a combination of mAbs 306 and 319 that recognizes human α7 subunits (Peng et al. 1994). Cell extracts were prepared as described above and aliquots were incubated in mAb-coated wells overnight at 4°C with 125I-α-BGT (20 nM; 0.48 Ci/μmol). For background determinations, unlabeled α-BGT (200 nM) was included in the incubation 1 h before addition of 125I-α-BGT. After overnight incubation, unbound material was removed as described above and the amount of bound 125I-α-BGT was determined by gamma counting.
Surface AChR Labeling
To measure surface AChRs, cells were collected from tissue culture flasks and gently pelleted by centrifugation followed by a wash in normal culture medium containing 10% FBS. Cells were resuspended in tissue culture medium and aliquoted (500 μl) into microcentrifuge tubes. Surface α3-containing AChRs were labeled by incubation (2–4 h) with 125I-mAb 210 (4 nM; 0.46 Ci/μmol) and surface β2-containing AChRs were labeled with 125I-mAb 295 (4–6 nM; 0.69 Ci/μmol). Background determinations were made in parallel by including at least 50-fold excess unlabeled mAb in a 1 h preincubation to block specific sites. Incubations were carried out at 37°C with constant mixing. After incubation, cells were diluted with an equal volume of tissue culture media, pelleted, and then washed twice more with clean media. Amount of bound 125I-mAb was then determined by gamma counting.
Western Blotting
Triton X-100 extraction of AChRs from IMR-32 and SH-SY5Y neuroblastoma cells were performed as described above. The extracts were incubated overnight at 4°C with mAb-coupled agarose to select and concentrate AChRs. AChRs were isolated using activated CH Sepharose 4B resin (Amersham Pharmacia Biotech) coupled with mAb 210 for α3 AChRs, coupled with mAb 295 for β2 AChRs, or coupled with mAb 337 for β4 AChRs (all at 2 mg mAb/ml of resin). After incubation, bound AChRs were eluted using 3% SDS sample buffer without reducing agents. The samples were loaded on 12% polyacrylamide gels containing SDS and electrophoresed. Transfer of the proteins was done in a semi-dry electroblotting chamber (Semi-Phor; Hoefer Scientific Instruments) to Trans-Blot® medium PVDF membrane (Bio-Rad). The blot membranes were then quenched for 1 h at room temperature with 5% dried milk in PBS containing 0.5% Tween 20 and 10 mM NaN3 (PBS/Tween) with constant agitation. Antisera against α3 or β2 subunits (Kuryatov et al. 2000) were then added at 1:200 dilution and incubated overnight at 4°C. After three washes with PBS/Tween, labeling was done by the addition of 125I-GART antibodies (2 nM, 0.81 Ci/μmol) for 3 h at room temperature, followed by a brief rinse and three washes (10 min each) with PBS/Tween. 125I-mAb 337 (2 nM, 0.83 Ci/μmol) was used to identify the β4 subunit in which case the primary labeling was followed by the wash steps with PBS/Tween. Autoradiography was performed at −80°C with exposures between 10 min and overnight using Kodak Biomax® MS film and intensifying screen.
Immunofluorescence with Confocal Microscopy
IMR-32 or permanently transfected cells were plated on either laminin-coated or collagen-coated glass coverslips, respectively, and fixed with 10% formalin/PBS for 1 h at room temperature for labeling with fluorescent antibody to track the presence of AChR subunit proteins. Fixed cells were then washed three times with PBS to remove fixative. Attachment of fluorophores to primary antibodies was done using a kit (Molecular Probes) to attach either Alexa fluor 488 or 594 to the antibodies. Primary labeling with unlabeled antibody was performed overnight at 4°C in PBS containing antibody with 5% goat serum. Secondary labeling was carried out after washing three times with PBS to remove the unbound primary antibody, followed by Alexa fluor 594-labeled GART (Molecular Probes) for mAb 295 or Alexa fluor 568-labeled goat anti–mouse (GAM) IgG (Molecular Probes) for mAb 337. Secondary antibodies were used at final concentrations of 4 μg/ml for GART or 3 μg/ml for GAM, and were applied for 3–4 h at room temperature in the presence of 5% goat serum. After washing three times with PBS, primary labeled mAb 210 (Alexa fluor 488) was applied overnight at 4°C at a final concentration of 5 μg/ml in the presence of 5% normal rat serum. To access intracellular AChR subunits, cells were permeabilized with 0.5% Triton X-100 for 1 h at room temperature before labeling with antibodies. To-Pro 3 iodide (Molecular Probes) was included as a nuclear counterstain at 0.5–1 μM for 20–60 min. Coverslips with labeled cells were rinsed twice with PBS and once with deionized water, then mounted on slides using Pro-Long antifade (Molecular Probes), dried, and stored at 4°C until viewed. Images of labeled cells were then acquired using a Leica TCS 4 D laser scanning confocal microscope.
RESULTS
Pharmacological Properties of Functional AChRs in IMR-32 Cells
Rapid application of nicotinic agonists to patch-clamped IMR-32 cells evoked responses ranging up to several hundred picoamps when clamped at −60 mV. An example of a concentration/response family of currents from a cell is shown in Fig. 1A using Cyt. The concentration/response relationships are shown for ACh, Nic, DMPP, and Cyt along with the curve fits used to determine the EC50 values (Fig. 1 B). The rank order of potency was DMPP > Nic ≥ Cyt = ACh. This rank order provided a profile for functional native AChR(s) present in IMR-32 cells for comparison with agonist profiles for AChRs of defined composition expressed in transfected cells as described below.
Figure 1.

Functional and pharmacological properties of AChRs expressed by the human neuroblastoma cell line IMR-32. (A) A family of currents recorded in response to increasing concentrations of Cyt for a cell clamped at −60 mV. In the inset, the scaled response of this cell to a maximally effective concentration of Cyt (300 μM) is shown relative to the response to ACh (300 μM) which was chosen to define agonist efficacy. (B). The full concentration/response relationships are shown for ACh (▪), Nic (•), Cyt (▴), and DMPP (⋄). With the exception of DMPP, all agonists had similar potencies. The EC50 values were 66 ± 5 μM for ACh, 48 ± 8 μM for Nic, 57 ± 3 μM for Cyt, and 16 ± 1 μM for DMPP. The relatively high efficacy of Cyt and the relatively high potency of DMPP were the most distinguishing pharmacological properties found for the IMR-32 AChR response. (C). The current/voltage relationship for IMR-32 cells recorded in response to application of 100 μM ACh. The currents were no longer inward at ∼0 mV, which was consistent with a nonselective cation channel. The currents exhibited strong inward rectification, but when depolarized beyond +60 mV, outward currents were observed. (D). Representative recording and exponential fit of the current decay for IMR-32 cells.
All of the agonists exhibited nearly full efficacy (i.e., ∼80%) with respect to ACh. The maximum responses to ACh and Cyt are shown for the cell in Fig. 1 A to illustrate the efficacy of Cyt, which was similar to that of Nic and slightly greater than the efficacy of DMPP. At high agonist concentrations, “rebound” currents were frequently observed, and the peak current amplitudes were often reduced compared with lower agonist concentrations. This was especially true for DMPP. This effect was likely the result of channel block by the agonist and the rebound current occurred as the channel block was relieved on washout.
Functional Properties of IMR-32 AChRs
The current-voltage relationship for ACh-activated currents from IMR-32 cells were studied between −120 and +80 mV (Fig. 1 C). The currents approached reversal when the holding potential was between −10 and 0 mV, which is expected for nonselective cation channels under these recording conditions. Additionally, strong inward rectification was found at positive holding potentials until +60 mV, where some reversal was observed.
Desensitization of currents recorded from IMR-32 cells was usually best described by a single exponential function which could be fit with a time constant of decay of τ = 368 ± 94 ms (n = 8) (Fig. 1 D). In some cases, the desensitization was fit best by a double exponential function with time constants of 73 ± 27 ms and 550 ± 136 ms (n = 2). In addition to desensitization during the application of agonist, the peak amplitude of the currents decreased slightly over the course of a full concentration/response experiment. This “rundown” was not prevented by inclusion of various agents in the recording electrode such as ATP, creatine phosphokinase, phosphocreatine, and magnesium, or various combinations of these substances. Additionally, using a recording solution that was based on CsCl also did not prevent the rundown (unpublished data).
Effects of Nicotinic Antagonists on IMR-32 Currents
Responses to 100 μM ACh were antagonized by coapplication of the antagonists d-tubocurare, mecamylamine, and hexamethonium with IC50 values of 0.4 ± 0.2, 3.2 ± 0.6, and 8.5 ± 3.0 μM, respectively (Fig. 2 A). The reversible α7 selective antagonist, methyllycaconitine (MLA), had no effect on Nic-activated (300 μM) currents when applied at 20 nM with a 10 min preincubation (Fig. 2 B). This concentration of MLA (20 nM) was almost three orders of magnitude higher than the IC50 reported for homomeric α7 AChRs (Palma et al. 1996) or α7 containing AChRs in rat hippocampal neurons (Alkondon et al. 1992). The lack of antagonism by MLA was consistent with the fact that the currents lacked the rapid desensitization that is a hallmark property of α7 AChRs. To validate the ability of our application system to activate α7 AChRs efficiently before they desensitized, we recorded the rapidly desensitizing Nic-activated currents that have been attributed to α7 AChRs from rat hippocampal neurons (unpublished data). Thus, it was concluded that any contribution of α7 AChRs to macroscopic currents from IMR-32 cells was negligible.
Figure 2.

Antagonist sensitivity of the AChR currents activated in IMR-32 cells. (A) The concentration response relationship is shown for d-tubocurare (d-TC; ▪), mecamylamine (Mec; ▾), and hexamethonium (C-6; •) on IMR-32 cells with IC50 values of 0.4 ± 0.2, 3.2 ± 0.6, and 8.5 ± 3.0 μM, respectively. All drugs were coapplied with 100 μM ACh at a holding potential of −60 mV. The peak amplitudes of the resulting currents were then plotted against concentration of the antagonist after normalizing to the peak amplitude of the current recorded in the absence of any antagonist. All three drugs completely blocked the currents at higher concentrations. (B) Representative currents are shown for testing the sensitivity of IMR-32 AChR currents to the reversible α7 selective antagonist MLA (20 nM). No inhibition was observed, which was consistent with the idea that the macroscopic current recorded in the cells reflects predominately the activity of α3 AChRs.
Agonist Potencies and Efficacies in Permanently Transfected α3 AChR Cell Lines
As reported previously (Wang et al. 1998), HEK tsA201 cells stably transfected with combinations of AChR subunits that are commonly expressed in ganglionic neurons (i.e., α3, α5, β2, and β4 subunits) responded robustly to nicotinic agonists. Peak currents ranged up to 15 nA when clamped at −60 mV. Here, we present a more thorough functional characterization of these cell lines so that we can compare, under like conditions, the properties of AChRs with known subunit composition to the properties IMR-32 AChRs as described above. Examples of currents activated by ACh (300 μM) or Cyt (300 μM) from each cell type (α3β4, α3β2, α3β4α5, and α3β2α5) are shown in Fig. 3. For α3β4 transfected cells, the agonist potency rank order was DMPP = Cyt > Nic > ACh, with EC50 values ranging from 15 μM for DMPP to 79 μM for ACh (Fig. 4). Cyt exhibited 60% efficacy, whereas DMPP had 50% efficacy and Nic had ∼85% efficacy (Fig. 4).
Figure 3.

Efficacy of Cyt on α3 AChRs expressed by permanently transfected HEK cells is determined by whether the AChR contains a β2 or β4 subunit. Representative currents are shown for permanently transfected cell lines that express α3β2, α3β2α5, α3β4, and α3β4α5 AChRs. For each cell, a pair of currents are superimposed that reflect current activated by a near saturating (300 μM) concentration of ACh and a maximally effective concentration (300 μM) of Cyt. Clearly, the efficacy of Cyt relative to ACh in the cell lines that express the β2 subunit is far lower (5% or less) than that found for the cell lines that express the β4 subunit (∼80% or more).
Figure 4.
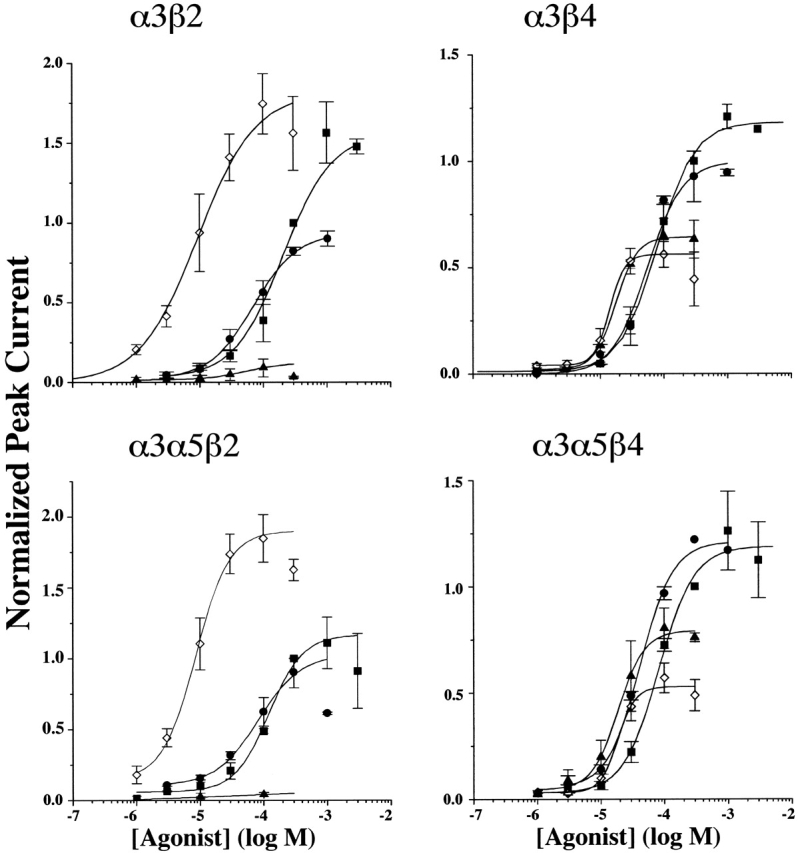
Concentration-response relationships for nicotinic agonists on α3 AChRs in permanently transfected HEK cells establish potency and efficacy profiles that allow for distinguishing β2 and β4 containing AChRs. Each panel depicts the concentration-response relationship for the indicated permanently transfected α3 cell line. The symbols reflect correspond to different agonists as follows: ACh (▪), Nic (•), Cyt (▴), and DMPP (⋄). The EC50 values are shown in Table . Each point in the figure represents the peak amplitude of the current evoked by that concentration of agonist normalized to the peak amplitude of the current evoked by 300 μM ACh in the same cell. The cells were clamped at −60 mV for α3β2 and α3α5β2 and at −30 mV for α3β4 and α3α5β4 cell lines. β4 AChRs were more sensitive to activation by ACh, whereas β2 AChRs were slightly more sensitive to activation by DMPP. Cyt had virtually no efficacy in activating β2 AChRs, whereas it had >50% efficacy on β4 AChRs. DMPP exhibited efficacy that was equal to, or higher than ACh on β2 AChRs, but it was <50% efficacious on β4 AChRs. Nic was poor in distinguishing between β2 and β4 AChRs. The data for ACh and Nic are taken from Wang et al. 1998.
The α3β2 and α3α5β2 cell lines expressed few surface AChRs unless the cells were incubated in Nic (Wang et al. 1998). Therefore, before performing functional studies, these cells were incubated overnight with 100 μM Nic. The medium was exchanged to remove the Nic at least 1 h before recording. The agonist sensitivity profile for α3β2 AChRs was markedly different from that for α3β4 AChRs, with the rank order of potency DMPP ≫ Cyt ≥ Nic ≫ ACh, which reflected EC50 values that ranged from 9 μM for DMPP to 209 μM for ACh. Consistent with previous studies of α3β2 AChRs (Papke and Heinemann 1994), the efficacy of Cyt was extremely poor (5%) on α3β2 AChRs (Fig. 3 and Fig. 4). This was in contrast to the much higher efficacy of Cyt on the α3β4 (75%), α3β4α5 (75%), or IMR-32 (80%) cell lines. In addition, DMPP exhibited efficacy greater than ACh. Thus, agonist efficacies for Cyt and DMPP relative to ACh are useful in predicting the presence of the β2 subunit in native α3 AChRs.
Reduced Temperature Increases Functional α3β2 AChRs
When incubated at reduced temperature (29°C) overnight, α3β2 cells increased their amount of functional AChRs as judged by the gain of responsiveness to agonist, much like when the cells were incubated in Nic overnight (Wang et al. 1998). The temperature-induced AChR increase was similar to a recent report for α4β2 AChRs expressed in HEK cells (Cooper et al. 1999). Since our functional studies with α3β2 or α3α5β2 AChRs required exposure to Nic before recording, we were unable to determine if the agonist exposure itself altered functionality when compared with naive AChRs. However, the temperature-induced increase in AChRs allowed us to characterize α3β2 AChRs without prior exposure to Nic (Fig. 5 A). The EC50 value for cells incubated at 29°C was 202 ± 14 μM, which compares well to the EC50 of 209 ± 26 μM for cells incubated in Nic (Fig. 5 B). Since the functional properties were the same regardless of treatment, it seems unlikely that Nic-treated α3β2 AChRs have properties that differ from α3β2 AChRs that have not been Nic-treated. Previous studies with α3β2 AChRs expressed in Xenopus oocytes showed that this Nic treatment did not alter the functional properties of the AChR (Wang et al. 1998). By contrast with the Nic-induced increase in surface α3β2 AChRs in the transfected cell line that was reflected by the appearance of substantial electrophysiological responses, overnight incubation of IMR-32 cells in Nic did not increase the amount of α3β2 AChRs on the surface of these cells (see last paragraph of Subunit Content of AChRs…) or alter their functional properties in a way that might reflect an increase in surface α3β2 AChRs (Fig. 5 C, compare with Fig. 1 A, inset). Similar results were found for IMR-32 cells incubated at 29°C (unpublished data).
Figure 5.

Culturing the α3β2 AChR cell line at 29°C increases the population of functional AChRs similar to overnight incubation in Nic. (A) A family of representative currents is shown for the concentration-response relationship for α3β2 cells that were placed at 29°C for at least 6 h and as long as 48 h. For comparison, currents recorded from cells that were incubated in Nic (100 μM) overnight are also shown (Wang et al. 1998). (B) The sensitivity to activation by ACh for the cells grown at reduced temperature were identical to those that had been incubated overnight in 100 μM Nic. The full concentration/response for both conditions is also shown. Responses were normalized to the response peak measured for 300 μM ACh for each cell. The EC50 for each condition was 202 ± 14 μM for cells grown at 29°C (□) and was 209 ± 26 μM for cells incubated overnight in Nic (▪). (C) Representative currents are shown for IMR-32 cells that were incubated overnight in Nic (100 μM). The responses to 300 μM ACh and 300 μM Cyt are nearly identical to currents recorded from naive IMR-32 cells (Fig. 1 A, inset).
Transfection of α3β4 and α3β2 Cell Lines with the α5 Subunit Had Little Effect on the Functional or Pharmacological Properties when Compared with the Parent Cell Line
α3α5β4 transfected cells exhibited pharmacological properties that paralleled closely those of the parent α3β4 cell line. The agonist rank order of potencies were as follows: DMPP = Cyt > Nic > ACh, and having EC50 values ranging from 20 μM for DMPP to 83 μM for ACh (Fig. 4). The agonist efficacies were nearly the same as for the α3β4 cell line with the exception of Nic (Table ). Previous immunoprecipitation studies of the α3α5β4 cell line showed that 14% of the total AChRs expressed the α5 subunit (Wang et al. 1998). This small percentage of total AChR might not be sufficient to contribute significantly to macroscopic currents. This is consistent with agonist sensitivities matching those of the α3β4 cell line. Additionally, the partial efficacies of both DMPP and Cyt were nearly identical for α3β4 and α3α5β4 cells.
Table 1.
Comparison of Agonist Potencies for Human AChRs in IMR-32 and in Transfected HEK Cells
| EC50 for Cell Line | |||||
|---|---|---|---|---|---|
| Agonist | IMR-32 | α3β4 | α3α5β4 | α3β2 | α3α5β2 |
| ACh | 59 ± 6 | 79 ± 8 | 81 ± 15 | 209 ± 26 | 121 ± 18 |
| n = 1.5 | n = 1.5 | n = 1.5 | n = 1.7 | n = 1.6 | |
| (100%) | (100%) | (100%) | (100%) | (100%) | |
| Nic | 48 ± 7 | 56 ± 15 | 42 ± 5 | 70 ± 6 | 83 ± 12 |
| n = 1.8 | n = 1.6 | n = 1.6 | n = 1.2 | n = 1.3 | |
| (85%) | (85%) | (100%) | (60%) | (85%) | |
| Cyt | 57 ± 3 | 18 ± 1 | 21 ± 3 | 47 ± 34 | ∼30 |
| n = 1.5 | n = 2.7 | n = 1.9 | n = 1.5 | n = ND | |
| (80%) | (75%) | (75%) | (5%) | (4%) | |
| DMPP | 16 ± 1 | 14 ± 1 | 20 ± 1 | 9 ± 3 | 8 ± 1 |
| n = 1.7 | n = 3.7 | n = 2.7 | n = 1.0 | n = 1.5 | |
| (80%) | (50%) | (55%) | (110%) | (170%) | |
Values for EC50 are measured in micromolar units.
The α3α5β2 cell line had pharmacological properties that differed only slightly from the parent α3β2 cell line, also. Nic was more potent than ACh on α3α5β2 cells, but the EC50 for ACh was significantly lower than for the α3β2 cell line (Fig. 4 and Table ). The rank order of agonist potencies for the α3α5β2 cell line (DMPP ≫ Cyt ≥ Nic > ACh) was essentially the same as that for the α3β2 cell line. Also, the efficacy of Nic was greater for α3α5β2 AChRs when compared with α3β2 AChRs. The similar pharmacological properties (Table ) between these cell lines suggest that the α5 subunit, present in 50% of total AChRs (Wang et al. 1998), has little impact on their agonist sensitivities. In Xenopus oocytes, incorporation of the α5 subunit in α3β2 AChRs increased both the desensitization rate and the Ca2+ permeability (Gerzanich et al. 1998).
Agonist Sensitivity and Efficacy Profiles for α3 Cell Lines and IMR-32 Cells
We used the pharmacological data for AChRs in transfected cells to establish prediction criteria for the presence of either β2 or β4 subunits in native human α3 AChRs that are expressed by IMR-32 cells. α3 AChRs that are relatively insensitive to activation by ACh (i.e., have EC50 values greater than 100 μM) and show full efficacy to activation by DMPP are likely to possess β2 subunits. AChRs where Cyt has significant efficacy and DMPP has less than full efficacy are likely to contain β4 subunits. The agonist potency profiles found for the α3β4 and α3α5β4 cell lines most closely resemble that found for IMR-32 cells (Fig. 6 A). Since the α3α5β4 cell line incorporates α5 subunit, the best match is between IMR-32 and the α3β4 cell line.
Figure 6.
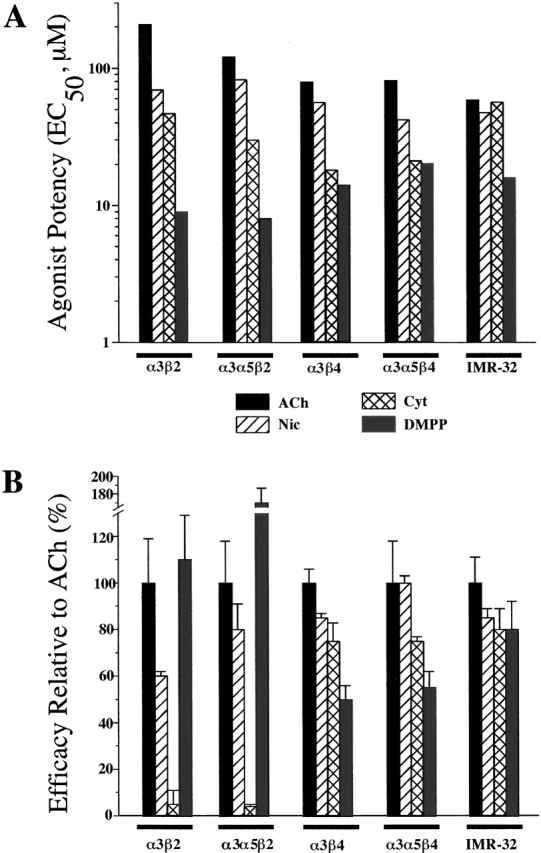
Agonist potency and efficacy profiles for HEK α3 cell lines and IMR-32 cells. (A) Bar graph depicting the EC50 values from Fig. 4 for ACh, Nic, Cyt, and DMPP on the HEK transfected cell lines and IMR-32 cells. The potency profile for IMR-32 cells best matches the profiles for the α3β4 and the α3β4α5 cell lines. The biggest discrepancy was seen for the potency of Cyt, which was higher on the β4 containing transfected cell lines than on IMR-32 cells, but the potencies of the other three agonists matched well. (B) Bar graph depicting the efficacies of the four agonists on the HEK transfected cell lines and IMR-32 cells. The efficacies of Cyt and DMPP serve as good predictors of the β subunit identity in unknown AChRs. Thus, for IMR-32 AChRs, the efficacy of both Cyt and DMPP along with the agonist potency profiles lead us to conclude that the predominant AChR in these cells is α3β4.
The agonist efficacy profiles provide additional support for the conclusion that IMR-32 cells express predominantly α3β4 AChRs (Fig. 6 B). Specifically, Cyt has >70% efficacy on the α3β4, α3α5β4, and IMR-32 cells and <10% efficacy on α3β2 and the α3α5β2 cell lines, whereas DMPP has >100% efficacy on α3β2 and α3α5β2, but <80% efficacy on α3β4, α3α5β4, and IMR-32 cells.
Effect of Holding Potential on Agonist Properties for α3β4 AChRs in HEK Cells
For ACh and Nic, the EC50 values were greater at −30 mV (79 ± 8 μM and 56 ± 10 μM, respectively) than at −60 mV (43 ± 10 μM and 35 ± 6 μM, respectively) whereas for DMPP the EC50 was little changed (15 ± 1 μM at −30 mV and 14 ± 1 μM at −60 mV). Reducing the holding potential also altered the efficacy of Nic and DMPP relative to ACh. The efficacy of Nic peaked between 300 μM and 1 mM at −30 mV, whereas the peak occurred at 100 μM at −60 mV. For DMPP, the efficacy relative to ACh increased from ∼50 to ∼65% by depolarizing from −60 to −30 mV. Thus, the effect of holding potential on the concentration-response relationship probably represents the impact of voltage on channel blockade by these agonists, but could also reflect voltage-dependent gating transitions.
All AChRs Exhibited Rectification Regardless of Subunit Composition
Strong inward rectification is a hallmark property of neuronal nicotinic AChRs. It was attributed to intracellular magnesium for α-BGT–sensitive currents in rat hippocampal neurons (Alkondon and Albuquerque 1994). The rectification was removed by mutations in amino acids that flank the channel forming region of α3β4 and α4β2 AChRs (Haghighi and Cooper 1998). Channel block by internal polyamines was also reduced by these mutations (Haghighi and Cooper 1998; Haghighi and Cooper 2000). All of the HEK α3 AChR cell lines exhibited inward rectification (Fig. 7). The current-voltage relations approach reversal between 0 and +5 mV, but failed to become outward at positive holding potentials up to +60 mV. Recordings at +100 mV with the α3α5β2 cell line had small outward currents that were ∼15% of the currents recorded at −100 mV (unpublished data). If polyamines caused the rectification, its persistence throughout 45–75-min recordings indicated that the large dilution by electrode solution was insufficient to relieve the channel block. Magnesium or another ion in the electrode solution could also be responsible for the rectification.
Figure 7.

Current-voltage relations for α3 AChRs expressed by permanently transfected HEK cells exhibit the strong inward rectification that is characteristic of neuronal nicotinic AChRs. For the four α3 AChR cells lines, currents were activated by 100 μM ACh at fixed holding potentials spanning −80 to +60 mV. The peak amplitudes of the currents are shown plotted against holding potential after normalizing to the amplitude of the peak current measured at −80 mV. In all cases the current ceased to be inward at a holding potential between ∼0 and +5 mV consistent with a nonselective cation channel. No outward currents were detected at holding potentials up to +60 mV.
Desensitization Rate Reflects the β Subunit Identity in Permanently Transfected HEK Cells
The α3β2 cells exhibited desensitization that was much faster than that found for α3β4 cells (Fig. 8). Coexpression of the α5 subunit had little effect on the desensitization of the α3β2 cell line and no effect on the α3β4 cell line. The α3β2 currents exhibited both single and double exponential decays. For the α3α5β2 cell line, desensitization was found to be slightly faster when compared with the α3β2 cell line. The α3β4 cell line and the α3α5β4 cell line had similar decay time constants and both were similar to the value found for IMR-32 AChRs.
Figure 8.

α3 AChRs containing the β2 subunit desensitize faster than those that contain the β4 subunit. Currents evoked by ACh (300 μM) in each α3 cell line are shown along with the fit of the current decay to either a single or double exponential function. Both α3β2 and α3α5β2 AChRs exhibited desensitization with a very rapid component having time constants of <100 ms. Some of the α3β2 cell currents could be fit by double exponential functions (5 out of 7) with the faster component matching the cells which had only a single exponential decay as well as a slower component that was several hundred milliseconds. Some of the α3α5β2 AChR currents (7 out of 12) had a small amplitude slower component. Both the α3β4 and the α3α5β4 AChRs had currents that desensitized much slower than the β2-containing AChRs and were always best fit by single exponential functions which had time constants of decay of ∼500 ms. All desensitization time constants were determined for currents that were recorded at −60 mV.
Single-channel Properties of α3β4 AChRs Expressed in HEK Cells Closely Match Those of AChRs Expressed by IMR-32
The HEK α3β4 AChRs had single-channel open times with time constants of 1.9 ± 0.6 and 6.0 ± 1.0 ms (Fig. 9). These values were similar to those obtained previously for α3β4 AChRs recorded from Xenopus oocytes under identical ionic conditions (1.4 ± 0.2 and 6.5 ± 0.8 ms) and similar to the values obtained for IMR-32 AChRs (1.5 ± 0.3 and 9.2 ± 1.2 ms; Nelson and Lindstrom 1999). The channel amplitudes for the HEK α3β4 AChRs were −2.3 ± 0.1 and −1.8 ± 0.1 pA at −80 mV. These amplitudes also were similar to those for both oocyte-expressed α3β4 AChRs (−2.3 ± 0.1 and −1.6 ± 0.1 pA) and IMR-32 AChRs (−2.2 ± 0.1 pA; Nelson and Lindstrom 1999). These data support the conclusion that the predominant functional AChR in IMR-32 cells is α3β4.
Figure 9.

The single-channel properties for α3β4 AChRs from transfected HEK cells are nearly identical to those for α3β4 AChRs expressed in Xenopus oocytes and for IMR-32. Representative recordings of the single-channel currents for α3β4 AChRs expressed in permanently transfected HEK cells are shown along with representative event amplitude and dwell time distributions. Superimposed on the event histograms are fits to either a double Gaussian function for the amplitude distribution or a double exponential function for the open time distribution. The recordings were made in the outside-out patch configuration using ACh (5 μM) as agonist at -80 mV. Mean channel open times were determined as described in materials and methods. The single-channel amplitudes and open times were nearly identical to those for oocyte-expressed AChRs and for IMR-32 AChRs recorded under the same conditions (Nelson and Lindstrom 1999; see preceding page).
Subunit Content of AChRs Expressed by IMR-32 and SH-SY5Y Cells
To estimate the fraction of IMR-32 AChRs that contained a particular subunit, mAb-coated microwells were used. AChRs that contained α3 (and/or α5) subunits (recognized by mAb 210), β2 subunits (recognized by mAb 295), or β4 subunits (recognized by mAb 337), were isolated from detergent extracts. mAbs 295 or 337 were tested for their efficiencies in immunoisolating AChRs from the transfected cells. mAb 295 (to β2) was found to isolate 109 ± 16% as many AChRs as did mAb 210 (to α3) from the α3α5β2 cell line (Table ). mAb 337 (to β4) was found to isolate 64 ± 2% as many AChRs as did mAb 210 (to α3) from the α3β4 cell line (Table ). The relatively low efficiency of mAb 337 was confirmed by saturation of immune precipitation using α3β4 AChRs in extracts from Xenopus oocytes where it bound 61 ± 2% as many AChRs as mAb 210. Increasing mAb 337 concentrations to 100-fold greater than the point of saturation failed to precipitate additional AChRs. This indicated that about one-third of total α3β4 AChRs existed in which the epitope to mAb 337 on all of the AChR's β4 subunits is either in the wrong conformation, obscured by another protein, or is consistently proteolyzed.
Table 2.
Relative Amounts of AChRs Containing Particular Subunits from Solid Phase RIAs
| Relative AChR amounts containing targeted subunit | ||||
|---|---|---|---|---|
| Source of AChR | α3 | β4 | β2 | α7 |
| IMR-32 | 100 | 65 ± 5 | 18 ± 4 | 140 ± 22 |
| IMR-32 (after Nic) | 139 ± 22 | 71 ± 5 | 50 ± 13 | 90 ± 1 |
| SH-SY5Y | 100 | 40 ± 6 | 49 ± 2 | ND |
| SH-SY5Y (after Nic) | 179 ± 16 | 22 ± 1 | 116 ± 1 | ND |
| α3β4 | 100 | 64 ± 2 | — | — |
| α3α5β2 (after Nic) | 100 | — | 109 ± 16 | — |
For IMR-32 cell extracts, [3H]epibatidine binding revealed that 65 ± 5% (n = 5) of AChRs bound contained the β4 subunit when compared with those which contained the α3 subunit, whereas 18 ± 4% contained the β2 subunit (Fig. 10 and Table ). Additionally, the amount of AChR that could be labeled by [3H]epibatidine on intact IMR-32 cells (4.7 ± 0.8 fmol per 35-mm dish) was not significantly different from the amount of AChR that could immunoisolated from detergent extracts from IMR-32 cells (4.0 ± 0.1 fmol per 35-mm dish). Previously, we found that prolonged incubation of another neuroblastoma cell, SH-SY5Y, in Nic increased total α3 AChRs (Peng et al. 1997). Subsequently, we determined that Nic incubation increased the amount of AChR in α3β2-transfected cells, but had no effect on α3β4-transfected cells, and that in SH-SY5Y, α3β2, but not α3β4 AChRs were upregulated (Wang et al. 1998). With this in mind, we tested Nic incubation on IMR-32 cells. Overnight Nic (100 μM) treatment increased total α3-containing AChRs by ∼40% (Fig. 10 and Table ), most of which could be attributed to an increase in β2-containing AChRs. Similar to previous findings in SH-SY5Y cells, Nic incubation increased the amount of α3 AChRs by ∼80%, most of which could be attributed to an increase in β2-containing AChRs.
Figure 10.

Incubation in Nic increases the amount of α3β2 AChRs in IMR-32 and SH-SY5Y cells, but not α3β4 AChRs. The bar graph represents the relative amounts of [3H]epibatidine binding to AChRs immunoisolated on mAb-coated microwells from Triton-X100 extracts of either IMR-32 or SH-SY5Y cells in the presence and absence of Nic (100 μM) overnight. mAb 210 binds α3 and α5 AChRs, mAb 295 binds β2 AChRs, and mAb 337 binds β4 AChRs. For both cell lines, Nic caused an increase in binding to AChRs isolated on 210-coated wells which was the same in magnitude as the amount of increase in binding to AChRs isolated on mAb 295-coated wells. AChRs isolated on mAb 337-coated wells were not altered significantly by Nic. For IMR-32 (untreated), mAb 210 isolated AChRs bound ∼12 fmol of [3H]epibatidine, which corresponds to roughly 1,800 AChRs per cell when assuming two binding sites per AChR, whereas for SH-SY5Y, mAb 210 isolated AChRs bound ∼3 fmol of [3H]epibatidine corresponding to ∼600 AChRs per cell.
The effects of Nic incubation on surface AChRs in intact cells was determined also. Surface α3-containing AChRs were measured using 125I-mAb 210 and surface β2-containing AChRs were measured using 125I-mAb 295. In aliquots of IMR-32 cells that bound 10 ± 0.4 fmol of 125I-mAb 210 on their surface, negligible (0.1 ± 0.5 fmol) 125I-mAb 295 binding was detected. Incubation in Nic (100 μM overnight) did not change the amount of binding of either mAb (125I-mAb 210, 10 ± 2 fmol; 125I-mAb 295, 0.4 ± 0.5 fmol). For SH-SY5Y cells, Nic incubation caused no change in the amount of 125I-mAb 210 (4.5 ± 1.1 fmol in control and 4.5 ± 0.4 fmol after Nic incubation) or 125I-mAb 295 binding to the surface of intact SH-SY5Y cells (2.8 ± 0.9 fmol in control and 2.7 ± 0.8 fmol after Nic incubation). Nic incubation had no effect on the properties of currents recorded from IMR-32 cells (Fig. 5 C), which is consistent with the surface binding results. Thus, the increase in α3β2 AChRs found in detergent extracts must reflect an increase in intracellular AChRs.
Immunofluorescence and Confocal Microscope
Confocal microscopy was used to visualize the distribution of α3 AChRs in IMR-32 neuroblastoma cells and the α3β2 or the α3β4 transfected cells. For the transfected cells, intense, clustered labeling was observed with antibody directed against the α3 subunit. The labeling of α3 largely overlapped with the labeling observed with antibody directed against the β4 subunit in the α3β4 cells and the β2 subunit in the α3β2 cells (Fig. 11). In the case of IMR-32 cells, the labeling of α3 subunit was also extensive and appeared in clusters that overlapped with the labeling observed for antibody directed at the β4 subunit (Fig. 11). Since mAb 337 (to β4) targets a cytoplasmic epitope, all images are shown for permeabilized cells. The clustered appearance of the α3 label was also observed on nonpermeabilized cells, indicating that surface AChRs were expressed in concentrated densities (unpublished data). The α3β4 cells seem to have the largest size clusters, which probably reflects the fact that these cells express the greatest levels of AChR (Wang et al. 1998).
Figure 11.

Confocal microscopy of α3 AChRs expressed by IMR-32 neuroblastoma cells and HEK cells transfected with α3β4 and α3β2 AChRs. Cells were plated, fixed, permeabilized, and labeled as described in materials and methods. For each cell type, green reflects labeling of α3 subunits by Alexa 488-labeled mAb 210. For IMR-32 cells and α3β4 cells, red reflects secondary labeling by Alexa 568-labeled GAM antibody against mAb 337 which bound β4 subunits. For α3β2 cells, red reflects secondary labeling by Alexa 594-labeled GART antibody against mAb 295 which bound β2 subunits. Yellow is a result of overlap in the labeling of colocalized subunits. For all cells, ToPro-3 iodide (blue) was included as a nuclear counterstain. The scale bars in the α3β4 panel represent 1, 2, 5, and 10 μm, respectively.
Western Blot Analysis of Immunoisolated AChRs
AChRs from IMR-32 cells, SH-SY5Y cells, the α3α5β2 cell line, and the α3β4 cell line were subjected to immunoisolation followed by Western blotting to establish subunit associations. α3 or β2 proteins were bound with subunit specific rat antisera raised against the α3 subunit or the β2 subunit and then labeled with 125I-GART. β4 protein was labeled directly with 125I-mAb 337. AChRs isolated by mAb 210 from IMR-32 cells or from the α3α5β2 transfected cell line gave strong bands when labeled with α3 subunit antiserum (Fig. 12). AChRs isolated by mAb 295 from the α3α5β2 cell line revealed strong labeling on Western blots with α3 or β2 (unpublished data) subunit antisera. However, with IMR-32 cells, relatively weak labeling by α3 or β2 (not shown) subunit antisera was observed for AChRs isolated by mAb 295 when compared with α3 subunit isolated by mAb 210, which indicated low levels of α3 subunit associated with β2 subunit. AChRs isolated by mAb 337 resin exhibited strong labeling from both the α3β4 cell line and from IMR-32 cells relative to α3 subunit isolated by mAb 210, which was consistent with most α3 subunits forming AChRs with β4 subunits (Fig. 12). 125I-mAb 337 labeling of AChR isolated with mAb 210 resin gave stronger signals than for AChR isolated from the same extract by mAb 337 resin, which reflects the difference in mAb efficiencies. These results confirm that IMR-32 cells express AChRs consisting largely of α3 and β4 subunits and that only very low levels of the β2 subunit are associated with the α3 subunit. Previous attempts to immune isolate AChRs from IMR-32 extracts with antisera to α4 or α6 subunits or mAb 299 (for α4) failed, reflecting their absence (Nelson and Lindstrom 1999).
Figure 12.
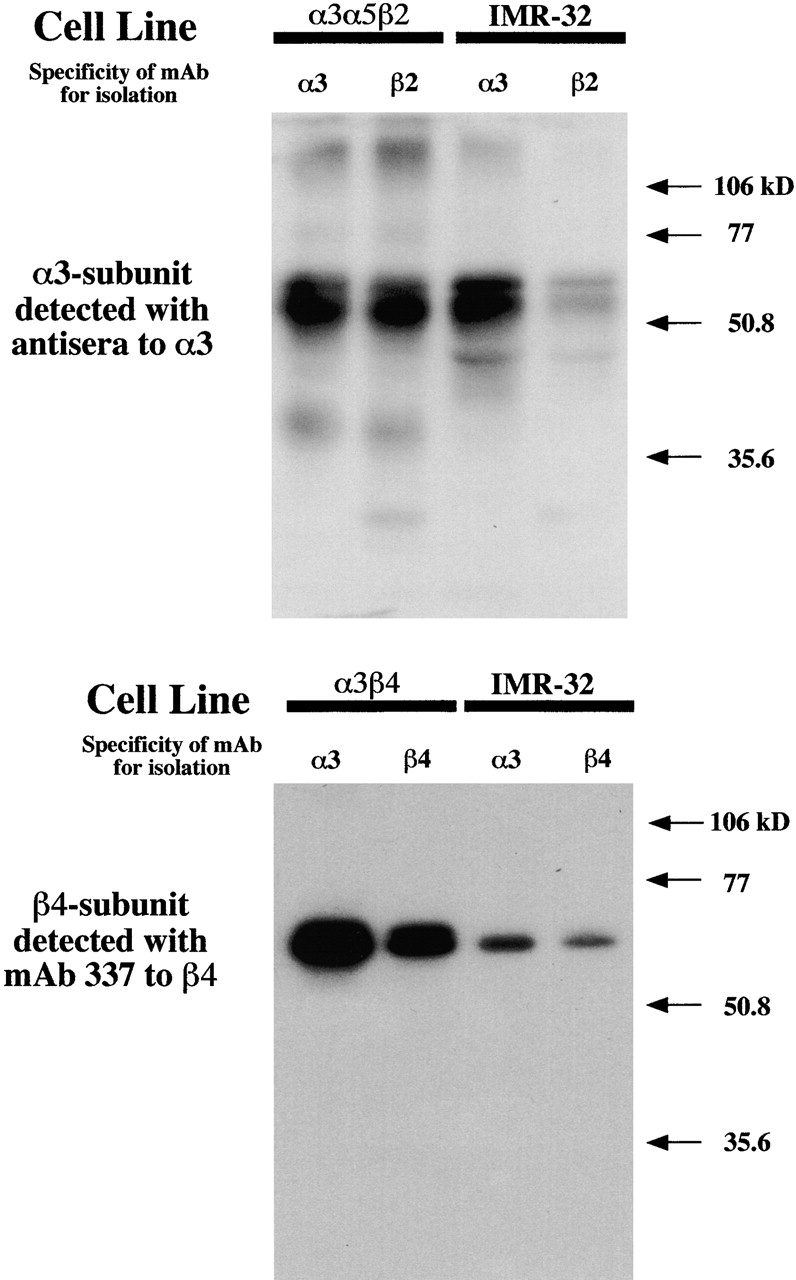
Western blots of neuroblastoma AChRs compared with AChRs expressed in permanently transfected HEK cells. AChRs in Triton X-100 extracts were immunoisolated on mAb-coated resin. α3-containing AChRs were isolated by mAb 210. β2-containing AChRs were isolated by mAb 295. β4-containing AChRs were isolated by mAb 337. Proteins were separated by electrophoresis in SDS on a 12% polyacrylamide gel. α3 subunit protein was bound with rat antisera raised against the α3 subunit and then labeled with 125I-GART. β4 subunit protein was labeled directly with 125I-mAb 337.
Presence of a-BGT Binding AChRs in IMR-32 Cells That Do Not Contain α3, α5, or β2 Subunits
IMR-32 cells have 2.6 ± 0.6 (n = 4) times more total α-BGT binding sites than epibatidine binding sites as measured by RIAs of detergent extracts. In a typical experiment, 34 ± 0.6 fmol of 125I-αBGT binding sites of α7-containing AChRs were immunoisolated on microwells coated with mAbs 306 and mAb 319, whereas 14 ± 0.6 fmol of [3H]epibatidine binding sites of α3-containing AChRs were immunoisolated on mAb 210-coated microwells from equal volumes of the same extract. Furthermore, on the cell surface, there are 1.7 ± 0.2 (n = 3) times more binding sites for 125I-α-BGT when compared with 125I-mAb 210. Thus, it was unclear why the functional properties failed to reflect the presence of α7 containing AChRs. However, if the ratio of α-BGT binding sites per AChR is 5:1 and the ratio of mAb 210 binding sites per AChR is 2:1, then there would be similar amounts of α7 and α3 AChRs on the surface. For an AChR with the rapid kinetics of a homomeric α7 AChR, this amount of AChR might not generate sufficient current for detection (see discussion).
It is possible that IMR-32 cells express heteromeric α7-containing AChRs with slower kinetics that are insensitive to inhibition by MLA. To detect such an AChR we used microwells coated with either mAb 210 (to α3 or α5 subunits) or mAb 295 (to β2 subunits) and tested for α-BGT binding. For AChRs isolated with either mAb, no 125I-α-BGT binding was detected (Fig. 13), but wells coated with α7-selective mAbs 306 and 319 bound 40 ± 4 fmol of 125I-α-BGT from these same extracts. We conclude that in IMR-32 cells, no α-BGT binding AChRs are formed by the coassembly of α3, α5, or β2 subunits with the α7 AChR subunit.
Figure 13.

Only IMR-32 AChRs immunoisolated with mAbs to α7 AChR subunit bind α-BGT. The bar graph depicts the relative amounts of 125I-α-BGT binding that occurred on mAb-coated microwells after isolation of IMR-32 AChRs solubilized in Triton X-100. Microwells coated with a combination of mAbs 306 and 319 which both recognize α7 subunits bound 35-45 fmol of 125I-α-BGT, whereas microwells coated with mAbs 210 (which recognizes α3 or α5 subunits) or 295 (which recognizes β2 subunits) exhibited virtually no binding above background for the same AChR extracts. Extracts from IMR-32 cells that had been treated overnight with Nic (100 μM) bound slightly less 125I-α-BGT than cells that were not treated with Nic when bound on wells coated with mAbs 306/319.
We also tested the effect of overnight Nic (100 μM) treatment and nerve growth factor treatment (0.1 μg/ml for 2 d) on the amount of α7 AChR expressed by IMR-32 cells. Nic treatment reduced slightly the total amount of α7 AChR in IMR-32 cells, whereas NGF had no effect (Fig. 13).
DISCUSSION
IMR-32 cells were derived from an abdominal tumor (Tumilowicz et al. 1970) and express nicotinic AChR subunits that are typical of ganglionic neurons: α3, α5, α7, β2, and β4 (Gotti et al. 1997). Although several AChR subtypes could be formed from these subunits, our data show that the functional properties of IMR-32 AChRs resemble those of α3β4 AChRs expressed in transfected HEK cells. No evidence was found for α3β2 or α7 AChR-mediated currents from IMR-32 cells. α3β2 AChRs represented a small percentage of total IMR-32 α3-AChRs and were absent from the cell surface. However, the amount of surface α7 AChRs appeared to be similar to the amount of surface α3 AChRs. The absence of function for α3β2 or α7 AChRs could result from differential downregulation of these AChRs in the present developmental state of these cells. Incubation in Nic or culturing at reduced temperature increased the amount of α3β2 AChRs in IMR-32 cells, but not on their surface, in spite of the dramatic increase in surface expression observed for α3β2 AChRs expressed in HEK cells after these treatments.
Transgenic deletions in mice of the α3, β2, and β4 subunits provided evidence that each of the subunits can contribute to AChRs that mediate autonomic transmission (Xu et al. 1999a,Xu et al. 1999b). However, the overlap in expression of β subunits and possible compensatory upregulation of other subunits in knockout animals prevents isolation of the specialized roles that particular α3 AChR subtypes might play. Also, the morbidity induced by α3 deletion shows the inability of α7 AChRs alone to sustain adequate autonomic synaptic function and provides indirect evidence for distinct roles played by these AChRs. A better understanding of functional differences among AChR subtypes is key to clarifying their roles played in the physiology of the autonomic nervous system.
Comparisons between the Functional and Pharmacological Properties of α3 AChRs Expressed by Permanently Transfected HEK Cells and IMR-32 Cells
The rank order of agonist potencies on IMR-32 cells (DMPP > Nic ≥ Cyt = ACh) was similar to reports for ganglionic preparations from both rat and chicken (Sargent 1993; McGehee and Role 1995). All of the agonists on IMR-32 cells had nearly full efficacies (80%). Currents from IMR-32 cells exhibited strong inward rectification and a relatively slow rate of desensitization. They were insensitive to α7-selective concentrations of methyllycaconitine, the potent reversible competitive antagonist of α7-containing AChRs.
In transfected cells, the rank order of potencies for α3β4 AChRs was DMPP = Cyt > Nic > ACh, which was similar to the rank order for IMR-32 AChRs. Moreover, with the exception of Cyt, the EC50 values found for agonists on α3β4 AChRs were nearly identical to those found for IMR-32 AChRs (Table ). Although the rank order of potencies for α3α5β4 cells was also similar to IMR-32 cells, both of these cell lines express few α5-containing AChRs (Wang et al. 1998; Nelson and Lindstrom 1999). Cyt clearly differentiates between β2- and β4-containing AChRs (Papke and Heinemann 1994) with ∼70% efficacy on both α3β4 cells and IMR-32 cells, but 5% or less on α3β2 or α3α5β2 transfected cells. Desensitization kinetics were similar between α3β4 AChRs and IMR-32 AChRs, both of which were much slower than the decay kinetics of α3β2 AChRs. Finally, the single-channel open times and channel amplitudes for the HEK-expressed α3β4 AChRs were nearly identical to those recorded from oocytes and very similar to those recorded from IMR-32 cells (Nelson and Lindstrom 1999).
These functional data failed to eliminate the possibility that IMR-32 AChRs consist of α3 and β4 subunits coassembled with β2 and/or α5 subunits since all possible subunit combinations were not tested in permanently transfected cell lines. It is conceivable, for example, that coassembly with β4 might mask the presence of β2 subunits in IMR-32 AChRs. However, the present and past molecular evidence help to dispel this concern. Here, we have shown that mAb 295 (specific for the β2 subunit) isolated <20% as many AChRs as mAb 210 (specific for α3 AChRs) from IMR-32 cell detergent extracts and it labeled no AChRs on the cell surface (see last paragraph of Subunit Content of AChRs… and next section). Previously, we showed that the amount of α5-containing AChRs in IMR-32 cells is 5% when compared with the amount of α3-containing AChRs (Nelson and Lindstrom 1999). These data taken together support the conclusion from our functional studies that the main AChR expressed by IMR-32 cells is composed of α3 and β4 subunits. Furthermore, the similarities in functional properties between IMR-32 AChRs and α3β4 AChRs expressed in HEK cells illustrate two important points: (1) the population of functional IMR-32 AChRs is homogenous, and (2) the expression environment does not appear to modify the functional properties of α3β4 AChRs.
Effects of Chronic Nic Exposure on AChRs of IMR-32 and SH-SY5Y
Previous studies demonstrated that Nic (micromolar range) incubation increased total α3 AChRs while the number of surface AChRs was unchanged in SH-SY5Y cells (Peng et al. 1997) or increased in HEK cells expressing α3β2 AChRs (Wang et al. 1998). Nic upregulation appears to be selective for human α3β2 AChRs since no effect was seen on human α3β4 AChRs (Wang et al. 1998). Nic concentrations required to upregulate α3β2 AChRs are substantially greater than the concentrations that are typically achieved in serum of cigarette smokers (Benowitz et al. 1990). Upregulation of α3β2 AChRs by high concentrations of Nic reveals a mode of modulation that could be common among AChRs that contain β2 subunits. The upregulation has been attributed to promoting AChR assembly coupled with reduced AChR turnover (Peng et al. 1997; Wang et al. 1998). Competitive antagonists did not block upregulation of α3β2 AChRs in HEK cells, but they caused low levels of upregulation in the absence of Nic (Wang et al. 1998). Mecamylamine, a noncompetitive antagonist, blocked upregulation and had no effect on amount of AChR by itself. Clearly, a drug-induced change in AChR conformation must occur, but the actual signaling pathway remains unclear. Nic increased α3-containing AChRs in IMR-32 cells to a lesser extent than those in SY-SY5Y cells. The difference in the extent of upregulation is consistent with the relative abundance of the β2 subunit in each of the neuroblastoma cells. In fact, β2-containing AChRs accounted for the majority of the AChR increase caused by Nic, whereas β4-containing AChRs were unaffected in both IMR-32 and SH-SY5Y cells.
No significant surface β2 AChRs were detected by antibody in IMR-32 cells, with, or without, Nic treatment. Also, Nic caused no change in the amount of antibody labeling of surface α3 AChRs. For SH-SY5Y, incubation in Nic caused no significant change in the number of α3- or β2-containing surface AChRs. Because the total number of AChRs expressed by these cells increased, whereas the number of surface AChRs was unchanged after incubation in Nic, AChRs must accumulate intracellularly as suggested previously (Peng et al. 1997).
Why might α3β2 and α3β4 AChRs within ganglionic neurons have distinct posttranslational regulatory mechanisms? IMR-32 and SH-SY5Y cells represent noninnervated human ganglionic neurons in a suspended state of maturity. The fact that they express different proportions of α3β2 and α3β4 AChRs and that α3β2 levels can be selectively increased by exposure to Nic or by reducing culture temperature shows that mechanisms exist that specifically regulate expression of α3β2 AChRs in ganglia at certain times of development, stress, or activity. It is interesting that our α3β2 cell line increases surface AChRs by these treatments, whereas the neuroblastoma cell lines do not. This could mean that other factors such as chaperone proteins or cytoskeletal-associated proteins (analogous to rapsyn for muscle AChRs [Bloch and Froehner 1987], gephyrin for glycine receptors [Betz et al. 1991], or GABA receptor–associated protein [Wang et al. 1999]) regulate surface expression, and that the presence of these components differs between HEK and the neuroblastoma cells. From the knockout mice studies, it seems that ganglionic AChRs composed of either β2 or β4 subunits are at least minimally sufficient to maintain autonomic function. Although the simplest conclusion would be that both of these AChRs mediate synaptic transmission, it is more likely a reflection of the limitations of nonconditional knockouts where compensatory substitution of one subunit for the missing subunit occurs. The significance of the particular β subunit that is expressed could play a role in subcellular signaling that controls synaptic establishment, maintenance, or control of expression of yet another target protein. Specific regulation of α3β2 AChR expression could be used to achieve these different goals. Comparisons between α3β2 and α3β4 cell lines reveal clear functional differences between these AChRs. α3β2 AChRs are less sensitive to activation and desensitize much more quickly than α3β4 AChRs. Additionally, α3 AChRs containing the β2 subunit have higher Ca2+ permeability than α3 AChRs containing the β4 subunit (Gerzanich et al. 1998). It is possible that regulation of β subunit expression is analogous to the developmental regulation of γ and ɛ subunits in fetal and adult muscle AChR, respectively. AChRs having the γ subunit exhibit smaller conductance, longer gating periods, and are not confined to the neuromuscular junction when compared with ɛ-containing AChRs (Hall and Sanes 1993). This difference in functional properties reflects differing needs of the muscle fiber at different stages of development. Although the conductances of the α3β2 and α3β4 AChRs are not significantly different, the gating kinetics differ significantly with the β4-containing AChRs exhibiting much longer open and burst times than α3β2 AChRs (Nelson and Lindstrom 1999). Their unique gating properties, along with their differential regulation, allow these α3 AChR subtypes to play specialized roles in ganglionic neurons.
Both neuroblastoma cell lines produce low amounts of α5-containing AChRs. For IMR-32 cells, ∼5% of α3 AChRs contained the α5 subunit. Such low levels would be insufficient to alter macroscopic currents. However, the same reasoning that was applied to regulated expression of β2-containing AChRs in ganglia could also be applied to α5-containing AChRs. Considering the impact of the α5 subunit on α3-AChR gating, conductance, and permeability (Gerzanich et al. 1998; Nelson and Lindstrom 1999), this would give ganglionic neurons yet another way to alter nicotinic activity to address the needs of the cell.
Expression of α7 AChRs in IMR-32 Cells with No Detectable Function
α-BGT–sensitive currents have been reported in IMR-32 cells previously (Gotti et al. 1995). However, because of current rundown and the relative irreversibility of α-BGT blockade, these data are difficult to interpret. In the present study, no IMR-32 currents had rapid kinetics like those of an α7-type response (Alkondon et al. 1992; Gopalakrishnan et al. 1995). We have recorded rapidly desensitizing, Nic-activated currents from rat hippocampal neurons that have been attributed to α7 AChRs to validate the efficiency of our application system (unpublished data). Additionally, we failed to inhibit Nic-evoked currents in IMR-32 cells with the reversible selective α7 AChR antagonist, MLA. Thus, the presence of α-BGT binding sites measured on the cell surface in IMR-32 cells was perplexing. However, if α7 AChRs have an α-BGT binding site for each subunit of a homopentameric AChR, there would be similar numbers of α7 AChRs when compared with α3 AChRs on the cell surface. Considering that the channel closing rate for homomeric α7 AChRs is at least 10,000 s−1 ([Anand et al. 1998] compared with ∼150 s−1 for α3β4 AChRs [Fig. 9]) and its desensitization rate is at least 3000 s−1 (unpublished data; compared with ∼2 s−1 for α3β4 AChRs [Fig. 8]) this number of α7 AChRs might fail to generate measurable current. Consistent with this reasoning, we estimate that there are sevenfold fewer α-BGT binding sites in IMR-32 cells than have been reported for transfected HEK cells from which human α7 AChRs have been characterized electrophysiologically (Gopalakrishnan et al. 1995). However, we cannot eliminate the possibility that the α7 AChRs expressed by IMR-32 cells are, indeed, nonfunctional. To test for the presence of nonhomomeric α7 AChRs with unexpected functional properties, we immunoisolated AChRs that contained either α3, α5, or β2 subunits, and then attempted to label them with 125I-α-BGT. No labeling was observed on these AChRs, but α7-selective mAbs 306 and 319 bound 40 ± 4 fmol of 125I-α-BGT from these same extracts confirming the presence of an α7 AChR with high affinity for toxin.
Conclusion
The physiological significance of the potential for such functional diversity among AChRs within individual neurons remains unclear. α7 AChRs are highly permeable to Ca2+, which makes them good candidates for triggering Ca2+-dependent events to alter expression of proteins involved in morphological or functional changes. The same may also be true of α3β2 AChRs as it has been shown previously that α3β2 AChRs have higher Ca2+ permeability than α3β4 AChRs. In fact, the more Ca2+-permeable AChRs might mediate development and plasticity, whereas α3β4 AChRs mediate excitatory synaptic transmission. In any case, it is possible that relative amounts of AChR subtypes expressed might vary in neurons from one ganglion to the next. Our data indicate that IMR-32 cells express α3β4 AChRs as the predominant functional AChRs. What remains to be elucidated is what mechanisms are used by ganglionic neurons to alter the proportions of AChR subtypes in response to changing cellular needs. Both incubation in Nic and temperature reduction provide useful models through which to study at least part of these regulatory mechanisms.
Acknowledgments
The authors thank John Cooper and Ben McNeil for their technical assistance.
This work supported by grants to J. Lindstrom from the National Institutes of Health (NS11323) and the Smokeless Tobacco Research Council, Inc., USA, Inc.
Footnotes
The current address of F. Wang is DuPont Central Research and Development, Experimental Station, Wilmington, DE 19880. The current address of V. Gerzanich is Department of Neurosurgery, University of Maryland School of Medicine, Baltimore, MD 21201.
Abbreviations used in this paper: AChR, acetylcholine receptor; Cyt, cytisine; HEK, human embryonic kidney; MLA, methyllycaconitine; Nic, nicotine; RIA, radioimmune assay.
References
- Alkondon M., Albuquerque E.X. Presence of alpha-bungarotoxin-sensitive nicotinic acetylcholine receptors in rat olfactory bulb neurons. Neurosci. Lett. 1994;176:152–156. doi: 10.1016/0304-3940(94)90070-1. [DOI] [PubMed] [Google Scholar]
- Alkondon M., Pereira E.F., Wonnacott S., Albuquerque E.X. Blockade of nicotinic currents in hippocampal neurons defines methyllycaconitine as a potent and specific receptor antagonist. Mol. Pharmacol. 1992;41:802–808. [PubMed] [Google Scholar]
- Anand R., Nelson M.E., Gerzanich V., Wells G.B., Lindstrom J. Determinants of channel gating located in the N-terminal extracellular domain of nicotinic α7 receptor. J. Pharmacol. Exp. Therap. 1998;287:469–479. [PubMed] [Google Scholar]
- Benowitz N., Porchet H., Jacob P. Pharmocokinetics, metabolism, and pharmacodynamics of nicotine. In: Wonnacott S., Russell M., Stolerman I., editors. Nicotine Psychophamacology. Oxford Science Publications; Oxford, England: 1990. pp. 112–157. [Google Scholar]
- Betz H., Kuhse J., Schmieden V., Malosio M.L., Langosch D., Prior P., Schmitt B., Kirsch J. How to build a glycinergic postsynaptic membrane. J. Cell Sci. Suppl. 1991;15:23–25. doi: 10.1242/jcs.1991.supplement_15.4. [DOI] [PubMed] [Google Scholar]
- Bloch R.J., Froehner S.C. The relationship of the postsynaptic 43K protein to acetylcholine receptors in receptor clusters isolated from cultured rat myotubes. J. Cell Biol. 1987;104:645–654. doi: 10.1083/jcb.104.3.645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper S.T., Harkness P.C., Baker E.R., Millar N.S. Up-regulation of cell-surface α4β2 neuronal nicotinic receptors by lower temperature and expression of chimeric subunits. J. Biol.Chem. 1999;274:27145–271452. doi: 10.1074/jbc.274.38.27145. [DOI] [PubMed] [Google Scholar]
- Covernton P.J., Kojima H., Sivilotti L.G., Gibb A.J., Colquhoun D. Comparison of neuronal nicotinic receptors in rat sympathetic neurones with subunit pairs expressed in Xenopus oocytes. J. Physiol. 1994;481:27–34. doi: 10.1113/jphysiol.1994.sp020416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschini D., Orr-Urtreger A., Yu W., Mackey L.Y., Bond R.A., Armstrong D., Patrick J.W., Beaudet A.L., De Biasi M. Altered baroreflex responses in α7 deficient mice. Behav Brain Res. 2000;113:3–10. doi: 10.1016/s0166-4328(00)00195-9. [DOI] [PubMed] [Google Scholar]
- Gerzanich V., Wang F., Kuryatov A., Lindstrom J. α5 subunit alters desensitization, pharmacology, Ca2+ permeability, and Ca2+ modulation of human neuronal α3 nicotinic receptors. J. Pharmacol. Exp. Therap. 1998;286:311–320. [PubMed] [Google Scholar]
- Gopalakrishnan M., Buisson B., Touma E., Giordano T., Campbell J.E., Hu I.C., Donnelly-Roberts D., Arneric S.P., Bertrand D., Sullivan J.P. Stable expression and pharmacological properties of the human α7 nicotinic acetylcholine receptor. Eur. J. Pharmacol. 1995;290:237–246. doi: 10.1016/0922-4106(95)00083-6. [DOI] [PubMed] [Google Scholar]
- Gotti C., Briscini L., Verderio C., Oortgiesen M., Balestra B., Clementi F. Native nicotinic acetylcholine receptors in human imr32 neuroblastoma cellsfunctional, immunological and pharmacological properties. Eur. J. Neurosci. 1995;7:2083–2092. doi: 10.1111/j.1460-9568.1995.tb00630.x. [DOI] [PubMed] [Google Scholar]
- Gotti C., Fornasari D., Clementi F. Human neuronal nicotinc receptors. Prog. Neurobiol. 1997;53:199–237. doi: 10.1016/s0301-0082(97)00034-8. [DOI] [PubMed] [Google Scholar]
- Haghighi A.P., Cooper E. Neuronal nicotinic acetylcholine receptors are blocked by intracellular spermine in a voltage-dependent manner. J. Neurosci. 1998;18:4050–4062. doi: 10.1523/JNEUROSCI.18-11-04050.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haghighi A.P., Cooper E. A molecular link between inward rectification and calcium permeability of neuronal nicotinic acetylcholine α3β4 and α4β2 receptors. J. Neurosci. 2000;20:529–541. doi: 10.1523/JNEUROSCI.20-02-00529.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall Z.W., Sanes J.R. Synaptic structure and developmentthe neuromuscular junction Cell. 72Suppl1993. 99 121 [DOI] [PubMed] [Google Scholar]
- Hamill O.P., Marty A., Neher E., Sakmann B., Sigworth F.J. Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Kuryatov A., Olale F.A., Choi C., Lindstrom J. Acetylcholine receptor extracellular domain determines sensitivity to nicotine-induced inactivation. Eur. J. Pharmacol. 2000;393:11–21. doi: 10.1016/s0014-2999(00)00027-3. [DOI] [PubMed] [Google Scholar]
- Lane R.D., Crissman R.S., Ginn S. High efficiency fusion procedure for producing monoclonal antibodies against weak immunogens. Methods Enzymol. 1986;121:183–192. doi: 10.1016/0076-6879(86)21017-4. [DOI] [PubMed] [Google Scholar]
- Mandelzys A., Pie B., Deneris E.S., Cooper E. The developmental increase in ACh current densities on rat sympathetic neurons correlates with changes in nicotinic ACh receptor α-subunit gene expression and occurs independent of innervation. J. Neurosci. 1994;14:2357–2364. doi: 10.1523/JNEUROSCI.14-04-02357.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGehee D.S., Role L.W. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu. Rev. Phys. 1995;57:521–546. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- Nelson M.E., Lindstrom J. Single channel properties of human α3 AChRsimpact of β2, β4 and α5 subunits. J. Physiol. 1999;516:657–678. doi: 10.1111/j.1469-7793.1999.0657u.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A., Goldner F.M., Saeki M., Lorenzo I., Goldberg L., De Biasi M., Dani J.A., Patrick J.W., Beaudet A.L. Mice deficient in the α7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J. Neurosci. 1997;17:9165–9171. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palma E., Bertrand S., Binzoni T., Bertrand D. Neuronal nicotinic α7 receptor expressed in Xenopus oocytes presents five putative binding sites for methyllycaconitine. J. Physiol. 1996;491:151–161. doi: 10.1113/jphysiol.1996.sp021203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papke R.L., Heinemann S.F. Partial agonist properties of cytisine on neuronal nicotinic receptors containing the β2 subunit. Mol. Pharmacol. 1994;45:142–149. [PubMed] [Google Scholar]
- Peng X., Katz M., Gerzanich V., Anand R., Lindstrom J. Human α7 acetylcholine receptorcloning of the α7 subunit from the SH-SY5Y cell line and determination of pharmacological properties of native receptors and functional α7 homomers expressed in Xenopus oocytes. Mol. Pharmacol. 1994;45:546–554. [PubMed] [Google Scholar]
- Peng X., Gerzanich V., Anand R., Wang F., Lindstrom J. Chronic nicotine treatment up-regulates α3 and α7 acetylcholine receptor subtypes expressed by the human neuroblastoma cell line SH-SY5Y. Mol. Pharmacol. 1997;51:776–784. doi: 10.1124/mol.51.5.776. [DOI] [PubMed] [Google Scholar]
- Ross R.A., Spengler B.A., Biedler J.L. Coordinate morphological and biochemical interconversion of human neuroblastoma cells. J. Natl. Cancer Inst. 1983;71:741–747. [PubMed] [Google Scholar]
- Rust G., Burgunder J.M., Lauterburg T.E., Cachelin A.B. Expression of neuronal nicotinic acetylcholine receptor subunit genes in the rat autonomic nervous system. Eur J. Neurosci. 1994;6:478–485. doi: 10.1111/j.1460-9568.1994.tb00290.x. [DOI] [PubMed] [Google Scholar]
- Sargent P.B. The diversity of neuronal nicotinic acetylcholine receptors. Ann. Rev. Neurosci. 1993;16:403–443. doi: 10.1146/annurev.ne.16.030193.002155. [DOI] [PubMed] [Google Scholar]
- Tumilowicz J.J., Nichols W.W., Cholon J.J., Greene A.E. Definition of a continuous human cell line derived from neuroblastoma. Cancer Res. 1970;30:2110–2118. [PubMed] [Google Scholar]
- Ullian E.M., McIntosh J.M., Sargent P.B. Rapid synaptic transmission in the avian ciliary ganglion is mediated by two distinct classes of nicotinic receptors. J. Neurosci. 1997;17:7210–7219. doi: 10.1523/JNEUROSCI.17-19-07210.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang F., Gerzanich V., Wells G.B., Anand R., Peng X., Keyser K., Lindstom J. Assembly of the human neuronal nicotinic receptor α3 subunit with β2, β4, and α5 subunits. J. Biol. Chem. 1996;271:17656–17665. doi: 10.1074/jbc.271.30.17656. [DOI] [PubMed] [Google Scholar]
- Wang F., Nelson M.E., Kuryatov A., Olale F., Cooper J., Keyser K., Lindstrom J. Chronic nicotine treatment upregulates human α3β2, but not α3β4 AChRs stably transfected in human embryonic kidney cells. J. Biol. Chem. 1998;273:28721–28732. doi: 10.1074/jbc.273.44.28721. [DOI] [PubMed] [Google Scholar]
- Wang H., Bedford F.K., Brandon N.J., Moss S.J., Olsen R.W. GABA(A)-receptor-associated protein links GABA(A) receptors and the cytoskeleton. Nature. 1999;397:69–72. doi: 10.1038/16264. [DOI] [PubMed] [Google Scholar]
- Whiting P.J., Lindstrom J.M. Characterization of bovine and human neuronal nicotinic acetylcholine receptors using monoclonal antibodies. J. Neurosci. 1988;8:3395–3404. doi: 10.1523/JNEUROSCI.08-09-03395.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Gelber S., Orr-Urtreger A., Armstrong D., Lewis R.A., Ou C.N., Patrick J., Role L., De Biasi M., Beaudet A.L. Megacystis, mydriasis, and ion channel defect in mice lacking the α3 neuronal nicotinic acetylcholine receptor Proc. Natl. Acad. Sci. USA. 96 1999. 5746 5751a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W., Orr-Urtreger A., Nigro F., Gelber S., Sutcliffe C.B., Armstrong D., Patrick J.W., Role L.W., Beaudet A.L., De Biasi M. Multiorgan autonomic dysfunction in mice lacking the β2 and the β4 subunits of neuronal nicotinic acetylcholine receptors J. Neurosci. 19 1999. 9298 9305b [DOI] [PMC free article] [PubMed] [Google Scholar]