

Abstract
The ATP-sensitive potassium (KATP) channel is named after its characteristic inhibition by intracellular ATP. The inhibition is a centerpiece of how the KATP channel sets electrical signaling to the energy state of the cell. In the β cell of the endocrine pancreas, for example, ATP inhibition results from high blood glucose levels and turns on electrical activity leading to insulin release. The underlying gating mechanism (ATP inhibition gating) includes ATP stabilization of closed states, but the action of ATP on the open state of the channel is disputed. The original models of ATP inhibition gating proposed that ATP directly binds the open state, whereas recent models indicate a prerequisite transition from the open to a closed state before ATP binds and inhibits activity. We tested these two classes of models by using kinetic analysis of single-channel currents from the cloned mouse pancreatic KATP channel expressed in Xenopus oocytes. In particular, we combined gating models based on fundamental rate law and burst gating kinetic considerations. The results demonstrate open-state ATP dependence as the major mechanism by which ATP speeds exit from the active burst state underlying inhibition of the KATP channel by ATP.
Keywords: ATP, ADP, Kir6.2, SUR, gating mechanism
INTRODUCTION
The ATP-sensitive potassium (KATP) channel couples electrical activity to metabolism in a variety of cells, and thus plays an important physiological role (Noma 1983; Ashcroft et al. 1984; Cook and Hales 1984; Jovanovic et al. 1998; Aguilar-Bryan and Bryan 1999). The KATP channel is assembled from four each of two distinct types of subunit. The pore-forming Kir6.x subunits (Inagaki et al. 1995) are likely the primary seat of ATP-dependent inhibition gating (Tucker et al. 1997, Tucker et al. 1998; Drain et al. 1998; John et al. 1998). The sulfonylurea receptor SURx subunits (Aguilar-Bryan et al. 1995) mediate inhibition by sulfonylureas and activation by MgADP and by potassium channel openers (Nichols et al. 1996; Gribble et al. 1997, Gribble et al. 1998; Shyng et al. 1997; Babenko et al. 2000).
The complex control of the KATP channel by its multiple ligands is still poorly understood. For example, in the β cell of the endocrine pancreas, only a handful of active KATP channels out of hundreds suffices to prevent cell electrical activity when energy is low, and closure of these few channels when energy is high initiates electrical signaling leading to insulin secretion (Cook et al. 1988). The need for such complete inhibition of the active states of the KATP channel suggests that multiple inhibitory mechanisms might be at work. One such inhibitory action is ATP binding to an inactive state of the channel stabilizing it (Noma 1983; Cook and Hales 1984; Ashcroft and Kakei 1989; Qin et al. 1989). Additional ATP ligands can bind to further stabilize the inactive state. A second inhibitory action is ATP binding to the active open state (Ashcroft et al. 1988; Drain et al. 1998; Ashcroft and Kakei 1989; Fan and Makielski 1999; Gillis et al. 1989; Nichols et al. 1991; Trapp et al. 1998). ATP-dependent gating from the open state of the KATP channel, however, has been claimed to be so energetically unfavorable as to be virtually nonexistent at any ATP concentration (Shyng et al. 1997; Babenko et al. 1999; Koster et al. 1999; Enkvetchakul et al. 2000, Enkvetchakul et al. 2001; Loussouarn et al. 2000).
Therefore, in this study, we focused our single-channel kinetic analysis on whether ATP directly affects the open state of single KATP channels. The activity of the KATP channel occurs in bursts of brief openings and briefer closings, separated by long-lived inactive interburst intervals. The rate of transition from the active burst state to the inactive interburst state is relatively slow in the absence of ligand (ligand-independent gating), and greatly accelerated by ATP (ATP-dependent gating; Drain et al. 1998; Tucker et al. 1998; Li et al. 2000; and this paper). ATP destabilization of the open state and speeding burst exit to the long-lived interburst would stably shut down activity contributing to inhibition. Here, we explored basic kinetic tests for destabilization of the open state by ATP occupancy as a mechanism explaining how ATP shortens the burst durations of the KATP channel.
Our results support mechanisms of KATP channel gating with an open state whose stability is greatly decreased when ATP is bound but is not decreased when MgADP bound in the presence of ATP or when ATP is absent. Models of KATP channel inhibition gating by ATP that do not account for ATP-dependent transitions from the open state are mechanistically incomplete.
MATERIALS AND METHODS
Mutagenesis
Mouse Kir6.2 and mouse SUR1 cloned from the βHC9 cell line (Drain et al. 1998) were used in this study. The Kir6.2::G334D/SUR1 and Kir6.2ΔC26 (Tucker et al. 1997) channel construction and characterization is as described previously (Drain et al. 1998).
Expression in Oocytes and Electrophysiology
Preparation and injection of Xenopus oocytes, patch pipet fabrication, and recording techniques were as described previously (Drain et al. 1994, Drain et al. 1998). Briefly, recordings, unless indicated otherwise, were obtained from single-channel current recording by using the inside-out configuration of the patch clamp at −80 mV with symmetrical 150 mM KCl, with Ca2+ buffered to 10 nM and 15 mM creatine phosphate and 10 U/ml creatine kinase (Sigma-Aldrich; Dzeja and Terzic 1998; Bienengraeber et al. 2000) and ATP concentration, as indicated in the bath and superfusate solutions. Bath solution was the same as the pipet solution but with 0.6 mM MgATP. ATP was added as the magnesium salt to minimize rundown (Trube and Hescheler 1984). The pipet solution contained the following (in mM): 150 KCl, 10 NaCl, 1 CaCl2, 10 EGTA, and 10 HEPES, pH 7.4 ± 0.05. Constant superfusion of the cytoplasmic face of patches was performed using a Biologic RSC-160 9-sewer pipe syringe–pressurized system (Molecular Kinetics Inc.). Recordings were always begun within 30 s after excision with the patch pipet partially inserted into one of the sewer pipes. Sufficient ATP dose–response data were obtained as rapidly as possible, typically, in about 10 min—most of that time being for the 0.6 mM ATP dose, where event frequency is very low. Experiments that showed rundown, characterized by a significant decrease in PO (open channel probability at 0 ATP) were discarded. Most attempts to record at all three ATP concentrations were incomplete due to rundown or insufficient number of events for fitting, but when parts of the dose–response data were of sufficient number in such experiments for fitting, or when simple arithmetic means were determined, the mean durations were statistically indistinguishable from those of the nine dose–response experiments presented here. Patch-clamp currents were obtained at −80 mV and amplified using an Axopatch 200A (Axon Instruments, Inc.) or EPC-9 (HEKA Elektronik) patch amplifier, low-pass filtered with an 8-pole Bessel filter (Frequency Devices) at a corner frequency of 4 kHz, and sampled at 20–50 kHz using HEKA PULSE v.8.4 (HEKA Elektronik). Square wave pulses input by a wave function generator (model Hm-8030–4; Hameg) were recorded by our recording systems to estimate the maximum dead time at 180 μs, where >95% of pulses could be measured.
Data Analysis
Analysis and display were done using TAC v.4.0 (Bruxton, Inc.), IGOR Pro v.4.0 (WaveMetrics, Inc.), and PageMaker v.6.5 (Adobe Systems, Inc.). Single-channel current events were detected using the time of the half amplitude of transitions between current levels with TAC v.4.0. Durations were corrected for missed events during construction of duration histograms based on the filter corner frequency of the recording by the method of Colquhoun and Sigworth 1995. Duration analysis was done with TAC-FIT v4.0 (Bruxton, Inc.), which uses the transformations of Sigworth and Sine 1987 to construct and fit duration histograms. Data are presented as mean ± SEM. Statistically significant differences (P < 0.001) between means at all different ATP concentrations for both burst and open lifetimes reported here were found by a series of tests including the Kolmogorov-Smirnov statistic, which does not assume any particular shape to the distribution of lifetime means (Conover 1980).
RESULTS
We studied ligand-dependent and independent gating transitions from the open state of the wild-type mouse pancreatic KATP channel expressed in Xenopus oocytes. The inside-out configuration of the patch clamp was used to control adenine nucleotide ligand concentrations at the cytoplasmic face of the membrane. Adenine nucleoside tri- and diphosphates were added as magnesium salts. When indicated, any MgADP generated was removed by the creatine phosphate/kinase scavenger system (Dzeja and Terzic 1998; Bienengraeber et al. 2000). The membrane was held at −80 mV in symmetrical 150 mM KCl. Gating behavior was stable as evidenced by the consistently high open probability (PO > 0.60) of all channels studied, and required using only fresh membrane patches (within 30 s of excision), rapid measurement of relevant dose responses, and buffering Ca2+ to 10 nM. The bath contained 0.6 mM ATP and with a 9-sewer pipe superfusion system, we rapidly measured single-channel gating in 0, 0.2, or 0.6 mM ATP at the cytoplasmic face of the channel. Typically, a complete dose response was acquired in about 10 min. In between these doses and at the end of the experiment, the PO was determined.
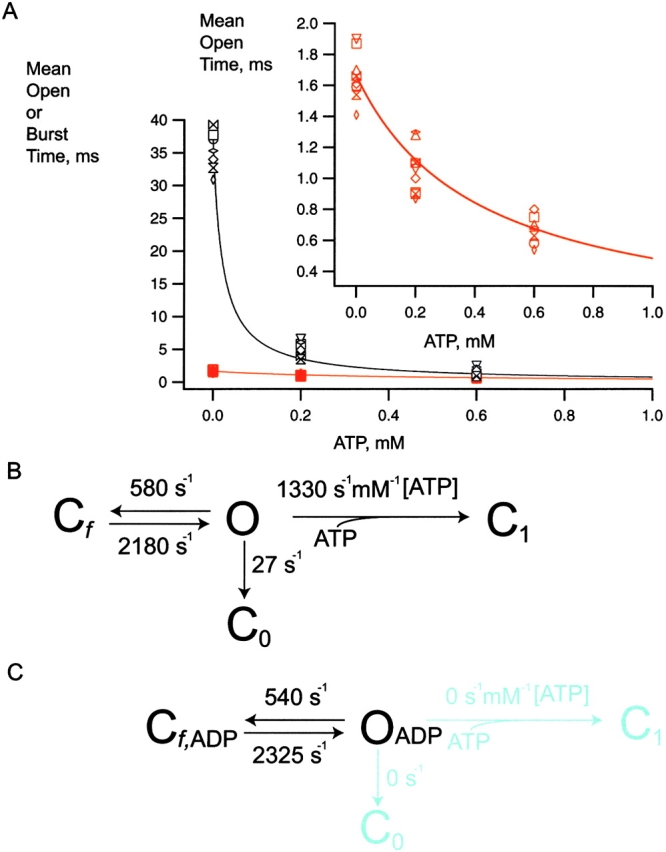
Fig. 1 shows the two major classes of gating mechanisms for KATP channel inhibition by ATP tested in this study. In Fig. 1 A, ATP binds to the open state O, destabilizing it and speeding transition to C1. (Subscript indicates number of ATP ligands bound.) The lower mechanism prohibits direct binding of ATP to O, but rather assumes between O and C1 a new long-lived inactive interburst state C0 to which ATP can bind, and to which the channel transits with a fixed, ATP-insensitive rate constant. Thus, Fig. 1 B predicts constant mean open durations that are independent of ATP concentration. This fundamental critical distinction between the two mechanisms is easily testable. The lifetime of a given state is the reciprocal of the sum of the rate constants for exit from that state. Therefore, we focused the single-channel kinetic analysis on whether ATP directly affects the open state lifetime of the KATP channel.
Figure 1.

Two classes of KATP channel ATP inhibition gating mechanism make different kinetic predictions for the ATP dependence of open state duration. (A) ATP concentration decreases open-state duration and thereby burst duration. The simple ATP-dependent burst scheme makes specific quantifiable predictions about how decremental ATP destabilization of the open state necessarily will account for dramatic decreases in burst durations. (B) ATP concentration is excluded from determining open state or burst durations. ATP binding to the open state is so energetically unfavorable that there is essentially no open-state occupancy at any ATP concentration. The hypothesis we tested here is that ATP binds the open state of the KATP channel, which destabilizes it relative to the inactive interburst state, thus providing the mechanism by which increasing ATP speeds the rate of burst exit. The ATP dependence of the open times in the top model exactly depends not only on rate constant 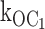 [ATP], but also on rate constant
[ATP], but also on rate constant 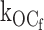 . C, closed state; O, open state. Subscripts: f, fast; 0, 0 ATP ligands bound; and 1, 1 ATP ligand bound. Cf is the short-lived or fast intraburst closed state. C0 and C1 are long-lived or slow interburst closed states that differ by whether ATP is bound and thereby their mean duration.
. C, closed state; O, open state. Subscripts: f, fast; 0, 0 ATP ligands bound; and 1, 1 ATP ligand bound. Cf is the short-lived or fast intraburst closed state. C0 and C1 are long-lived or slow interburst closed states that differ by whether ATP is bound and thereby their mean duration.
Open-State ATP Dependence
Fig. 2 shows representative single-channel currents of the KATP channel within 30 s after patch excision and the effect of increasing concentrations of ATP at the cytoplasmic face. Any MgADP generated was removed by the creatine phosphate/kinase scavenger system. We first considered what determines the open durations of the KATP channel in the absence of ATP. By studying KATP channel burst durations in the absence of ATP, we obtained an estimate of the ligand-independent gating rate constant for O to C0 (open state to inactive interburst state) 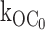 as 27 ± 2 s−1 (n = 9). (It is plausible that the ligand-independent transitions from the open state might arise from residual ATP or ATP synthesis in the excised patch, however, later in Fig. 5 we will show that this can be discounted.) From this result, together with the mean open durations, we obtained the gating rate constant for O to Cf (open state to fast intraburst closed state)
as 27 ± 2 s−1 (n = 9). (It is plausible that the ligand-independent transitions from the open state might arise from residual ATP or ATP synthesis in the excised patch, however, later in Fig. 5 we will show that this can be discounted.) From this result, together with the mean open durations, we obtained the gating rate constant for O to Cf (open state to fast intraburst closed state) 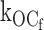 as 575 ± 18 s−1 (n = 9). In the absence of ATP, the open durations were determined mainly by the relatively very fast
as 575 ± 18 s−1 (n = 9). In the absence of ATP, the open durations were determined mainly by the relatively very fast 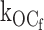 , which dwarfs the
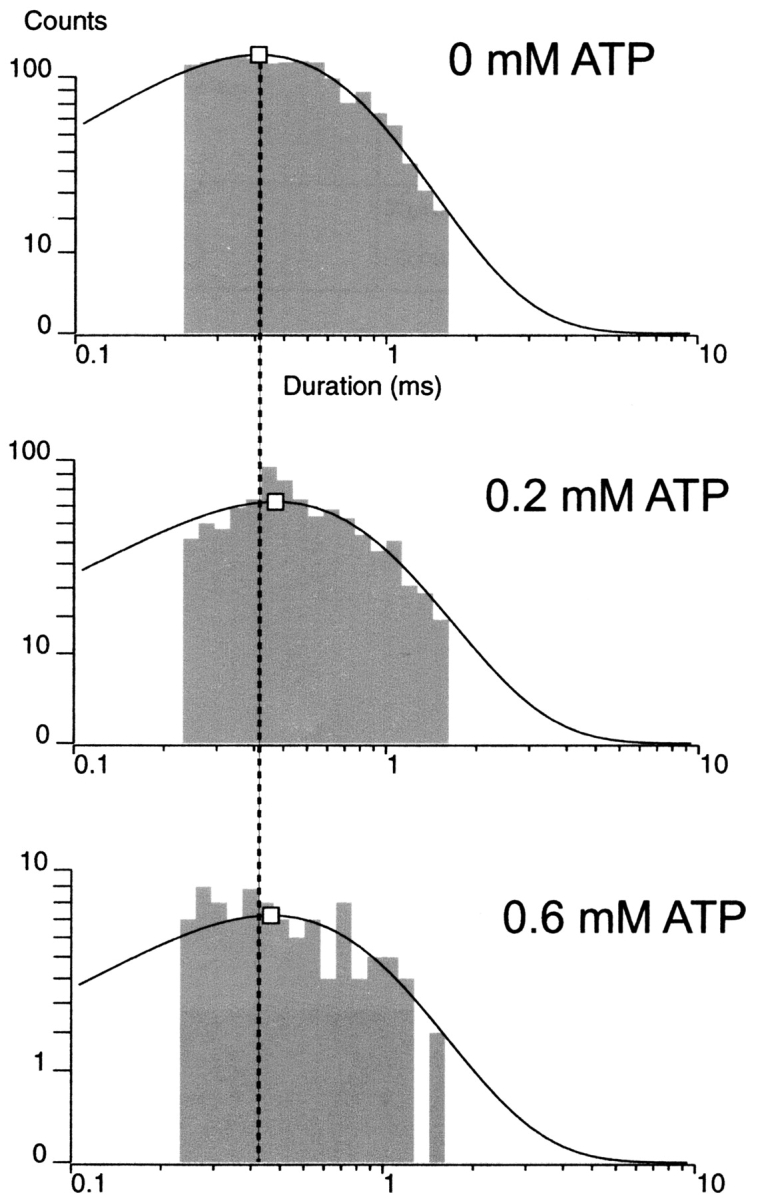
, which dwarfs the 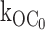 rate. Any ATP-dependent rate constant from the open state clearly would have to approach or exceed kOC to significantly decrease mean open durations. Accordingly, ATP concentrations (0.2 and 0.6 mM) that are 16- and 48-fold Ki for ATP inhibition were used because it is in this range that the burst kinetics of the KATP channel predict significant decrements in open durations. The current records show the dual action of ATP characteristic of the KATP channel, where burst durations dramatically decrease and interburst durations increase with increasing ATP. In Fig. 2 B, the effect on the open durations seen in the temporally expanded segments are subtle. Therefore, we constructed and fitted the relevant duration histograms from the entire dataset from this patch.
rate. Any ATP-dependent rate constant from the open state clearly would have to approach or exceed kOC to significantly decrease mean open durations. Accordingly, ATP concentrations (0.2 and 0.6 mM) that are 16- and 48-fold Ki for ATP inhibition were used because it is in this range that the burst kinetics of the KATP channel predict significant decrements in open durations. The current records show the dual action of ATP characteristic of the KATP channel, where burst durations dramatically decrease and interburst durations increase with increasing ATP. In Fig. 2 B, the effect on the open durations seen in the temporally expanded segments are subtle. Therefore, we constructed and fitted the relevant duration histograms from the entire dataset from this patch.
Figure 2.

ATP dependence of single KATP channel gating kinetics at 0, 0.2, and 0.6 mM ATP. (A) Effect of ATP on burst durations. (B) 10-fold expanded time scale to reveal effects of ATP on open durations. Single-channel currents from a representative inside-out patch within 30 s after patch excision continuously perfused with 0 ATP for 15 s. The patch was then exposed to 0.2 mM MgATP for 90 s, and finally 0.6 mM MgATP for 10 min. In between these [MgATP], the channel is exposed briefly to 0 MgATP (to check for rundown in activity) and to 0.6 mM MgATP to help maintain the initial high PO activity typical of the KATP channel in 0 MgATP. For each [ATP], single-channel currents are shown by using a slow time scale to emphasize burst durations reduction by increasing ATP (left) and by using a 10-fold faster time scale a segment is expanded to emphasize open durations reduction by ATP (immediately to the right).
Figure 5.

Evidence for ligand-independent gating transitions from the open state O to the inactive interburst C0. (A) In the absence of ATP, the Kir6.2::G334D/SUR1 mutant channel, which virtually eliminates ATP-dependent inhibition gating, exhibits gating transitions from the active burst to the inactive interburst, indicating little or no effect on ligand-gating to the inactive interburst, slow time scale. (B) Same as above except 10-fold faster time scale, emphasizing the open durations within these long bursts. (C) The distribution of burst durations for the G334D channel is single-exponential with mean duration of 39.8 ms. The reciprocal of the mean burst duration of the ATP-refractory Kir6.2::G334D/SUR1 is the first order rate constant 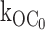 equal to 25.1 s−1, which is comparable to the
equal to 25.1 s−1, which is comparable to the 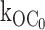 value 27 s−1 determined for the ATP-sensitive wild-type KATP channel. (D) The distribution of open durations for the G334D channel is single-exponential with mean duration of 1.78 ms, which is similar to the 1.68-ms value determined for the ATP-sensitive KATP channel. The G334D burst and open duration results provide strong support for the ligand-independent gating transition O to C0, and quantitatively exclude the possibility that the transition to C0 may be explained by ligand-dependent transitions driven by nominal ATP in the patch.
value 27 s−1 determined for the ATP-sensitive wild-type KATP channel. (D) The distribution of open durations for the G334D channel is single-exponential with mean duration of 1.78 ms, which is similar to the 1.68-ms value determined for the ATP-sensitive KATP channel. The G334D burst and open duration results provide strong support for the ligand-independent gating transition O to C0, and quantitatively exclude the possibility that the transition to C0 may be explained by ligand-dependent transitions driven by nominal ATP in the patch.
Fig. 3 shows the distribution of burst durations and open durations at 0, 0.2, and 0.6 mM ATP. The KATP channel showed dramatic reductions in burst durations and concomitant decrements in open durations in all nine patches tested. At 0, 0.2, and 0.6 mM ATP and −80 mV, the KATP channel gated with a mean burst duration of 36.8 ± 1.4, 3.64 ± 0.42, and 1.23 ± 0.20 ms (n = 7–9), respectively. Mean open durations decreased as well from 1.66 ± 0.08 ms and 1.14 ± 0.03 ms to 0.70 ± 0.02 ms (n = 7–9), respectively. Fig. 3 illustrates that the mean durations were each from single-exponential components, which is consistent with a single open conformation that is being destabilized (i.e., has its lifetime shortened) by ATP. For each of the burst and open duration histograms, dashed vertical lines are positioned at the long mean duration at 0 ATP for easier comparison with the intermediate and short mean durations at increasing ATP. The decrease of mean burst duration is accompanied by a significant decrease in mean open duration made clear at these ATP concentrations (P < 0.001). The burst, but not the open lifetime, data at 0.6 mM ATP could be fit with an additional minor fast component, with the mean value right up against the temporal resolution limit of our measurements. It may arise from an ATP-dependent decrease in the mean burst time of the fast component, which would further support our conclusions, from an ATP-dependent increase in the amplitude of an otherwise undetectable ATP-independent component or both. Because of its minor contribution and occurrence, this component was ignored. The data indicate KATP channel open state occupancy by ATP, observed as significantly decrementing mean open durations with increasing ATP. The result also leads to the important question whether the decrement in open durations by ATP accounts for the dramatic decrease in burst durations as the major mechanism for ATP-dependent burst exit. This question is addressed later (see Fig. 10).
Figure 3.

Burst and open duration histograms. (A) Distribution of burst durations as a function of ATP concentration. The log-scale abscissa of the Sine-Sigworth plots results in the peak of these single-exponential distributions positioned at the value of the time constant. The dashed line is placed at the higher time constant (0 ATP) to facilitate comparison with the intermediate and lower time constants at other ATP concentrations. (B) Distribution of open times as a function of ATP concentration. Note the decrement in open time with increasing ATP concentration correlates with a dramatic decrease in the corresponding burst durations.
Figure 10.
ATP-dependent open state mechanism for speeding burst exit, supported by single-channel kinetic data reported here. (A) Mean open time (red) or burst time (black) as a function of increasing [ATP]. The measured mean durations from individual patch experiments are shown by the position of individual symbols. The smooth curves through these symbols were generated as indicated in the text. Note that by using first order rate constants, together only with the decrementing mean open durations at increasing [ATP], we solved for the second order, ATP-dependent rate constant, 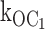 . This second order rate constant then was used to calculate the ATP dependence of not only the open, but also the burst durations shown by the smooth curves. The main plot shows that the rates determined from the decrementing open times quantitatively account for the mean open and burst duration data. The insetted plot shows the mean open duration ATP dependence with the y-axis expanded to emphasize that the mean open durations decrease with increasing ATP, as predicted by the determined rate constants. (B) The burst gating model supported by the results includes a second order, ATP-dependent rate constant,
. This second order rate constant then was used to calculate the ATP dependence of not only the open, but also the burst durations shown by the smooth curves. The main plot shows that the rates determined from the decrementing open times quantitatively account for the mean open and burst duration data. The insetted plot shows the mean open duration ATP dependence with the y-axis expanded to emphasize that the mean open durations decrease with increasing ATP, as predicted by the determined rate constants. (B) The burst gating model supported by the results includes a second order, ATP-dependent rate constant, 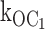 [ATP], from the open state (O) to the interburst state bound by one ATP ligand, C1. Cf is the short-lived intraburst closed state, and C0 is the long-lived inactive interburst closed state, each with no ATP bound. The relative values of the rate constants determined indicate that about twofold decrease in mean open duration requires ∼500 μM ATP, or tens of Ki units for ATP inhibition. Note that in the new gating mechanism, the inactive interburst state C0 is not an obligatory state to which the open channel must transit before ATP is permitted to bind. The open channel either first transits to the inactive interburst (e.g., in low ATP) or first binds ATP, which speeds its transition to C1. Although beyond the scope of this study, C0 and C1 can interconvert by ATP binding and dissociation reactions where bound ATP would stabilize the closed allosteric gating conformation(s) without transitions from the single-channel closed current level. For both mean open durations and mean burst durations, differences between values at 0, 0.2, and 0.6 mM ATP were each statistically significant (P < 0.001). (C) The burst gating model for the KATP channel in the presence of 150 μM MgADP. The gating transitions are effectively directed by bound MgADP to those within the burst by reducing to little or nothing the two burst exit pathways (transition and rate constants in aqua). When MgADP is bound to the channel, presumably at SUR1, the open state is precluded from binding inhibitory ATP, and gating transitions are constrained to gating transitions within the active burst. Gating transitions to the long-lived interburst by ligand-independent gating or ATP-dependent gating are permitted only upon unbinding of the MgADP, as above.
[ATP], from the open state (O) to the interburst state bound by one ATP ligand, C1. Cf is the short-lived intraburst closed state, and C0 is the long-lived inactive interburst closed state, each with no ATP bound. The relative values of the rate constants determined indicate that about twofold decrease in mean open duration requires ∼500 μM ATP, or tens of Ki units for ATP inhibition. Note that in the new gating mechanism, the inactive interburst state C0 is not an obligatory state to which the open channel must transit before ATP is permitted to bind. The open channel either first transits to the inactive interburst (e.g., in low ATP) or first binds ATP, which speeds its transition to C1. Although beyond the scope of this study, C0 and C1 can interconvert by ATP binding and dissociation reactions where bound ATP would stabilize the closed allosteric gating conformation(s) without transitions from the single-channel closed current level. For both mean open durations and mean burst durations, differences between values at 0, 0.2, and 0.6 mM ATP were each statistically significant (P < 0.001). (C) The burst gating model for the KATP channel in the presence of 150 μM MgADP. The gating transitions are effectively directed by bound MgADP to those within the burst by reducing to little or nothing the two burst exit pathways (transition and rate constants in aqua). When MgADP is bound to the channel, presumably at SUR1, the open state is precluded from binding inhibitory ATP, and gating transitions are constrained to gating transitions within the active burst. Gating transitions to the long-lived interburst by ligand-independent gating or ATP-dependent gating are permitted only upon unbinding of the MgADP, as above.
The dramatic decrease in burst duration by increasing [ATP] could also have a significant contribution by similar effects of ATP on the intraburst closed state Cf. Fig. 4 demonstrates that there is little or no effect on the intraburst closed state Cf by ATP. In 0, 0.2, and 0.6 mM ATP, the mean intraburst closed time actually slightly increased from 0.44 ± 0.01 and 0.47 ± 0.02, to 0.48 ± 0.02 ms (n = 7–9; P < 0.01). Because the burst comprises the open and fast closed states, the results indicate that the decrease in burst duration by ATP involves solely the open state.
Figure 4.
ATP has no effect on the intraburst shut times at concentrations where open times are reduced by greater than twofold. In general, the experiments showed if anything a slight increase in closed time durations with increasing ATP.
The analysis so far indicates direct open-state ATP binding and speeding of burst exit, obviating any obligatory transition from the open state via the C0 state on the way to the ATP-inhibited C1 state, which questions the status of C0. We tested for the C0 state as an additional open to interburst gating transition. Rigorous support for ligand-independent gating to such a C0 state would require excluding the plausible explanation for such gating as being due to residual ATP generated in the excised patch. This can be excluded by using a previously characterized mutant KATP channel (Kir6.2::G334D/SUR1) whose apparent affinity for ATP is reduced >500-fold (Drain et al. 1998). If wild-type KATP channel gating from the open to the inactive interburst state C0 were due to residual ATP present in the excised patch, then the G334D mutation should essentially eliminate these transitions in nominally 0 ATP conditions altogether, and cause burst durations to become extraordinarily long.
Fig. 5 shows the gating of a single Kir6.2::G334D/SUR1 channel in 0 ATP. Duration histograms were used to quantify the O to C0 rate of the G334D channel and found to average 1/40.4 ms or 25 ± 1 s−1 (n = 4), which is comparable to the 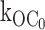 27 ± 2 s−1 (n = 9) rate of the wild-type channel. The transitions to the inactive interburst C0 is evident by burst durations of tens of ms duration suggesting
27 ± 2 s−1 (n = 9) rate of the wild-type channel. The transitions to the inactive interburst C0 is evident by burst durations of tens of ms duration suggesting 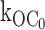 transition rates similar to the wild-type KATP channel. The results provide strong support for an additional, ligand-independent transition from the open state to an inactive interburst state, which is referred to as C0, where the 0 indicates 0 ATP ligands bound and distinguishes the inactive interburst from the faster intraburst closed state Cf and the slower ATP-bound inhibited interburst state C1.
transition rates similar to the wild-type KATP channel. The results provide strong support for an additional, ligand-independent transition from the open state to an inactive interburst state, which is referred to as C0, where the 0 indicates 0 ATP ligands bound and distinguishes the inactive interburst from the faster intraburst closed state Cf and the slower ATP-bound inhibited interburst state C1.
Open-state ATP Dependence and the “Ligand-insensitive” KATP Channel
We also studied the response of open times of the KATP channel in conditions where creatine phosphate and kinase were not added. Physiologically, similar conditions may occur where creatine kinase is downregulated or absent. Fig. 6 shows that without creatine phosphate and kinase that KATP channel activity is increased with the appearance of long duration bursts, even though 0.6 mM ATP is present. The long bursts, evidently refractory to the high ATP, are similar to the “ligand-insensitive” gating previously reported for the cardiac channel KATP channel in the presence of 2 mM MgUDP (Alekseev et al. 1998). They have demonstrated that MgUDP can constrain single cardiac KATP channels to gating transitions within the active intraburst, with infrequent transitions to the interburst. When MgUDP is bound, presumably at NBD2 of SUR2A, the cardiac channel exhibits extraordinarily long burst durations, as if it were insensitive to the tri-phosphate nucleoside ligands. Although evidently still sensitive to diphosphate nucleoside ligands, the term ligand insensitivity thus was used only with respect to inhibition of the KATP channel by triphosphate nucleoside ligands. We confirmed that the long duration bursts of our pancreatic KATP channel were due to MgADP generated by the patch in high MgATP as follows.
Figure 6.
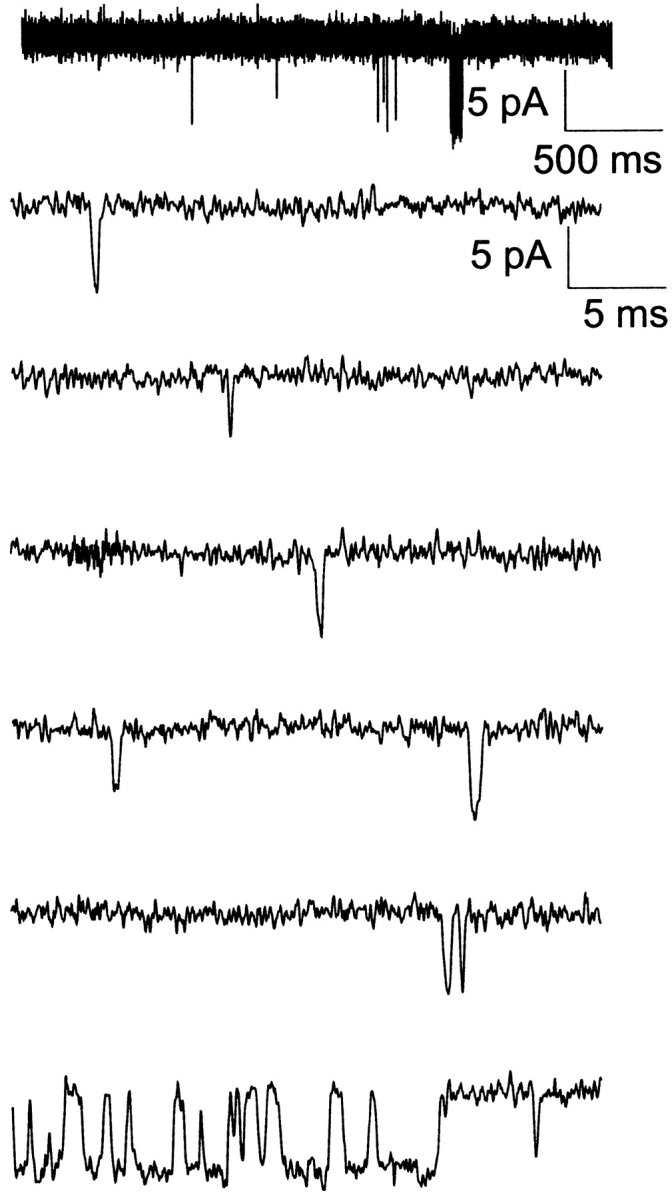
“Ligand-insensitive bursts” in the presence of 5 mM MgATP. Typically (92/97 patches) in the absence of creatine phosphate/kinase to scavenge any MgADP generated in the patch, tens and hundreds of one or two opening bursts would be accompanied by one to a few long bursts, as above. Note the occasional long bursts each with 10–100 openings with long mean durations, compared with the more frequent short bursts each with 1–2 openings with short mean durations. Thus, one long burst can have approximately the same number of openings as in 50 or more short bursts.
Fig. 6 shows current recordings demonstrating the occasional prominent long open duration burst (due to ligand-insensitive states in the presence of MgADP) amidst the frequent short duration bursts (with one or two openings) consistently observed in 0.6 mM ATP in the absence of creatine phosphate/kinase. In the absence of creatine phosphate and kinase, long open duration bursts are occasional yet each provides tens of open events, whereas the short open duration bursts each provides only one or two events. Duration histograms were used to compare KATP channel open lifetimes in 0.6 mM ATP in the absence of the scavenger system, in 0.6 mM ATP with the scavenger system, or simply in 150 μM MgADP.
Fig. 7 shows the open time histograms from each of the three nucleotide conditions. The kinetic analysis shows that in high ATP with MgADP generated (no creatine phosphate/kinase added) both short (0.35 ± 0.02; n = 5) and long (1.64 ± 0.02; n = 5) duration openings are observed. In high ATP with MgADP removed, only short (0.34 ± 0.01; n = 5) duration openings occur. When 150 μM MgADP alone is added, only long (1.81 ±0.05 ms; n = 5) duration openings are observed. At very high ATP, the long duration open state bound by MgADP likely represents the ATP-refractory state and was similar in duration to the long open duration bursts in the absence of both adenine nucleoside tri- and diphosphates. The short duration open state likely represents the unliganded, ATP-sensitive state in the presence of MgADP and high ATP, and was kinetically indistinguishable from the short open duration bursts in the presence of high ATP alone. The elimination of the long open durations by the presence of the creatine phosphate/kinase scavenging system suggests that they result from bound MgADP by MgATP hydrolysis at the SUR or elsewhere in the patch.
Figure 7.

At 5 mM MgATP, long mean open times are kinetically comparable to mean open times in either 0 MgATP or 150 μM MgADP. (A) Long and short mean open duration components are clearly detectable when the KATP channel is exposed to 5 mM MgATP. The value of the long time constant is indistinguishable from the single open time component observed for the KATP channel in 0 ATP in the absence of rundown. (B) Only short open duration component is observed for the KATP channel when the creatine phosphate/kinase system is used to scavenge any MgADP generated in the patch. (C) Only long open duration component is observed for the KATP channel when only 150 μM MgADP is added. Thus, in the absence of creatine kinase and creatine phosphate, at ≪5mM MgATP, the ATP-dependent open durations become kinetically indistinguishable from the ATP-refractory, constant long open durations, which should not but might lead to confusion over the ATP dependence of KATP channel open times.
In our previous study, without creatine phosphate/kinase, 92 out of 97 single-channel patches included the long duration bursts, and led to open duration histograms with comparable fractions of short and long openings observed here. In the remaining five patches of that study, no long bursts were observed, and ATP-dependent gating of the open state in the short bursts in high ATP was evident (Drain and Li 2000). Comparison of the histograms in Fig. 7 shows the relationships between short and long duration openings and nucleotide ligand. The short and long open time components in high ATP and moderate MgADP (without creatine phosphate/kinase) correspond to the short open time component in high ATP and no MgADP (with creatine phosphate/kinase), and the long open time component in high MgADP and no ATP (150 μM MgADP added without creatine phosphate/kinase), respectively. The ligand-insensitive bursts of long openings observed in 5 mM MgATP for the pancreatic KATP channel likely result from significant hydrolysis of the 5 mM MgATP to MgADP via NBD2 of SUR1, other cellular ATPases, and mass action (Dzeja and Terzic 1998; Bienengraeber et al. 2000; Carrasco et al. 2001; Zingman et al. 2001).
Taken together, the results thus far imply that the truncated Kir6.2ΔC26 channel expressed in the absence of SUR, and therefore devoid of the mechanism of MgADP antagonism of the ATP-inhibited state, should exhibit ATP-dependent mean open times even in the absence of the creatine phosphate/kinase scavenger system. Fig. 8 shows representative single-channel currents of the Kir6.2ΔC26 channel expressed without SUR and the effect of increasing concentration of ATP applied to the cytoplasmic face. Even in the absence of ATP, the truncated ΔC26 channel exhibited very brief bursts typically comprising one to a few openings. In Fig. 9, the detailed kinetic analysis of the truncated channel without SUR shows that the decrement of mean open duration by increasing ATP holds true. A single kinetic component accounted for the open time durations of the truncated Kir6.2ΔC26 channel without SUR and was significantly decreased by increasing ATP. In 0, 1, and 5 mM ATP, the mean open durations were 1.06 ± 0.03, 0.85 ± 0.03, and 0.47 ± 0.01 (n = 5; P < 0.005), respectively. Both the ATP-dependent open time component and absence of a long invariant long open time component in the truncated Kir6.2ΔC26 channel without SUR support the hypothesis that the long invariant open times require regulation by the presence of SUR. The results support the hypothesis that the long ATP-independent open times reflect MgADP antagonism of the ATP-inhibited state either by preventing ATP binding or its inhibitory action on the channel. Finally, it is worth noting that the 1.06 ± 0.03 ms (n = 5) mean open duration of the truncated Kir6.2ΔC26 channel without SUR is shorter than the 1.66 ± 0.08-ms (n = 9; P < 0.001) mean open duration of the wild-type KATP channel. The results indicates that, in the absence of ATP, the dramatically shortened bursts of the truncated Kir6.2ΔC26 channel without SUR, compared with the wild-type channel with SUR, is at least in part accounted for by a significant decrement in open times.
Figure 8.

ATP dependence of single truncated Kir6.2ΔC26 channels without SUR at 0, 1, and 5 mM ATP. (A) Effect of ATP on open durations. Single-channel currents from a representative inside-out patch within 30 s after patch excision continuously perfused with 0 ATP for 1 min. The patch was then exposed to 1 mM MgATP for 3 min, and finally 5 mM MgATP for 15 min. In between these [MgATP], the channel is exposed briefly to 0 MgATP (to check for rundown in activity) and to 0.6 mM MgATP to help maintain the initial PO activity in 0 MgATP.
Figure 9.
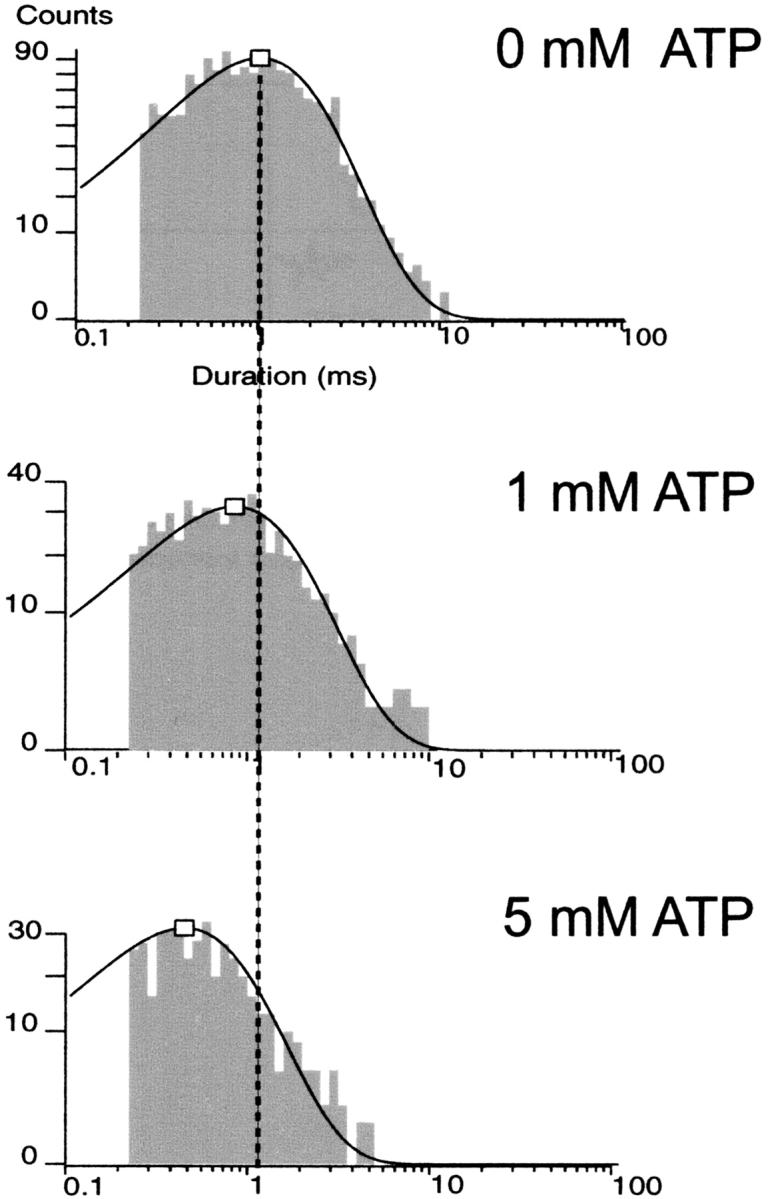
Open duration histograms of truncated Kir6.2ΔC26 channels without SUR at 0, 1, and 5 mM ATP. The log-scale abscissa of the Sine-Sigworth plots results in the peak of these single-exponential distributions positioned at the value of the time constant. The dashed line is placed at the higher time constant (0 ATP) to facilitate comparison with the intermediate and lower time constants at other ATP concentrations. Because the burst durations of the truncated ΔC26 channel without SUR even in 0 ATP are already very brief, the ATP dependence of burst times is not shown.
The Second Order Rate Constant Accounts for ATP Dependence of Both Open and Burst Durations
Fig. 10 reveals the kinetic relationship between open state and burst state of the KATP channel. The ATP dependence of the open state durations shown in this paper indicates unequivocally that ATP can bind and destabilize that state. The quantitative relationship between open state and burst state dependence of the KATP channel suggested to us that perhaps the decrement in open durations by ATP is the major mechanism for the dramatic reduction in burst duration by increasing ATP. We tested whether the measured decrements in mean open state durations quantitatively account for the dramatic decrease in burst durations measured by using simple rate law and burst gating mechanism kinetics (Neher and Steinbach 1978).
The ATP-dependent transition rates from the open to the inhibited interburst state can be calculated directly by the simple rate law that open state duration is equal to the reciprocal of the sum of the rates of all gating transitions from the open state. There are two ATP-independent rates from the open state: (1) the rate to the intraburst closed state (Cf), and (2) the inactive interburst state (C0), which must be considered by the analysis. The O to Cf rate (580 ± 18 s−1) and the O to C0 rate (27 ± 2 s−1) were determined at 0 ATP from mean open durations and mean burst durations, and mean number of openings per burst. By using these first order rate constants, together with the decrementing mean open durations at increasing ATP concentrations, we solved for the second order rate constant  (1,330 ± 75 s−1mM−1). We then used this second order rate constant to calculate the ATP dependence of not only open durations, but also burst durations. The clear result is that the ATP-dependent precipitous reduction in mean burst durations predicted by the measured decremental ATP dependence of mean open durations closely fits the measured reduction in mean burst durations. The solution is a special case of burst kinetics where each rate from the open state can be determined.
(1,330 ± 75 s−1mM−1). We then used this second order rate constant to calculate the ATP dependence of not only open durations, but also burst durations. The clear result is that the ATP-dependent precipitous reduction in mean burst durations predicted by the measured decremental ATP dependence of mean open durations closely fits the measured reduction in mean burst durations. The solution is a special case of burst kinetics where each rate from the open state can be determined.
DISCUSSION
The most important conclusion of these results is that ATP binds to destabilize the open state of the KATP channel as the mechanism of speeding burst exit rates underlying inhibition gating by ATP. There was little or no affect on the intraburst closed state Cf by ATP. Because the burst comprises the open and fast closed state, the results indicate that the decrease in burst duration by ATP involves solely the open state. A crucial result of our study is that the value of 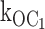 [ATP] 1,330 s−1mM−1 determined by the measured decrement in open duration accounts for the dramatic reduction in burst times with increasing ATP.
[ATP] 1,330 s−1mM−1 determined by the measured decrement in open duration accounts for the dramatic reduction in burst times with increasing ATP.
Any models of KATP channel inhibition gating by ATP that omit the ATP-dependent O to C1 gating transition are untenable. Indeed, the ATP dependence of the open gating conformation likely will be critical to understand the design and action of many physiological and pharmacological regulatory ligands. The mechanism explains why 10 Ki units or more ATP are required to appreciate the decrement in open times as the major mechanism underlying the profound reduction on burst times underlying KATP channel inhibition by ATP. In addition, the numerous regulatory gating states that alter ATP sensitivity, particularly the MgADP bound state, have to be better understood.
The decrement of open durations with increasing ATP of the wild-type pancreatic SUR1/Kir6.2 channel is supported by previously reported results with the truncated Δ26 KATP channel in the absence of SUR1 (Drain et al. 1998) as well as the cardiac SUR2/Kir6.2 channel (Fan and Makielski 1999). The truncated Kir6.2ΔC26 expressed in the absence of SUR1 exhibited mean open durations in 0, 0.5, and 1.0 mM MgATP of 1.1, 0.9, and 0.7 ms, respectively (Drain et al. 1998). We corroborated our earlier results in the present study by showing that in five patches tested, the truncated Kir6.2ΔC26 expressed in the absence of SUR1 exhibited significantly decrementing mean open durations in 0, 1, and 5 mM MgATP of 1.1, 0.8, and 0.5 ms, respectively. The absence of the SUR1 and its ATPases that convert MgATP to MgADP lead to consistent decrement in open durations by increasing ATP and suggested to us the need of the creatine phosphate and kinase scavenging system. The cardiac SUR2/Kir6.2 channel was well-studied for ATP-dependent gating in the inside-out configuration at 0 mV, 140 mM KCl bath, and 10 mM KCl pipet, and filtered at 2 kHz (Fan and Makielski 1999). These conditions and the distinct gating of the cardiac channel result in longer openings than observed for the pancreatic channel studied here. Nevertheless, similar ATP-dependent gating from the open state of the cardiac channel was demonstrated. Taken together, the results on these three channels unequivocally capture the ATP-bound open state of the KATP channel, which is observed as significant decrements in open durations at increasing ATP concentration.
Study of the ATP-dependent inhibition gating of KATP channels demands stationarity. To minimize rundown, we always buffer Ca2+ to 10 nM with EGTA and add adenine nucleotides as the magnesium salts to enable phosphoryl transfer reactions maintaining the initial high activity. Also, to maximize likelihood that each channel begins its recording lifetime in the same regulatory state, we do our experiments identically by using only freshly excised patches with the channel at PO > 0.6, and analyze only those that maintain this high open probability. At the start, during, and at the end of the experiments, we check the open probability briefly in 0 ATP. Obviously, if we also analyzed single KATP channels of low open probability in fresh patches that have short bursts of few openings in 0 ATP, together with those of high initial open probability as in this study, we would have a mixed ensemble and its mechanistic analysis then would be meaningless. Similarly, if the 0-ATP open probability of a channel runs down, even if it is reactivated, we are less assured that the same regulatory state of the channel is being studied.
In the quantitative analysis, we were as direct as possible by using the simple rate law that the open state lifetime is the reciprocal of the sum of the exit rates and membrane potentials where the pancreatic channel provides its regulatory role. Here, as in previous studies, we accounted for a minor but significant component contributed by a ligand-independent transition to the inactive interburst. The inactive interburst closed state designated C0 cannot be due to nominal ATP in the excised patch, as shown here by studying C0 in the G334D Kir6.2 mutant coexpressed with SUR1. This channel virtually eliminates ATP-dependent gating to the interburst with little or no effect on ligand-independent gating to C0. As shown here, the value of the ligand-independent, first order rate constant 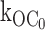 is 25 s−1, which is comparable to 27 s−1 for the wild-type KATP channel. Given the profound loss in ATP-dependent gating by the G334D mutation (Drain et al. 1998), the high similarity of the ligand-independent rates to the inactive interburst C0 for the mutant and wild-type channels excludes the explanation that transitions to the inactive interburst results from nominal ATP in the patch. We conclude that there is a true ligand-independent rate from the open to inactive interburst state
is 25 s−1, which is comparable to 27 s−1 for the wild-type KATP channel. Given the profound loss in ATP-dependent gating by the G334D mutation (Drain et al. 1998), the high similarity of the ligand-independent rates to the inactive interburst C0 for the mutant and wild-type channels excludes the explanation that transitions to the inactive interburst results from nominal ATP in the patch. We conclude that there is a true ligand-independent rate from the open to inactive interburst state 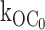 with a value ∼25 s−1.
with a value ∼25 s−1.
KATP channel inhibition gating by ATP, per se, is straightforward ligand-dependent burst kinetics, however, due to physiological MgATP levels and ATPases of SUR and elsewhere, this gating must be understood in the context of MgADP (Alekseev et al. 1998). Previous results indicate that MgUDP directs KATP channel gating to intraburst transitions, and make it insensitive to inhibitory ATP. The results presented here extend this observation by showing MgADP and inhibitory ATP directly compete for the open gating state of the channel. In particular, a KATP channel whose open state is occupied by MgADP features at least two kinetic changes. The rates O to C0 and the O to C1 rates decrease to little or nothing. Only upon unbinding of the MgADP would transitions to the long-lived interburst by ligand-independent gating or ATP-dependent gating be restored (Fig. 10). The simplest mechanisms underlying competition for the open state are direct physical interactions governed by steric hindrance or electrostatic repulsion between inhibitory ATP at the cytoplasmic COOH-terminal tail of Kir6.2 and its antagonist MgADP at NBD2 of SUR. Whether the nucleotides physically compete for the open state or functionally compete via an indirect physical mechanism remains to be determined.
The magnitude of the changes in ATP contribution in either the bulk cytosol or at the inhibitory ATP site of the KATP channel, for example, in glucose-stimulated β cells, are not well determined. We have considered the inhibition gating mechanism in a few of the possible combinations of adenine nucleotide levels. The results show that 5 mM MgATP likely helps to drive hydrolysis to generate ADP to physiological levels. Here, the regulatory response of the channel is to interconvert between ATP-sensitive short and ATP-refractory long open durations. A physiological view of these results is that the KATP channel can readily switch between functionally open states that differ in ATP sensitivity. In conditions of energy abundance, the open state of the KATP channel without MgADP bound can be destabilized by ATP occupancy of its site at the COOH-terminal cytoplasmic tail of Kir6.2 and inhibition prevails. In conditions of energy scarcity, however, the open state of the KATP channel with MgADP bound at its NBD2 site of SUR1, precludes ATP occupancy and inhibition.
Reversible “switching” between these ATP-sensitive states and an ATP-refractory open state likely play a major regulatory role in physiology. Here, open states that have dissociated MgADP can directly bind ATP and speed transition to the ATP-inhibited state, which would require ATP-dependent open state inhibition. The switching between ATP-sensitive and refractory open states provides a molecular mechanism for triggering glucose-stimulated insulin secretion and cardioprotection. For example, the plasma membrane KATP channel may act less like a graded regulator of insulin secretion than like a permissive on/off switch enabling graded insulin release via additional glucose-regulated mechanisms distal in the secretory signaling pathway. Perhaps, the plasma membrane KATP channel physiologically regulates not within the graded middle, but at high mM ATP concentration at the foot, of its ATP-dose inhibition curve.
Molecular and kinetic study of KATP channel states that are stabilized, destabilized, or unaffected by a given regulatory ligand, as done here for open state destabilization by ATP, will be necessary to adequately understand the physiological consequences of the complex functional interactions underlying KATP channel regulation. In the last few years, important regulatory roles of the enzymes adenylate kinase and creatine kinase that regulate the interconversion of adenine nucleotide phosphorylation, and thereby KATP channel gating, have been characterized and developed (Terzic et al. 1994; Dzeja and Terzic 1998; Bienengraeber et al. 2000; Carrasco et al. 2001; Zingman et al. 2001). We have captured the ATP-bound open state of the pancreatic KATP channel, unequivocally demonstrating ATP-dependent and ATP-refractory open states directly, and thereby adenine nucleotide ligand occupancy relationships between long-lived and short-lived open states underlying the metabolic regulation. Our kinetic analysis explains how the dramatically destabilized burst state of the KATP channel is a direct consequence of ATP destabilization of the open state that speeds burst exit to the inhibited interburst state. The open durations decrease significantly only when high ATP makes the variable, ATP-dependent, rates from the open to the inhibited interburst state approach or exceed the always fast transition rates to the closed intraburst state (Drain and Li 2000).
Acknowledgments
We thank Rick Aldrich for valuable discussions and comments, Paul DeWeer for critically improving the manuscript, and Guy Salama for suggesting the use of creatine kinase.
This work was supported by a grant from the National Science Foundation (MCB 9817116) to P. Drain.
Footnotes
Abbreviations used in this paper: KATP, ATP-sensitive potassium; PO, open channel probability.
References
- Aguilar-Bryan L., Nichols C.G., Weschler S.W., Clement J.P.T., Boyd A.E., III, Gonzalez G., Herrera-Sosa H., Nguy K., Bryan J., Nelson D.A. Cloning of the β-cell high-affinity sulfonylurea receptora regulator of insulin secretion. Science. 1995;268:423–426. doi: 10.1126/science.7716547. [DOI] [PubMed] [Google Scholar]
- Aguilar-Bryan L., Bryan J. Molecular biology of adenosine triphosphate-sensitive potassium channels. Endocr. Rev. 1999;20:101–135. doi: 10.1210/edrv.20.2.0361. [DOI] [PubMed] [Google Scholar]
- Alekseev A.E., Brady P.A., Terzic A. A ligand-insensitive state of cardiac ATP-sensitive K+ channels. Basis for channel opening. J. Gen. Physiol. 1998;111:381–394. doi: 10.1085/jgp.111.2.381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F.M., Kakei M. ATP-sensitive K+ channels in rat pancreatic beta-cellsmodulation by ATP and Mg2+ ions. J. Physiol. 1989;416:349–367. doi: 10.1113/jphysiol.1989.sp017765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashcroft F.M., Harrison D.E., Ashcroft S.J. Glucose induces closure of single potassium channels in isolated rat pancreatic β-cells. Nature. 1984;312:446–448. doi: 10.1038/312446a0. [DOI] [PubMed] [Google Scholar]
- Ashcroft F.M., Ashcroft S.J., Harrison D.E. Properties of single potassium channels modulated by glucose in rat pancreatic beta-cells. J. Physiol. 1988;400:501–527. doi: 10.1113/jphysiol.1988.sp017134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babenko A.P., Gonzalez G., Bryan J. Two regions of sulfonylurea receptor specify the spontaneous bursting and ATP inhibition of KATP channel isoforms. J. Biol. Chem. 1999;274:11587–11592. doi: 10.1074/jbc.274.17.11587. [DOI] [PubMed] [Google Scholar]
- Babenko A.P., Gonzalez G., Bryan J. Pharmaco-topology of sulfonylurea receptors. J. Biol. Chem. 2000;275:717–720. doi: 10.1074/jbc.275.2.717. [DOI] [PubMed] [Google Scholar]
- Bienengraeber M., Alekseev A.E., Abraham M.R., Carrasco A.J., Moreau C., Vivaudou M., Dzeja P.P., Terzic A. ATPase activity of the sulfonylurea receptora catalytic function for the KATP channel complex. FASEB J. 2000;14:1943–1952. doi: 10.1096/fj.00-0027com. [DOI] [PubMed] [Google Scholar]
- Carrasco A.J., Dzeja P.P., Alekseev A.E., Pucar D., Zingman L.V., Abraham M.R., Hodgson D.M., Bienengraeber M., Puceat M., Janssen E. Adenylate kinase phosphotransfer communicates cellular energetic signals to ATP-sensitive potassium channels. Proc. Natl. Acad. Sci. USA. 2001;98:7623–7628. doi: 10.1073/pnas.121038198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colquhoun D., Sigworth F.J. Fitting and statistical analysis of single-channel records. In: Sakmann B., Neher E., editors. Single-channel Recording. 2nd ed. Plenum Press; New York: 1995. pp. 483–587. [Google Scholar]
- Conover W.J. Practical nonparametric statistics 2nd ed 1980. John Wiley & Sons, Inc; New York: pp. 344–376 [Google Scholar]
- Cook D.L., Hales C.N. Intracellular ATP directly blocks K+ channels in pancreatic β-cells. Nature. 1984;311:271–273. doi: 10.1038/311271a0. [DOI] [PubMed] [Google Scholar]
- Cook D.L., Satin L.S., Ashford M.L., Hales C.N. ATP-sensitive K+ channels in pancreatic beta-cells. Spare-channel hypothesis. Diabetes. 1988;37:495–498. doi: 10.2337/diab.37.5.495. [DOI] [PubMed] [Google Scholar]
- Drain P., Li L. ATP-dependent and -independent transitions from the open state of KATP channels Biophys. J. 78 2000. 463A(Abstr.) [Google Scholar]
- Drain P., Dubin A.E., Aldrich R.W. Regulation of Shaker K channel inactivation gating by the cAMP-dependent protein kinase. Neuron. 1994;12:1097–1109. doi: 10.1016/0896-6273(94)90317-4. [DOI] [PubMed] [Google Scholar]
- Drain P., Li L., Wang J. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 1998;95:13953–13958. doi: 10.1073/pnas.95.23.13953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dzeja P.P., Terzic A. Phosphotransfer reactions in the regulation of ATP- sensitive K+ channels. FASEB J. 1998;12:523–529. doi: 10.1096/fasebj.12.7.523. [DOI] [PubMed] [Google Scholar]
- Enkvetchakul D., Loussouarn G., Makhina E., Shyng S.L., Nichols C.G. The kinetic and physical basis of K(ATP) channel gatingtoward a unified molecular understanding. Biophys. J. 2000;78:2334–2348. doi: 10.1016/S0006-3495(00)76779-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul D., Loussouarn G., Makhina E., Nichols C.G. ATP interaction with the open state of the KATP channel. Biophys. J. 2001;80:719–728. doi: 10.1016/S0006-3495(01)76051-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan Z., Makielski J.C. Phosphoinositides decrease ATP sensitivity of the cardiac ATP-sensitive K+ channel. A molecular probe for the mechanism of ATP-sensitive inhibition. J. Gen. Physiol. 1999;114:251–269. doi: 10.1085/jgp.114.2.251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gillis K.D., Gee W.M., Hammoud A., McDaniel M.L., Falke L.C., Misler S. Effects of sulfonamides on a metabolite-regulated ATPi-sensitive K+ channel in rat pancreatic B-cells. Am. J. Physiol. 1989;257:C1119–C1127. doi: 10.1152/ajpcell.1989.257.6.C1119. [DOI] [PubMed] [Google Scholar]
- Gribble F.M., Tucker S.J., Ashcroft F.M. The essential role of the Walker A motifs of SUR1 in KATP channel activation by Mg-ADP and diazoxide. EMBO J. 1997;16:1145–1152. doi: 10.1093/emboj/16.6.1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gribble F.M., Tucker S.J., Haug T., Ashcroft F.M. MgATP activates the β cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 1998;95:7185–7190. doi: 10.1073/pnas.95.12.7185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inagaki N., Gonoi T., Clement J.P.T., Namba N., Inazawa J., Gonzalez G., Aguilar-Bryan L., Seino S., Bryan J. Reconstitution of IKATPan inward rectifier subunit plus the sulfonylurea receptor. Science. 1995;270:1166–1170. doi: 10.1126/science.270.5239.1166. [DOI] [PubMed] [Google Scholar]
- John S.A., Monck J.R., Weiss J.N., Ribalet B. The sulphonylurea receptor SUR1 regulates ATP-sensitive mouse Kir6.2 K+ channels linked to the green fluorescent protein in human embryonic kidney cells. J. Physiol. 1998;510:333–345. doi: 10.1111/j.1469-7793.1998.333bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovanovic A., Jovanovic S., Carrasco A.J., Terzic A. Acquired resistance of a mammalian cell line to hypoxia-reoxygenation through cotransfection of Kir6.2 and SUR1 clones. Lab. Invest. 1998;78:1101–1107. [PubMed] [Google Scholar]
- Koster J.C., Sha Q., Shyng S., Nichols C.G. ATP inhibition of KATP channelscontrol of nucleotide sensitivity by the N-terminal domain of the subunit. J. Physiol. 1999;515:19–30. doi: 10.1111/j.1469-7793.1999.019ad.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L., Wang J., Drain P. The I182 region of Kir6.2 is closely associated with ligand binding in KATP channel inhibition by ATP. Biophys. J. 2000;79:841–852. doi: 10.1016/S0006-3495(00)76340-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loussouarn G., Makhina E.N., Rose T., Nichols C.G. Structure and dynamics of the pore of inwardly rectifying KATP channels. J. Biol. Chem. 2000;275:1137–1144. doi: 10.1074/jbc.275.2.1137. [DOI] [PubMed] [Google Scholar]
- Neher E., Steinbach J.H. Local anaesthetics transiently block currents through single acetylcholine-receptor channels. J. Physiol. 1978;277:153–176. doi: 10.1113/jphysiol.1978.sp012267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C.G., Lederer W.J., Cannell M.B. ATP dependence of KATP channel kinetics in isolated membrane patches from rat ventricle. Biophys J. 1991;60:1164–1177. doi: 10.1016/S0006-3495(91)82152-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C.G., Shyng S.L., Nestorowicz A., Glaser B., Clement J.P.T., Gonzalez G., Aguilar-Bryan L., Permutt M.A., Bryan J. Adenosine diphosphate as an intracellular regulator of insulin secretion. Science. 1996;272:1785–1787. doi: 10.1126/science.272.5269.1785. [DOI] [PubMed] [Google Scholar]
- Noma A. ATP-regulated K+ channels in cardiac muscle. Nature. 1983;305:147–148. doi: 10.1038/305147a0. [DOI] [PubMed] [Google Scholar]
- Qin D.Y., Takano M., Noma A. Kinetics of ATP-sensitive K+ channel revealed with oil-gate concentration jump method. Am. J. Physiol. 1989;257:H1624–H1633. doi: 10.1152/ajpheart.1989.257.5.H1624. [DOI] [PubMed] [Google Scholar]
- Shyng S., Ferrigni T., Nichols C.G. Control of rectification and gating of cloned KATP channels by the Kir6.2 subunit. J. Gen. Physiol. 1997;110:141–153. doi: 10.1085/jgp.110.2.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigworth F.J., Sine S. Data transformations for improved display and fitting of single-channel dwell time histograms. Biophys J. 1987;52:1047–1054. doi: 10.1016/S0006-3495(87)83298-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzic A., Findlay I., Hosoya Y., Kurachi Y. Dualistic behavior of ATP-sensitive K+ channels toward intracellular nucleoside diphosphates. Neuron. 1994;12:1049–1058. doi: 10.1016/0896-6273(94)90313-1. [DOI] [PubMed] [Google Scholar]
- Trapp S., Proks P., Tucker S.J., Ashcroft F.M. Molecular analysis of ATP- sensitive K channel gating and implications for channel inhibition by ATP. J. Gen. Physiol. 1998;112:333–349. doi: 10.1085/jgp.112.3.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trube G., Hescheler J. Inward-rectifying channels in isolated patches of the heart cell membraneATP-dependence and comparison with cell-attached patches. Pflügers Arch. 1984;401:178–184. doi: 10.1007/BF00583879. [DOI] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Zhao C., Trapp S., Ashcroft F.M. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 1997;387:179–183. doi: 10.1038/387179a0. [DOI] [PubMed] [Google Scholar]
- Tucker S.J., Gribble F.M., Proks P., Trapp S., Ryder T.J., Haug T., Reimann F., Ashcroft F.M. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 1998;17:3290–3296. doi: 10.1093/emboj/17.12.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zingman L.V., Alekseev A.E., Bienengraeber M., Hodgson D., Krager A.B., Dzeja P.P., Terzic A. Signaling in channel/enzyme multimersATPase transitions in SUR module gate ATP-sensitive K+ conductance. Neuron. 2001;31:233–245. doi: 10.1016/s0896-6273(01)00356-7. [DOI] [PubMed] [Google Scholar]