

Abstract
All members of the inward rectifiier K+ (Kir) channel family are activated by phosphoinositides and other amphiphilic lipids. To further elucidate the mechanistic basis, we examined the membrane association of Kir6.2 fragments of KATP channels, and the effects of site-directed mutations of these fragments and full-length Kir6.2 on membrane association and KATP channel activity, respectively. GFP-tagged Kir6.2 COOH terminus and GFP-tagged pleckstrin homology domain from phospholipase C δ1 both associate with isolated membranes, and association of each is specifically reduced by muscarinic m1 receptor–mediated phospholipid depletion. Kir COOH termini are predicted to contain multiple β-strands and a conserved α-helix (residues ∼306–311 in Kir6.2). Systematic mutagenesis of D307-F315 reveals a critical role of E308, I309, W311 and F315, consistent with residues lying on one side of a α-helix. Together with systematic mutation of conserved charges, the results define critical determinants of a conserved domain that underlies phospholipid interaction in Kir channels.
Keywords: K+ current, KATP, PIP2, Kir6.2, PH domain
INTRODUCTION
The opening and closing of ion channel pores is controlled by conformational changes in other parts of the protein, in response to changes in external stimuli. Activities of inward rectifier K+ (Kir) channels are modulated by diverse second messengers, including ATP (Kir6 sub-family) (Inagaki et al., 1995), pH (Kir1, Kir4 sub-family) (Fakler et al., 1996; Pearson et al., 1999; Yang and Jiang, 1999), and G-proteins (Kir3 sub-family) (Reuveny et al., 1994; Huang et al., 1995), but common to all is control of activity by polyphosphoinositides (PPIs),* particularly phosphatidylinositol-4,5-bisphosphate (PIP2) (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Rohacs et al., 1999; Zhang et al., 1999; Hilgemann et al., 2001). In essence, the “physiological” second messengers all act by augmentation of, or competition with, the activating effects of PPI, and when these are depleted from the membrane, Kir channel activity disappears (Baukrowitz et al., 1998a; Huang et al., 1998; Shyng and Nichols, 1998; Liou et al., 1999; Zhang et al., 1999). Evidence is accumulating to suggest that PPI, in particular PIP2, interact directly with the conserved cytoplasmic regions of the channel and that associated conformational changes are allosterically coupled to pore opening (Baukrowitz et al., 1998a; Huang et al., 1998; Shyng and Nichols, 1998; Rohacs et al., 1999; Enkvetchakul et al., 2000; Soom et al., 2001). Using site-specific mutagenesis, several sites, particularly within the COOH termini of Kirs, have been shown to be involved in activation by PPI (Baukrowitz et al., 1998a; Huang et al., 1998; Shyng and Nichols, 1998; Rohacs et al., 1999; Zhang et al., 1999; Shyng et al., 2000).
All cation channel superfamily members consist of a tetrameric arrangement of homomeric or similar domains, which may be in individual subunits, or covalently linked. Kir channels are composed of four subunits, each containing two transmembrane segments (M1, M2), a P-loop between M1 and M2, and cytoplasmic NH2 and COOH termini (Nichols and Lopatin, 1997; Doyle et al., 1998). The M2 helices line the inner vestibule of the channel and it has been proposed that this region behaves as a gate, governing the flow of ions through the channel and across the membrane (Liu et al., 1997; Perozo et al., 1999; Loussouarn et al., 2001). Approximately 40 amino acids preceding M1, and 150 amino acids after M2 are highly conserved in Kir channels, but the extreme NH2 and COOH termini are not. There is currently no secondary or tertiary structural information regarding Kir channels, so despite the detailed picture that is emerging of the pore itself (Doyle et al., 1998), and potential mechanisms of pore opening, the structural basis for the control of gating by intracellular domains is unknown. Using a combination of electrophysiological and biochemical approaches, we show that a conserved cytoplasmic region of Kir channels interacts with cell membranes, and propose that Kir channels contain a unique lipid interaction (KIRLI) domain that may conserve structurally significant features of phospholipid interacting PH domains.
MATERIALS AND METHODS
Molecular Biology
Point mutations were prepared by overlap extension at the junctions of the relevant residues by sequential PCR. Resulting PCR products were subcloned into pCMV6b vector. Before transfection, constructs were sequenced to verify the mutations.
Expression in COSm6 Cells
COSm6 cells were plated at a density of ∼2.5 × 105 cells/well (30 mm six-well dishes) and cultured in Dulbecco's modified Eagle medium plus 10 mM glucose (DMEM-HG), supplemented with FCS (10%). The following day, cells were transfected by adding FUGENE (Roche Applied Science) and 1 μg each of pCMV6b-Kir6.2 or mutant isoforms, pECE-SUR1 cDNA, and pECE–green fluorescent protein (GFP) directly to the media. The cells were split the next day onto coverslips for patching. For cell fractionation experiments, cells were plated on 60-mm diameter dishes. Transfection was performed using Lipofectamine (Invitrogen) and 12 μg of GFP fusion protein DNA per dish in DMEM-HG. The next day the DMEM-HG was replaced with complete media. All experiments were performed 36 h posttransfection.
Hypotonic Lysis and Fractionation
Transfected cells were grown to confluence in 60-mm dishes. The cells were washed twice with PBS (37°C) and scraped into PBS (4°C), pelleted by centrifugation, and resuspended in 1 ml hypotonic solution (5 mM Tris, pH 7.5, 1 mM MgCl2, 1 mM EGTA, 0.1 mM EDTA) containing mini COMPLETE protease inhibitors (Boehringer Mannheim). The cells swelled on ice for 45 min and were then passaged 60 times through a ball bearing homogenizer and centrifuged for 10 min at 1,000 g to remove nuclear debris and unbroken cells. The postnuclear lysate was centrifuged at 100,000 g (55,000 rpm in a TLA-100 rotor; Beckman) for 1 h. The pellet was resuspended in an equal volume of hypotonic solution with protease inhibitors and passed through a 21-gauge needle six times for resuspension. For cells treated with acetylcholine (ACh) in Fig. 5, 100 μM ACh was applied 15 min before swelling and lysis, and ACh was present throughout the remainder of the fractionation.
Figure 5.
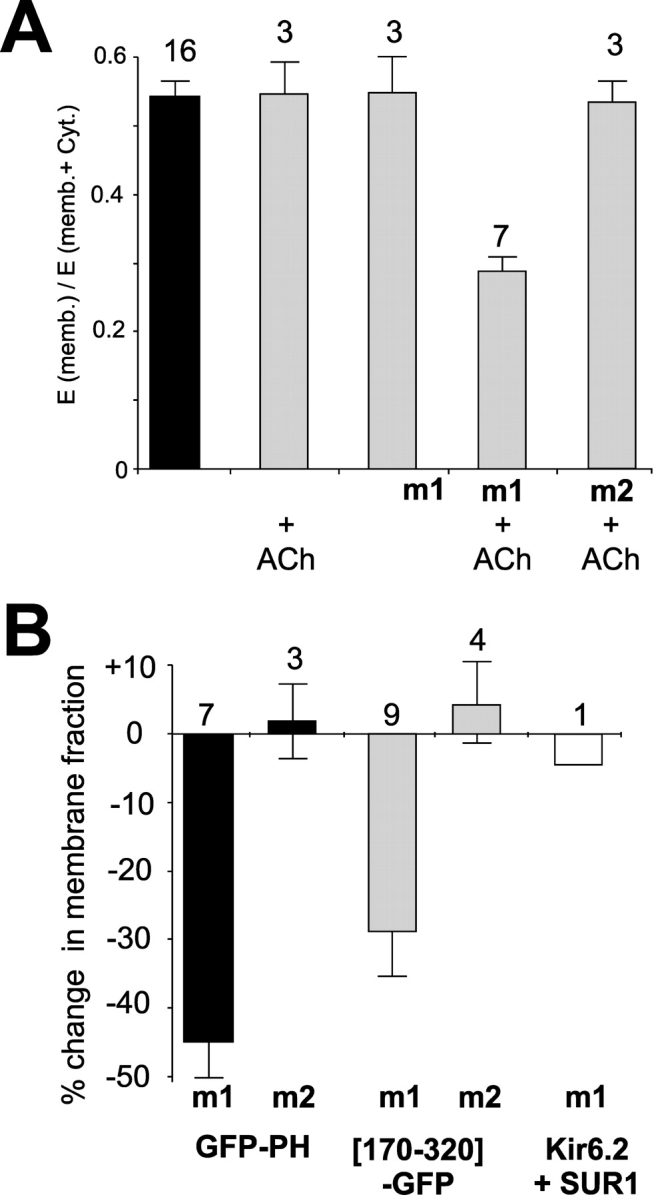
Specific decrease of membrane association following M1 receptor activation. (A) Fraction of fluorescence in membrane (mean ± SEM) from cells expressing GFP-PH and m1 or m2 receptors in the presence or absence of ACh, as indicated. (B) Percent change in fraction of fluorescence in the membrane upon addition of ACh for GFP-PH or channel constructs coexpressed with m1 or m2 receptors, as indicated (mean ± SEM, n indicated).
Fluorescence Measurements
Each 1 ml fraction of either supernatant or pellet was excited at 467 nm in a fluorometer. Emission spectra (480–600 nm) of buffer alone and of untransfected cells were collected for each experiment. GFP fluorescence spectra were obtained by subtraction of the appropriate spectrum from untransfected samples and normalized to protein density estimated from E480, i.e., adjusted spectra = (raw spectra – buffer spectra) − constant × (control spectra – buffer spectra). The fractional fluoresecence in the membrane (E[memb]/E[memb + Cyt]), reported in Figs. 4 and 5, was calculated from the emission at 518 nm, with excitation at 467 nm.
Figure 4.
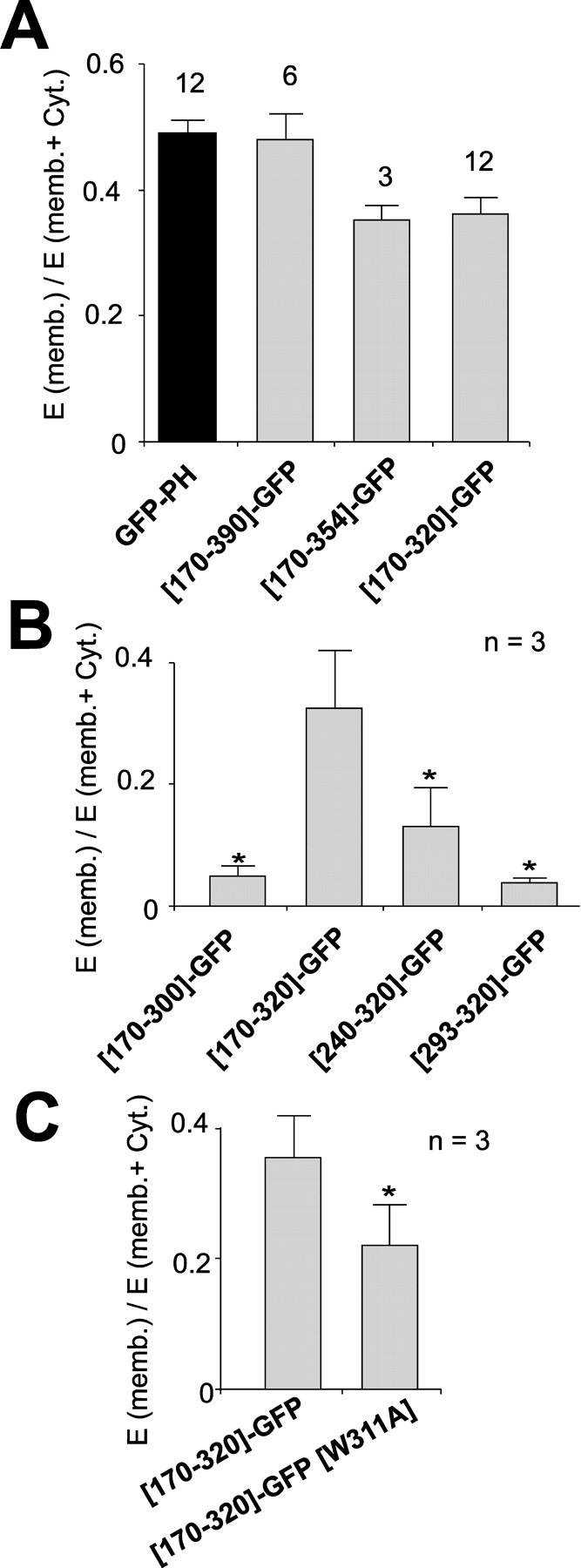
Membrane association of Kir6.2 COOH-terminal fragments. (A–C) Fraction of fluorescence in membrane (mean ± SEM) from cells expressing GFP-PH and GFP-tagged COOH-terminal fragments (mean ± SEM, n indicated). In B and C, results are shown from parallel transfections (n indicated, *, P < 0.05).
Confocal Microscopy
Cells were cultivated for 24–72 h after transfection and photographed in UV light under 1,000× magnification in a ZEISS microscope equipped with a 515-nm emission filter. Confocal analysis was performed using an Argon-Krypton laser (Bio-Rad Laboratories). Green fluorescence was detected at λ = 515 nm after excitation at λ = 488 nm. Cells were observed on thin coverslips, and digitized confocal images were prepared for presentation using Corel Photopaint (Corel Inc.).
Patch-clamp Measurements
Patch-clamp experiments were made at room temperature, in a chamber which allowed rapid exchange of bathing solution. Micropipettes were pulled from thin-walled glass (WPI, Inc.) on a horizontal puller (Sutter Instrument Co.). Electrode resistance was typically 0.5–1 MΩ when filled with K-INT solution (below). Inside-out patches were voltage-clamped with an Axopatch 1B amplifier (Axon Instruments, Inc.). Standard bath and pipette solutions (K-INT) had the following composition: 140 mM KCl, 10 mM K-HEPES, 1 mM K-EGTA, pH 7.3. PIP2 was diluted in K-INT and bath sonicated in ice for 30 min before use. ATP was added as the potassium salt. All currents were measured at a membrane potential of -50 mV. Data were filtered at 0.5–3 kHz, digitized at 22 kHz (Neurocorder; Neurodata) and stored on videotape. Experiments were replayed onto a chart recorder, or digitized into a computer using Axotape software (Axon Instruments, Inc.) and analyzed off-line using Microsoft Excel. Wherever possible, data are presented as mean ± SEM. Microsoft Solver was used to fit data by a least-square algorithm.
Secondary Structure Predictions
Secondary structure predictions were made for Kir6.2 using multiple sequence alignments, submitted through the Washington University Structural Genomics web site: http://www.biochem.wustl.edu/~StrucGen/.
RESULTS
The Kir6.2 COOH Terminus Associates with the Membrane
PIP2-interacting PH domains from various proteins have been tagged with GFP (Wang et al., 1996; Nagel et al., 1998; Varnai et al., 1999; Hodgkin et al., 2000) to study their interaction with the membrane. We took advantage of this approach to examine membrane association of isolated fragments of the Kir6.2 subunit of the KATP channel expressed in COSm6 cells. GFP-tagged constructs, transiently expressed in transfected cells, were analyzed by confocal microscopy (Fig. 1 A) and in a parallel cell fractionation assay (Fig. 1 B). The full-length channel Kir6.2-GFP is visible in the plasma membrane and punctate intracellular structures in intact cells (Fig. 1 A), and is >90% associated with the isolated membrane fraction (Fig. 1 B). In contrast, GFP alone shows a cytoplasmic distribution (Fig. 1 A) and is >90% associated with the isolated cytoplasmic fraction (Fig. 1 B), consistent with a soluble protein that does not interact with the membrane. The isolated COOH terminus of Kir6.2 (residues 170–390) tagged with GFP ([170–390]-GFP) also shows plasma membrane and intracellular localization under confocal microscopy (Fig. 1 A). In the fractionation assay, ∼45% of the fluorescence is associated with the membrane fraction (Fig. 1 B). The PH domain of phospholipase C δ1 (PLCδ1) tagged with GFP (GFP-PH) (gift of Tobias Meyer, Stanford University, Stanford, CA) was used as a positive control for a protein known to bind to phospholipids (specifically PI-4,5-bisphosphate) (Cifuentes et al., 1994; Lemmon et al., 1996) in the membrane. Cells transfected with this construct show GFP fluorescence in a thick band outlining the membrane of the cell. In the fractionation assay, the membrane association of the GFP-PH construct was very similar (∼45%) to that of [170–390]-GFP. Importantly, as shown in Fig. 1 C, all constructs are expressed as a single protein of appropriate molecular weight.
Figure 1.

Membrane association of Kir6.2 COOH terminus. (A) Confocal fluorescent images of COSm6 cells expressing Kir6.2-GFP+SUR1, GFP, [170–390]-GFP, or GFP-PH. Calibration bars represent 3 μm. (B, left) Representative emission spectra of membrane (black) and cytosolic (gray) fractions from transfected cells as in A (see materials and methods). (right) Fraction of fluorescence in membrane (mean ± SEM) from similar experiments, n indicated. (C) Western blot using polyclonal anti-GFP antibody of cytosolic (C) and membrane (M) fractions as in B. Arrows indicate expected size of tagged constructs. There is a probable contaminant protein at ∼40 kD in cytosolic fractions. Cytosolic anti-GFP reactivity at ∼30 kD may represent cleaved GFP in full-length Kir6.2-GFP assay.
Alanine Scan of Putative C-helix: Mutations of Conserved Residues Abolish Channel Activity
The cytoplasmic regions of Kir channels are highly conserved in the ∼40 amino acids preceding the M1 segment (residues 34–78 in Kir6.2) and ∼150 amino acids following M2 (residues 170–320 in Kir6.2), although they share no significant homology with any known protein domains. We performed predictions of the secondary structure of Kir channels using alignments of these primary sequences using various prediction algorithms (see Fig. 6 A, and discussion). The conserved COOH terminus is predicted to contain primarily β-strands and loops but only one significant α-helix (C-helix, approximately residues 306–311). To examine the likely structure and significance of this region, we performed a systematic mutagenesis of residues 307–315, replacing each residue with alanine. Functional channels were not obtained with mutations of residues E308, I309, W311, and F315, but essentially normal channel activity was observed for all other residues between D307 and R314 (Fig. 2, B and C). On a helical wheel projection (Fig. 2 D), residues E308, I309, W311, and F315 are all on the same face and E308, W311, and F315 are predicted to be separated by single turns of the helix.
Figure 6.
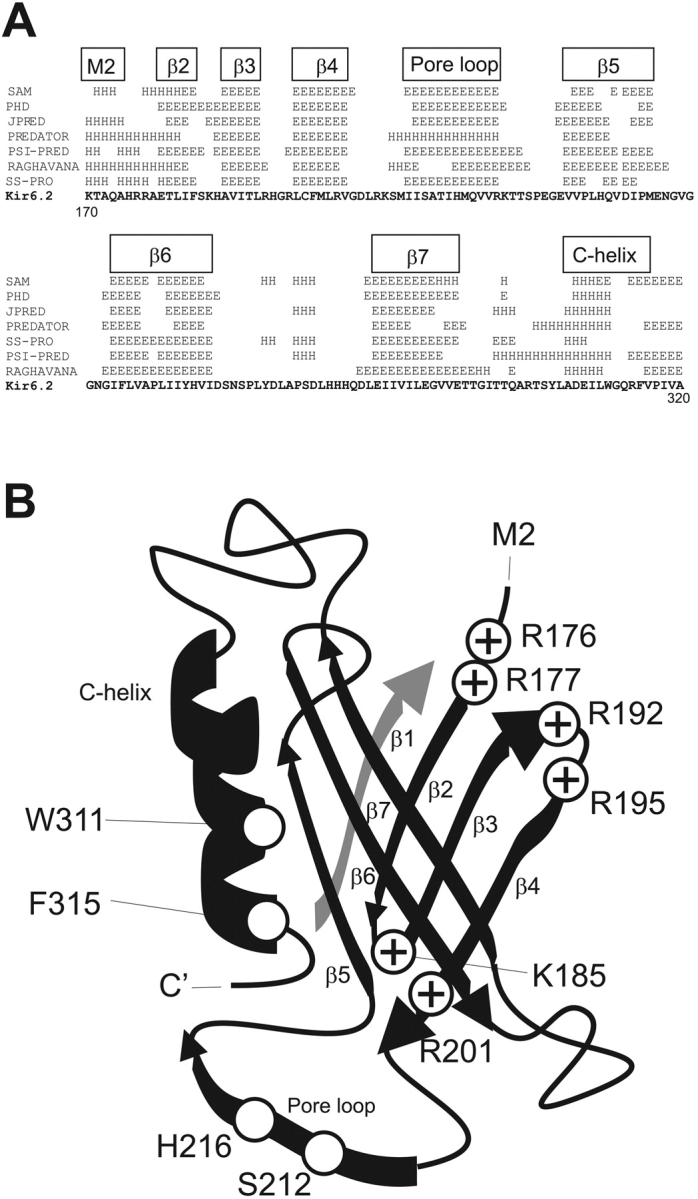
Predicted structure of conserved Kir cytoplasmic domain. (A) (above sequence) Computer-predicted secondary structures (E, β-strand, H, helix) of conserved COOH-terminal (residues 170–324) regions of Kir6.2 using multiple alignments with other inward rectifiers, generated by various algorithms indicated on left. The boxed indicators above correspond to the suggested secondary structures in B. (B) Cartoon ribbon diagram of suggested structural arrangement of the Kir6.2 KIRLI domain (see discussion).
Figure 2.

Conserved α-helix of Kir COOH terminus. (A) Conserved COOH-terminal α-helix of PH domain (boxed) of pleckstrin (Pleck), spectrin (Spec), and dynamin I (Dynam) aligned with predicted COOH-terminal α-helix of inward rectifiers (indicated by subfamily nomenclature). The conserved Trp (W311 in Kir6.2) and large hydrophobic (F315 in Kir6.2 or I) residues are boxed, upstream negative charges are shaded in gray. (B) Mean patch current for D307G and alanine-substituted mutants (E308A-F315A). No currents were recorded from E308A, I309A, W311A, and F315A mutants. (C) K1/2,ATP for mutant channels (mean ± SEM, n indicated). (D) Helical projection of residues D307–F315. Residues for which channel activity was abolished wen mutated are highlighted in black.
In a previous systematic mutagenesis of positive charges in the Kir6.2 COOH terminus (Shyng et al., 2000), it was striking that all 22 mutants could produce functional subunits, although in some cases, channel activity was observed only after application of PIP2, or after coexpression with high open state stability mutants such as Kir6.2[L157C], which expresses with high open probability and low ATP sensitivity (Enkvetchakul et al., 2000). Substitution of W311 or F315 with A, R, or D results in no detectable channel activity even after application of 100 μg/ml PIP2 (n = 3–5 patches in each case; unpublished data). Moreover, W311A and F315A mutants are not rescued by coexpression with L157C mutants (Fig. 3 A). W311A and F315A mutants actually cause a strong dominant-negative suppression of L157C subunits (Fig. 3 A). The only detectable currents correspond to those expected of pure L157C+SUR1 currents (Fig. 3 B), consistent with incorporation of even one W311A or F315A subunit into a tetramer being sufficient to abolish activity.
Figure 3.
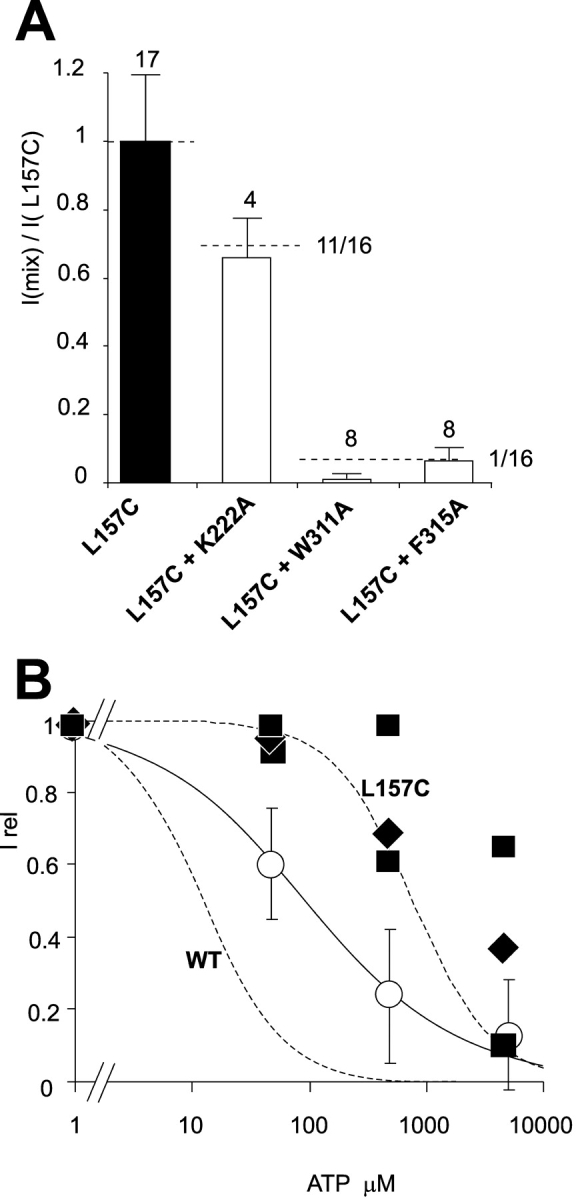
Dominant-negative inhibition by W311A and F315A mutants. (A) Current density (in zero ATP) in patches expressing Kir6.2[L157C]+SUR1 (L157C), or Kir6.2[L157C]+SUR1 coexpressed with other mutant Kir6.2 subunits, relative to currents in Kir6.2[L157C]+SUR1 patches. Dashed lines indicate predictions assuming that subunits are expressed at equal density, and that at least three K222A subunits are necessary to inhibit current, or only one W311A or F315A subunit is sufficient to inhibit activity. (B) Steady-state dependence of membrane current on [ATP] (relative to current in zero ATP, Irel) for wild type or L157C mutant channels (dashed, from [Shyng et al., 2000]). Superimposed data points are from individual patches in which L157C subunits were coexpressed with K222A (○), W311A ([▪), or F315A(♦) mutant subunits.
The Conserved Kir6.2 COOH Terminus Interacts with the Membrane
We next attempted to define structural boundaries of the KIRLI domain. The COOH-terminal 36 amino acids of Kir6.2 (residues 354–390) contain an endoplasmic reticular retention signal which affects channel trafficking to, but not function at, the membrane (Tucker et al., 1997; Zerangue et al., 1999; Ma et al., 2001). Fig. 4 A shows that the [170–354]-GFP fragment associates with the membrane slightly less effectively than the full length COOH terminus (i.e., [170–390]-GFP), but no further reduction of association is seen when the COOH terminus is reduced to the highly conserved (i.e., [170–320]-GFP) fragment (Fig. 4 A). However, truncation of a further 20 amino acids (i.e., [170–300]-GFP), which removes the putative C-helix, abolishes membrane association (Fig. 4 B). Deletions into the conserved domain from the NH2 terminus also considerably reduce association with the membrane and the short [293–320]-GFP has a similar cellular distribution to GFP alone (Fig. 4 B). Thus, we propose that residues 170–320 comprise the core of the KIRLI domain. To examine the role of W311 and F315 in membrane association, we introduced W311A and F315A mutants in the [170–320]-GFP construct. Each of these mutations affected expression level. In parallel transfections, fluorescence of W311A and F315A mutations were 28 ± 15% (n = 4) and 4 ± 3% (n = 3) of that of the wild type construct, respectively, suggesting instability of the expressed proteins. Only the fluorescence of the W311A mutant was high enough to quantify membrane association in the fractionation assay. As shown in Fig. 4 C, this point mutation significantly reduced the ability of this fragment to associate with the membrane.
m1 Receptor Activation Reduces Membrane Association of both the Kir6.2 KIRLI Domain and the PLCδ1 PH Domain
It has been demonstrated previously that PLC activation via m1 receptor stimulation causes a decrease of KATP current, consistent with reduced channel–PIP2 interactions (Xie et al., 1999). Other studies have dynamically monitored membrane PIP2 itself, making use of PLCδ1 PH domain tagged with GFP (GFP-PH) (Stauffer et al., 1998; Varnai and Balla, 1998) to provide a relative estimate of PIP2 and IP3 in response to m1 receptor stimulation and resulting PIP2 hydrolysis. Kobrinsky et al. (2000) used this approach to demonstrate redistribution of GFP-PH in transfected cells using confocal imaging. We have used the same principle to develop a quantifiable assay for dynamic redistribution of GFP-tagged channel fragments (Fig. 5). To quantify membrane association, cells were fractionated either in the presence or absence of ACh (see materials and methods). Fig. 5 A quantifies membrane association of GFP-PH expressed alone, and with muscarinic m1 or m2 receptors, from cells incubated in the presence or absence of 100 μM ACh. The membrane association of GFP-PH is specifically reduced only when the m1 receptor is expressed and activated by ACh. Similarly, [170–320]-GFP fluorescence is only reduced by m1 receptor activation (Fig. 5 B). In contrast, the distribution of fluorescence of full length Kir6.2-GFP (a transmembrane protein) was unaffected by m1 receptor activation (Fig. 5 B).
DISCUSSION
PIP2 Interaction with Kir Channels
It is now clear that PPIs, in particular PIP2, can have a profound activating effect on all inward rectifier K+ (Kir) channels (Hilgemann and Ball, 1996; Fan and Makielski, 1997; Baukrowitz et al., 1998b; Zhang et al., 1999; Hilgemann et al., 2001). Actin stabilizers such as phalloidin can inhibit channel rundown while depolymerizing agents such as cytochalasins can accelerate channel rundown (Furukawa et al., 1996; Harvey et al., 2000), leading to initial suggestions that the action of PIP2 was via cytoskeletal interactions. This idea has not been ruled out, but the weight of evidence indicates a direct action on Kir subunits, specifically involving interaction with the cytoplasmic COOH termini. Since most cytoskeletal proteins interact with phosphoinositides (PIP2 or PIP3) (Janmey, 1994), the demonstration that phosphatidylserine (PS) and other negatively charged lipids, such as acyl CoA, can also activate Kir channels, argues against a cytoskeletal intermediate. The most direct evidence is the finding that specific mutations in the COOH terminus can reduce both channel activity in cell membranes and binding of PIP2 to isolated fragments (Fan and Makielski, 1997; Huang et al., 1998; Shyng and Nichols, 1998; Soom et al., 2001) and the apparent antagonism of ATP inhibition and PPI activation of KATP channels (Baukrowitz et al., 1998b; Shyng and Nichols, 1998; Enkvetchakul et al., 2000).
A Cytoplasmic Membrane–interacting Domain in Kir Channels
Recent studies with PLCδ1 PH domain tagged with GFP (GFP-PH) have dramatically demonstrated localization of this domain to the cell membrane. One goal of the present study was to develop a physiological assay of membrane association of isolated Kir6.2 fragments, to test the hypothesis that a discrete domain exists and that it binds to the cell membrane in a PPI-dependent manner. Confocal fluorescence images indicate that GFP-tagged full-length Kir6.2 constructs are localized to the cell membrane, but are also retained in intracellular compartments (Makhina and Nichols, 1998) (Fig. 1 A). A visually similar pattern is observed for the isolated COOH terminus ([170–390]-GFP) that differs from the almost exclusively edge fluorescence observed for the PLCδ1 PH domain (GFP-PH). Importantly, this particular PH domain, but not others, has been reported to have high affinity and specificity for PI-4,5-P2 (Lemmon et al., 1995). Kir6.2 clearly interacts with multiple PPI species (Fan and Makielski, 1997), so while it is possible that the discrete intracellular fluorescence of [170–390]-GFP is due to retention within an enclosed compartment, it is also possible that the construct is associated with intracellular PPI to which PH-GFP has relatively low affinity.
The fractionation assay allows quantification of the association of GFP-tagged constructs with the membrane fraction, and hence a semiquantitative assay of the relative affinity of a given construct for binding to the membrane. The data indicate that while the conserved COOH terminus ([170–320]-GFP) associates with the membrane almost as well as the complete COOH terminus (170–390]-GFP) or the PLCδ1 PH domain (GFP-PH), truncations into this domain from either end (Fig. 4 B) greatly reduce association. Therefore, residues 170–320 form the essential core of the COOH-terminal lipid–interacting domain. Activation of muscarinic m1 receptors by ACh results in Gqα-mediated PLC activation and consequent PIP2 hydrolysis (Caulfield and Birdsall, 1998). In single COS-1 cells coexpressing GFP-PH and m1 receptors, ACh activation decreased membrane association of GFP-PH by 55% (Kobrinsky et al., 2000), consistent with a relative decrease in the PI-4,5-P2/IP3 ratio within the cell (Stauffer et al., 1998; Varnai and Balla, 1998). This shift in GFP-PH fluorescence was not seen with activation of coexpressed m2 receptors, which couple to alternative G-protein–mediated pathways. We have now used the same principle to further develop a quantifiable assay for association of fragments with native membranes. As shown in Fig. 5 A, membrane association of GFP-PH and of [170–320]-GFP are each specifically reduced only when the m1 receptor is expressed and activated by ACh.
Predicted Secondary Structure of Kir Channel COOH Terminus
Several different Kir COOH termini (Huang et al., 1998; Shyng and Nichols, 1998; Zhang et al., 1999; Shyng et al., 2000; Soom et al., 2001) have been shown to interact with membrane lipids, and homologous residues controlling PIP2 sensitivity have been identified in different channels (Baukrowitz et al., 1998b; Huang et al., 1998; Shyng and Nichols, 1998; Zhang et al., 1999; Shyng et al., 2000). Many of these assays are indirect, and cannot exclude the possibility that the interaction with the membrane lipid is mediated via an intermediate protein interaction. Nevertheless, together with the present results, the data suggest the presence of a common KIRLI domain formed from the conserved sequence in the channel COOH terminus.
We performed predictions of the secondary structure of Kir channels using alignments of the conserved ∼150 amino acids following M2 (residues 170–320 in Kir6.2, see Fig. 6 A). Following from the end of the M2 helix (predicted to end at ∼173), multiple β-strands and a COOH-terminal α-helix (residues 306–311) are predicted, a structural arrangement that is common to lipid-interacting PH domains (Rebecchi and Scarlata, 1998). PH domains invariably contain seven major antiparallel β-strands with a COOH-terminal α-helix that contributes to stability of the domain structure. Importantly, the COOH-terminal helix is the only region in PH domains over which primary sequence homology is typically found. The conserved Trp and the large hydrophobic residue at position +4 interact with hydrophobic amino acids in the β-sheets to provide stability to the whole PH domain (Pitcher et al., 1995; Touhara et al., 1995), and mutations of these residues severely impair PH domain function (Shaw, 1996; Fushman et al., 1998). Upstream negative charges and no helix-breaking prolines are also features of the PH domain C-helix, and these features are all present in the 307–315 region of Kir channels (Fig. 3 A). The data obtained from systematic mutation are consistent with this region being α-helical in Kir6.2, such that mutations on one face abolish channel activity (Fig. 2). The conserved W311 and F315 residues, located on this face, are not only critical for channel activity in the full-length construct (Fig. 3), but also for stability and membrane association of isolated COOH-terminal fragments (Fig. 4). We suggest that this helix may play a structural role in the integrity of the KIRLI domain, analogous to the COOH-terminal α-helix of PH domains.
A Possible Structure of the KIRLI Domain
Given the functional and structural similarities between the KIRLI domain and established PH domains, we have explored the possibility that the tertiary structure of the KIRLI domain might be similar to that of the PH domain. In the absence of any structural information, this speculative exercise may help to suggest further experiments. Fig. 6 B shows a cartoon illustrating a possible folding arrangement for the Kir6.2 COOH terminus cytoplasmic domain that would maintain an overall tertiary structure similar to that of the PH domain1 (two antiparallel β-sheets and COOH-terminal α-helix). The arrangement is of a “pocket” formed by the two β-sheets (residues 176–201 and 220–290) with the C-helix (residues 307–315) forming a “hinge” between the two sheets, W311 and F315 side-chains being predicted to protrude into the “pocket” (Rebecchi and Scarlata, 1998). There is now considerable evidence for interaction of the conserved regions of the NH2 terminus with the COOH terminus (Tinker et al., 1996; Koster et al., 1998; Tucker and Ashcroft, 1999) of Kir channels. This data may be reconciled by the suggestion that the conserved region of the NH2 terminus normally generates the β1 strand of the putative PH-like domain. Using protein fragments, Jones et al. (2001) have recently identified segments of the Kir6.2 COOH terminus that interact with the isolated NH2 terminus and defined three segments of interaction (residues 170–204, 214–222, and 279–323) that correspond to the same COOH-terminal regions identified by Soom et al. (2001) as important for binding to PIP2. Our suggested model includes critical roles for each of these regions in forming the KIRLI domain.
Charged residues clustered on one side of the “β-pocket” in the β1/β2 and β3/β4 loops of PH domains are involved in interaction with PIP2 (Harlan et al., 1995; Rebecchi and Scarlata, 1998; Soisson et al., 1998; Yagisawa et al., 1998; Carman et al., 2000). Systematic mutagenesis of Kir6.2 COOH-terminal–positive charges identified several residues (R176, R177, R192, R195) with significant effects on PIP2 sensitivity when neutralized (Shyng et al., 2000), consistent with them mediating electrostatic interactions with the negatively charged phosphates of PIP2. As shown in Fig. 6 B, placing the predicted COOH-terminal β-strands in the β2-β7 strand positions of the PH domain would cluster these critical residues in the β1/β2 and β3/β4 loops, i.e., on one side of the “β-pocket”. The R176 and R177 residues are conserved in other Kir channels, and neutralization of the equivalent K188 and R189 in Kir2.1 (Soom et al., 2001), or R188 in Kir1.1 (Huang et al., 1998) also reduces PIP2 sensitivity of channel activity.
Much evidence supports the idea of a negative allosteric interaction between PIP2 and ATP in regulating the activity of Kir6.2 channels (Baukrowitz et al., 1998a; Shyng and Nichols, 1998; Enkvetchakul et al., 2000), with functional competition between the two ligands, but separate binding sites. Several residues have emerged as candidate ATP binding residues (Drain et al., 1998; Gribble et al., 1998; Tucker et al., 1998; Shyng et al., 2000), and mutation of certain charged residues has large effects on ATP sensitivity, but minimal effects on PIP2 sensitivity. Intriguingly, two of these residues, K185 (Tucker et al., 1998) and R201 (Shyng et al., 2000), are located within the proposed KIRLI domain, and would be predicted to lie at the opposite side of the “β-pocket” to the PIP2-interacting residues (Fig. 6 B), consistent with the above hypothesis.
In addition to the computer-predicted β2-β7 strands and C-helix of Kir channels (Fig. 6 A), there is a region between putative β4 and β5 that is predicted to be either α-helix or β-strand (Fig. 6). Several studies on various Kir channels indicate that residues in this region actually form part of the cytoplasmic entrance to the pore. In Kir2.1, E224 (S212 in Kir6.2) controls the affinity for pore-blocking polyamines (Yang et al., 1995) and H216 in Kir6.2 controls the pH-dependence of polyamine block (Baukrowitz et al., 1999). Cysteine-substituents of several residues within this region of Kir2.1 are modified by methyl-thio-sulfydryl reagents, indicating water accessibility (Lu et al., 1999). PH domains can tolerate large insertions in the loops between β-strands (Rebecchi and Scarlata, 1998), and we propose that this region may form an insertion that lines the cytoplasmic entrance to the pore. As such it is likely to contribute to the control of open state stability of the channel, and, as with the M2 transmembrane segment (Enkvetchakul et al., 2000), mutations within it will affect open state stability and apparent PIP2 sensitivity of channel activity. Accordingly, neutralization of R206 or K222 in this region of Kir6.2 (Shyng et al., 2000), or mutations of residues in the equivalent 207–245 segment of Kir2.1 (Zhang et al., 1999), reduce apparent PIP2 sensitivity.
PIP2 activation is common to all Kir channels, suggesting a conserved mechanism of interaction. Previous studies have demonstrated direct interaction of isolated COOH-terminal fragments with PIP2 (Huang et al., 1998; Soom et al., 2001). We now demonstrate that the conserved COOH terminus (residues 170–320) associates with cellular membranes, and this association can be regulated by PLC activation and PIP2 hydrolysis. Systematic mutagenesis of Kir6.2 has identified positive charges in this domain that when mutated alter PIP2 sensitivity (Shyng et al., 2000); neutralization of any of the basic residues beyond the domain are without effect on channel activity (Shyng et al., 2000). Systematic alanine scanning now indicates the likely presence of a COOH-terminal α-helix; mutation of conserved Trp and Phe residues in this segment both abolish channel activity and interfere with domain expression and membrane association. The data provide evidence for the existence of a conserved KIRLI domain that is critically involved in physiological regulation of channel activity.
Acknowledgments
We are very grateful to Show-Ling Shyng for help with preliminary studies and constructs, and to Diomedes Logothetis and colleagues for m1 and m2 receptor clones.
This work was primarily supported by National Institutes of Health grant HL54171 (to C.G. Nichols), as well as by a National Institutes of Health Cardiovascular Training grant (Fellowship support of C.A. Cukras), and the Washington University National Institutes of Health DRTC (DK20579).
Footnotes
Abbreviations used in this paper: ACh, acetylcholine; KIRLI, Kir lipid interacting; PIP2, phosphatidylinositol-4,5-bisphosphate; PLC, phospholipase C; PPI, polyphosphoinositide; PS, phosphatidylserine.
Stable models of the KIRLI domain (unpublished data) can be generated by threading the Kir COOH terminus threaded onto the PH domain of pleckstrin as a template (Guex and Peitsch, 1997), but uncertainty in the correct alignment reduces the likely value of such models.
References
- Baukrowitz, T., U. Schulte, D. Oliver, S. Herlitze, T. Krauter, S.J. Tucker, J.P. Ruppersberg, and B. Fakler. 1998. a. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 282:1141–1144. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., U. Schulte, D. Oliver, S. Herlitze, T. Krauter, S.J. Tucker, J.P. Ruppersberg, and B. Fakler. 1998. b. PIP2 and PIP as determinants for ATP inhibition of KATP channels. Science. 282:1141–1144. [DOI] [PubMed] [Google Scholar]
- Baukrowitz, T., S.J. Tucker, U. Schulte, K. Benndorf, J.P. Ruppersberg, and B. Fakler. 1999. Inward rectification in KATP channels: a pH switch in the pore. EMBO J. 18:847–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carman, C.V., L.S. Barak, C. Chen, L.Y. Liu-Chen, J.J. Onorato, S.P. Kennedy, M.G. Caron, and J.L. Benovic. 2000. Mutational analysis of Gbetagamma and phospholipid interaction with G protein-coupled receptor kinase 2. J. Biol. Chem. 275:10443–10452. [DOI] [PubMed] [Google Scholar]
- Caulfield, M.P., and N. Birdsall. 1998. International Union of Pharmacology. XVII. Classification of muscarinic acetylcholine receptors. Pharmacol. Rev. 50:279–290. [PubMed] [Google Scholar]
- Cifuentes, M.E., T. Delaney, and M.J. Rebecchi. 1994. D-myo-inositol 1,4,5-trisphosphate inhibits binding of phospholipase C-delta 1 to bilayer membranes. J. Biol. Chem. 269:1945–1948. [PubMed] [Google Scholar]
- Doyle, D.A., J.M. Cabral, R.A. Pfuetzner, A. Kuo, J.M. Gulbis, S.L. Cohen, B.T. Chait, and R. MacKinnon. 1998. The structure of the potassium channel: molecular basis of K+ conduction and selectivity. Science. 280:69–77. [DOI] [PubMed] [Google Scholar]
- Drain, P., L. Li, and J. Wang. 1998. KATP channel inhibition by ATP requires distinct functional domains of the cytoplasmic C terminus of the pore-forming subunit. Proc. Natl. Acad. Sci. USA. 95:13953–13958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Enkvetchakul, D., G. Loussouarn, E. Makhina, S.L. Shyng, and C.G. Nichols. 2000. The kinetic and physical basis of K(ATP) channel gating: toward a unified molecular understanding. Biophys. J. 78:2334–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fakler, B., J.H. Schultz, J. Yang, U. Schulte, U. Brandle, H.P. Zenner, L.Y. Jan, and J.P. Ruppersberg. 1996. Identification of a titratable lysine residue that determines sensitivity of kidney potassium channels (ROMK) to intracellular pH. EMBO J. 15:4093–4099. [PMC free article] [PubMed] [Google Scholar]
- Fan, Z., and J.C. Makielski. 1997. Anionic phospholipids activate ATP-sensitive potassium channels. J. Biol. Chem. 272:5388–5395. [DOI] [PubMed] [Google Scholar]
- Furukawa, T., T. Yamane, T. Terai, Y. Katayama, and M. Hiraoka. 1996. Functional linkage of the cardiac ATP-sensitive K+ channel to the actin cytoskeleton. Pflugers Arch. 431:504–512. [DOI] [PubMed] [Google Scholar]
- Fushman, D., T. Najmabadi-Haske, S. Cahill, J. Zheng, H. LeVine III, and D. Cowburn. 1998. The solution structure and dynamics of the pleckstrin homology domain of G protein-coupled receptor kinase 2 (beta-adrenergic receptor kinase 1). A binding partner of Gbetagamma subunits. J. Biol. Chem. 273:2835–2843. [DOI] [PubMed] [Google Scholar]
- Gribble, F.M., S.J. Tucker, T. Haug, and F.M. Ashcroft. 1998. MgATP activates the beta cell KATP channel by interaction with its SUR1 subunit. Proc. Natl. Acad. Sci. USA. 95:7185–7190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guex, N., and M.C. Peitsch. 1997. SWISS-MODEL and the Swiss-PdbViewer: an environment for comparative protein modeling. Electrophoresis. 18:2714–2723. [DOI] [PubMed] [Google Scholar]
- Harlan, J.E., H.S. Yoon, P.J. Hajduk, and S.W. Fesik. 1995. Structural characterization of the interaction between a pleckstrin homology domain and phosphatidylinositol 4,5-bisphosphate. Biochemistry. 34:9859–9864. [DOI] [PubMed] [Google Scholar]
- Harvey, J., S.C. Hardy, A.J. Irving, and M.L. Ashford. 2000. Leptin activation of ATP-sensitive K+ (KATP) channels in rat CRI-G1 insulinoma cells involves disruption of the actin cytoskeleton. J. Physiol. 527:95–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilgemann, D.W., and R. Ball. 1996. Regulation of cardiac Na+,Ca2+ exchange and KATP potassium channels by PIP2. Science. 273:956–959. [DOI] [PubMed] [Google Scholar]
- Hilgemann, D.W., S. Feng, and C. Nasuhoglu. 2001. The complex and intriguing lives of PIP2 with ion channels and transporters. Sci STKE. 2001:RE19. [DOI] [PubMed]
- Hodgkin, M.N., M.R. Masson, D. Powner, K.M. Saqib, C.P. Ponting, and M.J. Wakelam. 2000. Phospholipase D regulation and localisation is dependent upon a phosphatidylinositol 4,5-biphosphate-specific PH domain. Curr. Biol. 10:43–46. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., S. Feng, and D.W. Hilgemann. 1998. Direct activation of inward rectifier potassium channels by PIP2 and its stabilization by Gbetagamma. Nature. 391:803–806. [DOI] [PubMed] [Google Scholar]
- Huang, C.L., P.A. Slesinger, P.J. Casey, Y.N. Jan, and L.Y. Jan. 1995. Evidence that direct binding of G beta gamma to the GIRK1 G protein-gated inwardly rectifying K+ channel is important for channel activation. Neuron. 15:1133–1143. [DOI] [PubMed] [Google Scholar]
- Inagaki, N., T. Gonoi, J.P. Clement, N. Namba, J. Inazawa, G. Gonzalez, L. Aguilar-Bryan, S. Seino, and J. Bryan. 1995. Reconstitution of IKATP: an inward rectifier subunit plus the sulfonylurea receptor. Science. 270:1166–1170. [DOI] [PubMed] [Google Scholar]
- Janmey, P.A. 1994. Phosphoinositides and calcium as regulators of cellular actin assembly and disassembly. Annu. Rev. Physiol. 56:169–191. [DOI] [PubMed] [Google Scholar]
- Jones, P.A., S.J. Tucker, and F.M. Ashcroft. 2001. Multiple sites of interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. FEBS Lett. 508:85–89. [DOI] [PubMed] [Google Scholar]
- Kobrinsky, E., T. Mirshahi, H. Zhang, T. Jin, and D.E. Logothetis. 2000. Receptor-mediated hydrolysis of plasma membrane messenger PIP2 leads to K+-current desensitization. Nat. Cell Biol. 2:507–514. [DOI] [PubMed] [Google Scholar]
- Koster, J.C., K.A. Bentle, C.G. Nichols, and K. Ho. 1998. Assembly of ROMK1 (Kir 1.1a) inward rectifier K+ channel subunits involves multiple interaction sites. Biophys. J. 74:1821–1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, M.A., K.M. Ferguson, R. O'Brien, P.B. Sigler, and J. Schlessinger. 1995. Specific and high-affinity binding of inositol phosphates to an isolated pleckstrin homology domain. Proc. Natl. Acad. Sci. USA. 92:10472–10476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemmon, M.A., K.M. Ferguson, and J. Schlessinger. 1996. PH domains: diverse sequences with a common fold recruit signaling molecules to the cell surface. Cell. 85:621–624. [DOI] [PubMed] [Google Scholar]
- Liou, H.H., S.S. Zhou, and C.L. Huang. 1999. Regulation of ROMK1 channel by protein kinase A via a phosphatidylinositol 4,5-bisphosphate-dependent mechanism. Proc. Natl. Acad. Sci. USA. 96:5820–5825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Y., M. Holmgren, M.E. Jurman, and G. Yellen. 1997. Gated access to the pore of a voltage-dependent K+ channel. Neuron. 19:175–184. [DOI] [PubMed] [Google Scholar]
- Loussouarn, G.P., L.R. Masia, R. Rose, and T. Nichols. 2001. Flexibility of the Kir6.2 inward rectifier K(+) channel pore. Proc. Natl. Acad. Sci. USA. 98:4227–4232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, T., Y.G. Zhu, and J. Yang. 1999. Cytoplasmic amino and carboxyl domains form a wide intracellular vestibule in an inwardly rectifying potassium channel. Proc. Natl. Acad. Sci. USA. 96:9926–9931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, D., N. Zerangue, Y.F. Lin, A. Collins, M. Yu, Y.N. Jan, and L.Y. Jan. 2001. Role of ER export signals in controlling surface potassium channel numbers. Science. 291:316–319. [DOI] [PubMed] [Google Scholar]
- Makhina, E.N., and C.G. Nichols. 1998. Independent trafficking of KATP channel subunits to the plasma membrane. J. Biol. Chem. 273:3369–3374. [DOI] [PubMed] [Google Scholar]
- Nagel, W., P. Schilcher, L. Zeitlmann, and W. Kolanus. 1998. The PH domain and the polybasic c domain of cytohesin-1 cooperate specifically in plasma membrane association and cellular function. Mol. Biol. Cell. 9:1981–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols, C.G., and A.N. Lopatin. 1997. Inward rectifier potassium channels. Annu. Rev. Physiol. 59:171–191. [DOI] [PubMed] [Google Scholar]
- Pearson, W.L., M. Dourado, M. Schreiber, L. Salkoff, and C.G. Nichols. 1999. Expression of a functional Kir4 family inward rectifier K+ channel from a gene cloned from mouse liver. J. Physiol. 514:639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perozo, E., D.M. Cortes, and L.G. Cuello. 1999. Structural rearrangements underlying K+-channel activation gating. Science. 285:73–78. [DOI] [PubMed] [Google Scholar]
- Pitcher, J.A., K. Touhara, E.S. Payne, and R.J. Lefkowitz. 1995. Pleckstrin homology domain-mediated membrane association and activation of the beta-adrenergic receptor kinase requires coordinate interaction with G beta gamma subunits and lipid. J. Biol. Chem. 270:11707–11710. [DOI] [PubMed] [Google Scholar]
- Rebecchi, M.J., and S. Scarlata. 1998. Pleckstrin homology domains: a common fold with diverse functions. Annu. Rev. Biophys. Biomol. Struct. 27:503–528. [DOI] [PubMed] [Google Scholar]
- Reuveny, E., P.A. Slesinger, J. Inglese, J.M. Morales, J.A. Iniguez-Lluhi, R.J. Lefkowitz, H.R. Bourne, Y.N. Jan, and L.Y. Jan. 1994. Activation of the cloned muscarinic potassium channel by G protein beta gamma subunits. Nature. 370:143–146. [DOI] [PubMed] [Google Scholar]
- Rohacs, T., J. Chen, G.D. Prestwich, and D.E. Logothetis. 1999. Distinct specificities of inwardly rectifying K(+) channels for phosphoinositides. J. Biol. Chem. 274:36065–36072. [DOI] [PubMed] [Google Scholar]
- Shaw, G. 1996. The pleckstrin homology domain: an intriguing multifunctional protein module. Bioessays. 18:35–46. [DOI] [PubMed] [Google Scholar]
- Shyng, S.L., C.A. Cukras, J. Harwood, and C.G. Nichols. 2000. Structural determinants of PIP(2) regulation of inward rectifier K(ATP) channels. J. Gen. Physiol. 116:599–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shyng, S.L., and C.G. Nichols. 1998. Membrane phospholipid control of nucleotide sensitivity of KATP channels. Science. 282:1138–1141. [DOI] [PubMed] [Google Scholar]
- Soisson, S.M., A.S. Nimnual, M. Uy, D. Bar-Sagi, and J. Kuriyan. 1998. Crystal structure of the Dbl and pleckstrin homology domains from the human Son of sevenless protein. Cell. 95:259–268. [DOI] [PubMed] [Google Scholar]
- Soom, M., R. Schonherr, Y. Kubo, C. Kirsch, R. Klinger, and S.H. Heinemann. 2001. Multiple PIP2 binding sites in Kir2.1 inwardly rectifying potassium channels. FEBS Lett. 490:49–53. [DOI] [PubMed] [Google Scholar]
- Stauffer, T.P., S. Ahn, and T. Meyer. 1998. Receptor-induced transient reduction in plasma membrane PtdIns(4,5)P2 concentration monitored in living cells. Curr. Biol. 8:343–346. [DOI] [PubMed] [Google Scholar]
- Tinker, A., Y.N. Jan, and L.Y. Jan. 1996. Regions responsible for the assembly of inwardly rectifying potassium channels. Cell. 87:857–868. [DOI] [PubMed] [Google Scholar]
- Touhara, K., W.J. Koch, B.E. Hawes, and R.J. Lefkowitz. 1995. Mutational analysis of the pleckstrin homology domain of the beta-adrenergic receptor kinase. Differential effects on G beta gamma and phosphatidylinositol 4,5-bisphosphate binding. J. Biol. Chem. 270:17000–17005. [DOI] [PubMed] [Google Scholar]
- Tucker, S.J., and F.M. Ashcroft. 1999. Mapping of the physical interaction between the intracellular domains of an inwardly rectifying potassium channel, Kir6.2. J. Biol. Chem. 274:33393–33397. [DOI] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, P. Proks, S. Trapp, T.J. Ryder, T. Haug, F. Reimann, and F.M. Ashcroft. 1998. Molecular determinants of KATP channel inhibition by ATP. EMBO J. 17:3290–3296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tucker, S.J., F.M. Gribble, C. Zhao, S. Trapp, and F.M. Ashcroft. 1997. Truncation of Kir6.2 produces ATP-sensitive K+ channels in the absence of the sulphonylurea receptor. Nature. 387:179–183. [DOI] [PubMed] [Google Scholar]
- Varnai, P., and T. Balla. 1998. Visualization of phosphoinositides that bind pleckstrin homology domains: calcium- and agonist-induced dynamic changes and relationship to myo-[3H]inositol-labeled phosphoinositide pools. J. Cell Biol. 143:501–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varnai, P., K.I. Rother, and T. Balla. 1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J. Biol. Chem. 274:10983–10989. [DOI] [PubMed] [Google Scholar]
- Wang, X., A.J. Levi, and A.P. Halestrap. 1996. Substrate and inhibitor specificities of the monocarboxylate transporters of single rat heart cells. Am. J. Physiol. 270:H476–H484. [DOI] [PubMed] [Google Scholar]
- Xie, L.H., M. Horie, and M. Takano. 1999. Phospholipase C-linked receptors regulate the ATP-sensitive potassium channel by means of phosphatidylinositol 4,5-bisphosphate metabolism. Proc. Natl. Acad. Sci. USA. 96:15292–15297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yagisawa, H., K. Sakuma, H.F. Paterson, R. Cheung, V. Allen, H. Hirata, Y. Watanabe, M. Hirata, R.L. Williams, and M. Katan. 1998. Replacements of single basic amino acids in the pleckstrin homology domain of phospholipase C-delta1 alter the ligand binding, phospholipase activity, and interaction with the plasma membrane. J. Biol. Chem. 273:417–424. [DOI] [PubMed] [Google Scholar]
- Yang, J., Y.N. Jan, and L.Y. Jan. 1995. Control of rectification and permeation by residues in two distinct domains in an inward rectifier K+ channel. Neuron. 14:1047–1054. [DOI] [PubMed] [Google Scholar]
- Yang, Z., and C. Jiang. 1999. Opposite effects of pH on open-state probability and single channel conductance of kir4.1 channels. J. Physiol. 520:921–927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerangue, N., B. Schwappach, Y.N. Jan, and L.Y. Jan. 1999. A new ER trafficking signal regulates the subunit stoichiometry of plasma membrane K(ATP) channels. Neuron. 22:537–548. [DOI] [PubMed] [Google Scholar]
- Zhang, H., C. He, X. Yan, T. Mirshahi, and D.E. Logothetis. 1999. Activation of inwardly rectifying K+ channels by distinct PtdIns(4,5)P2 interactions. Nat. Cell Biol. 1:183–188. [DOI] [PubMed] [Google Scholar]