

Abstract
Mutations in the dystrophin gene causing Duchenne’s muscular dystrophy (DMD), lead to pre-mature stop codons. In mdx mice, a model for DMD, they can be suppressed by aminoglycosides such as gentamicin. Dystrophin is likely to play a role in flow (shear stress) mediated endothelium-dependent dilation (FMD) in arteries. Thus we investigated the effect of gentamicin on vascular, structure and function in mdx mice.
Mice carotid and mesenteric resistance arteries (450 and 85 μm diameter, respectively) were mounted in vitro in arteriographs allowing continuous diameter measurements. In mdx mice, NO-dependent FMD and endothelial NO-synthase expression were lower than in control mice. In mdx mice treated with gentamicin, dystrophin was recovered in vascular cells, FMD and NO-synthase expression were identical to control in mdx mice treated with gentamicin. Smooth muscle-dependent contractions as well as dilation to acetylcholine (endothelium-dependent) and sodium nitroprusside (endothelium-independent) were not affected by the absence of dystrophin and/or by gentamicin. FMD, attenuated in vimentin-null mice, was not restored by gentamicin.
These findings open important perspectives in the mechanism involved in the pathophysiology of genetic diseases related to pre-mature stop codons such as DMD.
Keywords: Animals, Carotid Arteries, drug effects, physiopathology, ultrastructure, Codon, Nonsense, Coronary Vessels, pathology, Dystrophin, deficiency, physiology, Endothelium, Vascular, drug effects, physiopathology, Gene Expression Regulation, drug effects, Gentamicins, pharmacology, therapeutic use, Hemorheology, Male, Mesenteric Arteries, drug effects, physiopathology, ultrastructure, Mice, Mice, Inbred C57BL, Mice, Inbred mdx, Mice, Knockout, Muscle, Skeletal, blood supply, NG-Nitroarginine Methyl Ester, pharmacology, Nitric Oxide, physiology, Nitric Oxide Synthase, biosynthesis, genetics, Nitric Oxide Synthase Type II, Nitric Oxide Synthase Type III, Signal Transduction, Stress, Mechanical, Vasoconstriction, drug effects, Vasoconstrictor Agents, pharmacology, Vimentin, deficiency, genetics
Keywords: Duchenne’s muscular dystrophy, flow-mediated dilation, vasodilation
Introduction
Flow (shear stress) is the main physiological stimulus inducing the release of vasoactive agents by vascular endothelial cells (1, 2). Flow-mediated dilation (FMD) allows the adaptation of feeding arteries to the metabolic needs of each organ (1, 2). Mechanotransduction of shear stress involves the extracellular matrix and cell structure proteins (2). Depolymerization of F-actin into G-actin is rapid upon shear stress stimulation (3–5) and the absence of the intermediate filament vimentin markedly lowers FMD (6). Dystrophin is involved in skeletal and cardiac muscle cells mechanotransduction (7–11). Although dystrophin is present in vascular smooth muscle cells (12–14), no obvious functional abnormality of the smooth muscle has been found in mice lacking dystrophin (mdx mice) (14). In both mice and human endothelial cells the presence of dystrophin has been recently shown (14). Endothelium-dependent dilation, due to mechanical stimulation by flow (shear stress), is markedly and selectively attenuated in mdx mice, suggesting a role for dystrophin in the endothelial mechanotransduction of shear stress (14). This specific vascular dysfunction might disturb local blood flow supply to end-organs and would now required further investigation in patients with Duchenne’s muscular dystrophy (DMD). Indeed, ischemia has been described in skeletal and cardiac muscles in patients suffering dystrophy (15–17). As shear stress generated by flow is the main physiological stimulus triggering endothelium-dependent dilation, a defect in flow-mechanotransduction might have serious consequences in the short-term (acute local blood flow control) and in the long-term (arteries structure, protein expression). As flow regulates the expression of endothelial NO-synthase (eNOS), we measured the level of this protein in arteries isolated from mdx mice. In addition, eNOS expression and NO production have a key role in the structural adaptation of the vessel wall in response to changes in hemodynamic environment (18).
A chronic treatment of mdx mice with the aminoglycoside gentamicin can suppress stop codons in the gene encoding for dystrophin, thus inducing the recovery of the protein in skeletal muscles (19). We tested the hypothesis that vascular mechanotransdution of flow could be improved in mdx mice chronically treated with gentamicin. Indeed, the recovery of dystrophin should restore the capacity of the endothelium to transduce shear stress into dilation, thus supporting the hypothesis that the dystrophin has a key role in shear stress mechanotransduction in vascular endothelium. This study could also open new perspectives in the pathophysiology of genetic diseases such as DMD.
METHODS
Animals
Twelve-week old male mdx mice and their control (C57-B110; Iffa-Credo, L’Arbresle, France) were treated with gentamicin (Sigma, St-Louis) using a dose inducing the recovery of dystrophin in mdx mice (19) (34mg/kg per day, 14 days, Alzet minipumps implanted subcutaneously). Mice were then anesthetized for blood pressure measurement through a catheter placed in the left carotid artery (14). In another series of experiments twelve-week old male vimentin-null mice were treated with gentamicin (34mg/kg per day, 14 days) in order to test the selectivity of the treatment. Indeed, we have previously reported that FMD is markedly attenuated in vimentin-null mice (6). This decrease in FMD is quantitatively equivalent to that observed in mdx mice (14).
Isolated arteries
After anesthesia with pentobarbital the right carotid artery and mesenteric resistance arteries were isolated and cannulated at both ends in a video monitored perfusion system (20) (LSI, Burlington, VT) as previously described (6, 14). Briefly, cannulated arteries were bathed in a physiological salt solution. Diameter changes were measured when intraluminal pressure was increased step by step from 10 to 150 mmHg. Pressure we then set at 75 mmHg and flow increased by steps. At the end of each experiment arteries were bathed in a Ca2+-free physiological salt solution containing EGTA (2 mmol/L) plus sodium nitroprusside (10 μM) and pressure steps were repeated in order to determine the arteries passive diameter (6, 14). Contraction to phenylephrine, KCl (80 mmol/L) and dilation to acetylcholine and sodium nitroprusside were tested in another arterial segments from the same mouse and mounted in an arteriograph under an intraluminal pressure of 75 mmHg (6, 14). Flow-mediated and acetylcholine-induced dilations were repeated after NO synthesis blockade with L-NAME (10 μmol/L), as previously described (6, 14).
Immunolocalization of dystrophin in isolated arteries
Immunostaining of dystrophin was performed, as previously described (14). Briefly, arteries were mounted in embedding medium and frozen. Immunostaining was then performed on transverse cross section (5 μm thin) with anti-dystrophin antibodies (anti dys2, 1:200, Novacastra). Secondary antibodies were anti-rabbit antibodies conjugated to peroxidase (Amersham) (14) or anti-rabbit antibodies conjugated to biotin, which was then amplified by streptavidin-Texas-red reagent (Amersham).
In another group of experiments immunostaining of dystrophin was performed in isolated mesenteric arteries cannulated in arteriographs (pressure = 75 mm Hg, flow = 50 μl/min). Cell membranes were permeabilized with β-escin (90 mg/ml, 10 min). Anti-dystrophin antibodies (dys2) were then perfused for 30 min. A secondary antibody, bound to streptavidine and Texas-red, was used to labeled anti-dystrophin antibodies (14). Fluorescence staining was visualized using an Axiophot inverted microscope (Nikon, Tokyo, Japan) and an Odyssey XL confocal scanning system (Noran, Midleton, WI, USA).
Western-blot of dystrophin in isolated arteries
Samples of carotid and mesenteric arteries were collected and homogenized (Ultrasonic Processor, Bioblock Scientific, France). Proteins were separated by SDS-PAGE (Mini gel protean II system, Bio-Rad, 100V, using 300 ml 25 mM Tris, 192 mM glycine, 0.1% SDS) using a 4% stacking gel followed by a 4.5% running gel. After migration, proteins were transferred (50 V, overnight, 4°C using 800 ml 25mM Tris, 192 mM glycine, 10% methanol) to PVDF blotting membranes (Immobilon-P, Millipore). Membranes were then washed in TBS-T buffer (composition: 10 mM Tris/base pH 7.5, 0.1 M NaCl, 1 mM EDTA, 0.1% Tween 20) and blocked for 2 hr at room temperature (5% fat free dry milk in TBS-T). Membranes incubated 90 min with the primary antibody (anti dys1, anti dys2 or anti dys3, 1:1000), washed again (3 times for 10 min) and incubated with HRP-conjugated secondary antibody (Santa Cruz, 90 min RT, 1:2000). Membranes were washed (3 times for 10 min) and Dystrophin was visualized using the ECL-Plus Chemiluminescence kit (Amersham).
Western-blot of eNOS in isolated arteries
Samples of carotid and mesenteric arteries were collected and homogenized (Ultrasonic Processor, Bioblock Scientific, France). Proteins were separated by SDS-PAGE (Mini gel protean II system, Bio-Rad, 100V, using 300 ml 25 mM Tris, 192 mM glycine, 0.1% SDS) using a 4% stacking gel followed by a 7% running gel. After migration, proteins were transferred (50 V, overnight, 4°C using 800 ml 25mM Tris, 192 mM glycine, 10% methanol) to PVDF blotting membranes (Immobilon-P, Millipore). Membranes were then washed in TBS-T buffer (composition: 10 mM Tris/base pH 7.5, 0.1 M NaCl, 1 mM EDTA, 0.1% Tween 20) and blocked for 2 hr at room temperature (5% fat free dry milk in TBS-T). Membranes incubated 2 hours with the primary antibody (anti eNOS 1:5000), washed again (3 times for 10 min) and incubated with HRP-conjugated secondary antibody (Amersham, 1 hours RT, 1:2000). Membranes were washed (3 times for 10 min) and eNOS was visualized using the ECL-Plus Chemiluminescence kit (Amersham).
Data analysis
Results are expressed as means ± standard error (s.e.mean). EC50 or IC50 (concentration of agonist required to induce half the maximum response) and Emax (maximal response) were calculated for phenylephrine, SNP, and achetylcholine in each artery (6, 14). Significance of the differences between groups was determined by analysis of variance (ANOVA) and paired t test or by Bonferroni’s test. P values less than 0.05 were considered to be significant.
Results
Body weight and blood pressure (n=10 per group) were not affected by the absence of dystrophin and/or by gentamicin (table 1).
Table 1.
Body weight, mean arterial blood pressure (MAP) and pharmacological profile of carotid arteries and mesenteric resistance arteries isolated from mdx mice and their control treated with gentamicin. Contraction to phenylephrine (PE) and potassium (KCl (80 mmol/L) and dilation to acetylcholine (ACh) and sodium nitroprusside (SNP) were performed.
| Mice: | mdx | mdx | control | control | ||
|---|---|---|---|---|---|---|
| gentamicin: | − | + | − | + | ||
| Body weight | 32±2 | 30±2 | 34±3 | 31±2 | g | |
| MAP | 84±3 | 85±3 | 86±2 | 84±4 | mmHg | |
| Pharmacological profile of carotid and mesenteric arteries: | ||||||
| Carotid artery: | ||||||
| SNP: | IC50 | 65±14 | 74±18 | 88+13 | 80±17 | nmol/L |
| Imax | 98±2 | 95±4 | 99±1 | 96±3 | % | |
| ACh: | IC50 | 647±77 | 724±83 | 605±67 | 671±80 | nmol/L |
| Imax | 78±5 | 74±3 | 84±5 | 80±6 | % | |
| PE: | EC50 | 417±106 | 496±82 | 404±58 | 475±68 | nmol/L |
| Emax | 95±10 | 86±11 | 112±7 | 93±8 | μm | |
| Mesenteric resistance artery: | ||||||
| SNP: | IC50 | 52±6 | 66±9 | 48±8 | 63±10 | nmol/L |
| Imax | 99±2 | 97±3 | 99±1 | 98±2 | % | |
| ACh: | IC50 | 80±11 | 92±12 | 68±10 | 85±11 | nmol/L |
| Imax | 95±4 | 93±5 | 98±3 | 94±5 | % | |
| PE: | EC50 | 52±9 | 63±12 | 38±8 | 47±11 | nmol/L |
| Emax | 46±5 | 37±5 | 42±7 | 36±6 | μm | |
| Contraction to KCl (80 mmol/L): | ||||||
| Carotid arteries: | 97±9 | 90±9 | 113±9 | 95±10 | μm | |
| Mesenteric arteries: | 52±6 | 48±6 | 44±5 | 42±6 | μm | |
EC50 and IC50 represent the concentration necessary to reach 50% of the maximal effect; Emax and Imax give the maximal effect of the drug (n=10 per group). Maximal contraction is given as decrease in diameter (μm) for phenylephrine and as % dilation for SNP and acetylcholine.
No significant difference between groups was found.
Immunolocalization and protein expression of dystrophin
Dystrophin was present in both vascular smooth muscle and endothelial cells in control and gentamicin-treated mdx mice (fig. 1a and b). In mdx mice it was absent in both cell types in mesenteric and in carotid arteries (fig. 1a and b). Immunolabelling and confocal microscopy analysis were performed in cannulated arteries perfused under a pressure of 75 mmHg. As a consequence the confocal scanning was performed at high speed, avoiding movement artifacts and shading, but decreasing the image sharpness. Immunolabelling and confocal microscopy analysis of dystrophin showed that the protein was present in endothelial cells, at the level of the plasma membrane, in both control mdx mice treated with gentamicin (fig. 1c). Similarly, in smooth muscle cells, dystrophin was present at the level of the plasma membrane, in control mice and in mdx mice treated with gentamicin (data not shown). Dystrophin was not found in smooth muscle cells and endothelium cells in mdx mice.
Figure 1.
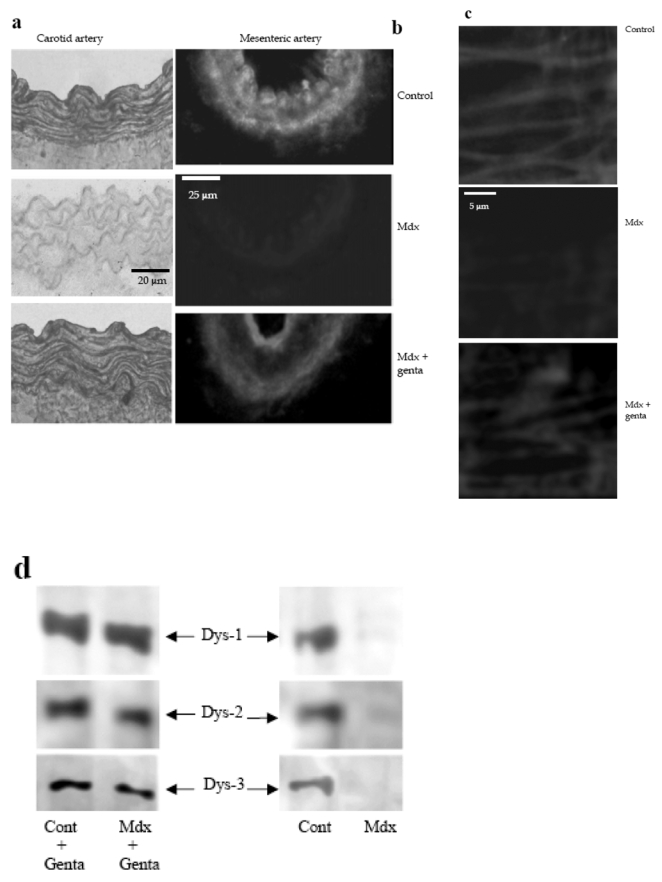
Immunolocalization of dystrophin in carotid arteries (revealed by peroxidase, a) and in mesenteric resistance arteries (Texas-red immunofluorescence, b) was performed using Dys2 anti-dystrophin antibodies. Arteries were isolated from control, mdx mice or mdx mice treated for two weeks with gentamicin. In mesenteric resistance arteries dystrophin (Dys2) was localized to the plasma membrane of endothelial cells, using immunofluorescence and confocal microscopy. This was performed in arteries perfused under a pressure of 75 mmHg and a flow of 50 μl/min (c). (n=6 mice per group). Due to the movement generated by the perfusion of the arteries in physiological conditions, the scanning of the vascular wall was performed at a speed of 10 images/sec.
The expression of dystrophin was determined in carotid arteries, using western-blot analysis. Dys1, Dys2 and Dys3 antibodies were directed against C-terminus, N-terminus and mid-rod domains of dystrophin, respectively (d and e). (n=8 mice per group).
*P <0.001; two-factor ANOVA, versus control.
#P <0.001; two-factor ANOVA, non-treated versus gentamicin-treated mdx mice.
Western-blot analysis in carotid arteries showed that a full-length dystrophin was recovered after treatment of mdx mice with gentamicin. Indeed, antibodies directed against the carboxy-terminus (dys2), the N-terminus (Dys3) or the mid rod domain (dys1) of dystrophin were used (fig. 1d). Quantitatively, approximately 40% of dystrophin were recovered in gentamicin-treated mdx mice (fig. 1e).
Structural analysis of mesenteric and carotid arteries
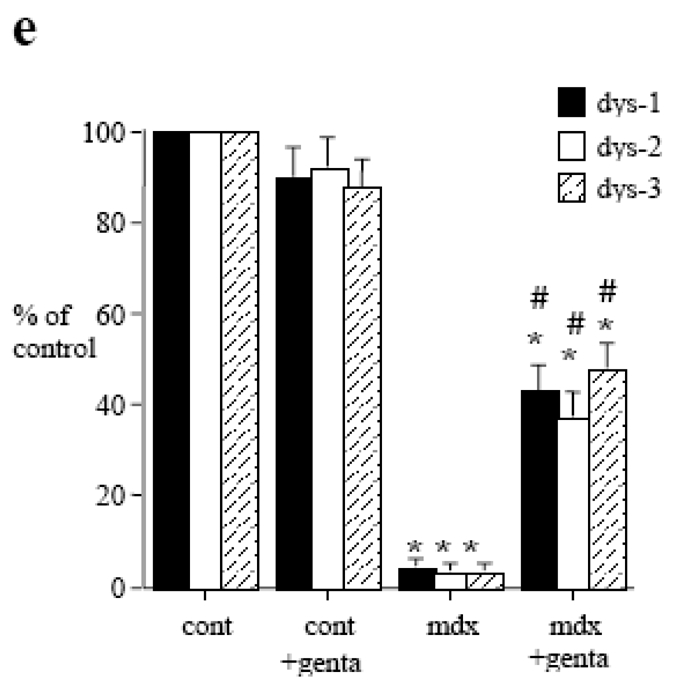
In isolated carotid and mesenteric resistance arteries bathed in a 0-calcium PSS containing EGTA and sodium nitroprusside, increasing pressure by step induced a rise in diameter. In mdx mice this passive diameter was similar than in control mice in both carotid and mesenteric arteries (fig. 2a, b). Passive arterial diameter was not affected by gentamicin in mesenteric and carotid arteries (fig. 2a, b). Wall thickness (fig. 2c, d) and cross sectional compliance (fig. 2e, f) were significantly lower in mdx than in control mice (fig. 2). In mdx mice treated with gentamicin, wall thickness and compliance were not different from those in control mice (table 2c–f).
Figure 2.
Passive vascular responses to pressure in carotid (left panel) and mesenteric resistance arteries (right panel) isolated from control or mdx mice treated for two weeks with gentamicin (genta, 34 mg/kg/day/14days). Arteries were submitted to stepwise increases in pressure when bathed in a Ca2+-free physiological salt solution containing EGTA (2 mmol/L) and sodium nitroprusside (10 μM), thus defining the passive arterial diameter, expressed as active tone (passive diameter - active diameter 6,14 (a, b). Arterial wall thickness was determined the same arteries (c, d). Cross-sectional compliance (e and f) was calculated from the diameter values shown in a and b. n=10 per group.
*P <0.05; two-factor ANOVA.
Arterial responses to pressure and flow
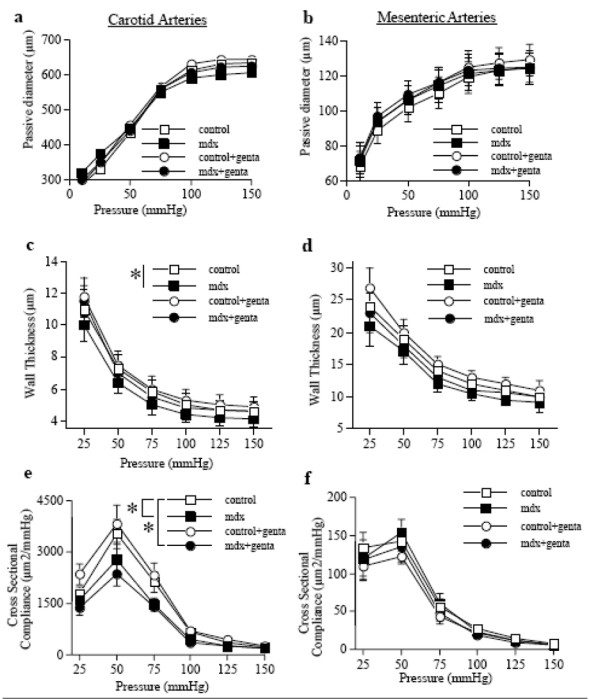
Pressure-induced myogenic tone was not affected by the absence of dystrophin and/or by gentamicin (fig. 3a and b). Myogenic tone was antagonized by flow-mediated dilation (FMD). Stepwise increases in flow induced a significant arterial dilation (fig. 3c,d,e). In both carotid and mesenteric arteries FMD was lower in mdx than in control mice (fig. 3c,e). Conversely, in gentamicin-treated mdx mice FMD was similar to control (fig. 3c,d). In vimentin-null mice FMD was lower than in the corresponding control mice, but FMD in vimentin-null mice was not improved by gentamicin (fig. 3e).
Figure 3.

Myogenic tone determined in carotid (a) and mesenteric resistance arteries (b) isolated from control or mdx mice treated for two weeks with gentamicin (genta, n=10 per group). Responses to flow were then determined in mesenteric (c) and carotid arteries (d) under a pressure of 75 mmHg.
In another group of experiments, the same experiments (flow-mediated dilation) were performed in vimentin-null mice mesenteric arteries (e).
*P <0.05; two-factor ANOVA, mdx versus control (c and d) or vim+/+ versus vim−/− (e)
NO-dependent dilation and NO-synthase in mesenteric and carotid arteries
Inhibition in NO-synthesis decreased FMD in mesenteric (fig. 4a,b) and carotid arteries (not shown). The effect of L-NAME was lower in mdx (fig. 4c) than in control mice (fig 4d). In gentamicin-treated mice the inhibitory effect of L-NAME on FMD was similar to that in control mice (fig. 4c,d).
Figure 4.
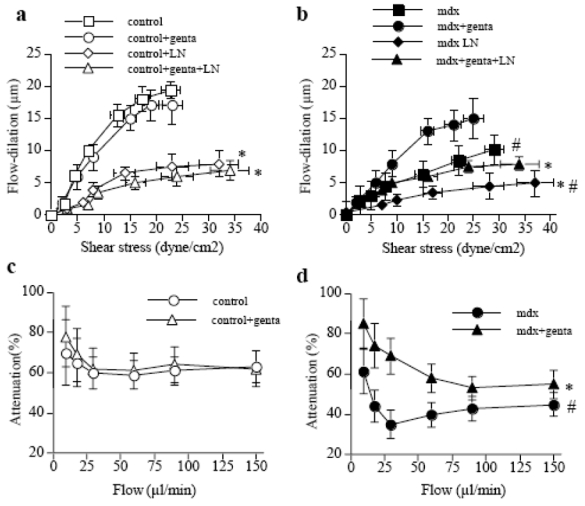
Effect of NO-synthesis blockade with L-NAME (LN) on flow-mediated dilation in mesenteric resistance arteries isolated from control (a) or mdx mice (b) treated for two weeks with gentamicin (genta). Data are represented as μm dilation (a and b) and as percentage inhibition of flow-mediated dilation by L-NAME (in control, c, and mdx, d, mice) (n=10 per group). NO-synthase expression, determined in carotid arteries is shown in e.
#P <0.001; two-factor ANOVA, non-treated versus gentamicin-treated mice.
*P <0.01; two-factor ANOVA, versus control.
The expression of eNOS in carotid (fig. 4e) and mesenteric arteries (not shown) was lower in mdx than in control mice, whereas in gentamincin-treated mdx mice it was equivalent to control.
Pharmacological profil of mesenteric and carotid arteries
Phenylephrine-induced contraction, as well as endothelium dependent (acetylcholine) and independent (sodium nitroprusside) dilation were not modified in mdx mice, relative to control mice (table 1). Similarly, KCl (80 mmol/L)-induced contraction was not affected by the absence of dystrophin (table 1). These parameters were not significantly affected by gentamicin, in either control or mdx mice (table 1).
Discussion
This study demonstrates that the selective decrease in flow-mediated endothelium-dependent dilation found in mdx mice was associated to a decreased of NO-dependent-dilation and a decreased NO-synthase expression. Both structural and functional vascular defects found in mdx mice could be recovered after a 2 week-long treatment with the aminoglycoside gentamicin.
Although dystrophin has been clearly shown to play a key role in force mechanotransduction in striated muscles, it is only our recent study that we has suggested its potential role in the mechanotransduction of flow (shear stress) in arteries (14). Other endothelium-dependent (acetylcholine) and independent (sodium nitroprusside) forms of dilation, as well as the arterial contractility were not affected by the absence of dystrophin. Flow (shear stress at the surface of the endothelial cells) is a major stimulus for vascular cell growth, remodeling and angiogenesis (2, 21). Thus, a defect in flow-mechanotransduction due to the absence of dystrophin could be deleterious and affect blood flow supply to organs, especially when an increase in blood flow is required in situations such as growth. Indeed ischemia occurs in skeletal and cardiac muscles of dystrophin deficient patients (17) and a defect in FMD might be a cause of this deficiency. This is supported by our finding showing that eNOS expression was decreased in mdx mice. The low responsiveness to flow (shear stress) in mdx mice arteries might be the cause of the decrease in eNOS expression. Indeed, flow is the main stimulus for eNOS expression (22, 23), and NO-dependent and independent production and the corresponding dilation are normal or increased when activated by other stimuli than flow, as shown in the present study and in a previous one (14). Thus the decrease of eNOS expression is more likely the consequence of the low sensitivity to flow in arteries from mdx mice. Nevertheless, the level of eNOS expression might be of importance for vascular adaptation to chronic changes in blood flow (18). A micro vascular dysfunction was initially suspected in DMD. Although no definitive evidence could be provided, a deficiency in the capacity of skeletal muscular cells to produce NO through neuronal NOS (or NOS-I) activation has been found. Skeletal muscle contraction induces a NOS-I-dependent arteriolar dilation, which is decreased in mdx mice (24). Similarly, disruption of the sarcoglycan-sarcospan complex, inducing cardiomyopathy, is associated to a deficiency of the coronary vasculature (25). In addition, the occurrence of ischemia has been shown in skeletal and cardiac muscles of dystrophin deficient patients (15–17). These observations are in agreement with the existence of an endothelial dysfunction in the microcirculation. Nevertheless, no functional study has been performed in arteries, especially concerning the endothelial function. Our actual observations provide direct evidence that, in physiological conditions, arterial responses to flow are blunted in dystrophin deficient mice. Together with previous reports showing that NO production by skeletal muscle during exercise (through the activation of the neuronal isoform of NOS) is decreased (17), our findings provides a rational for a role of a micro vascular dysfunction in the progression of DMD.
Gentamicin, after 2 weeks, restored FMD to control level. In addition, eNOS expression in arteries was normalized. This allows postulating that blood flow supply might also be normalized by gentamicin in any situation, rest or exercise. Although 40% of the dystrophin was recovered after the treatment, the endothelial response to flow was fully recovered. Dystrophin, in mice treated with gentamicin was probably a full-length protein as it was detected using antibodies direct against the C-terminal, the N-terminal and the mid rod domain of the protein. This study seems at variance with a recent work performed in 4 patients suffering DMD. In these 4 patients a similar treatment (2 weeks, same dose) failed to induce the recovery of the protein (26). Nevertheless, as pointed out by the authors, the dose and the duration of the treatment used in mice may not apply to human. Indeed, the choice of a dose is crucial, as previously shown (19). Considering the seriousness of the disease and the diversity of the mutations causing DMDs, and other genetic diseases with nonsense mutations, it might be important to plan other studies in patients. In this perspective the vascular side of the disease might also be considered, or reconsidered, in view of the novel findings provided by our study. In addition, other molecules, related or not to the aminoglycosides gentamicin, might be investigated in order to find drugs with less toxicity. Gentamicin is a highly nephro- and oto-toxic drug. The recovery of a normal vascular endothelial function and structure in mesenteric and carotid arteries after only 2-week long treatment with gentamicin is in agreement with previous observations showing a rapid (significant after 2 days) vascular remodeling due to a chronic blood flow change (18). Gentamicin in mdx mice induced the recovery of NO-dependent FMD and eNOS expression. As stated above, eNOS expression was probably recovered due to the normal shear stress sensing process due to the recovery of dystrophin in endothelial cells. Shear stress stimulates membrane-bound proteins such as integrins located in the focal adhesion sites (2, 27). In resistance arteries integrins such as the α3β5 integrin are involved in FMD in rat coronary arteries (28). The signal is then transmitted to eNOS through the activation of proteins such as Akt or HSP90 (29–31). The dissociation of eNOS from caveolin is necessary for its activation (32). Although no direct evidence is available showing the involvement of these proteins in the acute dilatory response of arteries to flow, dystrophin could interact in this process and be located in the region where eNOS and caveolin interact. Nevertheless, in skeletal muscle cells a relation between the neuronal form of NOS (nNOS), caveolin and dystrophin has been shown (33, 34).
In conclusion we found that dystrophin can be recovered after a short treatment with the aminoglycoside gentamicin. Gentamicin allows the recovery of dystrophin by suppressing a pre-mature stop codon in the gene encoding for dystrophin in mdx mice (19). This recovery of dystrophin restored flow-mediated endothelial-dependent dilation and eNOS expression. These findings open important perspectives in the mechanism involved in the pathophysiology of genetic diseases related to pre-mature stop codons such as DMD.
Acknowledgments
This work was supported in part by a grant from the French Association against Myopathies (AFM: Association France-Myopathies), Paris, France. Laurent Loufrani was a fellow of the AFM.
References
- 1.Bevan JA, Laher I. Pressure and flow-dependent vascular tone. Faseb J. 1991;9:2267. doi: 10.1096/fasebj.5.9.1860618. [DOI] [PubMed] [Google Scholar]
- 2.Davies PF. Flow-mediated endothelial mechanotransduction. Physiol Rev. 1995;75(3):519. doi: 10.1152/physrev.1995.75.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hutcheson IR, Griffith TM. Mechanotransduction through the endothelial cytoskeleton: mediation of flow- but not agonist-induced EDRF release. Br J Pharmacol. 1996;118(3):720. doi: 10.1111/j.1476-5381.1996.tb15459.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Welling WL, Zupka MY, Welling DJ. Mechanical properties of basement membrane. News Physiol Sci. 1995;10:30. [Google Scholar]
- 5.Oike M, Schwarz G, Sehrer J, Jost M, Gerke V, Weber K, Droogmans G, Nilius B. Cytoskeletal modulation of the response to mechanical stimulation in human vascular endothelial cells. Pflugers Arch. 1994;428(5–6):569. doi: 10.1007/BF00374579. [DOI] [PubMed] [Google Scholar]
- 6.Henrion D, Terzi F, Matrougui K, Duriez M, Boulanger CM, Colucci-Guyon E, Babinet C, Briand P, Friedlander G, Poitevin P, Levy BI. Impaired flow-induced dilation in mesenteric resistance arteries from mice lacking vimentin. J Clin Invest. 1997;100(11):2909. doi: 10.1172/JCI119840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Brown SC, Lucy JA. Dystrophin as a mechanochemical transducer in skeletal muscle. Bioessays. 1993:15. doi: 10.1002/bies.950150608. [DOI] [PubMed] [Google Scholar]
- 8.Ibraghimov-Beskrovnaya O, Ervasti JM, Leveille CJ, Slaughter CA, Sernett SW, Campbell KP. Primary structure of dystrophin-associated glycoproteins linking dystrophin to the extracellular matrix. Nature. 1992;355(6362):696. doi: 10.1038/355696a0. [DOI] [PubMed] [Google Scholar]
- 9.Ohlendieck K. Towards an understanding of the dystrophin-glycoprotein complex: linkage between the extracellular matrix and the membrane cytoskeleton in muscle fibers. Eur J Cell Biol. 1996;69(1):1. [PubMed] [Google Scholar]
- 10.Miyatake M, Miike T, Zhao J, Yoshioka K, Uchino M, Usuku G. Possible systemic smooth muscle layer dysfunction due to a deficiency of dystrophin in Duchenne muscular dystrophy. J Neurol Sci. 1989;93(1):11. doi: 10.1016/0022-510x(89)90157-3. [DOI] [PubMed] [Google Scholar]
- 11.Chien KR. Stress pathways and heart failure. Cell. 1999;98:555. doi: 10.1016/s0092-8674(00)80043-4. [DOI] [PubMed] [Google Scholar]
- 12.Lees D, Fabbrizio E, Mornet D, Pugnere D, Travo P. Parallel expression level of dystrophin and contractile performances of rat aortic smooth muscle. Exp Cell Res. 1995;218(1):401. doi: 10.1006/excr.1995.1172. [DOI] [PubMed] [Google Scholar]
- 13.Rivier F, Robert A, Hugon G, Mornet D. Different utrophin and dystrophin properties related to their vascular smooth muscle distributions. FEBS Lett. 1997;408(1):94. doi: 10.1016/s0014-5793(97)00398-0. [DOI] [PubMed] [Google Scholar]
- 14.Loufrani L, Matrougui K, Gorny D, Duriez M, Blanc I, Levy BI, Henrion D. Flow (shear stress)-induced endothelium-dependent dilation is altered in mice lacking the gene encoding for dystrophin. Circulation. 2001;103(6):864. doi: 10.1161/01.cir.103.6.864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Towbin JA, Bricker JT, Garson A., Jr Electrocardiographic criteria for diagnosis of acute myocardial infarction in childhood. Am J Cardiol. 1992;69(19):1545. doi: 10.1016/0002-9149(92)90700-9. [DOI] [PubMed] [Google Scholar]
- 16.Radda GK. Of mice and men: from early NMR studies of the heart to physiological genomics. Biochem Biophys Res Commun. 1999;266:723. doi: 10.1006/bbrc.1999.1890. [DOI] [PubMed] [Google Scholar]
- 17.Sander M, Chavoshan B, Harris SA, Iannaccone ST, Stull JT, Thomas GD, Victor RG. Functional muscle ischemia in neuronal nitric oxide synthase-deficient skeletal muscle of children with Duchenne muscular dystrophy. Proc Natl Acad Sci U S A. 2000;97(25):13818. doi: 10.1073/pnas.250379497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuttle JL, Nachreiner RD, Bhuller AS, Condict KW, Connors BA, Herring BP, Dalsing MC, Unthank JL. Shear level influences resistance artery remodeling: wall dimensions, cell density, and eNOS expression. Am J Physiol Heart Circ Physiol. 2001;281(3):H1380. doi: 10.1152/ajpheart.2001.281.3.H1380. [DOI] [PubMed] [Google Scholar]
- 19.Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104(4):375. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Halpern W, Osol G, Coy GS. Mechanical behavior of pressurized in vitro prearteriolar vessels determined with a video system. Ann Biomed Eng. 1984;12(5):463. doi: 10.1007/BF02363917. [DOI] [PubMed] [Google Scholar]
- 21.Ando J, Kamiya A. Blood flow and vascular endothelial cell function. Front Med Biol Eng. 1993;5:245. [PubMed] [Google Scholar]
- 22.Nadaud S, Philippe M, Arnal JF, Michel JB, Soubrier F. Sustained increase in aortic endothelial nitric oxide synthase expression in vivo in a model of chronic high blood flow. Circ Res. 1996;79(4):857. doi: 10.1161/01.res.79.4.857. [DOI] [PubMed] [Google Scholar]
- 23.Noris M, Morigi M, Donadelli R, Aiello S, Foppolo M, Todeschini M, Orisio S, Remuzzi G, Remuzzi A. Nitric oxide synthesis by cultured endothelial cells is modulated by flow conditions. Circ Res. 1995;76(4):536. doi: 10.1161/01.res.76.4.536. [DOI] [PubMed] [Google Scholar]
- 24.Lau KS, Grange RW, Chang WJ, Kamm KE, Sarelius I, Stull JT. Skeletal muscle contractions stimulate cGMP formation and attenuate vascular smooth muscle myosin phosphorylation via nitric oxide. FEBS Lett. 1998;431(1):71. doi: 10.1016/s0014-5793(98)00728-5. [DOI] [PubMed] [Google Scholar]
- 25.Coral-Vazquez R, Cohn RD, Moore SA, Hill JA, Weiss RM, Davisson RL, Straub V, Barresi R, Bansal D, Hrstka RF, Williamson R, Campbell KP. Disruption of the sarcoglycan-sarcospan complex in vascular smooth muscle: a novel mechanism for cardiomyopathy and muscular dystrophy. Cell. 1999;98(4):465. doi: 10.1016/s0092-8674(00)81975-3. [DOI] [PubMed] [Google Scholar]
- 26.Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, Hoffman EP, Fischbeck KH. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49(6):706. [PubMed] [Google Scholar]
- 27.Burridge K, ChrzanowskaWodnicka M. Focal adhesions, contractility, and signaling. Annu Rev Cell Dev Biol. 1996;12:463. doi: 10.1146/annurev.cellbio.12.1.463. [DOI] [PubMed] [Google Scholar]
- 28.Muller JM, Chilian WM, Davis MJ. Integrin signaling transduces shear stress-dependent vasodilation of coronary arterioles. Circ Res. 1997;80(3):320. doi: 10.1161/01.res.80.3.320. [DOI] [PubMed] [Google Scholar]
- 29.Dimmeler S, Assmus B, Hermann C, Haendeler J, Zeiher AM. Fluid shear stress stimulates phosphorylation of Akt in human endothelial cells: involvement in suppression of apoptosis. Circ Res. 1998;83(3):334. doi: 10.1161/01.res.83.3.334. [DOI] [PubMed] [Google Scholar]
- 30.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399(6736):597. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brouet A, Sonveaux P, Dessy C, Moniotte S, Balligand JL, Feron O. Hsp90 and caveolin are key targets for the proangiogenic nitric oxide-mediated effects of statins. Circ Res. 2001;89(10):866. doi: 10.1161/hh2201.100319. [DOI] [PubMed] [Google Scholar]
- 32.Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81(1):209. doi: 10.1152/physrev.2001.81.1.209. [DOI] [PubMed] [Google Scholar]
- 33.Baum O, Planitzer G, Richter H, Gossrau R. Irregular costameres represent nitric oxide synthase-1-positive sarcolemma invaginations enriched in contracted skeletal muscle fibres. Histochem J. 2000;32(12):743. doi: 10.1023/a:1004153111532. [DOI] [PubMed] [Google Scholar]
- 34.Gossrau R. Caveolin-3 and nitric oxide synthase I in healthy and diseased skeletal muscle. Acta Histochem. 1998;100(1):99. doi: 10.1016/S0065-1281(98)80009-3. [DOI] [PubMed] [Google Scholar]