

Abstract
In Kv channels, an activation gate is thought to be located near the intracellular entrance to the ion conduction pore. Although the COOH terminus of the S6 segment has been implicated in forming the gate structure, the residues positioned at the occluding part of the gate remain undetermined. We use a mutagenic scanning approach in the Shaker Kv channel, mutating each residue in the S6 gate region (T469-Y485) to alanine, tryptophan, and aspartate to identify positions that are insensitive to mutation and to find mutants that disrupt the gate. Most mutants open in a steeply voltage-dependent manner and close effectively at negative voltages, indicating that the gate structure can both support ion flux when open and prevent it when closed. We find several mutant channels where macroscopic ionic currents are either very small or undetectable, and one mutant that displays constitutive currents at negative voltages. Collective examination of the three types of substitutions support the notion that the intracellular portion of S6 forms an activation gate and identifies V478 and F481 as candidates for occlusion of the pore in the closed state.
Keywords: Kv channel, voltage-dependent gating, scanning mutagenesis, pore occlusion, closed gate
INTRODUCTION
Voltage-activated K+ channels, or Kv channels, contain a K+-selective ion conduction pore that opens and closes in response to changes in membrane voltage. Most commonly, membrane depolarization opens these channels and hyperpolarization closes them. Several lines of evidence suggest that the activation gate, where the pore is occluded in the closed state, is located near the intracellular extent of the pore. Early experiments on squid giant axon Kv channels, and more recent studies in the Shaker Kv channel, show that quaternary ammonium blockers can only access their intracellular binding site within the pore when the channel is in the open state (Armstrong, 1969, 1971; Armstrong and Hille, 1972; Holmgren et al., 1997). Once these blockers reach their binding site, they can be trapped if the activation gate is closed by hyperpolarization (Holmgren et al., 1997), suggesting that a gate exists between the blocker binding site in the pore and the intracellular solution.
In the KcsA K+ channel, a proton-gated channel that serves as a structural model for the S5-S6 portion of Kv channels (Doyle et al., 1998; MacKinnon et al., 1998; Lu et al., 2001), the intracellular part of the pore is constructed from the four TM2 α-helices, corresponding to the S6 TMs in Kv channels. These TM2 helices form an inverted tepee-like structure with the smoke-hole pointing in an intracellular direction where the helices bundle together (Fig. 1 B). The crystal structure of KcsA complexed with an electron-dense quaternary antimony compound shows that these blocking ions reside within the cavity above the TM2 helical bundle crossing (Zhou et al., 2001), marking the position of the quaternary ammonium ion binding site studied in Kv channels and suggesting that the activation gate is constructed from the four TM2 helices positioned near the tepee's apex. Electron paramagnetic resonance studies suggest that pH-dependent gating in KcsA involves movements in the intracellular portions of the TM2 helices, consistent with a gate in this region (Perozo et al., 1999; Liu et al., 2001).
Figure 1.

Sequence comparison between KcsA and the pore domain of the Shaker Kv channel. (A) Sequence alignment of the KcsA K+ channel with the pore domain of the Shaker Kv channel. Diagram above indicates secondary structural motifs in KcsA. TM1 and TM2 correspond to S5 and S6 in Kv channels, respectively. Residues in bold are highly conserved between KcsA and Shaker. Circles below Shaker sequence diagram results of gated accessibility studies with thiosulfonates (Liu et al., 1997). Positions with open circles display more rapid reaction in the open state and those with closed circles indicate rapid reaction in either closed or open states. Half-filled circles display intermediate differences in reaction rate between closed and open states. Red underline highlights the regions examined in the present study with the PVP motif highlighted in yellow. (B) Backbone fold of KcsA shown from the side. Positions shown in red and yellow correspond to the residues in S6 that are the focus of the present study. (C) Illustration of several possible phenotypes that might be expected for mutation of residues located in a tightly packed region of the closed state. The hypothetical gate residue is represented with an isoleucine. Mutation to the smaller Ala might allow K+ flux through the closed gate, whereas mutation to a charged residue like Asp might trap the gate open. Substitution with large side chains (e.g., Trp) might occlude the open gate (not illustrated) or sterically trap the gate open.
In the Shaker Kv channel, the state-dependent reaction between water-soluble methanethiosulfonate (MTS)* reagents (applied to the intracellular side) and cysteine residues introduced in the intracellular part of the S6 segment suggest that this region of Kv channels forms an activation gate (Liu et al., 1997). As illustrated in Fig. 1 A, the accessibility of residues COOH-terminal to N482 does not depend strongly on the gating state of the channel, whereas residues NH2-terminal to I477 display much greater reactivity if the channel is open. Cysteine residues introduced at 478 and 479, located between these two stretches, show weaker state-dependent reactivity with MTS reagents. Interestingly, the state-dependence of reactivity for both negatively and positively charged MTS reagents with 474C is similar, arguing that the S6 gate does not act as an electrostatic barrier, but inhibits flux by physical occlusion (del Camino and Yellen, 2001). Relatively strong state-dependent accessibility has also been observed for reaction between 474C and silver, a cysteine-reactive ion that closely resembles a K+ ion, supporting the notion that K+ ions are gated in this region (del Camino and Yellen, 2001). Although the type of movement that occurs in the S6 gate region remains poorly defined, it most likely involves movement of the S6 helices relative to each other because a metal bridge between 476C and H486 in adjacent subunits locks the activation gate open (Holmgren et al., 1998).
Which residues within the intracellular S6 gate are located within the occluding part of the gate? One might expect the answer to be evident in the structure of the KcsA K+ channel since this simple K+ channel can substitute for the S5-S6 region of Kv channels (Lu et al., 2001). However, whether the structure of KcsA represents the open or closed conformation remains unclear, although it appears to be more closed than would allow quaternary ammonium blockers to access the cavity (Zhou et al., 2001), and molecular dynamic simulations suggest that barrier to ion permeation is high (Berneche and Roux, 2000). In addition, there are several reasons to suspect that the intracellular parts of KcsA and Kv channels differ enough to obscure the identity of pore-occluding residues. First, a unique feature of the COOH-terminal part of S6 in Kv channels is the PVP motif, consisting of two highly conserved proline residues separated by a small hydrophobic amino acid (Fig. 1 A). Cross-linking studies in Kv channels suggest that the S6 α-helix is kinked (Holmgren et al., 1998; del Camino et al., 2000), a distortion that may occur at the PVP motif since proline residues tend to destabilize α-helices by breaking the hydrogen bond network (O'Neil and DeGrado, 1990; MacArthur and Thornton, 1991; Blaber et al., 1993; Monne et al., 1999). Second, cysteine residues introduced at 478 in Shaker, a position at the level of the narrow bundle crossing in KcsA, show relatively rapid reaction with even large MTS reagents (del Camino and Yellen, 2001), suggesting that the pore is quite wide at this level in Kv channels. Thus, even though the structure of KcsA and the S5-S6 region of Kv channels are probably quite similar, there may be subtle, yet important, differences in the intracellular gate region.
We reasoned that if side-chains in the COOH-terminal part of S6 occlude the pore in the closed state of the channel, as illustrated in Fig. 1 C, then it might be possible to replace this side chain with a different one that can either no longer serve this function, or that might disrupt the local structure enough to prevent normal gate function. We set out to find mutants with fundamental gating alterations by systematically mutating individual residues in the cytoplasmic gate region of the Shaker Kv channel to small, bulky, and charged residues. Remarkably, we find that many residues are rather insensitive to radical substitution and that mutations at only a few positions result in fundamental gating alterations, making them candidates for occlusion of the pore in the closed state. Preliminary reports of these findings have appeared in (Hackos and Swartz, 2000, 2001).
MATERIALS AND METHODS
Molecular Biology and Channel Expression
All experiments were performed using the Shaker H4 Kv channel (Kamb and et al., 1988) with a deletion of residues 6–46 to remove fast inactivation (Hoshi et al., 1990; Zagotta et al., 1990). Point mutations in Shaker-IR in pBlu-SK+ were generated through sequential PCR, ligation into appropriately digested vectors, and their sequence verified by automated DNA sequencing. All cDNA constructs were linearized with HindIII and transcribed using T7 RNA polymerase. Xenopus laevis oocytes were removed surgically and incubated with agitation for 1–1.5 h in a solution containing (mM): 82.5 NaCl, 2.5 KCl, 1 MgCl2, 5 HEPES, and 2 mg/ml collagenase (Worthington Biochemical Corp.), pH 7.6 with NaOH. Defolliculated oocytes were injected with cRNA and incubated at 17°C in a solution containing (in mM): 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, and 50 μg/ml gentamicin (Invitrogen/GIBCO BRL), pH 7.6 with NaOH for 1–7 d before electrophysiological recording or harvesting of channel protein.
Electrophysiological Recording
Macroscopic ionic and gating currents were recorded from expressed channels using two-electrode voltage clamp recording techniques between 1 and 5 d after cRNA injection using an OC-725C oocyte clamp (Warner Instruments). Unless otherwise noted, oocytes were studied in a 160-μl recording chamber that was perfused with a solution containing (mM): RbCl (50), NaCl (50), MgCl2 (1), CaCl2 (0.3) and HEPES (5), pH 7.6 with NaOH. Data were filtered at 2 kHz (8-pole Bessel) and digitized at 10 kHz. Microelectrode resistances were between 0.2–1.2 MΩ when filled with 3 M KCl. All experiments were performed at room temperature (∼22°C). For most of the conducting mutants, conductance (G)-voltage (V) relations were obtained by measuring tail current amplitude for various strength depolarizations using 50 mM Rb+ as the charge carrier to slow deactivation. The reversal potential under these ionic conditions was approximately −25 mV. Holding voltages were chosen where no steady-state inactivation could be detected, typically −130 to −80 mV. Inward tail currents were elicited by repolarization to negative voltages where the channel closes completely and tail current amplitude measured 1–3 ms after repolarization. For mutants with large rightward shifts in the voltage-activation relationship, outward tail currents were elicited by repolarization to voltages between 0 and 30 mV. In the case of F481W, where gating currents contaminate the tail current, G-V relations were obtained using steady-state currents and calculating G according to G = I/(V − Vrev). For the P475D mutant, which displayed a large leftward shift in the G-V and a large standing conductance, we were unable to hold cells at negative enough voltages (e.g., −150 mV). In these instances we used holding voltages between −110 and −130 mV and then gave 200–400-ms prepulses to −150 mV before depolarizing to obtain voltage-activation relations. For ionic current measurements, the linear capacity, leak, and endogenous currents were subtracted by obtaining voltage-activation relations in the absence and presence of Agitoxin-2 (100 nM to 1 μM). For nonconducting mutants, where gating currents were measured, linear capacity and background currents were subtracted using a P/−4 protocol (Armstrong and Bezanilla, 1974).
Voltage-activation relations were fit with single Boltzmann functions (see Fig. 2) according to:
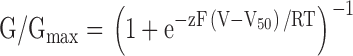 |
where G/Gmax is obtained from normalized tail current amplitudes, z is the equivalent charge, V50 is the half-activation voltage, F is Faraday's constant, R is the gas constant, and T is temperature in Kelvin.
Figure 2.
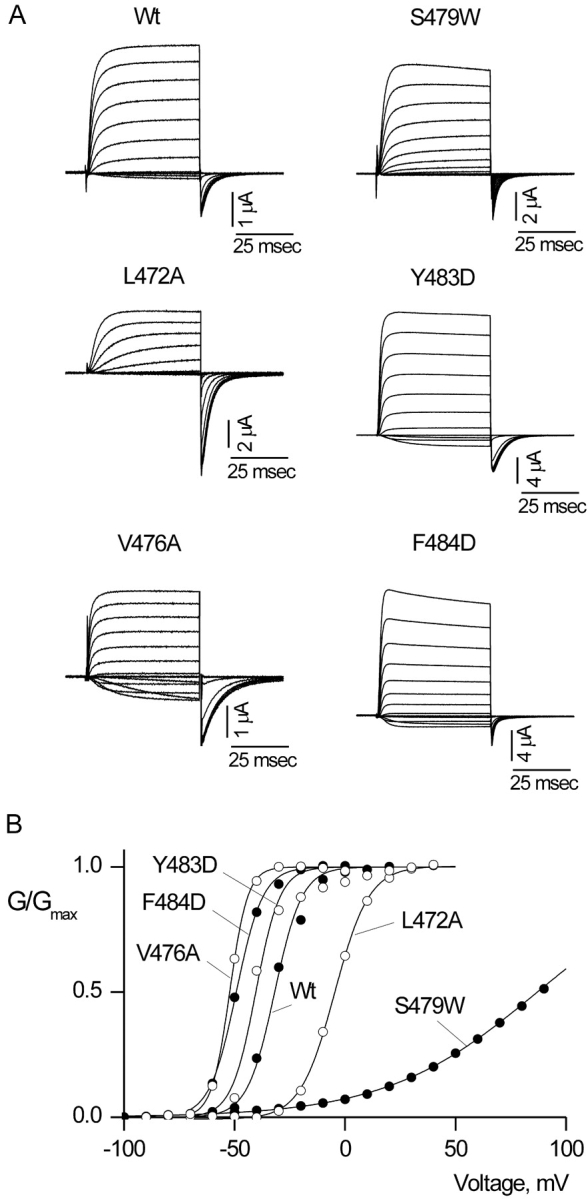
Shaker Kv channel mutants with intact voltage-dependent gating. (A) Families of ionic current records for Wt and five mutant Shaker channels. For Wt Shaker, holding voltage was −90 mV, tail voltage was −70 mV, and depolarizations were from −70 to 50 mV, in 10-mV increments. For S479W, holding voltage was −100 mV, tail voltage was −100 mV, and depolarizations were from −100 to 90 mV, in 10-mV increments. For L472A holding voltage was −100 mV, tail voltage was −70 mV, and depolarizations were from −70 to 40 mV, in 10-mV increments. For Y483D, holding voltage was −90 mV, tail voltage was −70 mV, and depolarizations were from −70 to 40 mV, in 10-mV increments. For V476A holding voltage was −100 mV, tail voltage was −130 mV, and depolarizations were from −90 to 40 mV, in 10-mV increments. For F484D, holding voltage was −100 mV, tail voltage was −80 mV, and depolarizations were from −80 to 50 mV, in 10-mV increments. In all cases, depolarizations were 50 ms in duration. Linear capacity and background conductances were identified and subtracted by blocking the Shaker channel with Agitoxin-2. (B) G-V relations for Wt and five mutants. Normalized tail current amplitudes, measured at voltages indicated in A, are plotted versus the voltage of the preceding depolarization. Smooth curves are single Boltzmann fits to the data with parameters as follows: Wt: V50 = −31.8 mV, z = 4.1; V476A: V50 = −52.2 mV, z = 6.1; F484D: V50 = −49.2 mV, z = 4.2; Y483D: V50 = −40.4 mV, z = 4.6; L472A: V50 = −4.7 mV, z = 3.3; S479W: V50 = 87.7 mV, z = 0.7.
Gating charge (Q)-V relations were obtained by integrating both On and Off components of gating current and then fitting the average Q versus V relations with single Boltzmann functions (see Fig. 3) according to:
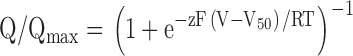 |
where Q/Qmax is the normalized average charge.
Figure 3.

Gating currents and Q-V relations for Shaker Kv channels with nonconducting phenotypes. (A) Families of gating current records for W434F and four other nonconducting mutant Shaker channels. In all cases, membrane voltage was depolarized to various test voltages and then repolarized to the holding voltage. For W434F, holding voltage was −90 mV and depolarizations were from −90 to 0 mV, in 10-mV increments. For V478W, holding voltage was −90 mV and depolarizations were from −80 to 20 mV, in 10-mV increments. For P473A, holding voltage was −100 mV and depolarizations were from –100 mV to −5 mV, in 5-mV increments. For P473W, holding voltage was –110 mV and depolarizations were from −110 to −20 mV, in 10-mV increments. For P475A, holding voltage was −100 mV and depolarizations were from −100 to 0 mV, in 10-mV increments. A P/−4 protocol was used to subtract leak and linear capacitive currents. (B) Normalized Q-V relations for W434F and three other nonconducting mutant Shaker channels. Q was obtained by integrating both the ON and OFF components of gating current, taking their average and normalizing to Qmax measured at depolarized voltages. Smooth curves are single Boltzmann fits to the data with parameters as follows: W434F: V50 = −47.1 mV, z = 4.1; P473W: V50 = −64.1 mV, z = 2.5; P473A: V50 = −55.9 mV, z = 7.0; V478W: V50 = −48.6 mV, z = 4.1; P475A: V50 = −41.1 mV, z =1.7.
Examination of Channel Maturation
We examined the extent of glycosylation (at Asn259 and Asn263) for 10 mutants for which we failed to detect either ionic or gating currents to determine if these mutants are retained in the endoplasmic reticulum (Santacruz-Toloza et al., 1994; Schulteis et al., 1995; Papazian et al., 1995). To detect Shaker protein for the maturation assay a tagged Shaker-IR H4 construct was generated that contained c-myc epitopes (Glu-Gln-Lys-Leu-Ile-Ser-Glu-Glu-Asp-Leu) inserted at the NH2 terminus (after Ala5) and the COOH terminus (after Val638). In addition, a sequence encoding for Arg, Gly, Ser, and 6 His residues was inserted at the COOH terminus immediately after the c-myc epitope. 4 d after cRNA injection a crude membrane fraction was harvested from oocytes using procedures similar to those described previously (Kobertz et al., 2000). Briefly, batches of 30–40 oocytes were homogenized in 1 ml of HEDP buffer (100 mM HEPES, 1 mM EDTA, pH 7.6 with NaOH) plus the following cocktail of protease inhibitors: 0.5 mM phenylmethanosulfonyl fluoride (Sigma-Aldrich), 50 μg/ml antipain (Sigma-Aldrich), 25 μg/ml (4-amidinophenyl) methanesulfonyl fluoride (Sigma-Aldrich), 40 μg/ml bestatin (Sigma-Aldrich), 4 TIU/ml aprotinin (ICN Biomedicals), 0.5 μg/ml leupeptin (Sigma-Aldrich), 0.7 μg/ml pepstatin A (Sigma-Aldrich). Homogenization and all subsequent steps were performed at 4°C. Oocyte homogenates in HEDP buffer were centrifuged at 3,000 g for 10 min. The supernatant was then overlaid on a 15% sucrose cushion prepared in ice-cold HEDP buffer and centrifuged at 175,000 g for 1.5 h. The crude membrane pellet was then solubilized in NuPAGE LDS sample buffer (Invitrogen) with 50 mM DTT and subjected to electrophoresis and Western analysis. Samples (each consisting of ∼4–10 oocytes equivalents of protein) were heated to 70°C for 10 min and loaded onto 10% Nu-PAGE Bis-Tris gel (Invitrogen) using the following solution as running buffer: 50 mM MOPS, 50 mM Tris base, 3.46 mM SDS, and 1 mM EDTA. See Blue Plus 2 (Invitrogen) was used as the protein molecular weight marker. Protein in the gel was then transferred onto nitrocellulose membrane (Amersham Pharmacia Biotech) using a semidry electrotransfer apparatus (E and K Scientific Products), where the transfer buffer consisted of 25 mM bicine, 25 mM bis-tris, 1 mM EDTA, and 10% methanol. After probing the nitrocellulose membrane with mouse antimyc antibody (Invitrogen), protein expression was detected using ECL Western blotting detection reagents (Amersham Pharmacia Biotech).
RESULTS
Our objective in scanning the intracellular gate of the Shaker Kv channel was to establish where substitutions are well tolerated and where they interfere with the ability of the gate to prevent ion flux when closed or to support ion flux when open. To do this, we systematically mutated individual residues from T469 to Y485 (Liu et al., 1997) to a small residue (alanine), a bulky residue (tryptophan), and a charged residue (aspartate). cRNAs encoding the 50 mutant channels were injected into Xenopus oocytes and two-electrode voltage clamp recording techniques were used to evaluate the functional properties of each mutant.
Mutants with Intact Voltage-dependent Activation
We first determined which mutants have intact voltage-dependent gating, which we defined as displaying undetectable macroscopic current at negative voltages and activation of ion selective macroscopic current following membrane depolarization. To identify ionic currents arising from the expressed channel we used Agitoxin-2 (Garcia et al., 1994) to block the Shaker Kv channel so that leak and endogenous currents could be subtracted. Fig. 2 shows current records and G-V relations for five representative mutants and Table I summarizes the gating properties for all 33 mutants with intact voltage-dependent gating. Although the most frequently observed phenotype was a moderate leftward shift in the voltage range over which the channel activates, several mutants (e.g., L472A and S479W) give rise to rightward shifts with dramatic decreases in slope, and one mutation (A471D) displays two distinct components in the voltage-activation relation (Fig. 2, Table I). Even though many of these mutations have quite dramatic effects on voltage-dependent gating, they do not disrupt the ability of the cytoplasmic activation gate to support ion flux when open or for the gate to minimize ion conduction when closed. For one functional mutant (V476D), voltage-dependent activation occurs at extremely negative voltages (V50 ∼ −200 mV), and thus we could not determine whether this mutant has intact voltage-dependent gating.
TABLE I.
Voltage Activation Relations for S6 Mutant Shaker K+ Channels
| Residue | V50 | z | n | Residue | V50 | z | n |
|---|---|---|---|---|---|---|---|
| mV | mV | ||||||
| Wt | −30.5 ± 0.6 | 3.8 ± 0.1 | 37 | ||||
| T469A | −36.3 ± 0.3 | 5.4 ± 0.2 | 12 | V478A | −41.9 ± 0.5 | 4.1 ± 0.2 | 13 |
| T469W | −36.7 ± 1.3 | 5.6 ± 0.3 | 6 | V478W | NC | ||
| T469D | NE | V478D | NE | ||||
| I470A | −17.9 ± 1.2 | 4.3 ± 0.1 | 16 | S479A | −30.3 ± 0.7 | 4.7 ± 0.1 | 11 |
| I470W | −47.1 ± 1.4 | 7.0 ± 0.5 | 6 | S479W | 55.0 ± 9.9 | 1.1 ± 0.2 | 5 |
| I470D | NE | S479D | −58.0 ± 0.8 | 7.0 ± 0.3 | 5 | ||
| A471W | −23.8 ± 0.5 | 3.0 ± 0.08 | 8 | N480A | 50.0 ± 8.5 | 1.0 ± 0.8 | 7 |
| A471Da | −37.0 ± 0.4 | 3.8 ± 0.2 | 11 | N480W | 5.3 ± 2.1 | 1.4 ± 0.07 | 7 |
| 40.5 ± 0.6 | 4.7 ± 0.2 | N480D | −48.5 ± 0.6 | 3.5 ± 0.2 | 12 | ||
| L472A | −4.9 ± 0.3 | 3.4 ± 0.06 | 20 | F481A | NE | ||
| L472W | 50.4 ± 3.6 | 0.5 ± 0.01 | 7 | F481W | −43.7 ± 0.4 | 6.0 ± 0.2 | 16 |
| L472D | NE | F481D | NE | ||||
| P473A | NC | N482A | −36.6 ± 0.2 | 4.8 ± 0.1 | 10 | ||
| P473W | NC | N482W | −47.8 ± 0.3 | 5.4 ± 0.2 | 11 | ||
| P473D | NE | N482D | −32.5 ± 0.4 | 4.3 ± 0.1 | 9 | ||
| V474A | −47.6 ± 0.7 | 5.0 ± 0.5 | 12 | Y483A | −35.4 ± 0.9 | 3.3 ± 0.2 | 8 |
| V474W | NE | Y483W | −31.1 ± 0.6 | 3.8 ± 0.1 | 11 | ||
| V474D | NE | Y483D | −42.1 ± 0.7 | 5.8 ± 0.2 | 8 | ||
| P475A | NC | F484A | −38.8 ± 0.5 | 3.6 ± 0.2 | 12 | ||
| P475W | −49.4 ± 1.8 | 4.0 ± 0.3 | 14 | F484W | −39.0 ± 0.5 | 4.4 ± 0.2 | 12 |
| P475D | −98.5 ± 2.3 | 3.3 ± 0.4 | 10 | F484D | −52.6 ± 0.3 | 4.9 ± 0.1 | 6 |
| V476A | −53.4 ± 1.2 | 6.4 ± 0.1 | 11 | Y485A | −26.1 ± 1.0 | 1.1 ± 0.02 | 11 |
| V476W | −53.4 ± 1.2 | 2.7 ± 0.08 | 11 | Y485W | −43.9 ± 0.7 | 6.3 ± 0.3 | 9 |
| V476Db | Y485D | 67.5 ± 3.4 | 0.9 ± 0.03 | 10 | |||
| I477A | −47.7 ± 0.4 | 3.8 ± 0.09 | 18 | ||||
| I477W | −60.4 ± 0.07 | 6.5 ± 0.6 | 9 | ||||
| I477D | NE |
NE indicates mutants that failed to display ion currents, gating currents, and in the maturation assay were only core glycosylated.
NC indicates mutants that were nonconducting but where gating currents could be observed at high expression levels.
For the A471D mutant, there are two kinetic components evident in the activation time course and two components in the tail current amplitude versus voltage relation. Relatively short depolarizing pulses (≤200 ms) were used to avoid slow inactivation. At some voltages the slow component did not reach steady state, resulting in overestimation of the z value for the second component.
For V476D, we were unable to hold cells at negative enough voltages to define the voltage-activation relation or to determine if the macroscopic conductance becomes minimal at negative voltages. Our rough estimate of the midpoint for the voltage-activation relation for this mutant is approximately −200 mV.
Nonconducting Mutants
There are four mutants that traffic to the plasma membrane, but do not conduct Rb+ or K+ ions over the range of voltages examined (±200 mV). Even though these mutants do not conduct ionic currents, they produce large nonlinear capacitive currents, or gating currents, that result from charge translocation within the voltage sensors. Gating currents obtained for W434F, a previously described nonconducting mutant (Perozo et al., 1993), are shown in Fig. 3 along with gating currents for V478W, P473A, P473W, and P475A. Table II shows the results obtained by fitting Q vs. V relations with single Boltzmann functions. It is interesting that the Q-V relations for V478W and W434F are indistinguishable, whereas the Q-V relations for the other three mutants are very different. For P473A and P473W, the Q-V relations are shifted to negative voltages, whereas in the case of P475A, the Q-V is shifted to positive voltages. All three proline mutants result in significant changes in the slope of the Q-V relation.
TABLE II.
Gating Charge-Voltage Relations for MutantShaker K+ Channels
| Residue | V50 | z | n |
|---|---|---|---|
| mV | |||
| W434F | −47.1 ± 0.4 | 4.2 ± 0.13 | 10 |
| P473W | −61.1 ± 0.64 | 3.2 ± 0.16 | 8 |
| P473A | −55.5 ± 0.36 | 5.3 ± 0.17 | 6 |
| P475A | −35.9 ± 3.1 | 1.5 ± 0.10 | 6 |
| V478W | −47.1 ± 0.12 | 3.8 ± 0.04 | 20 |
| F481W | −50.1 ± 0.37 | 3.5 ± 0.01 | 17 |
A Weakly Conducting Mutant
In the wild-type Shaker K+ channel, macroscopic ionic currents are ∼500 times larger in amplitude than gating currents. For mutation of F481 to tryptophan we observe a very different situation, where the gating currents and macroscopic ionic currents have similar amplitudes, a phenotype that we refer to as weakly conducting. As shown in the leak-subtracted current records in Fig. 4 A, immediately after the depolarizing voltage pulse a fast gating current is observed, followed by a slower ionic current. The ionic current can be blocked with the pore-blocking toxin Agitoxin-2, leaving the gating currents unaffected (Fig. 4 B). Weakly conducting phenotypes have been reported for several mutants in S5 (Kanevsky and Aldrich, 1999; Li-Smerin et al., 2000b). In these previous reports, the G-V relations are shifted toward more depolarized voltages, one of several findings suggesting that the final concerted opening transition has been greatly altered. However, in the case of F481W, the G-V relation has a modest leftward shift (<15 mV) and the Q-V relation is nearly indistinguishable from the control channel (Fig. 4, C and D). This suggests that both the early voltage-dependent transitions and the final weakly voltage-dependent concerted transition (Hoshi et al., 1994; Zagotta et al., 1994a,b; Schoppa and Sigworth, 1998a, 1998b,c) remain relatively unaltered in the F481W mutation.
Figure 4.

Ionic and gating currents for a weakly conducting mutant Shaker Kv channel. (A) Family of ionic current records. Holding voltage was −100 mV, tail voltage was −100 mV, and depolarizations were from −100 to 50 mV, in 10-mV increments. A P/−4 protocol was used to subtract leak and linear capacitive currents. (B) Family of gating currents recorded in the presence of 1 μM Agitoxin-2. Membrane voltage was depolarized for 30 ms to various test voltages (−100 to 10 mV) and then repolarized to the holding voltage (−100 mV). A P/−4 protocol was used to subtract leak and linear capacitive currents. (C) G-V relations for Wt and F481W. Conductance was calculated from the amplitude of steady-state current before repolarization, normalized and plotted versus voltage. Smooth curves are single Boltzmann fits to the data with parameters as follows: Wt: V50 = −32.8 mV, z = 3.7; F481W: V50 = −41.3 mV, z = 4.4. (D) Normalized Q-V relations for W434F and F481W. Q was obtained by integrating both the ON and OFF components of gating current, taking their average and normalizing to Qmax measured at depolarized voltages. Smooth curves are single Boltzmann fits to the data with parameters as follows: W434F: V50 = −47.2 mV, z = 4.1; F481W: V50 = −50.2 mV, z = 3.6.
A Constitutively Conducting Mutant
The final remaining functional mutant is unique in that it displays robust constitutive conduction at negative membrane voltages. In contrast to the wild-type channel, where macroscopic currents become negligible at negative voltages, P475D channels conduct K+ and Rb+ at all voltages examined (±200 mV). Fig. 5, C and D, show current records for P475D with K+ as charge carrier in the external solution and endogenous K+ as the charge carrier internally. The large standing current at −150 mV can be blocked with Agitoxin-2, indicating that it arises from the Shaker Kv channel rather than from endogenous channels (Fig. 5 A). (The currents shown in Fig. 5 A were obtained by subtracting currents recorded in the presence of Agitoxin-2 from those obtained in the absence of toxin.) Although there is a standing current at negative voltages, the P475D mutant does display voltage-dependent changes in macroscopic conductance. When fit with a single Boltzmann function, this voltage-dependent increase in conductance has a midpoint at −98 mV and a slope of 3.3, implying that this phenomenon is coupled to movement of gating charge. Thus, although the channel cannot prevent ion conduction at negative voltages, some form of gating remains intact.
Figure 5.

Constitutive conduction in the P475D mutant of the Shaker Kv channel. (A) Families of ionic currents for Wt, P475W, and P475D. For Wt, holding voltage was −90 mV, tail voltage was −70 mV, and depolarizations were from −70 to 90 mV, in 10-mV increments. For P475W, holding voltage was −100 mV, tail voltage was −100 mV, and depolarizations were from −100 to 50 mV, in 10-mV increments. For P475D, the holding voltage was −110 mV and membrane voltage was stepped to −150 mV for 200 ms before recording the traces shown. Tail voltage was −150 mV, and depolarizations were from −150 to 20 mV, in 10-mV increments. In the case of P475D, the extracellular solution contained 50 mM K+ instead of Rb+. Linear capacity and background conductances were identified and subtracted by blocking the Shaker channel with Agitoxin-2. Dashed line indicates the position of zero current. (B) G-V relations for Wt, P475W, and P475D. Normalized tail current amplitudes are plotted versus the voltage of the preceding depolarization. Smooth curves are single Boltzmann fits to the data with parameters as follows: Wt: V50 = −31.0 mV, z = 4.2; P475W: V50 = −50.9 mV, z = 3.2; P475D: V50 = −103.2 mV, z = 3.0.
Mutants with Defective Maturation
There are a total of 10 mutant channels that show no functional activity. We examined whether these mutants are retained in the endoplasmic reticulum or whether they traffic to the plasma membrane but are nonfunctional. The extent of glycosylation has been used as a marker for retention of mutant Shaker channels in the endoplasmic reticulum, with the retained channels only showing core glycosylation (Santacruz-Toloza et al., 1994; Papazian et al., 1995; Schulteis et al., 1995). Fig. 6 shows a Western blot of SDS polyacrylamide gels where the extent of glycosylation is revealed. The wild-type protein has two dominant forms, a core-glycosylated species (band ∼70 kD) and a more heavily glycosylated mature form (broad band at ∼100 kD). The N259Q/N263Q double mutant shows only a single band at ∼65 kD, marking the position of the unglycosylated protein. The nonconducting mutants (i.e., P473W, V478W) and F481W show quantities of mature protein that are similar to the wild-type channel. Although the levels of mature protein are low for P473A, they are readily evident. All 10 electrophysiologically silent mutants display only the 70-kD form, suggesting that they are retained within the endoplasmic reticulum and explaining why they do not produce ionic or gating currents.
Figure 6.

Mutant Shaker Kv channels with defects in maturation. Western blots from SDS poylacrylamide gels of c-myc–tagged Shaker protein obtained from crude oocyte membrane preparations. Each lane contains between 4 to 10 oocyte equivalents of Shaker protein. Wild-type protein has two dominant forms, a core-glycosylated species (band ∼70 kD) and a more heavily glycosylated mature form (broad band at ∼100 kD). The N259Q/N263Q double mutant shows only a single band at ∼65 kD and marks the position of the unglycosylated protein. Numbers to the left are molecular weight markers in kD. See materials and methods for details.
DISCUSSION
In the present study we mutated 17 residues in the cytoplasmic activation gate region in the Shaker Kv channel to small, bulky, and charged residues to better understand where ion conduction is prevented in the closed state. To view the results collectively we categorized the mutations by phenotype and then examined both their position and substitution dependence (Fig. 7). It is apparent that there are a large number of positions that are relatively insensitive to at least two of the three types of radical substitutions. At positions T469, I470, L472, V476, and I477, only mutations to aspartate are disruptive, in most cases preventing maturation of the channel, and at positions A471, S479, N480, N482, Y483, F484, and Y485, all three substitutions are well tolerated. Although these mutants have intact voltage-dependent activation gating, it is interesting that most of them display G-V relations that are shifted to more negative voltages. Since most of these mutants can be expected to disrupt local side chain packing, the trend whereby mutations shift the closed-open equilibrium in favor of the open state suggests that the closed conformation of the S6 gate is more tightly packed (and thus is more disrupted) than the open state. The most sensitive positions within the closed gate structure are likely to be those that lie within the occluding region of the pore at the axis of fourfold symmetry, where the channel must accommodate four substituted residues in a relatively small volume. Thus, although the tolerant positions identified above probably influence the packing of the closed gate, they are probably not located in the occluding part of the gate structure because they can open and close with only relatively small energetic perturbations. Although it is conceivable that additional gates elsewhere in the protein might mask a more pronounced defect in some of these S6 gate mutants, the tolerant positions stand in contrast to three residues in the PVP motif and both V478 and F481, where we see phenotypes of the sort one might predict for mutations in the occluding part of the gate structure (see residues within red boxes in Fig. 7).
Figure 7.

Summary of phenotypes observed for mutations in the intracellular part of the S6 segment. Sequence alignment between TM2 of KcsA K+ channel and S6 of the Shaker Kv channel with highly conserved residues shown in black. As in Fig. 1, the circles below Shaker sequence diagram results of gated accessibility studies with thiosulfonates (Liu et al., 1997). Functionally expressing mutant phenotypes are indicated with gray circles containing different color filling and nonexpressing mutants are shown with solid maroon circles.
The PVP motif is clearly an important region within the S6 TM, as most substitutions result in rather dramatic phenotypes. For example, three mutants in this motif (P473D, V474D/W) result in retention of the protein in the endoplasmic reticulum and three other mutants (P473A, P473W, and P475A) result in nonconducting channels where the Q-V relations are significantly different than the control channel. It is possible that these mutants appear to be nonconducting because their G-V relations are shifted to very depolarized voltages, although this seems remote since we see no evidence of ionic currents following depolarization to voltages as positive as 200 mV. One possible explanation for these nonconducting mutants is that the channel is predominantly found in an inactivated state, similar to what has been shown for the W434F mutation (Yang et al., 1997). This mechanism is intriguing given the extracellular position of W434 and the intracellular position of the PVP motif. Another possibility is that the gate no longer supports ion conduction even when in the open conformation, perhaps because it is occluded. Although it seems unlikely that the gate has been directly plugged by the substituted residue because mutations to both smaller (P473A, P475A) and larger (P473W) side chains gives rise to similar phenotypes, it is possible that the structure of a nearby occluding region has been rendered nonconducting by propagated structural changes.
One of the most remarkable phenotypes found for mutations in the PVP motif is the constitutive conduction seen in P475D. Although the macroscopic conductance in P475D does not depend on voltage between −200 and −150 mV (unpublished data), at more positive voltages there are small, but clear, voltage-dependent increases in macroscopic conductance. This suggests that although the gate cannot prevent ion conduction at negative voltages, there are voltage-dependent changes in the structure of the ion-conduction pore. What is the mechanism underlying the constitutive conduction in P475D mutant channels? In the wild-type Shaker Kv channel, the gate has a high open probability (∼0.85) when the voltage sensors are activated, and an exceedingly low open probability (<10−7) at negative voltages where the voltage-sensors are at rest, indicating very strong coupling between conformational changes in the voltage sensors and the gate (Islas and Sigworth, 1999). Thus, one attractive explanation for the constitutive conduction is that the strength of coupling between the two domains of the protein is much weaker in P475D. An alternate possibility is that the cytoplasmic activation gate may be leaky to ions, perhaps because the structure of the “closed” activation gate has been perturbed in such a way that it is no longer able to limit ion conduction. Even though the PVP motif probably serves an important role in the structure of the gate and perhaps in the way it couples to the voltage-sensors, it seems unlikely that side chains in the PVP motif directly participate in occlusion of the pore in the closed state. Substitution of cysteine residues at V474, the middle residue in the PVP motif, coordinate Cd2+ with high affinity (arguing for the participation of multiple cysteine residues in coordination) with little change in affinity between open and closed states, suggesting that V474 does not move very much during activation (Webster and Yellen, 2000). It seems unlikely that side chains in the PVP motif could be involved in pore occlusion if the S6 TM moves about a pivot point at V474.
If occlusion occurs below the level of the PVP motif, the present results suggest that V478 and F481, two of the most conserved residues in S6, are prime candidates for residues located at the occluding part of the pore. At 478 the aspartate mutant is retained in the endoplasmic reticulum, the tryptophan mutant is nonconducting, and only the relatively conservative substitution to alanine gives rise to channels with intact voltage-dependent gating. Since the nonconducting phenotype in V478W is observed with a mutation that increases side chain volume, an interesting possibility for V478W is that the gate is occluded even when in the open conformation. At F481, mutations to alanine or aspartate result in channels that do not mature, and the relatively conservative substitution to tryptophan results in a weakly conducting channel that has essentially unperturbed gating charge movement and voltage-dependent activation (Fig. 4). One very interesting explanation for this mutant is that the single channel conductance in the open state is ∼500-fold lower than for the wild-type channel, as if the gate is largely occluded when open. Although our results are consistent with the involvement of V478 and F481 in occlusion of the pore, another recent study shows that quite large MTS reagents react rapidly with cysteine residues introduced at V478 with only relatively weak state-dependence (del Camino and Yellen, 2001). If the relatively unfettered accessibility of position 478 reflects a wide intracellular entrance to the pore immediately below this level, as these authors suggest, then perhaps V478 occludes this end of the pore in the closed state. Alternately, the weak state-dependence for accessibility at V478 might result from complex access pathways for MTS reagents or possibly from the rapid reaction between MTS reagents and additional gating states of the channel. In either of these cases, F481 would remain an interesting candidate for occlusion of the pore in the closed state.
Acknowledgments
We thank Miguel Holmgren, Mark Mayer, and Zhe Lu for helpful discussions, and J. Nagle and D. Kauffman in the National Institute of Neurological and Disorders and Stroke DNA sequencing facility for DNA sequencing.
Footnotes
Abbreviation used in this paper: MTS, methanethiosulfonate.
References
- Armstrong, C.M. 1969. Inactivation of the potassium conductance and related phenomena caused by quaternary ammonium ion injection in squid axons. J. Gen. Physiol. 54:553–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M. 1971. Interaction of tetraethylammonium ion derivatives with the potassium channels of giant axons. J. Gen. Physiol. 58:413–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and F. Bezanilla. 1974. Charge movement associated with the opening and closing of the activation gates of the Na channels. J. Gen. Physiol. 63:533–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong, C.M., and B. Hille. 1972. The inner quaternary ammonium ion receptor in potassium channels of the node of Ranvier. J. Gen. Physiol. 59:388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berneche, S., and B. Roux. 2000. Molecular dynamics of the KcsA K(+) channel in a bilayer membrane. Biophys. J. 78:2900–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blaber, M., X.J. Zhang, and B.W. Matthews. 1993. Structural basis of amino acid alpha helix propensity. Science. 260:1637–1640. [DOI] [PubMed] [Google Scholar]
- del Camino, D., and G. Yellen. 2001. Tight steric closure at the intracellular activation gate of a voltage-gated k(+) channel. Neuron. 32:649–656. [DOI] [PubMed] [Google Scholar]
- del Camino, D., M. Holmgren, Y. Liu, and G. Yellen. 2000. Blocker protection in the pore of a voltage-gated K+ channel and its structural implications. Nature. 403:321–325. [DOI] [PubMed] [Google Scholar]
- Doyle, D.A., J.M. Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Garcia, M.L., M. Garcia-Calvo, P. Hidalgo, A. Lee, and R. MacKinnon. 1994. Purification and characterization of three inhibitors of voltage-dependent K+ channels from Leiurus quinquestriatus var. hebraeus venom. Biochemistry. 33:6834–6839. [DOI] [PubMed] [Google Scholar]
- Hackos, D.H., and K.J. Swartz. 2000. Mutations of a conserved proline in the inner helix of the pore domain of the Shaker K+ channel with altered gating properties. Biophys. J. 78:398a. [Google Scholar]
- Hackos, D.H., and K.J. Swartz. 2001. Disrupting the cytoplasmic gate in the Shaker K+ channel with mutations in S6. Biophys. J. 80:182a. [Google Scholar]
- Holmgren, M., K.S. Shin, and G. Yellen. 1998. The activation gate of a voltage-gated K+ channel can be trapped in the open state by an intersubunit metal bridge. Neuron. 21:617–621. [DOI] [PubMed] [Google Scholar]
- Holmgren, M., P.L. Smith, and G. Yellen. 1997. Trapping of organic blockers by closing of voltage-dependent K+ channels: evidence for a trap door mechanism of activation gating. J. Gen. Physiol. 109:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1990. Biophysical and molecular mechanisms of Shaker potassium channel inactivation. Science. 250:533–538. [DOI] [PubMed] [Google Scholar]
- Hoshi, T., W.N. Zagotta, and R.W. Aldrich. 1994. Shaker potassium channel gating. I: Transitions near the open state. J. Gen. Physiol. 103:249–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Islas, L.D., and F.J. Sigworth. 1999. Voltage sensitivity and gating charge in Shaker and Shab family potassium channels. J. Gen. Physiol. 114:723–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamb, A., J. Tseng-Crank, and M.A. Tanouye. 1988. Multiple products of the Drosophila Shaker gene may contribute to potassium channel diversity. Neuron. 1:421–430. [DOI] [PubMed] [Google Scholar]
- Kanevsky, M., and R.W. Aldrich. 1999. Determinants of voltage-dependent gating and open-state stability in the S5 segment of Shaker potassium channels. J. Gen. Physiol. 114:215–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobertz, W.R., C. Williams, and C. Miller. 2000. Hanging gondola structure of the T1 domain in a voltage-gated K(+) channel. Biochemistry. 39:10347–10352. [DOI] [PubMed] [Google Scholar]
- Li-Smerin, Y., D.H. Hackos, and K.J. Swartz. 2000. b. A localized interaction surface for voltage-sensing domains on the pore domain of a K+ channel. Neuron. 25:411–423. [DOI] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Liu, Y.S., P. Sompornpisut, and E. Perozo. 2001. Structure of the KcsA channel intracellular gate in the open state. Nat. Struct. Biol. 8:883–887. [DOI] [PubMed] [Google Scholar]
- Lu, Z., A.M. Klem, and Y. Ramu. 2001. Ion conduction pore is conserved among potassium channels. Nature. 413:809–813. [DOI] [PubMed] [Google Scholar]
- MacArthur, M.W., and J.M. Thornton. 1991. Influence of proline residues on protein conformation. J. Mol. Biol. 218:397–412. [DOI] [PubMed] [Google Scholar]
- MacKinnon, R., S.L. Cohen, A. Kuo, A. Lee, and B.T. Chait. 1998. Structural conservation in prokaryotic and eukaryotic potassium channels. Science. 280:106–109. [DOI] [PubMed] [Google Scholar]
- Monne, M., M. Hermansson, and G. von Heijne. 1999. A turn propensity scale for transmembrane helices. J. Mol. Biol. 288:141–145. [DOI] [PubMed] [Google Scholar]
- O'Neil, K.T., and W.F. DeGrado. 1990. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 250:646–651. [DOI] [PubMed] [Google Scholar]
- Papazian, D.M., X.M. Shao, S.A. Seoh, A.F. Mock, Y. Huang, and D.H. Wainstock. 1995. Electrostatic interactions of S4 voltage sensor in Shaker K+ channel. Neuron. 14:1293–1301. [DOI] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Perozo, E., R. MacKinnon, F. Bezanilla, and E. Stefani. 1993. Gating currents from a nonconducting mutant reveal open-closed conformations in Shaker K+ channels. Neuron. 11:353–358. [DOI] [PubMed] [Google Scholar]
- Santacruz-Toloza, L., Y. Huang, S.A. John, and D.M. Papazian. 1994. Glycosylation of shaker potassium channel protein in insect cell culture and in Xenopus oocytes. Biochemistry. 33:5607–5613. [DOI] [PubMed] [Google Scholar]
- Schoppa, N.E., and F.J. Sigworth. 1998. a. Activation of shaker potassium channels. I. Characterization of voltage-dependent transitions. J. Gen. Physiol. 111:271–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa, N.E., and F.J. Sigworth. 1998. b. Activation of Shaker potassium channels. II. Kinetics of the V2 mutant channel. J. Gen. Physiol. 111:295–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoppa, N.E., and F.J. Sigworth. 1998. c. Activation of Shaker potassium channels. III. An activation gating model for wild-type and V2 mutant channels. J. Gen. Physiol. 111:313–342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulteis, C.T., S.A. John, Y. Huang, C.Y. Tang, and D.M. Papazian. 1995. Conserved cysteine residues in the shaker K+ channel are not linked by a disulfide bond. Biochemistry. 34:1725–1733. [DOI] [PubMed] [Google Scholar]
- Webster, S.M., and G. Yellen. 2000. Position V474 in the S6 of Shaker potassium channels may act as a pivot-point for movements associated with channel opening. Biophysical J. 80:16a. [Google Scholar]
- Yang, Y., Y. Yan, and F.J. Sigworth. 1997. How does the W434F mutation block current in Shaker potassium channels? J. Gen. Physiol. 109:779–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1990. Restoration of inactivation in mutants of Shaker potassium channels by a peptide derived from ShB. Science. 250:568–571. [DOI] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, and R.W. Aldrich. 1994. a. Shaker potassium channel gating. III: Evaluation of kinetic models for activation. J. Gen. Physiol. 103:321–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagotta, W.N., T. Hoshi, J. Dittman, and R.W. Aldrich. 1994. b. Shaker potassium channel gating. II: Transitions in the activation pathway. J. Gen. Physiol. 103:279–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, M., J.H. Morais-Cabral, S. Mann, and R. MacKinnon. 2001. Potassium channel receptor site for the inactivation gate and quaternary amine inhibitors. Nature. 411:657–661. [DOI] [PubMed] [Google Scholar]